

Abstract
Telomeres are elongated by the enzyme telomerase, which contains a template-bearing RNA (TER or TERC) and a protein reverse transcriptase. Overexpression of a particular mutant human TER with a mutated template sequence (MT-hTer-47A) in telomerase-positive cancer cells causes incorporation of mutant telomeric sequences, telomere uncapping, and initiation of a DNA damage response, ultimately resulting in cell growth inhibition and apoptosis. The DNA damage pathways underlying these cellular effects are not well understood. Here, we show that the ataxia-telangiectasia-mutated (ATM) protein is activated and forms telomeric foci in response to MT-hTer-47A expression. Depletion of ATM from two cancer cell lines, including the p53-mutant UM-UC-3 bladder cancer line, rendered the cells largely unresponsive to MT-hTer-47A. Relative to ATM-competent controls, ATM-depleted cells showed increased proliferation and clonogenic survival and reduced cell death following MT-hTer-47A treatment. In contrast, ATM depletion sensitized the cancer cells to treatment with camptothecin, a topoisomerase inhibitor which induces DNA double-strand breaks. We show that the effects of ATM depletion on the MT-hTer-47A response were not due to decreased expression of MT-hTer-47A or reduced activity of telomerase at the telomere. Instead, ATM depletion allowed robust cancer cell growth despite the continued presence of dysfunctional telomeres containing mutant sequence. Notably, the number of end-to-end telomere fusions induced by MT-hTer-47A treatment was markedly reduced in ATM-depleted cells. Our results identify ATM as a key mediator of the MT-hTer-47A dysfunctional telomere response, even in cells lacking wild-type p53, and provide evidence that telomere fusions contribute to MT-hTer-47A cytotoxicity.
Keywords: ATM, telomerase, mutant template, telomere dysfunction, cancer therapy
Introduction
Telomeres are nucleoprotein structures that protect the integrity of the chromosome ends (1). Telomeres consist of multiple tandem repeats of a short DNA sequence - TTAGGG in humans - that specifically bind a variety of proteins. The complexes composed of these proteins, which in mammals have been termed shelterin, prevent the chromosome ends from being treated as DNA breaks and undergoing inappropriate repair processes (2). Each time a cell divides, the telomeric DNA shortens slightly due to the inability of the DNA replication apparatus to completely copy the ends of the telomeres (3). Once a certain critical length is reached, shelterin complexes are no longer effective in preventing the chromosome ends from initiating a DNA damage response, ultimately leading to cellular senescence or apoptosis (4, 5).
The enzyme telomerase, which is minimally composed of a protein reverse transcriptase (hTERT) and a template-containing RNA (hTER), counteracts telomere shortening by adding new TTAGGG repeats to the 3′ ends of the chromosomes (1). Thus, telomerase is capable of circumventing the limit on cell division imposed by telomere attrition. Telomerase activity is downregulated in most adult human cells, but it is readily detectable in stem and progenitor cell populations, as well as in the large majority (∼80-90%) of cancer cells (6). The telomerase activity in these cell types underlies their ability to divide repeatedly without reaching replicative senescence or undergoing apoptosis. Telomerase is therefore a key factor in the progression and maintenance of most tumors.
Given the high level of telomerase expression in most cancer cells and the greatly diminished levels in the vast majority of adult human cells, telomerase is an attractive target for cancer therapy. One promising approach currently in clinical trials involves inhibition of telomerase activity, leading to telomere shortening and senescence (6). Our laboratory has developed an alternative approach for targeting telomerase in cancer cells that involves the expression of mutated hTER with an altered template sequence (7, 8). These mutant template hTERs, hereafter referred to as MT-hTers, complex with hTERT in tumor cells and direct the addition of mutant telomeric repeats that are predicted to disrupt binding of shelterin components (7, 9, 10). MT-hTer treatment rapidly induces a DNA damage response, as indicated by the presence of DNA damage foci (which include 53BP1, RIF1, and γ-H2AX) that colocalize with telomeres (11, 12). In diverse cancer cell types, MT-hTer treatment quickly results in apoptosis and growth inhibition both in vitro and in vivo (7, 13). Importantly, this growth inhibition does not rely on p53 and pRb status and only occurs in cells that also express hTERT (13). In this paper, we focus on the “47A” mutant version of hTER (MT-hTer-47A), which has two mutated base pairs in the partially-repeated hTER template region, and hence is predicted to direct the addition of TTTGGG repeats, instead of wild-type TTAGGG repeats, onto telomeres (9, 13). MT-hTer-47A has demonstrated robust anti-proliferative effects in a variety of different telomerase-positive cancer cell lines (9, 13).
We focus here on the role of ataxia-telangiectasia-mutated (ATM) protein in the response to MT-hTer-47A-induced telomere dysfunction. ATM is a phosphatidylinositol-3-like kinase that functions at both telomeres and DNA double-strand breaks (14, 15). Studies in yeast and mammalian cells have shown that disruption of ATM signaling causes telomere shortening, at least in part by decreasing telomerase recruitment to the telomeres (16-18). In addition, loss of ATM function affects the frequency of end-to-end telomere fusions. In experiments involving prolonged cell growth, ATM disruption causes an increase in the number of telomeric fusions detected, which may be due to the accelerated telomere shortening of ATM-deficient cells or to enhanced survival of cells with end-to-end fusions (19, 20). In contrast, in a more short-term experiment, ATM depletion protected against fusion of telomeres rendered dysfunctional by acute loss of TRF2, suggesting that ATM can promote fusion of deprotected telomeres in certain cases (21).
ATM also plays an important role in coordinating the cellular response to DNA double-strand breaks (DSBs). ATM is activated and becomes autophosphorylated in response to DSBs, and subsequently phosphorylates a large number of proteins which modulate the checkpoint and repair responses of the damaged cell (15, 22). Depending on the cellular context, the ATM-directed response to DNA damage can promote cell death by initiating an apoptotic program or, conversely, can enhance cell survival by activating checkpoints and coordinating DNA repair (23). In cancer cells, the role of ATM in the DSB response appears to be largely cell-protective, as ATM depletion or inhibition in cancer cells commonly augments the cytotoxic effects of ionizing radiation and chemotherapeutics which induce DSBs (24-26).
DSBs and dysfunctional telomeres share many similarities. First, both lesions involve exposure of a double-stranded DNA end (2). Second, both lesions acquire DNA damage foci, which are local accumulations of proteins including ATM, the MRE11-RAD50-NBS1 complex, 53BP1, RIF1, and γ-H2AX (5, 11, 12, 27). Third, the cellular response to both types of lesions can ultimately result in senescence or apoptosis, depending on the cellular context (5, 28). Given these similarities, we tested whether ATM depletion would sensitize cancer cells to the effects of MT-hTer-47A, just as it sensitizes them to treatments which induce intrachromosomal DSBs.
We show first that, as anticipated, ATM is activated in response to MT-hTer-47A overexpression. Surprisingly, depletion of ATM does not sensitize the cancer cells to subsequent MT-hTer-47A treatment. Instead, the cells become largely unresponsive to MT-hTer-47A-induced dysfunctional telomeres, which persist in the proliferating cells. Strikingly, ATM depletion significantly reduces the frequency of end-to-end fusion of MT-hTer-47A-induced dysfunctional telomeres. These results identify ATM as a key mediator of MT-hTer-47A-induced cytotoxicity, in marked contrast to the protective role of ATM in the response to damaging agents that cause intrachromosomal DSBs.
Materials and Methods
Cell Lines and Culture
LOX melanoma cells were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum. UM-UC-3 bladder cancer and human embryonic kidney 293T cells were grown in DMEM supplemented with 10% fetal bovine serum. Cells were grown at 37°C in 5% CO2.
Plasmids and Lentivirus
The lentiviral vector system was provided by D. Trono (University of Geneva, Geneva, Switzerland; (29)). Lentivirus was prepared as described previously (11). The shRNA expression lentivectors were constructed as described previously (13). The ATM shRNA target sequences, derived from previously-published siRNA sequences, are as follows: 5′-GGTGCTATTTACGGAGCTG-3′ (ATM shRNA #1) and 5′-GCAACATACTACTCAAAGA-3′ (ATM shRNA #2) (30, 31). The control scramble shRNAs had the following “target” sequences: 5′-GTTCTACAACGTAACGAGGTT-3′ (scramble #1) and 5′-GTCAAAGAACGTTCAGACA-3′ (scramble #2). WT-hTER and MT-hTer-47A expression lentivectors have been described (13).
Lentivirus was titered by plating appropriate dilutions on LOX or UM-UC-3 cells and counting the number of green fluorescent protein (GFP)-positive foci present 72 hours later. For flow cytometry and immunofluorescence experiments, lentivectors carrying the puromycin resistance gene in place of GFP were used since the strong GFP expression in LOX cells would interfere with these assays. Cells were infected with shRNA-expressing lentivirus at ∼75 transducing units per cell and with hTER-expressing plasmids at ∼125 transducing units per cell. At this virus level, > 95% of LOX or UM-UC-3 cells received the desired lentivirus as judged by GFP expression. Thus, since no selection step was required, experiments could be performed rapidly on the bulk cell population.
Cell Growth Assays
LOX or UM-UC-3 cells were infected with shRNA-expressing virus at day -2, followed by hTER-expressing virus (either WT-hTER or MT-hTer-47A) at day 0. Cells were split as needed to maintain logarithmic growth. Cells were harvested at indicated time points and stained with Trypan blue. Viable cells were enumerated by hemocytometer.
For the MT-hTer-47A clonogenic assays, LOX cells were infected with shRNA and then the hTER-expressing virus as above. Cells were then harvested 2 days later and replated at a density of 100 cells per well in 6-well plates. After 7 days of additional growth, colonies were fixed with 95% ethanol and stained with 0.1% crystal violet. Colonies containing more than ∼20 cells were counted. For camptothecin (CPT) clonogenic assays, LOX cells were infected with shRNA-expressing virus and were reseeded 4 days later at a density of 100 cells per well in 6-well plates. After 12 hours, CPT was added to a final concentration of 50 nM, and the medium was replaced 24 hours later. The cells were grown for an additional 6 days, and colonies were counted as above.
For experiments with KU-55933 (KuDOS Pharmaceuticals), LOX cells were infected with lentivirus expressing WT-hTER or MT-hTer-47A at day 0, with KU-55933 (in DMSO) added 8 hours after infection. The drug was replaced daily, and cells were split as needed to maintain logarithmic growth.
Immunofluorescence and Flow Cytometry
LOX cells were infected with shRNA-expressing virus at day -2 and hTER-expressing virus at day 0. For immunofluorescence, cells were seeded on sterile coverslips on day 4. On day 6, cells were fixed in 2% paraformaldehyde in PBS and permeabilized with 0.5% Nonidet P-40 in PBS. Immunostaining was performed with rabbit anti-Rap1 (Bethyl Laboratories) and mouse anti-ATM pS1981 (Rockland), followed by the appropriate secondary Alexa Fluor 488 or 568 antibody (Molecular Probes)(11, 15). DNA was visualized with DAPI. Analysis was performed using the Deltavision Restoration Microscopy System (Applied Precision) with a 60X objective and 1.5X magnifier. Images were acquired using the Deltavision SoftWorx 3D capture program. Images were subjected to deconvolution and are presented as Quick Projections. All images were captured using identical exposure times, and signal intensities were standardized to allow comparison of relative intensities.
For flow cytometry, cells were stained with annexin V-phycoerythrin and 7-amino-actinomycin D per manufacturer protocol (BD Pharmingen), and analysis was performed with a FACSCalibur system (BD Biosciences).
Western Blotting
Cells were resuspended in 50 mM Tris-HCl pH 7.4, 250 mM NaCl, 5 mM EDTA, and 0.1% Nonidet P-40 containing protease and phosphatase inhibitors and subjected to two freeze-thaw cycles. Lysates were cleared by centrifugation at 14,000 rpm for 10 minutes, and samples were run on SDS-PAGE gels. Western blot analysis was performed with the following antibodies: mouse anti-ATM pS1981 (Rockland), mouse anti-ATM (2C1, GeneTex, Inc.), and horseradish peroxidase (HRP)-conjugated mouse anti-β actin (Abcam). HRP-conjugated anti-mouse secondary antibodies were used for ATM blots, and immunostaining was detected using ECL Plus Detection Reagent (GE Healthcare).
Real-time PCR and Gene Sequencing
RNA was isolated using the RNeasy Mini Kit with on-column DNase digestion (Qiagen). Reverse transcription was performed with StrataScript III and random primers per manufacturer protocol (Invitrogen), and real-time quantitative PCR (qPCR) was performed using Brilliant SYBR Green qPCR Master Mix on the M×3000P real-time system (Stratagene). The following primers were used for qPCR: 5′-TCACGTCATCCAGCAGAGAATGGA-3′ and 5′-CACACGGCAGGCATACTCATCTTT-3′ (β2-microglobulin); 5′-TTGCGGAGGGTGGGCCT-3′ and 5′-CGGGCCAGCAGCTGACATT-3′ (hTER). Data was analyzed using the MXPro software (Stratagene).
For confirmatory sequencing of the UM-UC-3 p53 mutation, amplification of a portion of p53 exon 4 from UM-UC-3 genomic DNA was performed with Pfu Turbo (Stratagene) using the following primers: 5′-GCCGTCCCAAGCAATGGATGATTT-3′ and 5′-AGGAAGCCAAAGGGTGAAGAGGAA-3′. Automated sequencing was then performed on both strands with the following nested primers: 5′-AGATGAAGCTCCCAGAATGCCAGA-3′ and 5′-ATACGGCCAGGCATTGAAGTCTCA-3′.
Telomeric Southern Blotting
UM-UC-3 genomic DNA was digested with HinfI and RsaI and run on a 0.6% agarose gel with 5 μg of DNA per lane. DNA was transferred to Hybond XL membrane (GE Healthcare), and wild-type telomeric sequence was detected using a 32P-labeled 5′-(CCCTAA)4-3′ probe. For detection of mutant repeats, a 32P-labeled 5′-(CCCAAA)4-3′ probe was used along with a 4-fold excess of unlabeled 5′-(CCCTAA)4-3′ and 5′-(CCCCAA)4-3′ oligonucleotides to block cross-hybridization to wild-type and subtelomeric sequences (9).
Telomeric Fluorescence in situ Hybridization (FISH)
Telomeric FISH was performed and imaged as described previously, except with a 4-hour colcemid treatment (32). Telomeric fusions were scored in a blinded fashion. Only fusions involving both sister chromatids were counted.
Results
MT-hTer-47A Expression Activates ATM
We first evaluated whether MT-hTer-47A expression in cancer cells induces activation of ATM, which is normally present in an inactive unphosphorylated form. An appropriate DNA damage stimulus leads to ATM autophosphorylation at serine 1981 (15). LOX melanoma cells were infected with lentivirus overexpressing either a wild-type version of hTER (WT-hTER) or MT-hTer-47A. In contrast to WT-hTER overexpression, MT-hTer-47A expression induced phosphorylated ATM foci, as shown by immunofluorescence using an antibody specific for ATM phosphorylated at serine 1981 (Fig. 1). The large majority of these activated ATM foci colocalized with telomeric RAP1 foci, consistent with prior results showing that most MT-hTer-47A-induced DNA damage foci are telomeric (11, 12). The phosphorylation of ATM in response to MT-hTer-47A expression was confirmed by Western blot analysis (Supplementary Fig. 1).
Fig. 1.

MT-hTer-47A expression activates ATM. LOX melanoma cells were infected with lentivirus expressing scramble #1 or ATM #2 shRNAs on day -2. The cells were subsequently infected with lentivirus expressing WT-hTER or MT-hTer-47A on day 0. Cells were harvested on day 4 and seeded on sterile coverslips. The cells were fixed and stained with the indicated antibodies on day 6. Multiple cells were examined from independent experiments, and representative images are shown. The scale bar at bottom right is 10 μm in length, and all images are at the same magnification.
ATM Depletion Abrogates the Cancer Cell Response to MT-hTer-47A Expression
In order to understand the role of ATM in the MT-hTer-47A response, we designed two lentiviral shRNA constructs targeting the ATM mRNA based on previously published siRNA sequences (30, 31). LOX melanoma cells were infected with lentivirus expressing these shRNAs, and depletion of ATM protein was analyzed by Western blot. Each ATM shRNA caused significant depletion of ATM, with shRNA #2 demonstrating more complete knockdown than shRNA #1 (Fig. 2A). As expected, ATM shRNA #2 almost completely eliminated the appearance of activated ATM in response to MT-hTer-47A expression, as demonstrated by both immunofluorescence and Western blot (Fig. 1 and Supplementary Fig. 1).
Fig. 2.
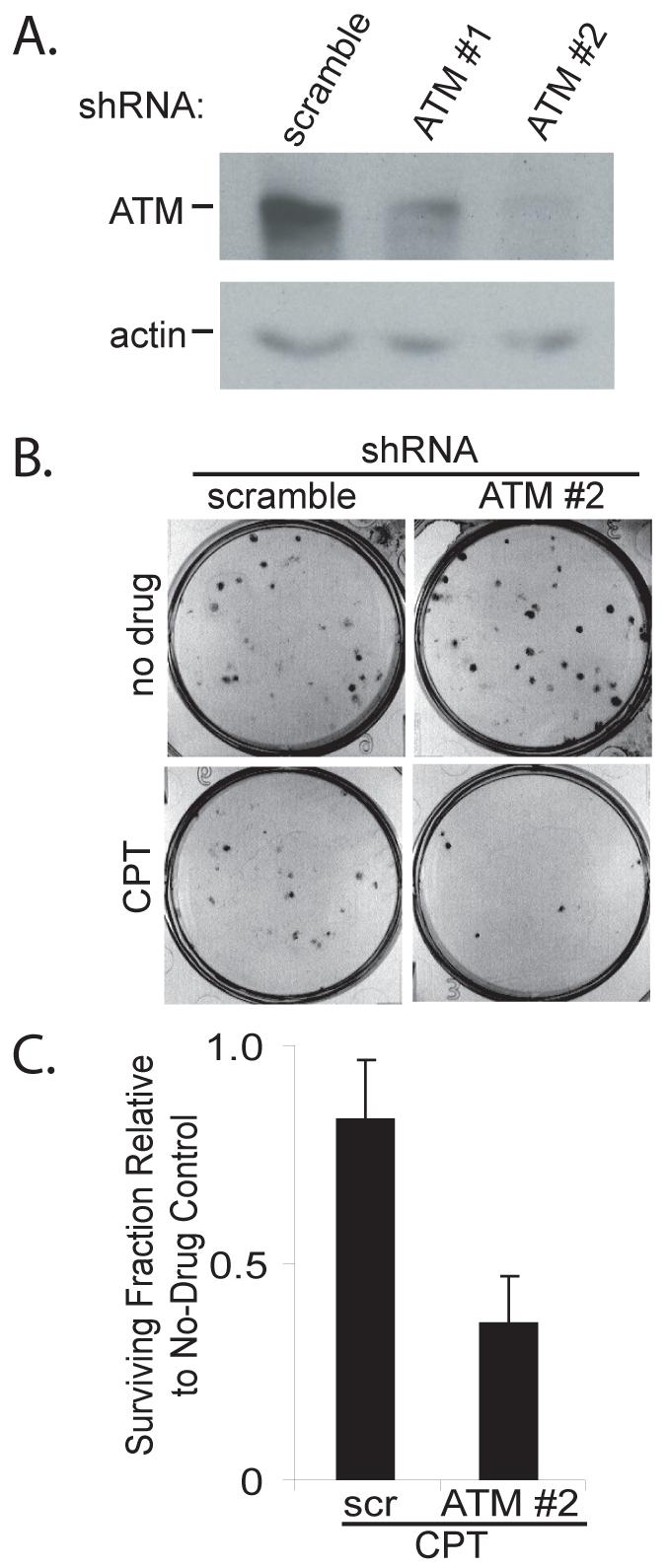
ATM depletion sensitizes LOX cells to camptothecin. A. LOX melanoma cells were infected with lentivirus expressing the indicated shRNAs and cells were harvested 4 days later. ATM knockdown was analyzed by Western blot, with actin serving as a loading control. B. LOX cells were infected with scramble #1 or ATM #2 shRNAs and seeded in 6-well plates 3 days later at a density of 100 cells per well. After twelve hours, the cells were treated with 50 nM camptothecin in DMSO (CPT) or DMSO alone (no drug). The medium was replaced 24 hours later and the cells were grown for an additional 6 days. Cells were fixed and stained with crystal violet to assess colony number, and images of representative wells are shown. C. The graph shows the fraction of LOX colonies surviving camptothecin treatment relative to the corresponding DMSO controls. The data are derived from three independent experiments. Error bars indicate the standard deviation, and the difference between the samples is statistically significant (p = 0.01).
Loss of ATM function in cancer cells commonly sensitizes them to ionizing radiation and other treatments which cause double-strand DNA breaks, thereby lowering their clonogenic survival (24-26). To confirm that the ATM depletion was effective in reducing ATM cellular function, we tested whether ATM knockdown sensitized LOX cells to DNA double-strand breaks induced by the type I topoisomerase inhibitor camptothecin (33). The ATM-depleted LOX cells displayed significantly reduced clonogenic survival after treatment with camptothecin, indicating that ATM knockdown was sufficient to alter a known response of cancer cells to DNA damage (Fig. 2B and 2C).
The effect of ATM depletion on the MT-hTer-47A response was evaluated first by measuring cell proliferation curves. As seen previously, expression of MT-hTer-47A in LOX melanoma cells led to rapid growth inhibition in comparison to a WT-hTER control (Fig. 3A) (13). Additional control experiments as well as previously-published work showed that WT-hTER overexpression itself had little or no effect on cell proliferation, confirming that the difference in the WT-hTER and MT-hTer-47A proliferation curves is due to growth inhibition by MT-hTer-47A rather than a stimulatory growth effect of WT-hTER or other effect of the lentiviral transfection (Supplementary Fig. 2) (13). To explore the role of ATM in the MT-hTer-47A response, LOX melanoma cells were first treated with anti-ATM shRNA or control “scramble” shRNA, followed 48 hours later by WT-hTER or MT-hTer-47A. ATM depletion had no significant impact on proliferation of cells overexpressing WT-hTER (Fig. 3A). In marked contrast, ATM depletion substantially abrogated the inhibition of LOX cell proliferation caused by MT-hTer-47A (Fig. 3A). The same experiment was performed in UM-UC-3 bladder cancer cells with equivalent results (Fig. 3B).
Fig. 3.

ATM depletion or inhibition rescues growth of MT-hTer-47A-treated cells. LOX (A) and UM-UC-3 (B) cells were infected with lentivirus expressing scramble #1 (closed symbols) or ATM #2 (open symbols) shRNAs at day -2, followed by infection with lentivirus expressing WT-hTER (squares) or MT-TER-47A (triangles) at day 0. Cells were harvested and counted by hemocytometer at indicated time points. C. LOX melanoma cells were infected with lentivirus expressing scramble #1 (black bars), scramble #2 (white bars), ATM #1 (grey bars), or ATM #2 (stippled bars) shRNAs at day -2, followed by lentivirus expressing WT-hTER or MT-hTer-47A at day 0. Cells were harvested and counted by hemocytometer at indicated time points. The graph depicts the percent cell proliferation of MT-hTer-47A-treated samples relative to matched WT-hTER-treated samples. Error bars indicate standard deviation for three separate hTER infections. Analysis of day 6 data from at least three independent experiments indicates that the increased proliferation in MT-hTer-47A-treated cells seen with the ATM #1 and ATM #2 shRNAs compared to the scramble controls is statistically significant (p < 0.01 and 0.0001 respectively). The difference in the growth of MT-hTer-47A-treated cells between the ATM #1 and ATM #2 shRNA samples is also statistically significant (p < 0.02). D. LOX cells were infected with lentivirus expression WT-hTER or MT-hTer-47A, and KU-55933 was added at the indicated concentrations 8 hours later. Cells were harvested and counted 7 days after infection. Proliferation of each sample is shown relative to the WT-hTER, no-drug control. The growth of MT-hTer-47A cells is significantly increased by KU-55933 at 1 μM and 3 μM (p < 0.005). The inset shows the growth of MT-hTer-47A-treated cells relative to the WT-hTer-treated cells at each drug concentration.
We performed control experiments to confirm that the effects seen on the MT-hTer-47A-expressing cells were due to the loss of ATM function. First, to address the possibility that shRNA off-target effects might be influencing our results, we analyzed the impact of two different ATM shRNAs, as well as two different scramble shRNA controls, on MT-hTer-47A-induced growth inhibition (34). Both of the ATM shRNAs reduced the impact of MT-hTer-47A on LOX cell proliferation relative to the scramble controls (Fig. 3C). Furthermore, the influence of the two shRNAs directly correlated with their ability to knockdown ATM. ATM shRNA #1, which was less effective at knocking down ATM, was correspondingly less effective at rescuing LOX cell growth after MT-hTer-47A expression (Fig. 3C). Finally, we analyzed the effect of the recently identified ATM inhibitor KU-55933 on MT-hTer-47A-induced growth inhibition. KU-55933 reportedly acts as a competitive inhibitor for the ATP binding site of ATM (24). As seen with the ATM shRNAs, KU-55933 partially rescued the growth of MT-hTer-47A-treated LOX cells in a dose-dependent manner (Fig. 3D). Together, these results indicate that it is inhibition of ATM activity which rescues LOX cell proliferation after MT-hTer-47A treatment.
Previous studies have shown that telomere dysfunction induced by various MT-hTers results in altered cellular morphology, increased cell death, and decreased clonogenic survival (7, 9, 10, 13, 35). ATM depletion in LOX melanoma cells prior to treatment with MT-hTer-47A significantly reduced all of these effects: the flattened, enlarged cell morphology induced by MT-hTer-47A was largely abrogated (Fig. 4A); the increase in annexin V-positive dead and dying cells seen after MT-hTer-47A expression was significantly reduced (Fig. 4B); and the drop in clonogenic survival in MT-hTer-47A-treated cells was largely eliminated (Fig. 4C and 4D).
Fig. 4.
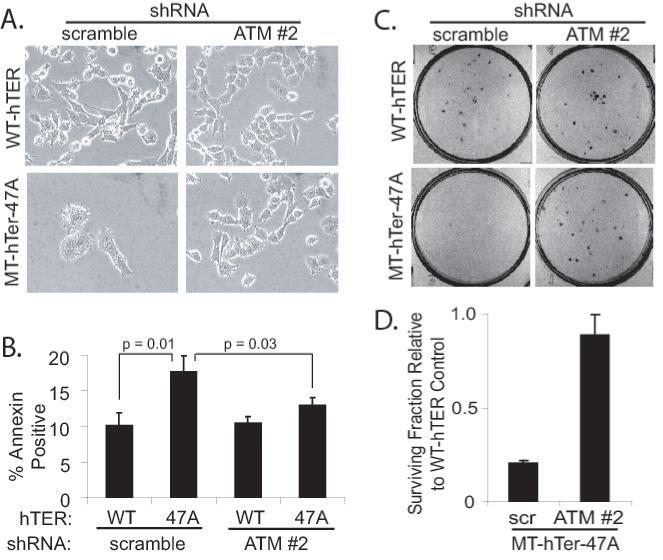
ATM depletion minimizes MT-hTer-47A effects in LOX cells. LOX melanoma cells were infected with scramble #1 or ATM #2 shRNAs at day -2, followed by WT-hTER or MT-hTer-47A at day 0. A. Phase-contrast microscope images were obtained at day 8. B. Day 6 cells were harvested and stained with annexin V-phycoerythrin and 7-amino-actinomycin D (7-AAD), and staining was analyzed by flow cytometry. The graph indicates the number of annexin positive cells (irrespective of 7-AAD status) in each sample. Error bars indicate the standard deviation for three separate hTER infections. C. Day 2 cells were harvested and replated in 6-well plates at a density of 100 cells per well. Cells were fixed and stained with crystal violet after 7 days of additional growth to assess colony number, and images of representative wells are shown. D. The graph shows the fraction of surviving colonies in MT-hTer-47A-treated wells relative to the corresponding WT-hTER-treated wells based on data from three independent experiments. Error bars indicate the standard deviation, and the difference between the samples is statistically significant (p < 0.001).
The rescue of clonogenic survival by ATM depletion was particularly interesting since prior studies and this work have shown that ATM depletion sensitizes cancer cells to drugs which cause DNA double-strand breaks (Fig. 2B and 2C) (24-26). Thus, we conclude that while ATM depletion decreases LOX clonogenic survival after treatment with camptothecin, it has just the opposite effect on cells treated with MT-hTer-47A, despite the fact that both treatments engage a robust ATM-dependent DNA damage response.
ATM Depletion Does Not Block hTER Overexpression
A recent report suggested that ATM inhibition blocks efficient transduction of HIV-1-derived lentiviral vectors by interfering with successful integration into the host genome (36). Therefore, we tested the possibility that ATM depletion might have prevented the MT-hTer-47A effects by blocking delivery and expression of the mutant template lentiviral construct. Using a real-time RT-PCR assay, we analyzed hTER expression four and six days after infection of LOX melanoma cells, the time period during which the ATM effect on cell proliferation first becomes evident. The lentiviral hTER constructs resulted in WT-hTER and MT-hTer-47A overexpression 20-40 fold higher than the endogenous hTER levels (data not shown). Importantly, ATM depletion did not lower hTER overexpression levels, indicating that ATM knockdown does not block delivery of the hTER constructs (Supplementary Fig. 3). The same result was also obtained with UM-UC-3 cells (data not shown). The drop in MT-hTer-47A expression by day 6 in LOX cells without ATM depletion is consistent with selection against cells with high MT-hTer-47A expression levels. Interestingly, this drop was not observed in ATM-depleted cells, consistent with their being protected from growth inhibition.
MT-hTer-47A Induces Marked Telomere Lengthening and Incorporation of Mutant Telomeric Repeats in ATM-Depleted Cells
Studies in yeast and mammalian cells have suggested that ATM plays a role in telomere length maintenance, at least in part by aiding in the recruitment of telomerase to the telomere (16-18, 37). We therefore tested the possibility that ATM depletion might have protected MT-hTer-47A-treated cells by decreasing the activity of the mutant telomerase complex at the telomere, thereby reducing incorporation of mutant repeats. Telomere length changes in LOX melanoma cells are difficult to detect since these cells have extremely long telomeres (>40 kb) (13). We therefore used the human bladder cancer cell line UM-UC-3 for these experiments (38). Previous work had shown that overexpression of WT-hTER in UM-UC-3 cells induces rapid, easily measurable telomere lengthening since this cell line has limiting hTER levels and short telomeres (32). To confirm cell line identity and p53-mutant status, we sequenced part of the p53 exon 4 from UM-UC-3 genomic DNA. As expected, our UM-UC-3 cells carried the phenylalanine (TTC) to cysteine (TGC) mutation at codon 113 of p53 as reported previously (data not shown)(38).
We first demonstrated that ATM depletion alone has no detectable effect on UM-UC-3 telomere length over our 10-day experimental period (Supplementary Fig. 4). To analyze the impact of ATM depletion on telomere lengthening induced by hTER overexpression, we performed Southern blotting for telomeric DNA eight days after infection with either WT-hTER or MT-hTer-47A lentivirus. As expected from previous results with this cell line, overexpression of WT-hTER led to marked telomere elongation, with the median telomere restriction fragment (TRF) length rising from ∼3.5 kb to ∼6.5 kb (Fig. 5A, compare lanes 1 and 2)(32). Notably, ATM depletion had no significant impact on this telomere elongation, with the median TRF length reaching ∼7.3 kb (Fig. 5A, compare lanes 2 and 4). This result clearly shows that in our overexpression system, depletion of ATM has no detectable impact on the ability of telomerase to act at the telomeres.
Fig. 5.
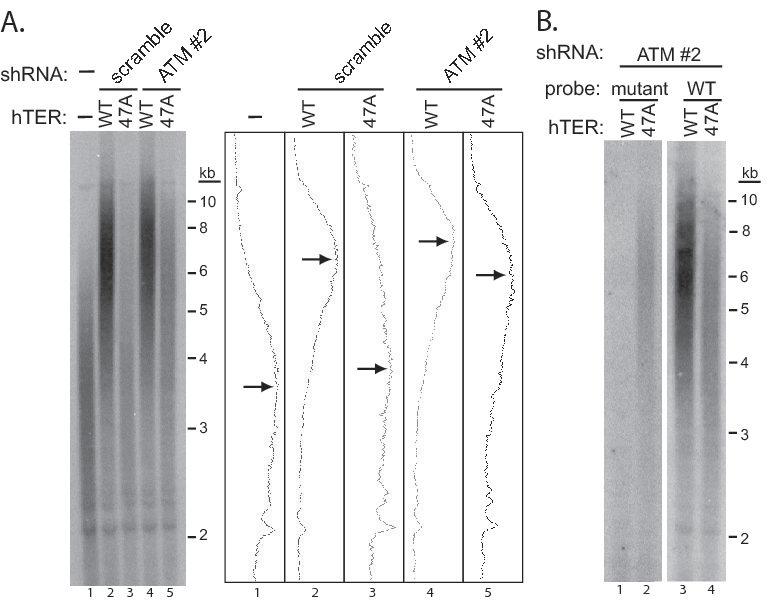
MT-hTer-47A expression induces telomere elongation in ATM-depleted UM-UC-3 cells. UM-UC-3 cells were infected with lentivirus expressing scramble #1 and ATM #2 shRNAs at day -2, followed by lentivirus expressing WT-hTER and MT-hTer-47A at day 0. Cells were harvested at day 8 and genomic DNA was isolated and digested with HinfI and RsaI. A. Southern blotting was performed using an oligonucleotide probe for wild-type telomeric DNA (5′-(CCCTAA)4-3′). Adjacent graphs show signal intensities for each lane, and arrows indicate the approximate peak signal in each. B. Southern blotting was performed with an oligonucleotide probe targeting the mutant telomeric DNA (5′-(CCCAAA)4-3′) (left panel). A four-fold excess of two unlabeled oligonucleotides (5′-(CCCTAA)4-3′ and 5′-(CCCCAA)4-3′) was added to the mutant probe hybridization to block nonspecific binding to wild-type and subtelomeric sequences. The blot was then stripped and hybridized to the wild-type telomeric probe (right panel).
In contrast to the results with WT-hTER, ATM depletion had a significant effect on the telomere length distribution observed in the bulk cell population following MT-hTer-47A expression. In the ATM-competent control UM-UC-3 cells, the median TRF length increased only slightly from ∼3.5 kb to ∼3.8 kb (Fig. 5A, compare lanes 1 and 3), while in the ATM-depleted cells the median TRF length of the bulk cell population increased markedly to ∼5.9 kb (Fig. 5A, compare lanes 1 and 5). To confirm that this lengthening was due to activity of MT-hTer-47A telomerase at the telomeres, we adapted a Southern blotting technique which specifically detects incorporated mutant repeats in human telomeres (9). Using this strategy, we detected mutant telomeric repeats in UM-UC-3 telomeres elongated by MT-hTer-47A expression, directly confirming substantial mutant repeat incorporation (Fig. 5B).
Collectively, these results rule out the possibility that ATM depletion protects cells from MT-hTer-47A either by blocking the expression of MT-hTer-47A, or by decreasing telomerase activity and thereby limiting mutant repeat incorporation at the telomeres. Instead, our findings indicate that ATM depletion allows survival of cells with mutant telomeric repeats in the bulk cell population by preventing an ATM-coordinated cellular response.
MT-hTer-47A treatment induces telomere fusions in an ATM-dependent manner
Expression of mutant telomerase templates in Kluyveromyces lactis and in a human tumor cell line has been shown previously to induce end-to-end telomere fusions (10, 39). Previous work in Tetrahymena and in LOX melanoma cells has shown that MT-hTer overexpression leads to the appearance of anaphase bridges, as predicted from telomere fusions (35, 40).
To explore the effect of MT-hTer-47A overexpression on telomere fusions in LOX cells, we analyzed metaphase spreads 3 days after infection with either WT-hTER or MT-hTer-47A, a time when the growth effects of MT-hTer-47A are just beginning to manifest. MT-hTer-47A treatment caused a substantial number of fusions (Fig. 6). Over 50% of the metaphase spreads from MT-hTer-47A-treated cells had at least one fusion event, with an average of 3.2 fusions per fusion-containing metaphase. In contrast, untreated or WT-hTER-treated LOX cells had no telomere fusions. Strikingly, depletion of ATM prior to MT-hTer-47A treatment reduced the percentage of metaphase spreads containing fusions more than three-fold, with no metaphase containing more than 2 fusions. These data show that MT-hTer-47A quickly and robustly induces telomere fusions in LOX melanoma cells, and that ATM depletion largely protects against these fusion events.
Fig. 6.
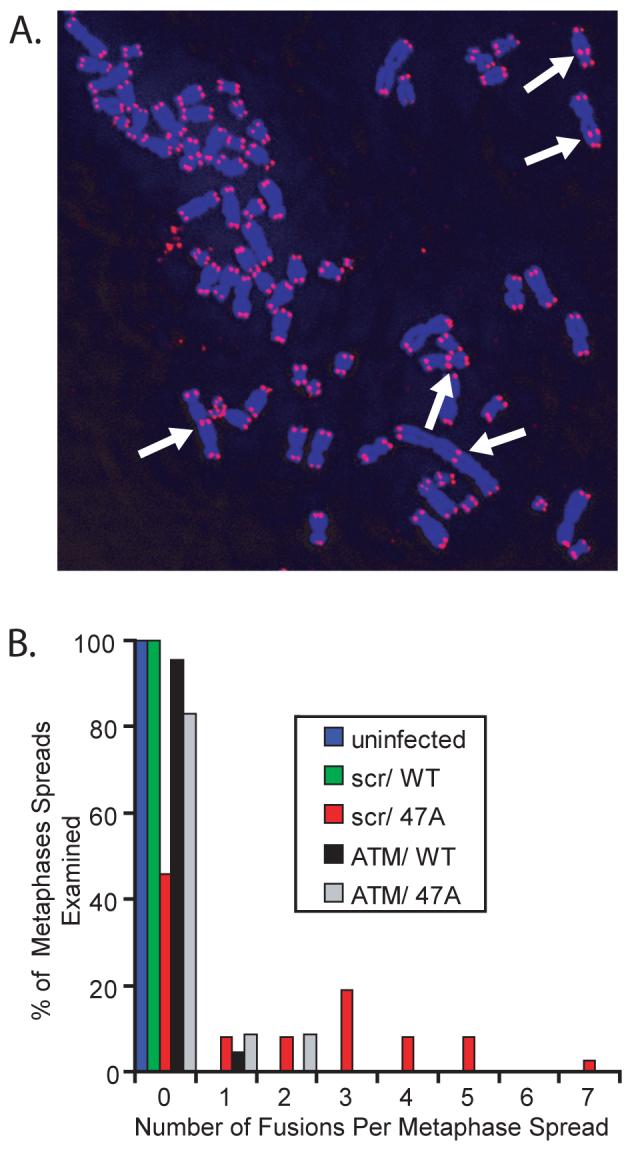
MT-hTer-47A expression induces telomere fusions in an ATM-dependent manner. LOX cells were infected with lentivirus expressing scramble #1 and ATM #2 shRNAs at day -2, followed by lentivirus expressing WT-hTER and MT-hTer-47A at day 0. Cells were treated with colcemid on day 3, and metaphase spreads were prepared. Telomeres were labeled by fluorescence in situ hybridization. A. A representative image showing multiple telomere fusions (arrows) in a cell treated with scramble #1 shRNA and MT-hTer-47A. B. A histogram showing the percentage of metaphase spreads containing different numbers of telomeric fusions. Between 22 and 37 metaphase spreads were analyzed for each sample type, and results were consistent across two independent experiments. In scramble #1-treated cells, MT-hTer-47A caused a significant increase in fusions compared to WT-hTER (p = 0.0001). In MT-hTer-47A-treated cells, ATM knockdown significantly reduced the number of fusions observed compared to the scramble #1 control (p < 0.0001).
Discussion
Telomere dysfunction, whether the result of replicative shortening or experimental disruption of the telomeric DNA-protein complex structure, leads to a rapid DNA damage response which can ultimately induce senescence and/or apoptosis (5, 28). Recent work has shown that the ATM and ATR (ataxia-telangiectasia-mutated and Rad3-related) kinases can act as key mediators of this response, with their relative importance determined in part by the exact nature of the telomeric lesion (5, 21, 27, 28). Here we have shown that depletion of ATM in two different types of human cancer cells renders them largely unresponsive to telomere disruption induced by expression of a particular mutant-template telomerase RNA, MT-hTer-47A. With the ATM response pathway blocked, the cancer cells are able to grow robustly even in the presence of the elongated, dysfunctional telomeres. Strikingly, ATM depletion largely blocks end-to-end fusion of the MT-hTer-47A-induced dysfunctional telomeres.
Our work parallels a recent study in SV40 large T antigen-immortalized mouse embryonic fibroblasts (MEFs) which examined the role of ATM in another type of dysfunctional telomere response (21). That study showed that ATM can promote the end-to-end fusion of telomeres rendered dysfunctional through knockout of the TRF2 protein, which is a key component of the protective shelterin complex (2). While our study uses a different method of telomere disruption (mutant repeat incorporation), we have likewise found that fusion of MT-hTer-47A-induced dysfunctional telomeres is largely ATM-dependent in human cancer cells. Furthermore, our work suggests that these telomere fusions at least partly underlie the ability of MT-hTer-47A treatment to inhibit cancer cell proliferation. Telomere shortening in cells with checkpoint deficiencies ultimately leads to what has been termed “M2 crisis,” in which numerous short telomeres undergo end-to-end fusions (41). These fusion events initiate breakage-fusion-bridge cycles, mitotic catastrophe, and apoptosis. Our data suggest that MT-hTer-47A treatment similarly initiates a “crisis-like” state, resulting in significant fusion of dysfunctional (although still long) telomeres and growth inhibition.
This “crisis-like” model fits well with several characteristics of MT-hTer-47A growth inhibition. First, MT-hTer-47A equally inhibits the growth of p53-positive and p53-negative HCT116 colon cancer cell lines (13). A p53-independent effect would be expected if MT-hTer-47A cytotoxicity is mediated through end-to-end telomere fusions. The high fusion levels observed are predicted to cause genomic havoc and consequent loss of cell viability even in the absence of a p53-directed response. Second, we have shown here that the ability of ATM depletion to rescue the growth of MT-hTer-47A-treated cancer cells directly correlates with a marked drop in end-to-end telomere fusion events. Finally, a role for fusions in MT-hTer-47A-induced cytotoxicity can explain why ATM depletion sensitizes cancer cells to agents which cause intrachromosomal DSBs but desensitizes them to MT-hTer-47A. In the case of intrachromosomal DSBs, ATM is required to activate damage checkpoints and assist in the repair of the broken chromosomes. By promoting DNA repair prior to resumption of cell division, ATM allows continued proliferation without the loss of acentric chromosomal fragments (42). In the case of MT-hTer-47A, ATM activity at the dysfunctional telomeres leads to numerous end-to-end fusions, which are aberrant “repair” events that will ultimately inhibit cell survival. Our results thus indicate that while ATM inhibition may enhance the effectiveness of therapeutics which cause DSBs throughout the genome, such an approach would be counterproductive in the case of MT-hTer-47A treatment (23, 42).
While end-to-end telomere fusions almost certainly underlie at least part of the MT-hTer-47A-induced cytotoxicity, it is also possible that direct signaling from unfused dysfunctional telomeres induces apoptosis. Prior work in cells overexpressing a dominant-negative form of TRF2 showed that the resulting dysfunctional telomeres could directly induce apoptosis through an ATM-dependent pathway, without requiring progression through S phase (28). However, that induction was also dependent on the presence of wild-type p53, while the MT-hTer-47A growth effect reported here is not.
While ATM depletion substantially reduces the cellular response to MT-hTer-47A expression in both LOX and UM-UC-3 cell lines, the rescue is only partial. Residual ATM activation may be responsible for the partial response to MT-hTer-47A, since overexposure of the immunofluorescence images in Fig. 1 shows weak foci of phosphorylated ATM visible at a subset of telomeres after MT-hTer-47A treatment, even in the ATM-depleted cells (data not shown). Alternatively, other DNA damage response pathways may also respond to MT-hTer-47A-induced dysfunctional telomeres. A good candidate is the ATR pathway, since diverse types of telomere dysfunction can activate ATR in addition to ATM (5, 21, 43).
There is increasing recognition that the response of individual tumors to a given treatment will vary as a result of genetic heterogeneity (44). The work presented here suggests that tumors with low-to-absent ATM, which include certain leukemias and a subset of breast cancers, may have a muted response to MT-hTer-47A treatment (45, 46). Prior work in both model organisms and mammalian cells has shown that the cellular consequences of MT-Ter expression varies depending on the exact mutations made in the template region (7, 47, 48). In multiple organisms, different MT-Ters activate different checkpoint pathways and lead to growth phenotypes that can vary from wild-type growth to senescence to rapid cell death (Smith and Blackburn, unpublished data)(48). Likewise, expression of different MT-hTers in cancer cells leads to different levels of growth inhibition for reasons that have not been defined (7). Given this heterogeneity, it is likely that other MT-hTers will not show the same level of dependency on ATM signaling and therefore work more effectively in the context of ATM-deficient tumors (Dana Smith and Elizabeth Blackburn, unpublished data)(7, 48). For example, some MT-hTers might instead act predominately through the ATR pathway by exposing the single-strand 3′ telomeric overhang (21). Thus, a more detailed understanding of how different MT-hTers disrupt the shelterin complex and engage the DNA damage machinery may ultimately allow more effective targeting of diverse tumor types.
Supplementary Material
Acknowledgments
The authors thank Lifeng Xu and Shang Li for their valuable scientific input and critical reading of the manuscript.
Financial Support: The Bernard Osher Foundation, NIH Breast Cancer SPORE CA58205, and NIH R01 CA096840 to E.H.B.; American Cancer Society Postdoctoral Fellowship PF-05-111-01 to B.A.S.
References
- 1.Blackburn EH. Telomere states and cell fates. Nature. 2000;408(6808):53–6. doi: 10.1038/35040500. [DOI] [PubMed] [Google Scholar]
- 2.de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19(18):2100–10. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 3.Levy MZ, Allsopp RC, Futcher AB, Greider CW, Harley CB. Telomere end-replication problem and cell aging. J Mol Biol. 1992;225(4):951–60. doi: 10.1016/0022-2836(92)90096-3. [DOI] [PubMed] [Google Scholar]
- 4.Hahn WC. Role of telomeres and telomerase in the pathogenesis of human cancer. J Clin Oncol. 2003;21(10):2034–43. doi: 10.1200/JCO.2003.06.018. [DOI] [PubMed] [Google Scholar]
- 5.Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a) Mol Cell. 2004;14(4):501–13. doi: 10.1016/s1097-2765(04)00256-4. [DOI] [PubMed] [Google Scholar]
- 6.Shay JW, Wright WE. Telomerase therapeutics for cancer: challenges and new directions. Nat Rev Drug Discov. 2006;5(7):577–84. doi: 10.1038/nrd2081. [DOI] [PubMed] [Google Scholar]
- 7.Kim MM, Rivera MA, Botchkina IL, Shalaby R, Thor AD, Blackburn EH. A low threshold level of expression of mutant-template telomerase RNA inhibits human tumor cell proliferation. Proc Natl Acad Sci U S A. 2001;98(14):7982–7. doi: 10.1073/pnas.131211098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blackburn EH. Telomerase and Cancer: Kirk A. Landon--AACR prize for basic cancer research lecture. Mol Cancer Res. 2005;3(9):477–82. doi: 10.1158/1541-7786.MCR-05-0147. [DOI] [PubMed] [Google Scholar]
- 9.Marusic L, Anton M, Tidy A, Wang P, Villeponteau B, Bacchetti S. Reprogramming of telomerase by expression of mutant telomerase RNA template in human cells leads to altered telomeres that correlate with reduced cell viability. Mol Cell Biol. 1997;17(11):6394–401. doi: 10.1128/mcb.17.11.6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guiducci C, Cerone MA, Bacchetti S. Expression of mutant telomerase in immortal telomerase-negative human cells results in cell cycle deregulation, nuclear and chromosomal abnormalities and rapid loss of viability. Oncogene. 2001;20(6):714–25. doi: 10.1038/sj.onc.1204145. [DOI] [PubMed] [Google Scholar]
- 11.Xu L, Blackburn EH. Human Rif1 protein binds aberrant telomeres and aligns along anaphase midzone microtubules. J Cell Biol. 2004;167(5):819–30. doi: 10.1083/jcb.200408181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li S, Crothers J, Haqq CM, Blackburn EH. Cellular and gene expression responses involved in the rapid growth inhibition of human cancer cells by RNA interference-mediated depletion of telomerase RNA. J Biol Chem. 2005;280(25):23709–17. doi: 10.1074/jbc.M502782200. [DOI] [PubMed] [Google Scholar]
- 13.Li S, Rosenberg JE, Donjacour AA, et al. Rapid inhibition of cancer cell growth induced by lentiviral delivery and expression of mutant-template telomerase RNA and anti-telomerase short-interfering RNA. Cancer Res. 2004;64(14):4833–40. doi: 10.1158/0008-5472.CAN-04-0953. [DOI] [PubMed] [Google Scholar]
- 14.Shay JW, Wright WE. Telomeres are double-strand DNA breaks hidden from DNA damage responses. Mol Cell. 2004;14(4):420–1. doi: 10.1016/s1097-2765(04)00269-2. [DOI] [PubMed] [Google Scholar]
- 15.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421(6922):499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 16.Goudsouzian LK, Tuzon CT, Zakian VA. S. cerevisiae Tel1p and Mre11p are required for normal levels of Est1p and Est2p telomere association. Mol Cell. 2006;24(4):603–10. doi: 10.1016/j.molcel.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 17.Wu Y, Xiao S, Zhu XD. MRE11-RAD50-NBS1 and ATM function as co-mediators of TRF1 in telomere length control. Nat Struct Mol Biol. 2007;14(9):832–40. doi: 10.1038/nsmb1286. [DOI] [PubMed] [Google Scholar]
- 18.Chan SW, Chang J, Prescott J, Blackburn EH. Altering telomere structure allows telomerase to act in yeast lacking ATM kinases. Curr Biol. 2001;11(16):1240–50. doi: 10.1016/s0960-9822(01)00391-8. [DOI] [PubMed] [Google Scholar]
- 19.Chan SW, Blackburn EH. Telomerase and ATM/Tel1p protect telomeres from nonhomologous end joining. Mol Cell. 2003;11(5):1379–87. doi: 10.1016/s1097-2765(03)00174-6. [DOI] [PubMed] [Google Scholar]
- 20.Metcalfe JA, Parkhill J, Campbell L, et al. Accelerated telomere shortening in ataxia telangiectasia. Nat Genet. 1996;13(3):350–3. doi: 10.1038/ng0796-350. [DOI] [PubMed] [Google Scholar]
- 21.Denchi EL, de Lange T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature. 2007;448(7157):1068–71. doi: 10.1038/nature06065. [DOI] [PubMed] [Google Scholar]
- 22.Matsuoka S, Ballif BA, Smogorzewska A, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316(5828):1160–6. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 23.Zhou BB, Anderson HJ, Roberge M. Targeting DNA checkpoint kinases in cancer therapy. Cancer Biol Ther. 2003;2(4 Suppl 1):S16–22. [PubMed] [Google Scholar]
- 24.Hickson I, Zhao Y, Richardson CJ, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64(24):9152–9. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 25.Fedier A, Schlamminger M, Schwarz VA, Haller U, Howell SB, Fink D. Loss of atm sensitises p53-deficient cells to topoisomerase poisons and antimetabolites. Ann Oncol. 2003;14(6):938–45. doi: 10.1093/annonc/mdg240. [DOI] [PubMed] [Google Scholar]
- 26.Collis SJ, Swartz MJ, Nelson WG, DeWeese TL. Enhanced radiation and chemotherapy-mediated cell killing of human cancer cells by small inhibitory RNA silencing of DNA repair factors. Cancer Res. 2003;63(7):1550–4. [PubMed] [Google Scholar]
- 27.Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol. 2003;13(17):1549–56. doi: 10.1016/s0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- 28.Karlseder J, Broccoli D, Dai Y, Hardy S, de Lange T. p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science. 1999;283(5406):1321–5. doi: 10.1126/science.283.5406.1321. [DOI] [PubMed] [Google Scholar]
- 29.Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol. 1997;15(9):871–5. doi: 10.1038/nbt0997-871. [DOI] [PubMed] [Google Scholar]
- 30.Li J, Stern DF. Regulation of CHK2 by DNA-dependent protein kinase. J Biol Chem. 2005;280(12):12041–50. doi: 10.1074/jbc.M412445200. [DOI] [PubMed] [Google Scholar]
- 31.Wang J, Wiltshire T, Wang Y, et al. ATM-dependent CHK2 activation induced by anticancer agent, irofulven. J Biol Chem. 2004;279(38):39584–92. doi: 10.1074/jbc.M400015200. [DOI] [PubMed] [Google Scholar]
- 32.Xu L, Blackburn EH. Human cancer cells harbor T-stumps, a distinct class of extremely short telomeres. Mol Cell. 2007;28(2):315–27. doi: 10.1016/j.molcel.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6(10):789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 34.Jackson AL, Bartz SR, Schelter J, et al. Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol. 2003;21(6):635–7. doi: 10.1038/nbt831. [DOI] [PubMed] [Google Scholar]
- 35.Goldkorn A, Blackburn EH. Assembly of mutant-template telomerase RNA into catalytically active telomerase ribonucleoprotein that can act on telomeres is required for apoptosis and cell cycle arrest in human cancer cells. Cancer Res. 2006;66(11):5763–71. doi: 10.1158/0008-5472.CAN-05-3782. [DOI] [PubMed] [Google Scholar]
- 36.Lau A, Swinbank KM, Ahmed PS, et al. Suppression of HIV-1 infection by a small molecule inhibitor of the ATM kinase. Nat Cell Biol. 2005;7(5):493–500. doi: 10.1038/ncb1250. [DOI] [PubMed] [Google Scholar]
- 37.Hector RE, Shtofman RL, Ray A, et al. Tel1p preferentially associates with short telomeres to stimulate their elongation. Mol Cell. 2007;27(5):851–8. doi: 10.1016/j.molcel.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 38.Cooper MJ, Haluschak JJ, Johnson D, et al. p53 mutations in bladder carcinoma cell lines. Oncol Res. 1994;6(12):569–79. [PubMed] [Google Scholar]
- 39.McEachern MJ, Iyer S, Fulton TB, Blackburn EH. Telomere fusions caused by mutating the terminal region of telomeric DNA. Proc Natl Acad Sci U S A. 2000;97(21):11409–14. doi: 10.1073/pnas.210388397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kirk KE, Harmon BP, Reichardt IK, Sedat JW, Blackburn EH. Block in anaphase chromosome separation caused by a telomerase template mutation. Science. 1997;275(5305):1478–81. doi: 10.1126/science.275.5305.1478. [DOI] [PubMed] [Google Scholar]
- 41.Shay JW, Wright WE. Senescence and immortalization: role of telomeres and telomerase. Carcinogenesis. 2005;26(5):867–74. doi: 10.1093/carcin/bgh296. [DOI] [PubMed] [Google Scholar]
- 42.Jeggo PA, Lobrich M. Contribution of DNA repair and cell cycle checkpoint arrest to the maintenance of genomic stability. DNA Repair (Amst) 2006;5(910):1192–8. doi: 10.1016/j.dnarep.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 43.Guo X, Deng Y, Lin Y, et al. Dysfunctional telomeres activate an ATM-ATR-dependent DNA damage response to suppress tumorigenesis. EMBO J. 2007;26(22):4709–19. doi: 10.1038/sj.emboj.7601893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Million RP. Impact of genetic diagnostics on drug development strategy. Nat Rev Drug Discov. 2006;5(6):459–62. doi: 10.1038/nrd2034. [DOI] [PubMed] [Google Scholar]
- 45.Hall J. The Ataxia-telangiectasia mutated gene and breast cancer: gene expression profiles and sequence variants. Cancer Lett. 2005;227(2):105–14. doi: 10.1016/j.canlet.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 46.Fang NY, Greiner TC, Weisenburger DD, et al. Oligonucleotide microarrays demonstrate the highest frequency of ATM mutations in the mantle cell subtype of lymphoma. Proc Natl Acad Sci U S A. 2003;100(9):5372–7. doi: 10.1073/pnas.0831102100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu GL, Bradley JD, Attardi LD, Blackburn EH. In vivo alteration of telomere sequences and senescence caused by mutated Tetrahymena telomerase RNAs. Nature. 1990;344(6262):126–32. doi: 10.1038/344126a0. [DOI] [PubMed] [Google Scholar]
- 48.Lin J, Smith DL, Blackburn EH. Mutant telomere sequences lead to impaired chromosome separation and a unique checkpoint response. Mol Biol Cell. 2004;15(4):1623–34. doi: 10.1091/mbc.E03-10-0740. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.