

Abstract
The X-ray crystal structures of mannose trimming enzyme Drosophila Golgi α–mannosidase II (dGMII) complexed with the inhibitors mannostatin A (1) and an N-benzyl analog (2) have been determined. Molecular dynamics simulations and NMR studies have shown that the five-membered ring of mannostatin A is rather flexible occupying pseudo-rotational itineraries between 2T3 and 5E, and 2T3 and 4E. In the bound state, mannostatin A adopts a 2T1 twist envelope conformation, which is not significantly populated in solution. Possible conformations of the mannosyl oxacarbenium ion and an enzyme-linked intermediate have been compared to the conformation of mannostatin A in the co-crystal structure with dGMII. It has been found that mannostatin A best mimics the covalent linked mannosyl intermediate, which adopts a 1S5 skew boat conformation. The thiomethyl group, which is critical for high affinity, superimposes with the C-6 hydroxyl of the covalent linked intermediate. This functionality is able to make a number of additional polar and non-polar interactions increasing the affinity for dGMII. Furthermore, the X-ray structures show that the environment surrounding the thiomethyl group of 1 is remarkably similar to the arrangements around the methionine residues in the protein. Collectively, our studies contradict the long held view that potent inhibitors of glycosidases mimic an oxacarbenium ion like transition state.
Keywords: Mannosidases, Glycosidase Inhibitors, Oxacarbenium ion, Mannostatin, X-ray, Thiomethyl, Methionine
Introduction
Glycosidases are enzymes that catalyze the cleavage of glycosidic bonds and play critical roles in a number of biological processes. Inhibitors of these enzymes have garnered much attention and are currently used for the treatment of diabetes, viral infections and Gauchers disease.1-5 Furthermore, inhibition of the mannose trimming enzyme human Golgi α–mannosidase II (GMII; mannosyl-oligosaccharide 1,3-1,6-α–mannosidase II; E.C. 3.2.1.114), which acts late in the N-glycan processing pathway, provides a route to blocking the oncogene-induced changes in cell surface oligosaccharide structures.6-9 GMII selectively cleaves α(1-3) and α(1-6) mannosyl residues present in its natural substrate GlcNAcMan5GlcNAc2.10 It is a retaining Family 38 glycosylhydrolase, which employs a two-stage mechanism involving two carboxylic acids positioned within the active site which act in concert: one as a catalytic nucleophile and the other as a general acid/base catalyst.11-15 Protonation of the exocyclic glycosyl oxygen of a substrate molecule leads to bond-breaking and simultaneous attack of the catalytic nucleophile to form a glycosyl enzyme intermediate. Subsequent hydrolysis of the covalent intermediate by a nucleophilic water molecule gives an α-mannose product with overall retention of configuration. Studies with 5-fluoro pseudo-substrates and deuterium labeled substrates have shown that the transition states on either side of the covalent intermediate have marked oxacarbenium ion character.11-16 Furthermore, X-ray crystal structures of the wild type and a mutant Drosophila melanogaster GMII (dGMII) in which the acid/base catalyst has been removed, with fluorinated sugar analogs have revealed that the glycosyl enzyme intermediate adopts a distorted 1S5 skew boat conformation.16 In this conformation, the leaving group is placed anti-periplanar to the lone pair of the ring oxygen, a requirement for the departure of the leaving group according to Deslongchamp's anti-periplanar lone-pair hypothesis.17 Furthermore, steric clashes between the syn-hydrogens at C-3 and C-5 and the attacking water are minimal in the 1S5 skew boat conformation.
Potent inhibitors of glycosidases are believed to mimic oxacarbenium ion-like transition states.11-15 The inhibitory activity of the natural product swainsonine has long been attributed to the resemblance of its 5-membered ring to a flattened six-membered ring mannosyl oxacarbenium ion. In fact, the crystal structure of swainsonine complexed with dGMII shows the inhibitor to be tilted in such a way as to bring the equivalent of its anomeric carbon close to the presumed catalytic nucleophile.18
Mannostatins A and B, which were isolated from the soil microorganism Streptoverticillus, are some of the most potent inhibitors of class II α-mannosidases reported thus far (Figure 1).19 They were the first non-azasugar type inhibitors to be discovered that possess an aminocyclopentitol structure. The inhibitors are of the reversible, competitive type and do not show the slow-binding phenomenon exhibited by swainsonine and its analogs. Mannostatin A effectively blocked the processing of influenza viral hemagglutin in cultured MDCK cells and caused the accumulation of hybrid type protein linked oligosaccharides, which is in agreement with blocking Golgi mannosidase II.8 The synthesis and biological evaluation of a small number of Mannostatin analogs has revealed that the basicity of the primary amine and the neighboring cis-diol are essential for inhibitory activity.20-26
Figure 1.

Cyclopentitol inhibitors of Golgi α-Mannosidase II. The inhibition constants in the parentheses indicate that the compound 3, which lacks the thiomethyl moiety, is much less potent compared with compounds 1 and 2.
While interest in mannostatin A continues to grow, the mode of inhibition of this compound is not well understood. In this respect, its carbocyclic structure represents a significant departure from common alkaloid-based glycosidase inhibitors, and it is unclear whether mannostatin resembles either d-mannose or the mannosyl oxacarbenium ion.
We have now determined the X-ray crystal structure of dGMII in complex with mannostatin A (1) and an N-benzyl analog (2). We have analyzed in detail key molecular interactions, in particular, the interaction of the thiomethyl ether with the protein to determine the structural basis for inhibition. Furthermore, molecular dynamics simulations of both the unbound mannostatin A and its complex with the enzyme have been performed to obtain information about the solution phase conformational properties and dynamic features of the enzyme-inhibitor interactions. Finally, possible conformations of the mannosyl oxacarbenium ion and the covalent-linked saccharide intermediate have been compared to the conformation of mannostatin A observed in the co-crystal structure.
Results and Discussion
Optically pure Mannostatin A (1), N-benzyl Mannostatin A (2) and 1-deoxyaminocyclopentitetrol (3) were prepared by literature procedures.20, 22, 27 The rate of hydrolysis of different concentrations of 4-methylumbelliferyl α-D-mannopyranoside alone and in the presence of different concentrations of inhibitor was measured fluorometrically and Ki values were determined from Dixon plots. As can be seen in Figure 1, Mannostatin A is a more potent inhibitor then its N-benzylated counterpart. On the other hand, 1-deoxyaminocyclopentitetrol 3, which lacks the thiomethyl ether, is a poor inhibitor indicating that the thiomethyl moiety of 1 makes important interactions with the binding site of the enzyme.
In order to rationalize the inhibition data, X-ray crystal structures of compounds 1 and 2 complexed with dGMII were solved at 1.3 Å. Data collection and refinement statistics are reported in supplemental data Table 1S. The quality of the density around the bound inhibitors is shown in Figure 2. The X-ray crystal structure of dGMII complexed with compound 1 did not show any significant changes in the backbone structure compared to the free enzyme (RMSD = 0.129 Å vs. PDB 1HTY). The five-membered ring of the inhibitor adopts a 2T1 twist envelope conformation (phase angle, φ = 201°), which is stacked against the aromatic ring of Trp95, a type of interaction seen in many carbohydrate-protein complexes.28-30 Important close contacts between the inhibitors and dGMII are detailed in Table 1. There are a number of contacts, which have been observed in previous dGMII crystal structures.16,18,31,32 The 3,4 cis-diol of 1 is complexed with a Zn2+ ion in the active site of dGMII resulting in T6 coordination geometry, while the 2-hydroxyl forms hydrogen bonds with Asp472 and Tyr727. Furthermore, the amine of 1 forms hydrogen bonds with catalytic acid residues Asp204 and Asp341, and Tyr269. The binding of the inhibitors within the context of selected active site residues is shown in Figure 3.
Figure 2.
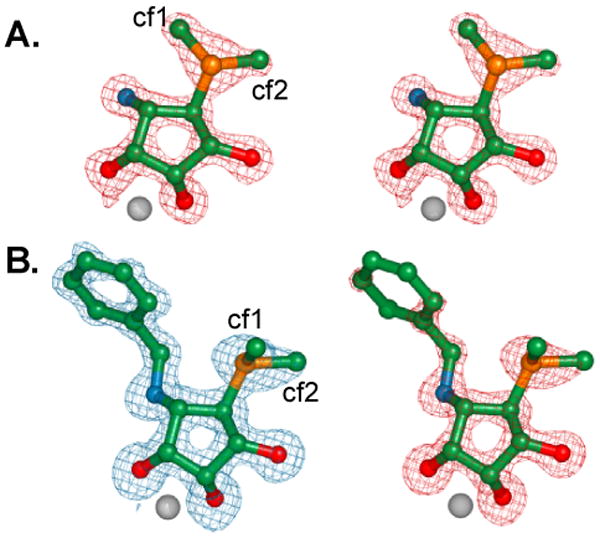
Stereoviews of electron density of bound inhibitors: Simulated annealing omit maps (Fo-Fc). A. Mannostatin A 1, contoured at 5 sigma B. N-benzyl mannostatin 2 contoured at 3 sigma (blue) or 5 sigma (red). This figure was created with Pymol. cf1 and cf2 refer to the two possible conformations of C7 which fit into the observed density.
Table 1.
Distances between different inhibitor atoms and protein atoms as observed in the complexes of swainsonine (PDB id 1HWW), 1 and 2 with dGMII.
| Swainsonine | 1 | 2 | |||
|---|---|---|---|---|---|
| Protein Atom | Inhibitor Atom | Distance
(Å) |
Inhibitor Atom | Distance
(Å) |
Distance
(Å) |
| H90 NE2 | O-2 | 2.97 | O-4 | 3.07 | 3.11 |
| D92 OD1 | O-1 | 3.04 | O-3 | 2.92 | 2.99 |
| D92 OD2 | O-2 | 2.54 | O-4 | 2.55 | 2.51 |
| O-1 | 2.83 | O-4 | 2.82 | 2.91 | |
| D204 OD2 | O-2 | 2.97 | O-3 | 2.92 | 2.95 |
| N-4 | 2.75 | N | 2.77 | 2.9 | |
| R228 NH2 | C-7 (cf1) | 3.11 | 5.08 | ||
| S | 4.01 | 3.54 | |||
| Y269 OH | N-4 | 4.17 | N | 2.78 | 2.96 |
| D341 OD2 | N | 2.99 | 3 | ||
| W415 CH2 | C-7 (cf2) | 3.36 | 3.17 | ||
| W415 CZ3 | C-7 (cf2) | 3.57 | 3.17 | ||
| H471 NE2 | O-1 | 3.12 | O-3 | 3.1 | 3.02 |
| D472 OD1 | O-8 | 2.56 | O-2 | 2.59 | 2.62 |
| D472 OD2 | O-1 | 2.6 | O-3 | 2.53 | 2.57 |
| Y727 OH | O-8 | 2.64 | O-2 | 2.72 | 2.68 |
| R876 O | ----- | ----- | C-7 (cf1) | 4 | 2.5 |
| S | 3.49 | 4.14 | |||
| N | 3.03 | ||||
| WATERS | C-7 (cf1) | 2.56, 2.85, 2.88 | |||
Figure 3.
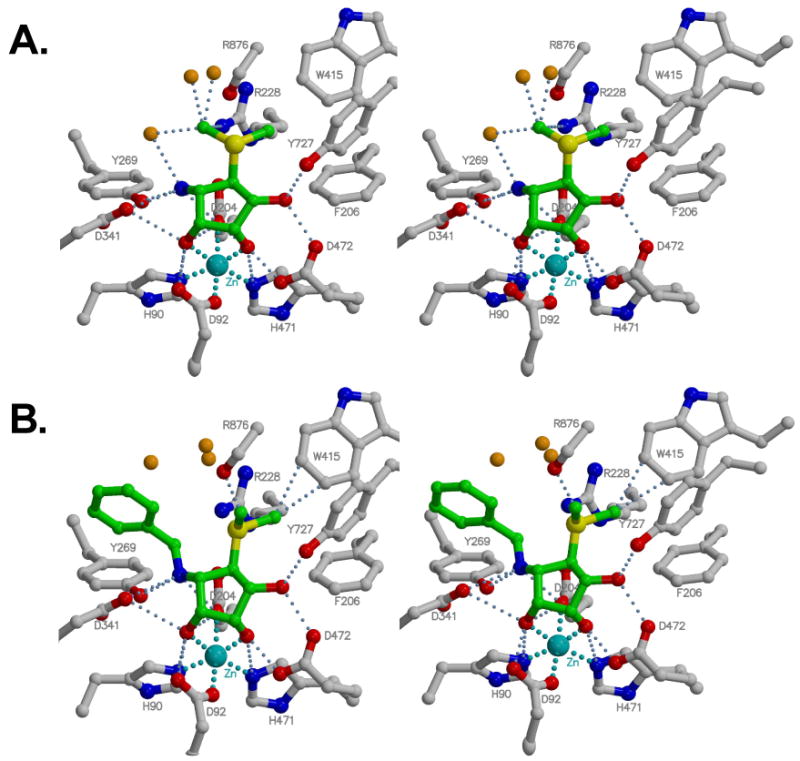
Stereoview of the interaction of Mannostatin A (1, A) and N-Benzyl Mannostatin (2, B) with residues in the active site of dGMII. Interactions closer than 3.2 Å are indicated. The interactions with zinc are indicated in cyan. Water molecules appear as orange balls. Distances are presented in Table 1.
Data from SAR experiments (referred to above) has pointed to the importance of the amine and cis-diols in the inhibitory activity of mannostain A and the crystal structure beautifully illustrates how these groups interact with the protein. Similar modes of interaction, with almost identical distances, were seen in the crystal structure of dGMII complexed with swainsonine (Table 1) although only a single interaction with the amine group is observed in that case.
The thiomethyl moiety of 1 and 2 is a feature that is not observed in any other glycosidase inhibitors and the data summarized in Figure 1 indicate that this structural feature is critical for potent inhibition. It must be noted that the thiomethyl moiety is structurally similar to the side chain of a methionine residue. It has been proposed that the sulfur atom and ε–CH3 group of methionine residues are involved in several different interactions important for protein stability.33-37 For example, aryl-sulfur interactions have been proposed favorable because of the observed proximity of the methionine residue to the aromatic side chains in protein X-ray structures and small molecules X-ray structures. In general, these interactions are either hydrophobic or electrostatic, of the types S–CH3---Ar or S---H–Ar, respectively. In addition, the sulfur atom, possessing an empty d-orbital is also known to be involved in S---X (X = O and N) interactions.37 Such an interaction is stabilized by the orbital interaction between the lone pair on O or N and the anti-bonding orbital of the sulfur atom. The strong dipole of the methionine residue also makes it an ideal hydrogen bond acceptor. However, an analysis of the NH---S and OH---S hydrogen bonds in proteins suggests that the sulfur atom in methionine has only a weak character of a hydrogen bond acceptor.35,38
In the context of dGMII at least one third of the methionines occur in a hydrophilic or surface environment, and a number bear a striking resemblance to the environment of the thiomethyl group of the inhibitors. In Figure 4, three different methionine residues whose methyl carbon occupies environments similar to 1 are presented, along with their respective distances from water (or a backbone carbonyl in the case of Met769). In two cases (Met264 and Met769) alternative conformations of the thiomethyl group are seen, similar to the situation observed with the inhibitors, while Met224 shows a single strong conformation that places the methyl group in close proximity to waters. In Met264 both confomers are in a hydrophilic environment. In Met769, one orientation places the methyl group in close proximity to 2 waters and the backbone oxygen of Pro684, while the other orientation results in the terminal methyl group finding itself in a more hydrophobic environment, similar to what is observed for 1. It should be noted that all the waters that are in close proximity to methionine have other strong hydrogen bond partners and the methionine residues do not appear to be their principal form of interaction with the protein.
Figure 4.
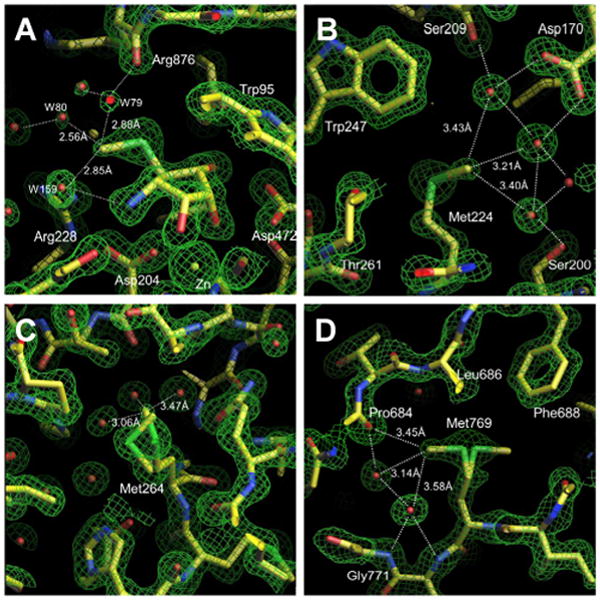
Comparison of the density of Mannostatin A cf1 with methionine residues of dGMII found in similar environments. Possible water interactions are shown with dotted lines. Distances between the thiomethyl group and proximal waters are indicated. The electron density (2Fo-Fc) is contoured at 1.5 -2 sigma. The figures are generated in O. A. Mannostatin A cf1 B. Methionine 224 C. Methionine264, showing both conformers D. Methionine769, showing both conformers.
The sulfur atom of the thiomethyl moiety of 1 seems to form a nonbonded S---O type of interaction with the main chain carbonyl oxygen of Arg876. Statistical analysis of protein structures performed by Iwaoka et al37 suggests that most S---O interactions are between the sulfur atom in the methionine residue and backbone carbonyl oxygens or side chain carboxylate oxygens. An analysis of the spatial placement of sulfur with respect to the oxygen revealed that the interaction between the π-orbital on the carbonyl oxygen and the antibonding σ* orbital of S-C bond is largely responsible for this observation. Ab initio calculations with small molecules indicate that these interactions could result in stabilization of up to 2.5 kcal mol−1 when these atoms are separated by 3.3 Å.37 It is, however, important to note that these calculations have been performed in vacuo and therefore the binding energies may be different in an aqueous environment due to solvation effects. In the 1-dGMII complex the sulfur atom is located at 3.49 Å from the Arg876O, suggesting that the interaction observed between the sulfur atom in the thiomethyl moiety and the carbonyl oxygen of Arg876 must be reasonably strong.
The most striking structural feature of these inhibitor complexes is the fact that the C-7 carbon (i.e. the methyl group in the thiomethyl functionality) can occupy two possible positions (Figures 2 and 3). Both these positions are clearly visible in the electron density maps (Figure 2) and, in the case of 1, have B-factors of 5.2 Å2 and 6.8 Å2 (conformer 1 (cf1) and conformer 2 (cf2), respectively) suggesting that cf1 conformer might be slightly favored.
The methyl group obviously forms favorable interactions in each of these positions, but the exact nature of the interactions is somewhat ambiguous and arguments can be made for both hydrophobic and hydrophilic bonds. In the case of the cf1 conformation of 1, the methyl carbon is 4.3 Å from the CZ carbon in Arg228. The interaction between the methyl and Arg228 could be a C–H---cation type interaction where the C–H acts as donor. A statistical analysis of 1154 protein structures for C–H---π interactions revealed that such interactions are possible in proteins and are mostly intrahelical interactions39 which could be classified as weak hydrogen bonds and which play an important role in the secondary structure stabilization. It was also found that the terminal methyl in methionine residue is one of the prominent donors for such interactions. The thiomethyl group of 1 can act as a donor to form such interaction with Arg228. There have been several theoretical studies, which estimate stabilization energies in other types of C–H---π interactions. However, to the best of our knowledge, energetic evaluation of the interaction between an aliphatic C–H and guanidinium moiety of an arginine has not been reported. Nevertheless, the relatively close proximity of the guanidinium moiety as observed in the 1-dGMII complex, indicate that there is strong possibility of formation of C–H---π type of interaction between the two functionalities.
It is tempting to speculate that the polarizable methyl group may hydrogen bond with neighboring water molecules. Three water molecules (W79, W80 and W159) are present at less than 3.2 Å from the methyl carbon in the cf1 conformation, while a fourth (W80) is 3.3 Å away. W159 also makes a strong hydrogen bond with the amino group of 1 and two other waters. Waters 78-80 show relatively weak electron density while W159 is quite well defined. Iwaoka et al37 showed that a single C–H---O hydrogen bond from the methyl group to a backbone oxygen was weakly stabilizing and one could surmise that 3 such weak hydrogen bonds to water would have an additive effect.
On the other hand, there are arguments for hydrophobic interactions being the predominant binding force.40,41 In this respect, it has been shown that methionines display mainly hydrophobic character,40 at least in the context of artificial hairpin peptides where their measurements were made. In fact, it could be that the water molecules are actually stabilizing the cf2 conformations rather than the cf1 conformation by binding to sulfur. In the cf2 conformation, sulfur can form a hydrogen bond with these water molecules via the lone pair electrons. Moreover, W79 is hydrogen bonded to Arg876O (2.7 Å) and W80 is hydrogen bonded to W79 and W81 suggesting that there may not be any interaction between the methyl in 1 cf1 and the water-sulfur cf2 interaction is what is visible in the electron density.
The X-ray crystal structure shows that in the cf2 conformation the thiomethyl ether of 1 makes two arene-sulfur interactions with Phe206 and Tyr727. In protein structures, methionine is often found in close proximity of aromatic side chain residues indicating that arene-sulfur interactions are important for protein structure stabilization.33-36 In general, arene-sulfur interactions are either hydrophobic of the S–CH3---Ar type or electrostatic of the S---H–Ar type. Ab initio calculations of a dimethyl sulfide-benzene complex has predicted a stabilization energy of −1.6 kcal mol-1 when methyl hydrogens point towards the centroid of an aromatic ring and when the distance between the sulfur atom and the centroid of the benzene ring (Rcen) is 5.8 Å.42 In an alternative model, where both methyl groups are at the edges of the aromatic ring and the sulfur is 4.9 Å from the centroid of the aromatic ring, a stabilization energy of −2.5 kcal mol-1 was calculated.42 Furthermore, mutational experiments with β-hairpin model peptides40 have indicated that arene-sulfur interactions are primarily hydrophobic in nature contributing up to −1 kcal mol−1 of stabilization energy.
Interestingly, the X-ray crystal structure shows that the thiomethyl functionality of 1 cf2 complexes with Phe206 of dGMII through a hydrophobic SCH3---Ar type interaction. In this case, the sulfur atom is placed slightly off-centered from the aromatic ring (Rcen = 5.8 Å) similar to a mode predicted by ab initio calculations. The Tyr727 also makes a hydrophobic interaction with the thiomethyl group but in this case the methyl group is placed at the edge of the aromatic ring (Rcen=5.3 Å) similar to the second mode of interaction predicted by ab initio calculations.43 Contact analysis shows that the thiomethyl group also makes van der Waals contacts with Trp415 and Arg876.
In the case of the 2-dGMII complex, the methyl carbon in the thiomethyl functionality (C-7) also occupies two positions, the cf2 position is similar between 1 and 2 but the cf1 has moved. (Figure 3B). In the cf1 conformation, the methyl carbon is close to the backbone carbonyl oxygen of Arg876 and the sulfur atom seems to interact with NH2 of Arg228, which is at 3.5 Å from the sulfur atom, via the lone pairs of the sulfur atom. This interaction has been studied in protein structures before and the structural preferences observed for this interaction suggest a contribution from the n(S)→σ*(NH) orbital interaction to the stability.37 Theoretical studies have shown that such interaction can contribute up to 2 kcal mol−1 to the stability with 3.6 Å separation between the two atoms.37 In the alternate cf2 conformation, this interaction may be weakened by the geometric position of the methyl group. However, this loss in the interaction may easily be compensated by the interaction between the sulfur atom and the Arg876O, which is also observed in 1-dGMII complex and has been explained earlier. In addition to this interaction, the thiomethyl moiety is also involved in sulfur-π type interactions with Phe206 and Tyr727 in the same way as in the case of 1. Additionally in the case of 2 cf2 the C-7 is perfectly centered at a distance of 3.17 Å with CH2 and CZ3 of Trp415. The close contacts between the thiomethyl group and water molecules observed in 1-dGMII complex are not seen in the 2-dGMII complex; there is no equivalent to W159 whose position has been taken up by the phenyl ring and the W79 equivalent (W361) has moved to 4 Å from the sulfur, and the W80 equivalent (W360) is now 3.8 Å away. There is now an intramolecular interaction between the phenyl ring and sulfur (see below), which displaces the water interactions.
It is important to note that additional modified derivatives of mannostatin (1) need to be synthesized and biologically evaluated to determine whether the polar or non-polar interactions of the thiomethyl moiety are responsible for its enhanced activity.
Docking studies44 had indicated that the aromatic substituent of 2 would be able to make a parallel, off-centered π-π stacking interaction with phenyl ring of Tyr269 and therefore, it was expected that 2 would be a better inhibitor than 1. The unexpected finding that 2 is a poorer inhibitor than 1 can, however, be rationalized by careful examination of the crystal structure of dGMII with 2 (Figure 3). In both 1 and 2, the five-membered rings are identically positioned in the binding site of dGMII (Figure 3B). The distance between the centroid of the aromatic ring of compound 2 and Tyr269 is 5.7 Å at an angle of 118° between the planes made by the aromatic rings, indicating that no edge to face binding interactions are made which require a distance of 5.0 Å and angle of 90°. The unexpected orientation of the aromatic ring of 2 can be rationalized by an intramolecular S---H–Ar type hydrogen bond between the thiomethyl moiety and the phenyl ring with P = 0.3 Å and L = 5.0 Å (where P is the vertical and L is the horizontal distance between the sulfur atoms and the centroid of the aromatic ring). Ab initio calculations have indicated that this special arrangement of phenyl ring and the sulfur atom can contribute up to −1.5 kcal mol−1 of bonding energy.45 These calculations do, however, not take into account entropic and solvation effects.
MD Simulations
Cyclopentane rings, such as in compounds 1-3, are inherently flexible due to their ability to assume several twist and envelope conformations (Figure 5), which can interconvert with relative ease via pseudo-rotational itineraries.46,47 Since ring substitutions may affect these pseudo-rotational itineraries,47 compounds 1-3 may have different conformational properties. These differences may, in part, account for differences in the inhibition constants.
Figure 5.

Pseudo-rotational itinerary for compounds 1-3. Different envelope (E) and twist (T) conformers shown in the figure are uniquely defined by the value of phase angle (φ) of puckering. The phase angles for compounds 1-3 were computed using the carnal module in the AMBER suite of programs by employing Cremer and Pople's parameters (reference 61). The bound conformation of 1 (blue) lies outside the preferred solution phase conformational families (red) as sampled in MD simulation of free ligand.
The conformational properties of 1 were probed by a 5-ns MD simulation of just the ligands in explicit water starting from the conformation of 1 observed in the X-ray crystal structure with dGMII. The most populated conformational family (80% occupancy) occupies pseudo-rotational itineraries between 3T4 and 5E (φ = 90-144°) whereas a second and minor conformational family (20% occupancy) represents conformers between 2T3 and 4E (φ = 234-288°) (Figures 5 and 6A). The 2T1 conformer observed in the crystal structure was not populated and, thus, it appears that 1 is complexed in a high-energy conformation. The bound conformation of 1 is probably stabilized by a coordination of 3- and 4-oxygens with Zn+2 ion and a hydrogen bond between the amine nitrogen and Asp204, which arrange atoms C-3, C-4 and C-5 in single plane resulting in an almost perfect 2T1 (φ = 198°) conformation. This conformation also allows interactions of the thiomethyl moiety with the aromatic residues of Phe206 and Tyr727.
Figure 6.

Plots of phase angles (φ) as a function of time in the MD simulation trajectories. A: 1 in water, B: 2 in water, C: 3 in water, D: 1 bound to dGMII, E: 2 bound to dGMII and F: 2 bound to dGMII with the phenyl ring in 2 making parallel off-center type of interaction with the phenyl ring in Tyr269. MD trajectories for ligands in a box of TIP3P water molecules (top row) indicate that compound 1 populates two clusters (A): the first consists of conformers 3T4, E4, 5T4 and 5E (φ = 90-144°) and the second consists of conformers 2T3, E3, 4T3 and 4E (φ = 234-288°). Compound 2 populates only one conformational cluster (B) with conformers 2T3, E3, 4T3 and 4E. In the case of 3, the interconversion barrier is reduced resulting in frequent conversion between different conformers (C). It can be noted that the bound conformation (2T1, φ = 201°) of the five-membered ring in 1 and 2 is different than the conformers populated in the 5-ns MD simulations and remains in the same conformational family comprising of E1, 2T1 and 2E conformers (φ = 180-216°) during the 10 ns trajectory (bottom row, D and E). Although, the alternate orientation of the phenyl ring in 2 induces some flexibility in the five-membered ring, during the entire simulation the five-membered ring is restricted to the same conformational family (F), indicating that this arrangement of phenyl ring is also possible.
The MD simulation of 2 (Figure 6B) showed only the cluster of conformers between 3T4 and 5E (φ = 90-144°) indicating that the benzyl substituent increases the barrier for interconversion between conformational states. Interestingly, the intramolecular S---H–Ar interaction between the thiomethyl moiety and the phenyl ring was not stable and after 200 ps the distance between the phenyl and sulfur was already 7 Å. Although the removal of the thiomethyl group of compound 3 decreased the interconversion barrier as evident from the frequent interconversions between the two clusters of conformers (Figure 6C), it populated the same conformational clusters as 1.
In order to validate the computational results, proton-proton J-coupling constants for compounds 1-3 were calculated over the entire trajectory using a Karplus type equation48 and the resulting values compared with experimental data from NMR solution experiments (Table 2). Gratifyingly, a good agreement was obtained between the computed and experimental J-values. The best agreements were obtained for compound 3, which is aided by a more effective statistical sampling of the two conformational states in 3.
Table 2.
Computed and experimental homonuclear J-coupling constants (in Hz) for compounds 1-3. The coupling constants were calculated using a karplus type equation proposed by Haasnoot et.al. (reference 48) over the entire 5-ns trajectory from MD simulations of ligands in a box of TIP3P water molecules. The values in the parentheses indicate the experimental values.
| 1 | 2 | 3 | ||
|---|---|---|---|---|
| JHb-H1 | 1.5 (1.5) | |||
| JH1-H2 | 6.2 (7.5) | 5.0 (6.3) | JHa-H1 | 7.9 (7.6) |
| JH2-H3 | 6.3 (4.8) | 6.2 (5.1) | JH1-H2 | 7.5 (6.2) |
| JH3-H4 | 4.8 (4.1) | 4.8 (4.3) | JH2-H3 | 5.9 (5.6) |
| JH4-H5 | 5.1 (6.3) | 5.0 (6.1) | JH3-H4 | 5.2 (5.6) |
| JH5-H1 | 7.0 (7.0) | 8.0 (7.9) | JH4-Ha | 7.6 (7.6) |
| JH4-Hb | 6.0 (5.7) | |||
MD Simulations of Protein-bound Ligands
While the X-ray crystal structures of 1 and 2 complexed with dGMII provided a great deal of insight into the binding modes of the inhibitors, MD simulations offer additional information about the dynamic properties of the complexes. These simulations also allow an unambiguous analysis of hydrogen bonding patterns and occupancies and make it possible to determine the dynamic nature of H-π type, sulfur-π type hydrogen bonds and π-π type stacking interactions.
MD simulations of the complexes of 1 and 2 with dGMII only provided stable complexes when started with inhibitors in which the amino groups were protonated. This observation supports the notion that protonation of an amine of a glycosidase inhibitor is crucial for binding.49 Furthermore, this behavior is in agreement with previously published SAR studies indicating the necessity of the amino group for inhibitory activity of the compounds.20-26 During the entire simulations no significant changes were observed in the protein backbone compared to the X-ray structure suggesting that MD simulations did not induce any conformational changes in the backbone. In addition to this, during the entire 10-ns simulation the cyclopentane rings of compounds 1 and 2 exhibit little flexibility as only one a small region of the pseudorotation wheel was occupied (E1, 2T1, 2E: φ = 180-216°, Figure 6D and 6E). This conformational family is different than the conformational states sampled in the simulations of ligands in explicit water, indicating that the cyclopentane rings do not revert to their low energy conformation.
Analysis of hydrogen bonding patterns and occupancies provides information about which interactions are the most important ones for complexation. Typically, the strongest hydrogen bonds have the highest occupancies, the smallest standard deviations and the shortest heavy atom separations. For compound 1, very strong hydrogen bonds were observed between the 2-hydroxyl oxygen and Asp472 OD1-OD2, 3-hydroxyl oxygen and Asp472 OD2 and 4-hydroxyl oxygen and Asp341 OD1 (Table 3). Moderately strong hydrogen bonds were assigned between 4-hydroxyl oxygen and Tyr269 OH, the amine nitrogen and Asp204 OD1-OD2, and Tyr269 OH and Asp341 OD1-OD2. The 2-hydroxyl oxygen also accepted a proton from Tyr727 to form a moderate hydrogen bond. In addition, the methyl group in the thiomethyl ether remained in cf2 conformation during the entire 10-ns period. The interactions between the thiomethyl moiety and the aromatic rings of Phe206 and Tyr727 were observed during the entire simulation further supporting the notion that these are important for ligand binding.
Table 3.
Hydrogen bonding interactions between 1 and 2, and dGMII over the 10-ns trajectories, computed using the carnal module in AMBER 7.0. The first value is the average distance between the specified atoms in Å, the values in parentheses indicate the percent occupancies.
| Donor atom | Acceptor atom | 1 | 2 | 2a |
|---|---|---|---|---|
| R228NH2 | S | 3.8 (5) | 3.9 (0.1) | 3.8 (32) |
| Y727OH | O-2 | 3.0 (40) | 3.1 (74) | 3.2 (51) |
| O-2 | D472OD1 | 3.2 (100) | 3.0 (100) | 3.0 (98) |
| O-2 | D472OD2 | 2.6 (100) | 2.9 (100) | 2.9 (100) |
| O-3 | D472OD1 | 3.9 (0.6) | 3.8 (29) | 3.9 (14) |
| O-3 | D472OD2 | 2.5 (100) | 2.5 (100) | 2.5 (100) |
| O-4 | Y269OH | 3.7 (54) | 3.8 (0.2) | 3.8 (1.5) |
| O-4 | D341OD1 | 2.5 (100) | 3.6 (90) | 3.6 (93) |
| O-4 | D341OD2 | 3.8 (7) | 2.6 (100) | 2.6 (100) |
| N | D204OD1 | 3.6 (33) | 3.6 (97) | 3.6 (83) |
| N | D204OD2 | 3.2 (49) | 3.4 (97) | 3.4 (81) |
| N | Y269OH | 2.9 (52) | 2.9 (91) | 3.0 (96) |
| N | D341OD1 | 3.0 (43) | 3.1 (100) | 3.4 (99) |
| N | D341OD2 | 3.5 (43) | 3.4 (99) | 3.3 (99) |
The properties of the hydrogen bonds of the complex of 2 with dGMII were very similar compared to those of 1. Furthermore, the intramolecular hydrogen bond between the thiomethyl moiety and phenyl ring was maintained throughout the simulation. In order to investigate in more detail the importance of the intramolecular S---H–Ar interaction of 2 in the complex with dGMII, a MD simulation was performed whereby the phenyl ring of 2 was stacked against Tyr269. Interestingly, the π-π stacking interaction was found to be stable (data not shown) with average R1 = 3.3 Å and R2 = 2.1 Å, where R1 is the vertical and R2 is the horizontal distance between the centroids of the phenyl rings. Theoretical study on benzene dimer indicates that this geometrical arrangement of the aromatic rings results in the stabilization energy of up to −1.5 kcal mol−1.50 Furthermore, the conformation of the cyclopentane ring and hydrogen bonding and other interactions were unchanged. This result is in agreement with our previous modeling studies,44 even though this interaction is not observed in the crystal structure. This discrepancy between the crystal structure and the modeling study may be a result of the fact that the docking program used does not explicitly consider conformational entropy.
Overlay studies
The mode of inhibition of GMII by azasugars such as swainsonine has been rationalized by their resemblance to the mannosyl oxacarbenium ion, a putative intermediate in the hydrolysis of glycosides. However, extension of this model to the aminocyclopentitols, such as mannostatin, has been a subject of debate.20,51-53 Previous attempts to predict the precise binding mode of mannostatin A and its analogs resulted in multiple binding models that differed in the conformation of the five-membered ring of these inhibitors.44 The X-ray structures of dGMII complexed with inhibitors 1 and 2 offers a unique opportunity to compare the three dimensional arrangement of the functional groups of 1 and 2 relative to that of the mannosyl oxacarbenium ion.
Previously, it has been reported that the mannosyl oxacarbenium ion can adopt at least two conformations in which C-4 is either above (flap-up) or below (flap-down) the plane of the C-2, C-1, O-5 and C-5 atoms.52,53 Optimized geometries of both the conformers were obtained by quantum mechanical calculations at the DFT/B3LYP/6-31G* level of theory. It was found that the flap-up conformer was more stable than flap-down conformer by 4.4 kcal mol−1, which is in agreement with previous calculations.52 Subsequently, the two conformers of the oxacarbenium ion were superimposed on the conformation of mannostatin A observed in the X-ray crystal structure. The zinc coordinating C-3, C-4 cis-diol of 1 mimics the C-3, C-2 cis-hydroxyl of the oxacarbenium ion and therefore the O-3, C-3, C-4 and O-4 of 1 were superimposed onto O-3, C-3, C-2 and O-2 of the oxacarbenium ion conformers (Figure 7).
Figure 7.
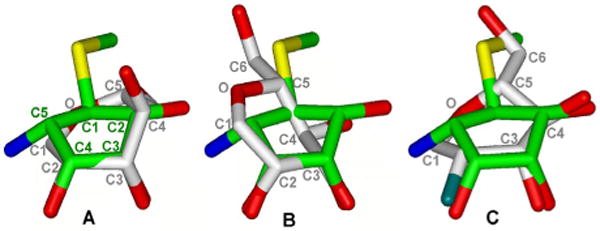
Superimposition of Mannostatin A (1) in bound conformation onto the flap-up (A) and flap-down (B) oxacarbenium ion, and covalent intermediate (C, PDB ID 1QX1). Mannostatin A mimics best the covalent intermediate.
In the case of the flap-up conformer, the cyclopentane ring of compound 1 seems to mimic the sugar ring with C-2, C-3 and C-4 carbon atoms lying very close to C-4, C-3 and C-2 carbon atoms in the sugar ring and the C-1 carbon is positioned very close to the ring oxygen. The zinc-coordinating 3- and 4-oxygen atoms in 1 display a nearly perfect overlap with corresponding hydroxyl oxygens with RMS deviation of 0.16 Å. The C-5 carbon is located above the anomeric carbon of the oxacarbenium ion placing the amino nitrogen in the region of the exo-cyclic anomeric oxygen of the substrate. The protonated form of this amine at physiological pH raises the possibility that in this arrangement the amine nitrogen mimics the positive charge on the exo-cyclic anomeric oxygen, which develops during the formation of the oxacarbenium ion. In the case of the inhibitor, swainsonine, the amine nitrogen is positioned very close to the ring oxygen suggesting that it probably mimics the positive charge on the ring oxygen in the transition state. The C-2 hydroxyl and thiomethyl moiety of 1 do not seem to mimic any functionality of the oxacarbenium ion. However, these moieties are involved in multiple interactions with protein residues enhancing the affinity of 1 for dGMII.
Attempts to superimpose 1 with the flap-down conformer were less successful. The C-3 and C-4 carbon atoms lie near the C-3 and C-2 atoms in the oxacarbenium ion, respectively, while all the other carbons are at a considerable distance. Moreover, the 3- and 4-hydroxyl groups display poorer overlap with the corresponding hydroxyl groups in the oxacarbenium ion with greater RMS deviation of 0.33 Å, compared to 0.16 Å in the flap-up conformer. The amine group now points away from the β-face of the oxacarbenium ion. It has been proposed that β-mannanase catalyzed hydrolysis proceeds through a transition state in which the oxacarbenium ion adopts a B2,5 conformation.54 Therefore, the geometries of the mannosyl oxacarbenium ion in the B2,5 and 2,5B conformation were optimized and the resulting structures superimposed on the bound conformation of 1. In each case, only the C-3, C-4 diol of 1 coordinating with Zn overlayed well with the C-2, C-3 diol of the mannosyl oxacarbenium ions (results not shown) demonstrating that 1 does not mimic a TS in the boat conformation.
Interestingly, the overlay of 1 with a covalently trapped fluorinated sugar analog,16 which mimics the covalently linked reaction intermediate and is found in a distorted 1S5 skew boat conformation, shows a good correspondence with all three hydroxyls of 1. Furthermore, in this overlay the sulfur is in a close proximity to the C-6 hydroxyl of the covalently linked sugar analog (Figure 7C). The thiomethyl group of 1 makes, however, additional polar and non-polar interactions with protein residues compared to the equivalent C-6 hydroxyl, thereby enhancing the affinity of compound 1 for dGMII. The amine is in close proximity to C-1 of the covalently linked intermediate and therefore is ideally positioned to interact with the catalytic nucleophile (Asp204).
Thus, the overlay studies indicate that mannostatin mimics best the enzyme-linked mannosyl intermediate, which has been proposed to adopt a 1S5 skew boat conformation. This conformation has been implicated in the reaction mechanism of α- as well as β-retaining glycosidases. For example, the three-dimensional structure of a β-mannanase from Pseudomonas cellulosa (endo β-retaining mannosidase) was determined at various points along its reaction pathway.54 Interestingly, the conformation of the Michaelis complex of the β-mannanase was identical to that of the fluorinated sugar analog linked to dGMII.
The five-membered ring of swainsonine is believed to resemble the flattened six-member ring of the mannosyl oxacarbenium ion. However, superimpositions of swainsonine onto the two mannosyl oxacarbenium ion conformers and the enzyme trapped intermediate showed that the best overlay is obtained with the latter intermediate (overlays shown in the supplementary material). It is generally believed that potent glycosidase inhibitors mimic an oxacarbenium-ion like transition state. The results of our studies highlight that they may also mimic reaction intermediates such as the Michaelis complex or enzyme-linked intermediate.
In his superimposition study,53 Winkler reported that Mannostatin A mimics the flap-up conformer with the functional groups present in mannostatin A mimicking different hydroxyl groups in the oxacarbenium ion. Winkler used the X-ray structure of Mannostatin A tetraacetate for these superimposition studies.55 It must be noted that conformation of the five-membered ring in Mannostatin B tetraacetate is 3T4 (φ = 93.5°), which is different than the bound conformation of the cyclopentane ring (2T1) in both the X-ray structures reported here. Moreover, 2- and 3-hydroxyl groups in mannostatin A were superimposed onto the zinc-coordinating 3- and 2-hydroxyl groups in the oxacarbenium ion, respectively. However, from the X-ray structure of 1-dGMII complex, it is revealed that 3- and 4-hydroxyl groups in 1 coordinate with the Zn2+ ion and thus mimic the zinc-coordinating cis-diol in the actual substrate. The important role of the zinc ion in the catalytic mechanism of dGMII has recently been discussed.32
Conclusions
Glycosidases are enzymes that play crucial roles in the biosynthesis of glycoproteins. Inhibitors of these enzymes have garnered much attention as lead compounds for drug discovery for diseases such as viral- and bacterial infections, diabetes, Gauchers disease and cancer. In many cases, the mode of inhibition by these compounds is not well understood, complicating efforts to design and synthesize more potent- and/or selective inhibitors. As a result, only a very small number of glycosidase inhibitors have been successfully developed as therapeutics.
The studies reported here indicate that Mannostatin A mimics the covalently linked mannosyl intermediate, which has been shown to adopt a 1S5 skew boat conformation. In particular, the zinc-coordinating 3- and 4-hydroxyls of 1 display a good overlap with the corresponding cis-diol of the mannosyl residue. Furthermore, the amine nitrogen of 1 is positioned close to the C-1 carbon of enzyme-linked mannosyl intermediate and therefore can interact with the carboxylic acid of the catalytic nucleophile Asp204. The thiomethyl group of mannostatin A, which is required for high affinity binding, showed a good overlay with the C-6 hydroxyl of the covalently linked intermediate. This functionality is, however, able to make a number of additional interactions increasing the affinity for dGMII. The thiomethyl moiety of 1 is structurally similar to the side chain of methionine residues. Interestingly, the environment surrounding the thiomethyl group of 1 is remarkably similar to the arrangements around methionine residues in the protein structure. Another important finding is that the methyl carbon of the thiomethyl moiety of 1 in the complex with GMII adopts two different conformations. In both conformations, the sulfur interacts with the backbone oxygen of Arg876 through the π-orbital on the carbonyl oxygen and the antibonding σ* orbital of the S-C bond. In cf1 conformation, the polarizable methyl group is in a hydrophilic environment interacting with three water molecules and the π-system of the Arg228 side chain. Whereas, the methyl group of the cf2 conformation is in a hydrophobic environment where it forms C–H---π type interactions with the phenyl rings of Phe206 and Tyr727.
The MD simulations and NMR studies of the uncomplexed inhibitor showed that the five-membered ring can adopt two different conformational families. In the bound state, however, it is restricted to a single conformation, which is different from the conformations observed for the free ligand. This finding is surprising because it is generally believed that potent inhibitors are complexed in their ground state conformations. Probably, the flexibility of five membered rings renders the energy barrier for the conformational change relatively low.
Compound 2 was designed in such a way that its benzyl moiety can interact with aromatic residues of the binding site of the enzyme thereby increasing its affinity. However, it was found that this compound has a slightly reduced affinity indicating that the benzyl group cannot make favorable interactions with the enzyme. The unexpected finding could be rationalized by careful examination of the crystal structure of dGMII with 2. Thus, the five-membered ring of compounds 1 and 2 were identically positioned in the binding site of dGMII. The distance between the centroid of the aromatic ring of compound 2 and Tyr269 was such that no edge to face binding interactions could be made. The unexpected orientation of the aromatic ring of 2 could be rationalized by an intramolecular S---H–Ar type hydrogen bond between the thiomethyl moiety and the phenyl ring.
Experimental Procedures
Compounds 1-3 were prepared by reported procedures.20,22,27 Inhibition constants were determined as detailed in Li et al.44
Crystallization, Data Collection and Structure Refinement
Measurement of inhibition, crystallization, data collection and structural refinement were carried out essentially as outlined by Kuntz et al31 with the exceptions noted below. Inhibitors were dissolved at 100 mM in water (1) or methanol (2) Crystals of dGM2 were grown up overnight, washed with phosphate buffered reservoir solution and soaked with 10 mM 1 for at least 3-6 hrs. 2 was co-crystallized with dGMII in Tris-buffered reservoir solution without phosphate washing.56 Crystal quality was assessed on our home source and data was collected on Beamline F1 at the Cornell High Energy Synchrotron Source. Approximately 400 frames with 0.5 degree oscillation/frame were collected for each dataset. Each of the structures was refined independently. The last stage involved addition of alternate conformations and low quality waters followed by energy minimization and individual B-factor refinement. The quality of the final model was assessed using a number of structure validation programs including MolProbity, WhatIf, and hetze.
Molecular Dynamics Simulations
The sander57 module of AMBER 7.058 in conjunction with PARM99 parameter set for proteins, was utilized in molecular dynamics simulations. A modified GLYCAM59 parameter set for glycosides and glycoproteins was used for all three ligands. The X-ray crystal structures were protonated using the leap module in AMBER and were solvated by a 25 Å droplet of TIP3P60 waters around the Zn+2 ion. Initially the solvent positions were optimized by energy minimization followed by energy minimization of whole system. This was followed by MD simulations at 300 K for 10-ns. In each simulation the system was heated gradually to 300 K in 100-ps and then held at 300 K for the entire simulation. For all simulations, protein residues within a sphere of 10 Å (with Zn+2 ion as the center of the sphere), all waters and ligands were allowed to move. All other atoms were frozen to their positions in the crystal structures.
Simulations of free ligands were performed for 5-ns, under periodic boundary conditions at constant pressure. These simulations followed similar protocols for energy minimizations as used for the simulations of protein-bound ligands. The initial conformations of ligands in these simulations were kept same as observed in the crystal structure. In order to determine hydrogen bond occupancies, hydrogen bonding interactions were assumed to be present if the participating heavy atoms were 4 Å apart and the angle between the heavy atoms and the donating hydrogen was 60°. All MD simulations used an integration time step of 2 fs and scaling of 1-4 electrostatic and van der Waal interactions by standard values of 1/1.2 and 1/2.0, restraint of all hydrogen-containing bonds through the SHAKE algorithm57 and a cutoff of 8-Å for all non-bonded interactions. The MD trajectories were analyzed using the carnal module in AMBER 7.0. The phase angles of puckering were computed using Cremer and Pople's parameters.61
Geometry optimization of flap-up and flap-down conformers of mannosyl oxacarbenium ion and superimpositon
All the geometry optimizations were performed with Gaussian 98 program62 using density functional theory (B3LYP63-65) and 6-31G* basis set.66 The optimized conformers of flap-up and flap-down oxocarbenium ions were determined by constraining the dihedral angle made by C-2, C-1, O-5 and C-5 to 0° and allowing all other parameters to optimize at BLYP/6-31G* level of theory. The hydroxyl groups of the resulting structures were placed in three different orientations representing three rotamers and the conformers were reoptimized without any restraints at B3LYP/6-31G* level. For flap-up conformer two- and for flap-down conformer three unique conformers were identified with different orientations of hydroxy oxygen atoms. For each conformer, the C-2, C-1, O-5 and C-5 were in one plane with a distance between C1 and endocyclic oxygen of 1.27 Å, indicating oxacarbenium-like character. The lowest energy conformation of flap-up and flap-down conformers was used for superimposition with inhibitors 1 and 2. The covalent intermediate geometry was obtained from PDB 1QX1. The superimpositions were performed using Insight II 2000.
Supplementary Material
Protein coordinates and structure factors are available from the Protein Data Bank (www.rcsb.org). The PDB IDs are 2F7O (Mannostatin A) and 2F7P (benzylated Mannostatin A). The statistics for data collection and refinement, plots of MD trajectory analysis and figure of overlays of swainsonine with the oxacarbenium ions and the covalent intermediate are available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
The authors wish to thanks the staff of CHESS for data collection time and support. The authors are also grateful for financial support for this work by NCI of NIH (5UO1CA91295) and the Canadian Institutes of Health Research.
References
- 1.Asano N, Nash RJ, Molyneux RJ, Fleet GWJ. Tetrahedron-Asymmetry. 2000;11:1645–1680. [Google Scholar]
- 2.Berecibar A, Grandjean C, Siriwardena A. Chem Rev. 1999;99:779–844. doi: 10.1021/cr980033l. [DOI] [PubMed] [Google Scholar]
- 3.Lillelund VH, Jensen HH, Liang XF, Bols M. Chem Rev. 2002;102:515–553. doi: 10.1021/cr000433k. [DOI] [PubMed] [Google Scholar]
- 4.vonItzstein M, Thomson RJ. Curr Med Chem. 1997;4:185–210. [Google Scholar]
- 5.Yuasa H, Saotome C, Kanie O. Trends Glycosci Glycotechnol. 2002;14:231–251. [Google Scholar]
- 6.Dennis JW, Granovsky M, Warren CE. Biochim Biophys Acta. 1999;1473:21–34. doi: 10.1016/s0304-4165(99)00167-1. [DOI] [PubMed] [Google Scholar]
- 7.Dennis JW, Koch K, Yousefi S, Vanderelst I. Cancer Res. 1990;50:1867–1872. [PubMed] [Google Scholar]
- 8.Elbein AD. FASEB J. 1991;5:3055–3063. doi: 10.1096/fasebj.5.15.1743438. [DOI] [PubMed] [Google Scholar]
- 9.Goss PE, Baker MA, Carver JP, Dennis JW. Clin Cancer Res. 1995;1:935–944. [PubMed] [Google Scholar]
- 10.Tulsiani DRP, Harris TM, Touster O. J Biol Chem. 1982;257:7936–7939. [PubMed] [Google Scholar]
- 11.Vasella A, Davies GJ, Bohm M. Curr Opin Chem Biol. 2002;6:619–629. doi: 10.1016/s1367-5931(02)00380-0. [DOI] [PubMed] [Google Scholar]
- 12.Heightman TD, Vasella AT. Angew Chem, Int Ed. 1999;38:750–770. doi: 10.1002/(SICI)1521-3773(19990315)38:6<750::AID-ANIE750>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 13.Zechel DL, Withers SG. Acc Chem Res. 2000;33:11–18. doi: 10.1021/ar970172+. [DOI] [PubMed] [Google Scholar]
- 14.Zechel DL, Withers SG. Curr Opin Chem Biol. 2001;5:643–649. doi: 10.1016/s1367-5931(01)00260-5. [DOI] [PubMed] [Google Scholar]
- 15.McCarter JD, Withers SG. Curr Opin Struct Biol. 1994;4:885–892. doi: 10.1016/0959-440x(94)90271-2. [DOI] [PubMed] [Google Scholar]
- 16.Numao S, Kuntz DA, Withers SG, Rose DR. J Biol Chem. 2003;278:48074–48083. doi: 10.1074/jbc.M309249200. [DOI] [PubMed] [Google Scholar]
- 17.Deslongchamps P. Stereoelectronic effects in organic chemistry. Vol. 1 Pergamon Press; Oxford: 1983. [Google Scholar]
- 18.van den Elsen JMH, Kuntz DA, Rose DR. EMBO J. 2001;20:3008–3017. doi: 10.1093/emboj/20.12.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aoyagi T, Yamamoto T, Kojiri K, Morishima H, Nagai M, Hamada M, Takeuchi T, Umezawa H. J Antibiot. 1989;42:883–889. doi: 10.7164/antibiotics.42.883. [DOI] [PubMed] [Google Scholar]
- 20.King SB, Ganem B. J Am Chem Soc. 1994;116:562–570. [Google Scholar]
- 21.Ogawa S, Morikawa T. Bioorg Med Chem Lett. 1999;9:1499–1504. doi: 10.1016/s0960-894x(99)00224-3. [DOI] [PubMed] [Google Scholar]
- 22.Ogawa S, Morikawa T. Bioorg Med Chem Lett. 2000;10:1047–1050. doi: 10.1016/s0960-894x(00)00162-1. [DOI] [PubMed] [Google Scholar]
- 23.Kleban M, Hilgers P, Greul JN, Kugler RD, Li J, Picasso S, Vogel P, Jager V. Chembiochem. 2001;2:365–368. doi: 10.1002/1439-7633(20010504)2:5<365::AID-CBIC365>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 24.Boss O, Leroy E, Blaser A, Reymond JL. Org Lett. 2000;2:151–154. doi: 10.1021/ol991252b. [DOI] [PubMed] [Google Scholar]
- 25.Popowycz F, Gerber-Lemaire S, Demange R, Rodriguez-Garcia E, Asenjo ATC, Robina I, Vogel P. Bioorg Med Chem Lett. 2001;11:2489–2493. doi: 10.1016/s0960-894x(01)00477-2. [DOI] [PubMed] [Google Scholar]
- 26.Ogawa S, Washida K. Eur J Org Chem. 1998:1929–1934. [Google Scholar]
- 27.Uchida C, Kimura H, Ogawa S. Bioorg Med Chem. 1997;5:921–939. doi: 10.1016/s0968-0896(97)00030-8. [DOI] [PubMed] [Google Scholar]
- 28.Boraston AB, Nurizzo D, Notenboom V, Ducros V, Rose DR, Kilburn DG, Davies GJ. J Mol Biol. 2002;319:1143–1156. doi: 10.1016/S0022-2836(02)00374-1. [DOI] [PubMed] [Google Scholar]
- 29.Chavez MI, et al. Chem Eur J. 2005;11:7060–7074. doi: 10.1002/chem.200500367. [DOI] [PubMed] [Google Scholar]
- 30.Fernandez MD, Canada FJ, Jimenez-Barbero J, Cuevas G. J Am Chem Soc. 2005;127:7379–7386. doi: 10.1021/ja051020+. [DOI] [PubMed] [Google Scholar]
- 31.Kuntz DA, Ghavami A, Johnston BD, Pinto BM, Rose DR. Tetrahedron-Asymmetry. 2005;16:25–32. [Google Scholar]
- 32.Kuntz DA, Liu H, Bols M, Rose DR. Biocatalysis Biotransform. 2006;24:1–7. [Google Scholar]
- 33.Reid KSC, Lindley PF, Thornton JM. Febs Letters. 1985;190:209–213. [Google Scholar]
- 34.Pal D, Chakrabarti P. J Biomol Struct Dyn. 1998;15:1059–1072. doi: 10.1080/07391102.1998.10509001. [DOI] [PubMed] [Google Scholar]
- 35.Pal D, Chakrabarti P. J Biomol Struct Dyn. 2001;19:115–128. doi: 10.1080/07391102.2001.10506725. [DOI] [PubMed] [Google Scholar]
- 36.Samanta U, Pal D, Chakrabarti P. Proteins. 2000;38:288–300. [PubMed] [Google Scholar]
- 37.Iwaoka M, Takemoto S, Okada M, Tomoda S. Bull Chem Soc Jpn. 2002;75:1611–1625. [Google Scholar]
- 38.Ippolito JA, Alexander RS, Christianson DW. J Mol Biol. 1990;215:457–471. doi: 10.1016/s0022-2836(05)80364-x. [DOI] [PubMed] [Google Scholar]
- 39.Brandl M, Weiss MS, Jabs A, Suhnel J, Hilgenfeld R. J Mol Biol. 2001;307:357–377. doi: 10.1006/jmbi.2000.4473. [DOI] [PubMed] [Google Scholar]
- 40.Tatko CD, Waters ML. Protein Sci. 2004;13:2515–2522. doi: 10.1110/ps.04820104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alber F, Kuonen O, Scapozza L, Folkers G, Carloni P. Proteins. 1998;31:453–459. doi: 10.1002/(sici)1097-0134(19980601)31:4<453::aid-prot11>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 42.Pranata J. Bioorg Chem. 1997;25:213–219. [Google Scholar]
- 43.Cheney BV, Schulz MW, Cheney J. Biochim Biophys Acta. 1989;996:116–124. [Google Scholar]
- 44.Li B, Kawatkar SP, George S, Strachan H, Woods RJ, Siriwardena A, Moremen KW, Boons GJ. Chembiochem. 2004;5:1220–1227. doi: 10.1002/cbic.200300842. [DOI] [PubMed] [Google Scholar]
- 45.Duan GL, Smith VH, Weaver DF. Mol Phys. 2001;99:1689–1699. [Google Scholar]
- 46.Kilpatrick JE, Pitzer KS, Spitzer R. J Am Chem Soc. 1947;69:2483–2488. doi: 10.1021/ja01202a070. [DOI] [PubMed] [Google Scholar]
- 47.Pitzer KS, Donath WE. J Am Chem Soc. 1959;81:3213–3218. [Google Scholar]
- 48.Haasnoot CAG, Deleeuw F, Altona C. Tetrahedron. 1980;36:2783–2792. [Google Scholar]
- 49.Varrot A, Tarling CA, Macdonald JM, Stick RV, Zechel DL, Withers SG, Davies GJ. J Am Chem Soc. 2003;125:7496–7497. doi: 10.1021/ja034917k. [DOI] [PubMed] [Google Scholar]
- 50.Sinnokrot MO, Sherrill CD. J Phys Chem A. 2004;108:10200–10207. [Google Scholar]
- 51.Knapp S, Dhar TGM. J Org Chem. 1991;56:4096–4097. [Google Scholar]
- 52.Winkler DA, Holan G. J Med Chem. 1989;32:2084–2089. doi: 10.1021/jm00129a011. [DOI] [PubMed] [Google Scholar]
- 53.Winkler DA. J Med Chem. 1996;39:4332–4334. doi: 10.1021/jm960237z. [DOI] [PubMed] [Google Scholar]
- 54.Ducros VMA, Zechel DL, Murshudov GN, Gilbert HJ, Szabo L, Stoll D, Withers SG, Davies GJ. Angew Chem Int Ed. 2002;41:2824–2827. doi: 10.1002/1521-3773(20020802)41:15<2824::AID-ANIE2824>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 55.Morishima H, Kojiri K, Yamamoto T, Aoyagi T, Nakamura H, Iitaka Y. J Antibiot. 1989;42:1008–1011. doi: 10.7164/antibiotics.42.1008. [DOI] [PubMed] [Google Scholar]
- 56.Shah N, Kuntz DA, Rose DR. Biochemistry. 2003;42:13812–13816. doi: 10.1021/bi034742r. [DOI] [PubMed] [Google Scholar]
- 57.Ryckaert JP, Ciccotti G, Berendsen HJC. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 58.Case DA, et al. Amber 7.0. University of California; San Francisco, CA: 2002. [Google Scholar]
- 59.Woods RJ, Dwek RA, Edge CJ, Fraserreid B. J Phys Chem. 1995;99:3832–3846. [Google Scholar]
- 60.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J Phys Chem. 1983;79:926–935. [Google Scholar]
- 61.Cremer D, Pople JA. J Am Chem Soc. 1975;97:1354–1358. [Google Scholar]
- 62.Frisch MJ, et al. Gaussian 98. Revision A.11.3. Gaussian, Inc.; Pittsburgh, PA: 2002. [Google Scholar]
- 63.Becke AD. Phys Rev A. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 64.Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 65.Lee CT, Yang WT, Parr RG. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 66.Hariharan PC, Pople JA. Theor Chim Acta. 1973;28:213–222. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Protein coordinates and structure factors are available from the Protein Data Bank (www.rcsb.org). The PDB IDs are 2F7O (Mannostatin A) and 2F7P (benzylated Mannostatin A). The statistics for data collection and refinement, plots of MD trajectory analysis and figure of overlays of swainsonine with the oxacarbenium ions and the covalent intermediate are available free of charge via the Internet at http://pubs.acs.org.