

Abstract
The identification of proteins by tandem mass spectrometry relies on knowledge of the products produced by collision-induced dissociation of peptide ions. Most previous work has focused on fragmentation statistics for ion trap systems. We analyzed fragmentation in MALDI TOF/TOF mass spectrometry, collecting statistics using a curated set of 2459 MS/MS spectra, and applying bootstrap resampling to assess confidence intervals. We calculated the frequency of 18 product ion types, the correlation between both mass and intensity with ion type, the dependence of amide bond breakage on the residues surrounding the cleavage site, and the dependence of product ion detection on residues not adjacent to the cleavage site. The most frequently observed were internal ions, followed by y ions. A strong correlation between ion type and the mass and intensity of its peak was observed, with b and y ions producing the most intense and highest-mass peaks. The amino acids P, W, D and R had a strong effect on amide bond cleavage when situated next to the breakage site, whereas residues including I, K and H had a strong effect on product ion observation when located in the peptide but not adjacent to the cleavage site, a novel observation.
Introduction
A cornerstone of protein identification is the analysis of peptides by tandem mass spectrometry (MS/MS)1, which relies on mass analysis of product ions produced by collision-induced dissociation (CID) (Fig. 1). The pattern of product ions produced by CID is analyzed by software to either derive a de novo peptide sequence2–9 or to match the pattern to peptides in a sequence database 10–21. The ions produced by CID are strongly dependent upon the sequence of the peptide being analyzed and the instrument used, producing great variability in the mass and intensity of peaks in a spectrum. This variability in fragmentation presents a challenge for peptide sequence identification algorithms.
Figure 1.
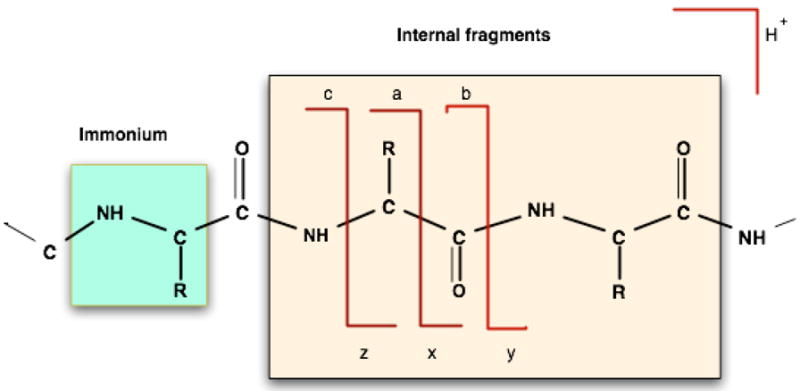
Bond cleavages in MS/MS fragmentation. The direct cleavage of peptide bonds results in b ions (containing the N-terminus of the peptide) and y ions (containing the C-terminus of the peptide). The other backbone bond cleavages result in a, x, c, and z ions. The breakage of two peptide bonds simultaneously gives rise to internal fragments along with b and y ions. Another common type is an immonium ion, comprised of an internal fragment of just one residue, formed by a combination of a-type and y-type cleavages.
Many common peptide identification algorithms rely on hand-coded fragmentation rules, using models that assume simple intensity and mass distributions for each of the ion types. Statistical characterization of fragmentation patterns for a given instrument can be used to improve identification results. Most previous analyses have been performed for fragmentation in an electrospray ionization (ESI) ion trap instrument, such as those of Tabb et al. who examined the correlation of ion type with mass and intensity22, and several groups who reported the effect of specific residues adjacent to the cleavage site on fragmentation23–25. To provide similar information for another commonly used mass spectrometry platform – matrix assisted laser desorption ionization (MALDI) coupled to a dual time of flight analyzer (TOF/TOF) – we performed a comprehensive analysis of fragmentation for a curated set of 2459 spectra from trypsin digested proteins that were separated by either 2D SDS PAGE or liquid chromatography (LC). Our analysis examined: (1) the frequency of 18 different ion types; (2) the correlation of peak mass and intensity to each ion type; (3) the effect of each of the 20 residues when adjacent to the cleavage site in either the N- or C- terminal position; and (4) the effect of intermediate residues on the observation of product ions produced by bond cleavage. To assess the ability of our data set to produce statistically valid results, we applied bootstrap resampling26,27, a commonly used method for obtaining descriptive statistics from limited data sets.
Materials and Methods
Data Set
The data used in this analysis were derived from both our own analysis of E. coli proteins on a MALDI-TOF/TOF, and from an external reference data set generated on the same instrument. Our own set came from 314 E. coli samples, 144 of which were obtained from in-gel trypsin digestion of spots from a 2-dimensional polyacrylamide gel electrophoresis (2D PAGE) separation of the soluble protein fraction, and 170 of which were obtained by tryptic digestion of reversed phase liquid chromatography (RPLC) separated fractions of a ribosome enriched cellular extract. Digested protein samples were suspended in an α-Cyano-4-hydroxycinnamic acid matrix and analyzed by an Applied Biosystems 4700 TOF/TOF using a collisional energy of 1 keV.
For each of our protein samples, the 4700 performed a parent/precursor-ion scan producing a list of peptide masses, from which the 10 most intense peaks having a signal-to-noise (S/N) ratio above 35 were automatically selected for MS/MS fragmentation. As part of an automated analysis pipeline, the resulting MS and MS/MS data were analyzed using Mascot16. This produced peptide matches for about 1,000 MS/MS spectra with their corresponding sequences that had ion match scores above 10 (~33% of the total). To minimize false-positive matches, we further culled the data set, taking advantage of the fact that proteins were separated before digestion and MS/MS analysis, meaning that if a protein is present in sufficient abundance to produce one peptide match, we should observe other peptide matches to the same protein. Therefore, if one of the following conditions held, we included the peptide in the final data set: (1) there were at least two other peptides matching the same protein from the same set of parent ions (i.e. same sample spot/fraction), each with scores above 50, or (2) the total match score for the protein, including the parent ion list (peptide mass fingerprint) and the peptide in question, was above 200. This selection process produced 710 MS/MS spectra for which there were high confidence in the assignment. While curating the list in this way reduced the quantity of spectra, it had the benefit of decreasing the chance that false positive spectrum identifications skewed the statistics collected.
We then processed all MS/MS spectra for our data set using the Data Explorer software, version 4.3, (Applied Biosystems, Foster City, CA) to: (1) determine the monoisotopic peak for each observed ion using the Filter Peak List function; (2) internally smooth the spectra using the boundary conditions determined by the mass/resolution pairs (100/1000, 600/6000), using the spectrum resolution function; and (3) eliminate peaks with an S/N ratio below 5.
The 710 MS/MS spectra we generated and their corresponding peptide sequences are available at: http://bioinfo.med.unc.edu/GDownloads/page6.html. with the file name “KData.zip”. The complete E. coli data set comprising the ~3,000 original spectra generated at UNC have been deposited with ProteomeCommons.org, and can be accessed using the hash 741378e339eaf3d99bl90ccl3069fl78 or at https://dfs.proteomecommons.org/data/741378e339eaf3d99b190cc13069f178.
The externally-obtained data set was comprised of 1749 MS/MS spectra obtained on an Applied Biosystems 4700 TOF/TOF from over 300 known, purified proteins, published on Proteome Commons by Strahler et al http://www.proteomecommons.org. While acquired on the same instrument as our E. coli data, for these data the 29 most intense MS ions from each precursor scan were selected for MS/MS fragmentation. This led to more MS/MS spectra derived from low-abundance precursors than for the data set collected on our instrument. Further details of protein purification and spectrum generation are provided at http://www.proteomecommons.org/archive/1117680671827/index.html.
We combined the two data sets to produce a total of 2,459 MS/MS mass lists along with their corresponding peptide sequences. To reduce the effect of small noise peaks on the analysis, we filtered all MS/MS mass lists to retain only the 70 most intense peaks from the spectrum for analysis.
Data Analysis – The StatPackage Program
We developed a program, StatPackage, to assign the peaks in an MS/MS spectrum to a specific ion type when the corresponding peptide sequence is known, as is the case for our data. The program calculates the ion masses that might be produced by a peptide sequence S comprised of an amino acid sequence a1, a2, a3,… of length n. For each possible subsequence of S containing the N-terminus of the peptide (a, b, and c ions) and ending at position k (k runs from 1 to n−1), it calculates ion masses as:
| (1) |
where m(ai) is the mass of the residue at position i, H is the mass of hydrogen, and Δoffset is the mass offset for a particular ion series type. For b ions, Δoffset= 0, for a ions Δoffset = −21.995, and for c ions Δoffset = 17.0265.
For subsequences starting at position j (j=2,…, n) and ending with the C terminus (x, y, and z ions) it calculates ion masses as:
| (2) |
where O is the mass of oxygen. For y ions, Δoffset= 2.0156, for x-ions Δoffset= 27.995, and for z ions Δoffset= −12.995. Internal fragment ion masses (due to double amide bond breakage or secondary fragmentation of b or y-type ions) starting at position j (j =2,…,n−2) and ending at position h (h=3,…, n−1) are calculated by
| (3) |
For each of the ion types listed, neutral loss products are also calculated, by subtracting the masses of ammonia or water from the calculated ion mass according to equations 1–3. Immonium ions are calculated by subtracting 26.99 Da from each individual residue mass present in the peptide.
The theoretical mass for each ion produced by the sequence is compared to the mass list from the MS/MS spectrum, and if it is within 0.2 Da tolerance of an observed mass, the program assigns the peak to that corresponding ion type. Peaks are labeled ‘unassigned’ if they remain unmatched after this process is completed for all residues in the peptide. Though only rarely occurring, when the calculated masses of more than one theoretical ion produced by the peptide sequence are within 0.2 Da and correspond to the same experimentally observed mass, we use the following order of preference for choosing the matching one, based on the reported frequency of occurrence in past studies: y, b, y*, y°, b*, b°, a, a*, a°, immonium, internal, internal*, internalo, precursor*, and precursoro.
To calculate the intensity distribution of the 18 considered ion types, the program divides the relative intensity range into ten bins for each of the observed ion types, and calculates the frequency of each ion type in each of these bins. It does the same to calculate the mass distribution of ion types, dividing the relative mass range from 0 Da to the mass of the precursor ion into ten bins, then calculating the frequency of each of the 18 types in each bin for all spectra.
The program also calculates the effect of each of the 20 common amino acids on CID fragmentation. It calculates the effect that each residue has on bond breakage and product ion observation when adjacent to the cleavage site, positioned either on the N- or C-terminal side. To calculate frequency of cleavage fc for each amino acid A, on the side corresponding to the terminus T (one of {N, C}), it counts the number of times an ion is observed corresponding to cleavage for that amino acid ( ) then divides by the total number of times that residue is encountered in all sequences (NA):
| (4) |
We also calculated the effect of each type of amino acid on the directionality of fragmentation using a method that compares the intensity of the resultant product ion peaks with the residue type, as was done previously by Tabb et al.22 For each residue located in a peptide, the program finds in the spectrum the product ion peaks produced by fragmentation to both that residue’s C-terminal side and its N-terminal side. To calculate the N-bias, the program subtracts the intensity of the C-terminal peak from the N-terminal peak for a given product ion type (e.g. b ion or y ion), and divides by the sum of the two for normalization. For example, to calculate the N-bias for the amino acid alanine for b-type ions we have:
| (5) |
where is the intensity of the peak for a b ion produced by cleavage to the N-terminal side of an alanine occurring in a peptide, and is the intensity of the peak for the b ion produced by cleavage to the C-terminal side of the same alanine. If either the C-terminal or N-terminal fragment ion peaks can not be found in the spectrum for particular residue, then the N-bias for that amino acid is set to 1 or −1, respectively. The program then averages the results across all occurrences of each amino acid in the data set, and produces confidence intervals for the values using bootstrap resampling as described below.
Similarly, to calculate the effect each amino acid type on the fragmentation and/or observation of product ions containing them when not adjacent to the site of bond breakage, the program calculates the number of times each amino acid A is found present internally to a matched product ion ( ) of either b or y type (i.e. T equal to N or C, respectively), and it divides by the total number of times that amino acid occurs overall (NA):
| (5) |
Because the amino acid composition may play somewhat different roles in the observation of various ion types, for simplicity the calculation for the effect of internal amino acids on daughter ion observation is only performed for the predominant b-type and y-type ions.
Bootstrap Resampling
The program implements bootstrap resampling to assign confidence intervals to each result reported. Bootstrap resampling derives descriptive statistics from a limited sample set by repeatedly sub-sampling the original data26, 27. The program implements bootstrap resampling by random selection of subsets of the data with replacement, of specified size n, for the specified number of iterations m, then recalculating the statistics and reporting them for each of the iterations. Sub-sampling with replacement means that each spectrum from the original set might be selected more than once in the sub-sampled set.
We ran the program to use sub-sampled data sets of size n=2000, and repeating for m=20 iterations for all of the calculations reported herein. For each statistic, we then used the bootstrap resampling output to calculate the standard deviation and the 95% confidence intervals.
Program Availability
This command-line program is written in Objective-C under an open-source license, and is available with pre-compiled binaries for Windows, Mac OS X, and Linux. It inputs a list of files in peak list (.pkl) format along with a corresponding list of matching amino acid sequences, then calculates the described statistics and reports them to standard output. For bootstrap resampling, it repeatedly reports the results from sub-sampling the data sets. The program and some sample data are available at http://bioinfo.unc.edu/glabsoftware/StatPackage/index.html.
Results and Discussion
Characterization on the Basis of Ion Type
The frequency of observation for each of the 18 ion types analyzed is shown in Table 1. We were able to assign 79% of the fragment ion peaks to one of these 18 categories, with 21% remaining unassigned. Internal fragment ions (having both a b- and y-type cleavage) comprised 16% of the total ions observed, with y ions at 12% and b ions comprising 9%. These three ion types along with their neutral loss products totaled nearly 60%, indicating that the peptide bond is the most susceptible to cleavage in MALDI TOF/TOF. Immonium ions were also frequent, at 11%. Neutral losses of water or ammonia varied with ion type: 28% of the observed b ions lost water as opposed to about 16% of y ions. Conversely, for y ions, the probability of losing ammonia was higher, with about 38% of y ions losing ammonia compared to only about 18% of b ions.
Table 1.
Frequency of each ion type observed in the data set. The superscript ° represents the neutral loss of water (~18 Da), and * represents the neutral loss of ammonia (~17 Da). The CI’s column shows the 95% confidence intervals as the percentage of the corresponding reported values.
| Ion Type | Frequency | CI’s |
|---|---|---|
| Unassigned | 0.21 | ±8% |
| internal | 0.16 | ±4% |
| y ion | 0.12 | ±5% |
| immonium | 0.11 | ±5% |
| b ion | 0.091 | ±4% |
| internalo | 0.055 | ±8% |
| internal* | 0.05 | ±7% |
| y* | 0.045 | ±10% |
| a ion | 0.042 | ±5% |
| b° | 0.025 | ±10% |
| yo | 0.0201 | ±10% |
| b* | 0.017 | ±11% |
| a* | 0.013 | ±10% |
| a° | 0.0099 | ±10% |
| x ion | 0.0089 | ±14% |
| c ion | 0.0087 | ±15% |
| precursor* | 0.0069 | ±45% |
| z ion | 0.0056 | ±14% |
| precursoro | 0.00015 | ±28% |
Peptide bonds (CO-NH) are the most labile of the backbone bonds, and in low energy CID often used in ion trap systems, b and y ions tend to predominate, with a ions also being produced (loss of carbon monoxide from b ions). Our results indicate that the higher-energy CID used in the MALDI TOF/TOF results in more frequent peptide bond breakage, as measured by the large proportion of internal fragments observed. These may result from continuing absorption of energy by b and y ions after the first bond breakage, producing a secondary fragmentation that leads to an internal ion. We also observed breakage of the alpha-carbon to carbonyl-carbon bond (CH-CO) to produce a and x ions, and the bond between amide-nitrogen to alpha-carbon (NH-CH) to yield c and z ions, albeit at low frequency, with only a-type ions occurring above 1%. While the numbers are small, the bootstrap-produced confidence intervals indicate consistency amongst the rates at which these low-abundance peaks are found in the data, except for losses of ammonia or water from the precursor (precursor* and precursor0, respectively). Hence, these results appear to reflect true low levels of non-peptide-bond fragmentation rather than false-positive matches to noise peaks, except in the case of neutral losses from the precursor ion.
Mass and Intensity Dependence of Ion Types
We calculated the intensity distribution of ions by dividing the relative intensity range into ten bins, and calculated the frequency of each ion type in each of these bins, as shown in Figure 2(a). As has previously been observed, y ions are typically more intense than b ions, predominating the top two bins. The b ions were observed as frequently as y ions in the 20–40% intensity range, but dropped off in the high intensity bins.
Figure 2.
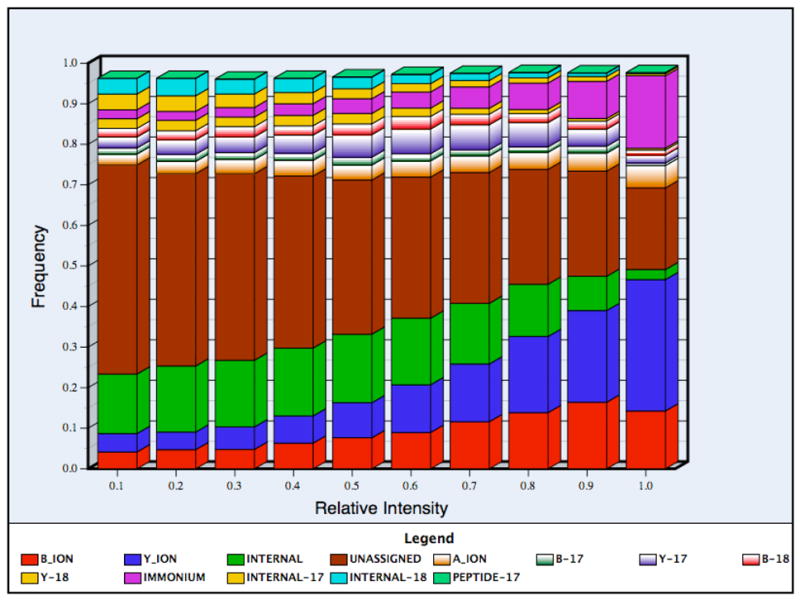
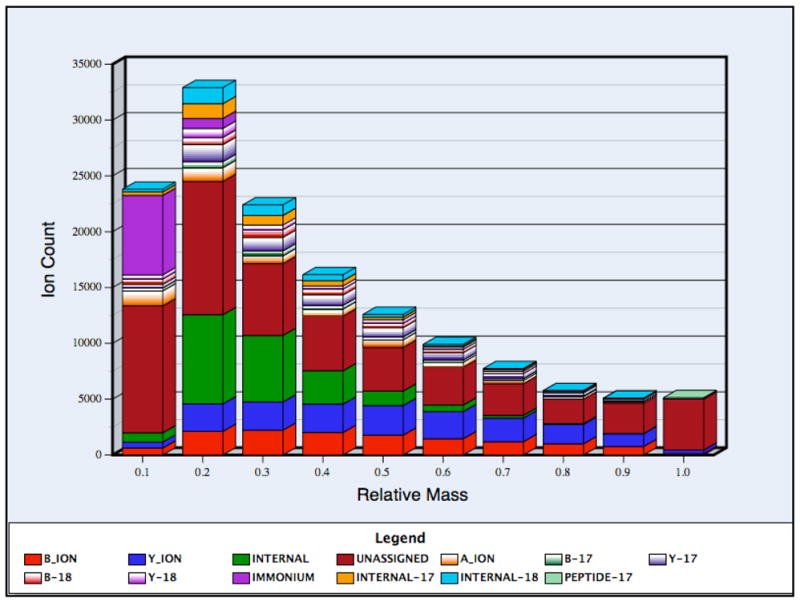
Figure 2(a). The frequency ratios of each type of product ion as a function of peak intensity. The intensity distribution was calculated as the relative frequency (listed as percentile ranges) of observing each ion type in a fraction of observed ions. The peaks were divided into ten bins according to their intensities and in each bin the relative frequency of the different ion types was calculated. The ions with very small probabilities (such as c, x, z ions) are not shown in the histogram. The 95% confidence intervals were very small (within 20% of the observed values) and were thus omitted for clarity.
Figure 2(b). The frequency ratios of each type of product ion as a function of relative mass. The horizontal axis represents the masses of fragment ions as a proportion of precursor mass. The observed mass range for product ions was divided into ten different bins according to their masses with respect to precursor ion and the relative frequency of observing each ion type was calculated for each bin. The 95% confidence intervals were small (within 20% of the reported values) and were thus omitted for clarity.
The large number of ‘unassigned’ ions, present even for the highest-intensity category of peaks (Figure 2(a)) at about 20%, suggests that there are other ion types produced during fragmentation that our program did not consider. This category might include side-chain cleavages, internal fragments due to the breakage of bonds other than amide, and/or the simultaneous loss of water and ammonia from each of the ion types. These peaks may also result from internal rearrangement of the fragment ions28. The larger proportion of unassigned peaks occurring in low-intensity bins may be due to low abundance reaction byproducts of fragmentation and/or noise.
We similarly examined the relationship between the ion type and frequency of occurrence in different mass ranges. We divided the relative mass range into ten bins, running from zero to the mass of each precursor ion, and calculated the frequency of each ion type for each bin. There was a clear dependence of ion type on the mass and intensity of observation; for example unassigned peaks occurred most frequently at masses near the precursor mass, even though we considered the loss of ammonia and water from the precursor ion. The high frequency of unassigned peaks in the highest mass bin, which includes the precursor, may be due to the simultaneous loss of water and ammonia from the precursor, which was not considered by our program. In addition, b ions were as likely as y ions in lower mass ranges, while in higher mass ranges the probability of observing y ions was greater and reached a maximum at about 70–80% of the precursor ion mass. Since the internal ions are the products of secondary fragmentation of b and y ions, they occurred primarily in the lower mass bins. The lowest mass bin mainly consisted of immonium ions and unassigned peaks (internal fragments were not observed in this bin because single amino acids were considered to be immonium ions). The large proportion of unassigned peaks in the lowest mass bin may be due to the various low-mass ions produced with just a single side chain by high energy collision-induced dissociation29,30.
Residue Effects on Fragmentation
We examined the correlation between each of the 20 amino acids and the propensity for bond cleavage on both their N-terminal and C-terminal sides. For each of the 20 amino acids, we divided the total number of N-terminal and C-terminal bond cleavage events (i.e. matched peaks for one of the ions produced) by the total number of the same residue present in all matched peptide sequences for the data set. For example, to calculate the N-terminal cleavage probability for alanine, the number of alanines present in all matched peptides was counted, along with the number of those corresponding to a cleavage towards the N-terminal side (i.e. the alanine becomes part of a y ion). We excluded both of the peptide’s terminal residues for this calculation. The C-terminal residue was excluded to avoid bias towards lysine and arginine from tryptic digestion, and the N-terminal residue was excluded because the bl ion is not often observed, which skews the resulting frequency calculations at this site. The results of the analysis excluding both termini are shown in Figure 3(a).
Figure 3.

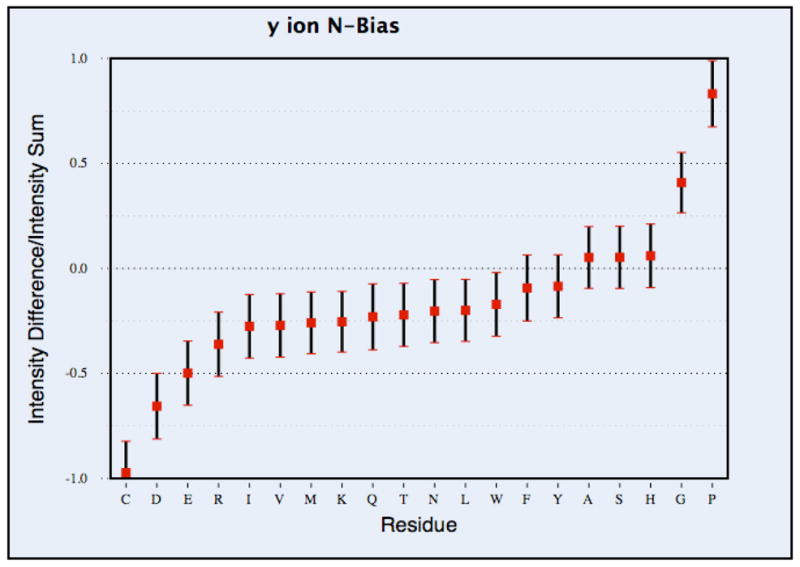
Figure 3(a). Directionality of bond cleavage. The blue bars represent the cleavage frequency at the N-terminal side while the red bars represent the cleavage frequency at the C-terminal side of each residue. The frequency was calculated by dividing the total number of cleavage events for a specific amino acid by the total number of amino acid of that type present in the dataset. The 95% confidence intervals are shown with black bars, calculated by bootstrap resampling as described in the methods.
Figure 3(b). The N-bias for y ions, corresponding to each residue type. The N-bias was calculated by taking the ratio of the intensity difference of the C-terminal peaks and the N-terminal peaks to the intensity sum, for all observed y ions produced from breakages adjacent to the given residue. The mean value of the N-bias is marked by the small square and the 95% confidence interval is shown with black bars, calculated by bootstrap resampling.
To facilitate comparison against previous papers, we also calculated the intensity-based N-bias as was done in the work of Tabb et al.22, the results of which are shown in Figure 3(b) for y ions (the b ion calculation is in Supplementary Figure 1). The difference with this approach is that it only calculates cleavage bias for a single ion type at a time, whereas in the frequency calculation of Figure 3(a), all ion types produced by a given cleavage event were counted. This can have an effect on the resulting statistics, because when only one ion type is considered at a time, results can be affected if one of the termini was not observed when a bond breakage preferentially produces ions of a different type, such as those involving loss of ammonia or water. Histidine provides an example of the difference between these calculation approaches. In the intensity-based N-bias calculation, it had a positive N-bias for y ions (Figure 3(b)) and a negative N-bias for b ions (Supplementary Figure 1). However, in the frequency-based calculation, taking account of all 12 ion types considered, histidine had an overall bias towards C-terminal cleavage (Figure 3(a)). For a number of other amino acids where the cleavage bias was prominent, such as proline, glycine, aspartic acid and cysteine, we found that both the results are consistent.
In both types of analysis, certain amino acids had a strong influence on the process of fragmentation. Proline had a strong bias towards fragmentation on its N-terminal side, as has been observed for other instrument types22, 23, 25. In frequency-based calculation, when all ion types were considered, residues with N-terminal cleavage bias were tryptophan, glycine, methionine and serine (Figure 3a). There were also amino acids that showed the opposite bias, with a higher frequency of cleavage on the C-terminal side (i.e. the residue itself becomes part of a b ion). This was greatest for cysteine, but also true for aspartic acid and glutamic acid, in both the y ion analysis (Fig. 3(b)) and the all-ion analysis (Fig. 3(a)). With all ion types considered, C-terminal bias was also displayed by phenylalanine, arginine, lysine and histidine.
Tabb et al. performed an analysis similar in methodology to that used here and found that for the non-polar amino acids isoleucine, leucine and valine, there was an increased C-terminal cleavage bias for y ions, whereas for proline, glycine, and serine there was increased N-terminal cleavage bias22. Similarities between their results and our own included the propensity for N-terminal cleavage bias associated with both proline and glycine, and C-terminal cleavage bias for valine, glutamine, and histidine. A substantive difference we found was the preferential C-terminal cleavage of residues cysteine, aspartic acid, glutamic acid, arginine and lysine. Another substantive difference we found was N-terminal cleavage bias for tryptophan, which was present only when all ion types were considered in the frequency-based calculation.
Kapp et al. also examined the cleavage effects of residue pairs, finding that the primary cleavage effect was C-terminal of aspartic acid for singly-charged peptide ions that had the proton localized to an arginine residue, while for peptide ions with a mobile proton the cleavage bias shifted to the N-terminal side of proline 25. Huang et al. reported bias toward C-terminal cleavage after acidic residues for y ions24. They noted that when arginine was at the C terminus of the peptide ion, the resulting CID cleavage occurred with high frequency on the C-terminal side of aspartic acid and glutamic acid. Our results indicate a preference for C-terminal cleavage following all charged amino acids, which includes the basic residues (K, R, and H), along with the acidic residues (E and D). In addition, we observed C-terminal cleavage bias following the polar residues cysteine and tyrosine, along with the non-polar valine and phenylalanine. The strong C-terminal cleavage bias for histidine, aspartic acid, glutamic acid, arginine, lysine, cysteine, and tyrosine might be due to each of them having a side chain with a nucleophile. When activated with collisional energy, it may donate an electron, preferentially to the C-terminal carbonyl carbon atom due to its proximity, resulting in amide bond breakage, as shown in the example for cysteine in Figure 4.
Figure 4.
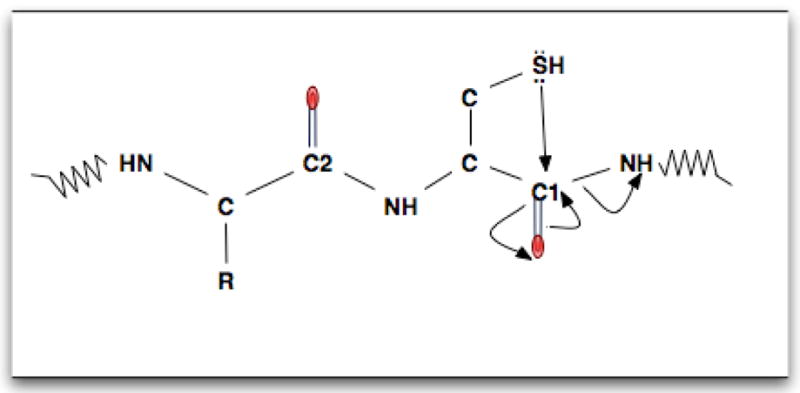
A model for the preferential C-terminal cleavage of cysteine. The formation of bond by the sulfhydryl group with the nearest neighbor carbonyl carbon atom (Cl) is more favorable than the formation of bond with the carbonyl carbon atom of the previous residue (C2). This facilitates the breakage of C-terminal cleavage of cysteine than the N-terminal cleavage. Electron transfer is shown by the arrows.
While our results agree with Kapp et al., Huang et al. and Breci et al.23 that the polar amino acids are likely to cleave C-terminally, we found that for the residue cysteine the C-terminal cleavage is preferable to the N-terminal cleavage, although cysteine showed a reduced tendency to produce ions of the type we analyzed from cleavage at either terminus. While enhanced C-terminal cleavage of cysteine for doubly charged peptides with a mobile proton was reported by Kapp et al., Breci and Huang et al., did not report statistics for cysteine. Since our data set had 548 occurrences of cysteine, there were enough data to produce small confidence intervals in bootstrap resampling, supporting the conclusion that enhanced C-terminal cleavage of cysteine occurs in MALDI TOF/TOF fragmentation. A putative explanation for this may be the nucleophilic sulfhydryl (SH) side chain of cysteine, that when collisionally activated, attacks the nearest carbonyl carbon atom to cause C-terminal cleavage (Fig. 4).
Though there were many similarities with previous results, the differences we observed may be due to several factors. One is that most of the peptides we analyzed were singly charged since this is the predominant ion type produced by MALDI, in contrast to Tabb et al. who reported results for doubly-charged peptides that are predominant in ESI. As well, the collision energy employed by the TOF/TOF is higher, and the ion source different than for the ion trap analyses. Finally, we performed an analysis that considered the frequency of cleavage when all ion types produced by a breakage were considered together, which may produce different results than the intensity-based approach used by Tabb et al., as discussed previously.
The Effect of Internal Residues on Ion Observation
We examined whether the presence of specific residues in the peptide ion that were not adjacent to a bond breakage had an effect on the observation of b or y ions containing them. The frequency of amino acids present was calculated for both b and y ions by dividing the number present in each of the types by the total number found for all peptides matched in the data set. Since y ions are typically larger than b ions (the average y ion size was 8 residues while the average b ion size was 7 residues), we normalized the probability to avoid size bias. To minimize bias due to the presence of K and R at the C-terminus of tryptic peptides, we excluded the residues from the peptide terminus for the calculation, so that the results for peptides containing K or R are only those due to sites of missed tryptic cleavage. The resulting frequencies are shown in Figure 5.
Figure 5.

The frequency of amino acid occurrences within b and y ions. The bars represent the frequency at which a specific amino acid was present in a b-type ion (blue) or y-type ion (red). This was calculated by dividing the number of times an amino acid was present within the given ion type by the total number of occurrences of that amino acid in the collection of peptides. The 95% confidence interval is shown, calculated by bootstrap resampling.
We found that the basic residues histidine, lysine and arginine are more likely to be in b ions than in y ions. We also found certain amino acids are more likely to be present in y ions, including: glycine, isoleucine, threonine, tryptophan glutamic acid, tyrosine, methionine, alanine and proline. Normally for tryptic digests, lysine and arginine are primarily present in y ions because they are at the C-terminus of the peptide. These basic residues often attract a proton during ionization, which may explain the greater intensities typically observed for y ions. However, in the case of a missed tryptic cleavage or occurrence of histidine, where one of these basic residues is located internally to the peptide, we find that the b ion frequencies are substantially greater than those of the y ions, agreeing with a previous observation for ion trap instruments31. While the effects of other residues were not as drastic, the results demonstrate that composition of the rest of the peptide is important in determining whether it will be fragmented and/or observed in MALDITOF/TOF MS.
We also found that cysteine produced a significant reduction in observation of either b or y ions containing it, possibly due the great influence of sulfhydryl side-chain on the fragmentation process as explained by O’Hair32. Briefly, the presence of cysteine in the N-terminal side of a peptide causes the loss of NH3, whereas the presence of cysteine in an intermediate position facilitates the loss of water through nucleophilic attack by the sulfhydryl group of the adjacent N-terminal carbonyl residue, which was not reported since we considered only b and y ions. Another possibility is that the reduced observation of these ions is due to the negative influence of cysteine on ionization of the precursor, which we have observed as part of a separate work (manuscript in preparation).
Conclusion
The characteristics of the fragment ion series generated from a peptide depend on the type of ion source and mass analyzer used. For CID in a MALDI TOF/TOF instrument, the frequency of observing each ion type is dependent upon the mass of the ion, the intensity of the ion, the residues adjacent to the backbone cleavage producing the ion, and the residue composition in the rest of the ion aside from its termini.
Our results showed substantive differences from those reported for ion trap instruments, especially notable in the presence of basic residues or cysteine, demonstrating the importance of considering instrument-specific fragmentation models. Ultimately, algorithms that are built using such statistics will need to be tailored for each instrument to produce optimal identification results based on statistics such as those presented here.
Supplementary Material
Acknowledgments
We thank Eric Hamlett for E. coli ribosomal protein samples, and the UNC/Duke Michael Hooker Mass Spectrometry Core Facility for the mass spectrometry on the MALDI TOF/TOF. We are also thankful to Prof. Gary Glish, Mark Holmes and Jameson Miller for critically reading the manuscript. The work was supported by NIH National Center for Research Resources R01 RR020823 to MCG, and NIH National Human Genome Research Institute R01HG003700 to MCG. Support for KR provided by American Heart Association 051537OU.
Literature Cited
- 1.Hernandez PMM, Appel RD. Mass Spectrom Rev. 2006;25:235–254. doi: 10.1002/mas.20068. [DOI] [PubMed] [Google Scholar]
- 2.Bartels C. Biomed Environ Mass Spectrom. 1990;19:363–368. doi: 10.1002/bms.1200190607. [DOI] [PubMed] [Google Scholar]
- 3.Dancik V, Addona TA, Clauser KR, Vath JE, Pevzner PA. J Comput Biol. 1999;6:327–342. doi: 10.1089/106652799318300. [DOI] [PubMed] [Google Scholar]
- 4.Frank A, Pevzner P. Anal Chem. 2005;77:964–973. doi: 10.1021/ac048788h. [DOI] [PubMed] [Google Scholar]
- 5.Horn DM, Zubarev RA, McLafferty FW. Proc Natl Acad Sci U S A. 2000;97:10313–10317. doi: 10.1073/pnas.97.19.10313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma B, Zhang K, Hendrie C, Liang C, Li M, Doherty-Kirby A, Lajoie G. Rapid Communications in Mass Spectrometry. 2003;17:2337–2342. doi: 10.1002/rcm.1196. [DOI] [PubMed] [Google Scholar]
- 7.Nickerson DA, Tobe VO, Taylor SL. Nucleic Acids Res. 1997;25:2745–2751. doi: 10.1093/nar/25.14.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taylor JA, Johnson RS. Anal Chem. 2001;73:2594–2604. doi: 10.1021/ac001196o. [DOI] [PubMed] [Google Scholar]
- 9.Bandeira N, Tang H, Bafna V, Pevzner P. Anal Chem. 2004;76:7221–7233. doi: 10.1021/ac0489162. [DOI] [PubMed] [Google Scholar]
- 10.Field HI, Fenyo D, Beavis RC. Proteomics. 2002;2:36–47. [PubMed] [Google Scholar]
- 11.Eng JK, McCormack AL, Yates JRI. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 12.Falkner J, Andrews P. Bioinformatics. 2005;21:2177–2184. doi: 10.1093/bioinformatics/bti362. [DOI] [PubMed] [Google Scholar]
- 13.Geer LY, Markey SP, Kowalak JA, Wagner L, Xu M, Maynard DM, Yang X, Shi W, Bryant SH. J Proteome Res. 2004;3:958–964. doi: 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- 14.LeDuc RD, Taylor GK, Kim YB, Januszyk TE, Bynum LH, Sola JV, Garavelli JS, Kelleher NL. Nucleic Acids Res. 2004;32:W340–345. doi: 10.1093/nar/gkh447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mann M, Wilm M. Anal Chem. 1994;66:4390–4399. doi: 10.1021/ac00096a002. [DOI] [PubMed] [Google Scholar]
- 16.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 17.Sadygov RG, Eng J, Durr E, Saraf A, McDonald H, MacCoss MJ, Yates JR., 3rd J Proteome Res. 2002;1:211–215. doi: 10.1021/pr015514r. [DOI] [PubMed] [Google Scholar]
- 18.Tabb DL, Saraf A, Yates JR., 3rd Anal Chem. 2003;75:6415–6421. doi: 10.1021/ac0347462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yates JR, 3rd, Eng JK, McCormack AL. Anal Chem. 1995;67:3202–3210. doi: 10.1021/ac00114a016. [DOI] [PubMed] [Google Scholar]
- 20.Yates JR, 3rd, Eng JK, McCormack AL, Schieltz D. Anal Chem. 1995;67:1426–1436. doi: 10.1021/ac00104a020. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Z, Sun S, Zhu X, Chang S, Liu X, Yu C, Bu D, Chen R. BMC Bioinformatics. 2006;7:222. doi: 10.1186/1471-2105-7-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tabb DL, Smith LL, Breci LA, Wysocki VH, Lin D, Yates JR., 3rd Anal Chem. 2003;75:1155–1163. doi: 10.1021/ac026122m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Breci LA, Tabb DL, Yates JR, 3rd, Wysocki VH. Anal Chem. 2003;75:1963–1971. doi: 10.1021/ac026359i. [DOI] [PubMed] [Google Scholar]
- 24.Huang Y, Triscari JM, Tseng GC, Pasa-Tolic L, Lipton MS, Smith RD, Wysocki VH. Anal Chem. 2005;77:5800–5813. doi: 10.1021/ac0480949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kapp EA, Schutz F, Reid GE, Eddes JS, Moritz RL, O’Hair RA, Speed TP, Simpson RJ. Anal Chem. 2003;75:6251–6264. doi: 10.1021/ac034616t. [DOI] [PubMed] [Google Scholar]
- 26.Davison, A. C. a. H., D.V Cambridge University Press 1998.
- 27.Zoubir AMaBB. IEEE signal Processing Mazagine. 1998:56–76. [Google Scholar]
- 28.Yague J, Paradela A, Ramos M, Ogueta S, Marina A, Barahona F, Lopez de Castro JA, Vazquez J. Anal Chem. 2003;75:1524–1535. doi: 10.1021/ac026280d. [DOI] [PubMed] [Google Scholar]
- 29.Falick AHWM, Medzihradszky KF, Baldwin MA, Gibson BW. J Am Soc Mass Spectrom. 1993;4:882–893. doi: 10.1016/1044-0305(93)87006-X. [DOI] [PubMed] [Google Scholar]
- 30.Papayannopoulos I. Mass Spectrom Rev. 1995;14:49–73. [Google Scholar]
- 31.Tabb DL, Huang Y, Wysocki VH, Yates JR., 3rd Anal Chem. 2004;76:1243–1248. doi: 10.1021/ac0351163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Hair RA. Journal of Mass Spectrometry. 2000;35:1377–1381. doi: 10.1002/1096-9888(200012)35:12<1377::AID-JMS83>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.