

Abstract
Investigations determined the critical amino acids for α-thrombin’s interaction with protease activated receptors 1 and 4 (PAR1 and PAR4) at the thrombin cleavage site. Recombinant PAR1-wild type (wt) exodomain was cleaved by α-thrombin with a Km of 28 μM, a kcat of 340 s-1 and kcat/Km of 1.2×107. When the P4 or P2 position was mutated to alanine, PAR1-L38A or PAR1-P40A, respectively, the Km was unchanged, 29 or 23 μM, respectively; however the kcat and kcat/Km were reduced in each case. In contrast, when Asp39 at P3 was mutated to alanine, PAR1-D39A, the Km and kcat were both reduced ~3-fold making the kcat/Km the same as PAR1-wt exodomain. Recombinant PAR4-wt exodomain was cleaved by α-thrombin with a Km of 61 μM, a kcat of 17 s-1 and kcat/Km of 2.8 ×105. When the P5 or P4 position was mutated to alanine PAR4-L43A or PAR4-P44A, respectively, there was no change in the Km (69 or 56 μM, respectively) however; the kcat was lowered in each case (9.7 or 7.7 s-1, respectively). Mutation of the P2 position (PAR4-P46A) also had no effect on the Km but markedly lowered the kcat and kcat/Km ~35-fold. PAR1-wt exodomain and P4 and P3 mutants were noncompetitive inhibitors of α-thrombin hydrolyzing Sar-Pro-Arg-pNA. However, PAR1-P40A displayed mixed type of inhibition. Mutation of P4, P3 or P2 had no effect on the Ki. All PAR4 exodomains were competitive inhibitors of α-thrombin. Mutation of P5, P4 or P2 had no effect on the Ki. These investigations show that Leu at P4 in PAR1 or P5 in PAR4 critically influences the kinetics of α-thrombin binding and cleavage of PAR1 and PAR4 exodomains. It also implies that factors other than the hirudin-like binding region on PAR1 exodomain predominate in influencing PAR1 cleavage on cells.
Keywords: Thrombin, Protease Activated Receptor, PAR1, PAR4
The serine protease thrombin is the terminal enzyme of the hemostatic system. During clot formation, thrombin converts fibrinogen to fibrin, activates factor XIII that cross links fibrin in the clot, and activates factors XI, VIII and V to generate more thrombin (1). Further, thrombin activates protein C which negatively regulates thrombin generation by inactivating thrombin-activated factors VIII and V (1). Thrombin is also a major activator of platelets by binding and cleaving protease activated receptors 1 and 4 (PAR1 and 4). The protease activated receptor family are a novel class of G-protein coupled receptors (GPCR) that are activated by the proteolysis of the N-terminal exodomain (2). Upon proteolysis, the newly formed N-terminus acts as a tethered ligand that activates the receptor and initiates multiple signaling cascades via heterotrimeric G-proteins (3,4). There are four PARs (PAR1-4). PAR1, 3 and 4 are activated by thrombin, whereas PAR2 is activated by trypsin or tryptase (5). Other serine proteases like FVIIa, FXa, plasmin, activated protein C, cathepsin G and MMP1 also activate PARs although less efficiently (6-10).
The exodomain of PAR1 has two binding sites for thrombin (11). In addition to binding thrombin’s active site, PAR1 has a hirudin-like sequence (K51YEPF55) that binds the exosite I of thrombin inducing allosteric effects on thrombin lowering the energy required for PAR1 cleavage (12-14). The importance of the hirudin-like sequence for more efficient PAR1 activation has been confirmed with thrombin exosite and PAR1 exodomain mutations (13-16). Much less focus has been given to thrombin’s interaction with PAR1 at its cleavage site. It has been proposed that the optimal sequence for thrombin substrates is Leu or Ile at P4 and Pro at P2 positions (17). PAR1 has the optimal residues in the P4 and P2 positions (L38DPR41). The P4 to P1 positions of PAR1 are the same as that of protein C which, in the absence of thrombomodulin, is a poor thrombin substrate due to the Asp167 at P3 interacting with thrombin’s Glu192 (18). Mutation of Asp167 to Gly in protein C or Glu192 to Gln in thrombin results in increased efficiency of cleavage of protein C by thrombin (18,19). Similarly, mutation of the hirudin-like sequence in PAR1 results in a shift in the EC50 of thrombin activation from 0.1 nM to 20 nM when PAR1 is expressed on COS7 cells (20). This phenotype was partially rescued by mutating Asp39 at P3 to Gly (20).
PAR4 does not contain a hirudin-like sequence; as a result, it interacts with thrombin primarily at the active site. It has been proposed that PAR4 interacts with thrombin via two proline residues at P4 and P2, Pro44 and Pro46, respectively. There is also a role for Leu43 at P5 for PAR4 cleavage. Ayala et al. show by molecular modeling with PAR4 peptides that Leu43 interacts with thrombin residues Leu99, Ile174 and Trp215 (13). Cleary et al. demonstrated in NMR studies with PAR4 peptides that Leu43 exhibits minor proton line broadening, suggesting Leu43 binds to thrombin but to a lesser extent than the Pro44-Arg47 region (21). Further, Leu43 can interact with Pro44 and Pro46 in two distinct conformations in the presence of thrombin, suggesting that there is flexibility where Leu43 interacts with thrombin (21). They demonstrate that the NMR structure of a PAR4 peptide (L43PAP46) had a similar 3-dimensional structure as a PAR1 peptide that was co-crystallized with thrombin (21, 22). These combined studies indicate that more needs to be known about how thrombin interacts with the substrate cleavage site of the exodomains of PAR1 and PAR4.
In an effort to define how thrombin interacts with the exodomains of PAR1 and PAR4 at the cleavage site, we sought to identify the amino acids on PAR1 and PAR4 that are important for thrombin’s interaction in the context of the full exodomain. The current report identifies the amino acids in the P4 (Leu38) and P2 (Pro40) positions of PAR1 and P5 (Leu43), P4 (Pro44) and P2 (Pro46) positions for PAR4 are important for efficient cleavage by thrombin since Ala substitution of each adversely affects the kcat. Parenthetically, none of these point mutations introduced into PAR1 or PAR4 influence the Km of thrombin cleavage. This investigation is the first functional evidence that the Leu38 at P4 for PAR1 and Leu43 at P5 for PAR4 have an important role for thrombin binding and cleavage. These data suggest that the Leu and Pro in these positions on these receptors cooperate to form a similar 3-dimensional structure that is recognized by thrombin.
Materials and Methods
Cloning and mutagenesis of PAR1 and PAR4 exodomains
Recombinant PAR1-wt, PAR4-wt, PAR4-P44A and PAR4-P46A exodomains have been previously described (23). Additional mutations were introduced into PAR1 or PAR4 exodomain by overlapping PCR with the same protocol. The mutagenizing primers for PAR1-L38A, PAR1-D39A, PAR1-P40A and PAR4-L43A are shown in Table 1. All constructs were verified by DNA sequencing and inserted into pET31b (Novagen, San Diego, CA) for expression in Escherichia coli.
Table 1.
PAR Exodomain Mutagenesis Primers
| Primer | Sequencea |
|---|---|
| PAR1-L38Ab | ACAAATGCCGCAGATCCCCGGTCATTT |
| PAR1-L38Ac | AAATGACCGGGGATCTGCGGTGGCATTTGT |
| PAR1-D39Ab | AATGCCACCTTAGCACCCCGGTCATTTCTT |
| PAR1-D39Ac | AAGAAATGACCGGGGTGCTAAGGTGGCAAT |
| PAR1-P40Ab | GCCACCTTAGATGCACGGTCATTTCTTCT |
| PAR1-P40Ac | GAGAAGAAATGACCGTGCATCTAAGGTGGC |
| PAR4-L43Ab | ACGCCCTCAATCGCACCTGCCCCCGCGGC |
| PAR4-L43Ac | GCCGCGGGGGCAGGTGCGATTGAGGGCGT |
mutagenized codon in bold
sense primer
antisense primer
Recombinant protein production
Purified recombinant exodomain of PAR1, PAR1 mutants, PAR4 or PAR4 mutants were prepared from 2 liter cultures of E. coli BLR(DE3) as previously desribed (23). The Mr of each expressed PAR exodomain was verified by MALDI-TOF mass spectrometry (Pro-TOF 2000 MALDI mass spectrometer, Perkin Elmer, Wellesley, MA) in the Center for Proteomics and Mass Spectrometry, Case Western Reserve University (Table 2). The concentration of purified recombinant exodomain was determined by UV absorption (ε 280 = 9970 M-1 cm-1 for PAR1-wt, PAR1-L38A, PAR1-D39A or PAR1-P40A) or (ε 280 = 8480 M-1 cm-1 for PAR4-wt, PAR4-L43A, PAR4-P44A or PAR1-P46A).
Table 2.
Mutant PAR1 and PAR4 Exodomains
| Exodomain | Sequence ab | Massc |
|---|---|---|
| PAR1-wt | …NATLDPRSFLL… | 8493 |
| PAR1-L38A | …NATADPRSFLL… | 8451 |
| PAR1-D39A | …NATLAPRSFLL… | 8449 |
| PAR1-P40A | …NATLDARSFLL… | 8467 |
| PAR4-wt | …SILPAPRGYGK… | 6435 |
| PAR4-L43A | …SIAPAPRGYGK… | 6394 |
| PAR4-P44A | …SILAAPRGYGK… | 6405 |
| PAR4-P46A | …SILPAARGYGK… | 6405 |
Mutations are shown in bold
P1 arginine is underlined
Mass as determined by mass spectrometry
Cleavage of soluble recombinant PAR exodomain
Cleavage of purified PAR1-wt, PAR4-wt or mutant exodomain (3-400 μM) in reaction buffer (10 mM Tris-HCl, 150 mM NaCl, pH 8.0) was initiated by the addition of α-thrombin (0.5 to 50 nM, specific activity = 3725 units/mg, Haematological Technologies, Essex Junction, VT). The optimal range of substrate concentration varied depending on the PAR exodomain mutant that was being tested. Reactions were quenched at given time intervals by removing 100 μL aliquots and placing them into tubes that contained 50 μL acetic acid. The reactions were monitored with RP-HPLC (Waters Delta 600, Waters Corporation, Milford, MA) on a Waters Atlantis PAA column. The rate of product formation was determined by calculating the peak areas for the N-terminal and C-terminal cleavage products and comparing them to a standard curve. The standard curve was generated with purified cleavage products from completely cleaved exodomains prepared by treating exodomain with molar excess of α-thrombin. Initial rates were determined at early time points where the progress curves were linear. The data were fit to the Henri-Michaelis-Menten equation (equation 1) using Sigmaplot (Systat, Inc.) and the kinetic constants were determined (see below).
| Equation 1 |
In other experiments, the catalytic efficiency (kcat/Km) was determined at substrate concentrations ≪ Km where equation 2 is valid; s is catalytic efficiency, eT is enzyme concentration, t is time (13).
| Equation 2 |
Inhibition of thrombin hydrolysis of Sar-Pro-Arg-pNA
Reactions were initiated by adding α-thrombin (0.5 nM) to Sar-Pro-Arg-pNA (35-1400 μM) in the absence or presence of PAR1-wt, PAR4-wt or mutant exodomain (5-200 μM) in Tris-HCl, 150 mM NaCl, pH 8.0. For some experiments, thrombin was preincubated with 10 μM hirugen (hirudin C-terminal fragment 53-64, American Diagnostica, Inc., Stamford, CT) for 2 min prior to initiation of the reaction. The Km for Sar-Pro-Arg-pNA hydrolysis by α-thrombin was determined to be 75 μM. Therefore, the range of concentrations used in these experiments (0.5 to ~20 times the Km) was adequate to characterize the inhibition by the PAR exodomains. Initial rates were determined by taking absorbance readings every 5 seconds with a Beckman DU-64 kinetic spectrophotomer at early time points where the progress curves were linear. Absorbance readings were converted to concentration terms using E405 = 9887 M-1cm-1 (24). The type of inhibition, Ki and, where applicable, α(ie. mixed inhibition, where α is the factor by which the Ki changes when the substrate is simultaneously bound to the enzyme) were determined as described below in the next section.
Data analysis
Initial velocity data were fit to Henri-Michaelis-Menten equation (equation 1) by nonlinear least squares regression analysis using with SigmaPlot kinetic analysis software (Systat, Inc.) (25). In the calculations, all data from all experiments were analyzed simultaneously (25,26). Kinetic constants were derived from these analyses and are reported ±95% confidence interval. The Ki for PAR exodomain inhibition of thrombin cleavage of Sar-Pro-Arg-pNA were determined by fitting initial velocity data to 8 models of enzyme inhibition also using a global analysis nonlinear least squares regression analysis with SigmaPlot enzyme kinetics module (Systat) (26). The best model was chosen based on the Akaike information criteria (AIC) (27). The Ki for each PAR exodomain is reported at ± 95% confidence interval. Double reciprocal plots are shown only for graphical representation of the model and were not used to determine the Ki or type of inhibition. P-values were determined using a two-tailed t-test with Prism software (Graphpad, San Diego, CA).
Results
Investigations were performed to identify the amino acids at the α-thrombin cleavage site of PAR1 and PAR4 that are important for α-thrombin to bind and cleave. To this end, a series of PAR1 and PAR4 exodomain alanine substitution mutants were generated (Table 2) and the efficiency with which α-thrombin binds and cleaves was determined. The homogeneity of each recombinant exodomain was assured between preparations by mass spectrometry (Table 2).
Initial studies directly determined the influence of α-thrombin on cleavage of PAR1-wt exodomain. The Km (28 μM), kcat (340 s-1) and kcat/Km (1.2 ×107 s-1M-1) for α-thrombin cleaved PAR1-wt were consistent with previously published data (Figure 1A and Table 3) (13,14). As an independent confirmation of this approach, we determined the kcat/Km (1.1 ×107 s-1M-1) at low substrate concentration where equation 2 is valid (13). The Km of the P4 mutant (PAR1-L38A) was unchanged from PAR1-wt (Figure 1B and Table 3). However, the kcat was decreased 7-fold to 47 s-1 (p = 0.013). The Km for PAR1-D39A was 10.1 μM (p = 0.02), 3 fold lower than PAR1-wt (Table 3, Figure 1C). However, the kcat for PAR1-D39A was also lowered 3 fold (p = 0.0002) (Table 3). The mutation of Asp39 at P3 influenced both kcat and Km yielding a catalytic efficency similar to PAR1-wt, which indicates nonproductive binding of this mutant to the active site of thrombin (28). Alternatively, the Km for the P2 mutant (PAR1-P40A) was unchanged from PAR1-wt but the kcat was lowered to 230 s-1 (p = 0.03) (Figure 1D and Table 3). Overall, the lack of effect on the Km of most of the PAR1 mutants was likely due to the hirudin-like sequence of the PAR1 exodomain (K51YEPF55) which bound to thrombin’s exosite I independent of the active site. As a result, thrombin’s dependence on the amino acids at the cleavage site was decreased for its binding to PAR1. The kcat/Km for each of the PAR1 mutants was confirmed at low substrate concentration where equation 2 is valid as described above for PAR1-wt. In each case the kcat/Km agreed with the data presented above (Table 3).
Figure 1.
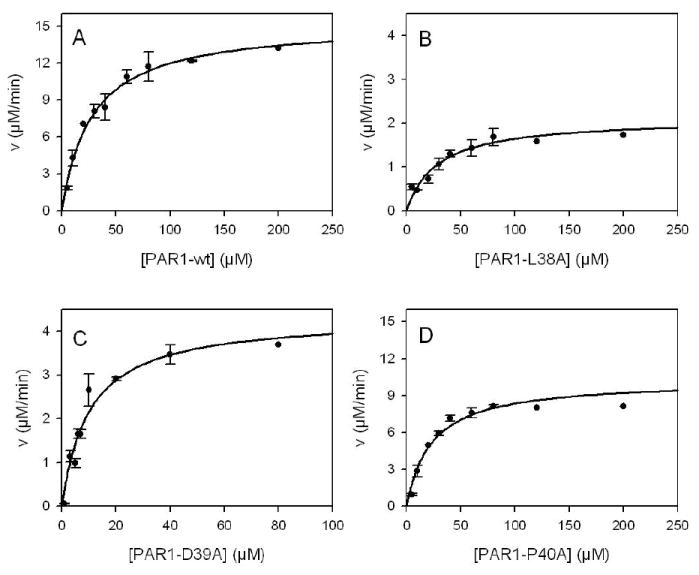
Cleavage of PAR1-wt or mutant exodomains with α-thrombin. Reactions with purified exodomains (3-200 μM) in reaction buffer (10 mM Tris-HCl, 150 mM NaCl, pH 8.0) were initiated by the addition of α-thrombin (0.75 nM for A, C and D or 0.5 nM for B). Acid quenched reactions were resolved on HPLC and initial rates were determined by calculating the peak areas at early time points where the progress curves were linear and compared to a standard curve generated by completely cleaved exodomain. Curve A is α-thrombin cleavage of PAR1-wt exodomain; curve B, PAR1-D39A; curve C, PAR1-L38A; and curve D, PAR1-P40A. The data were fit to the Henri-Michaelis-Menten equation with nonlinear least squares analysis to determine the kinetic constants. Error bars represent the standard deviation and the apparent absence of error bars indicates small standard deviation. Note the change in the scaling for the y-axis in panels B and C.
Table 3.
Kinetic constants for α-thrombin cleaving soluble PAR exodomains.
| Km (μM)a | kcat (s-1) a |
kcat/Km (M-1×s-1)b |
kcat/Km (M-1וs-1)c |
|
|---|---|---|---|---|
| PAR1-wt | 28 ± 8.8 | 340 ± 36 | 1.2 ×107 | 1.1 ×107 |
| PAR1-L38A | 29 ± 11 | 47 ± 9.2d | 1.7 ×106 | 3.5 ×106 |
| PAR1-D39A | 10 ± 3.4d | 140 ± 20d | 1.4×107 | 1.3 ×107 |
| PAR1-P40A | 23 ± 7.8 | 230 ± 26d | 9.9×106 | 9.1 ×106 |
| PAR4-wt | 61 ± 24 | 17 ± 2.3 | 2.8 ×105 | 4.1 ×105 |
| PAR4-L43A | 69 ± 28 | 9.7 ± 1.6d | 1.4 ×105 d | 2.1 ×105 d |
| PAR4-P44A | 56 ± 22 | 7.7 ± 1.1d | 1.5 ×105 d | 2.5 ×105 d |
| PAR4-P46A | 72 ± 32 | 0.64 ± .08d | 8.9 ×103 d | 7.9 ×103 d |
10 mM Tris·HCl, 150 mM NaCl, pH 8.0, 37°C
± 95% confidence interval
determined by direct measurement of kcat and Km
determined by: [S] = [S] 0exp(-seTt) at [S] ≪ Km
p < 0.05 compared to wild type sequence
Additional investigations determined the amino acids that were important for the α-thrombin-PAR4 interaction at the α-thrombin cleavage site as above for PAR1. Initial studies characterized the interaction of α-thrombin with the PAR4-wt exodomain. The kcat and Km for PAR4-wt was 61 μM and 17 s-1, respectively (Table 3 and Figure 2A). The mutations introduced at P5 (PAR4-L43A), P4 (PAR4-P44A), and P2 (PAR4-P46A) had no effect on the Km compared to PAR4-wt, (69 μM, p = 0.65), (56 μM, p = 0.57), (72 μM, p = 0.56) respectively, (Figure 2A-D and Table 3). However, the kcat was reduced (9.7 s-1, p = 0.001) for PAR4-L43A indicating that Leu43 at P5 is important for efficient PAR4 cleavage by thrombin (Figure 2B and Table 3). The PAR4-P44A also had a reduced kcat (7.7 s-1, p = 0.001) confirming a role for Pro44 in the PAR4 cleavage by thrombin (Figure 2C and Table 3). The kcat (0.64 s-1) was also significantly reduced for PAR4-P46A (Figure 2D and Table 3). Twenty-fold more α-thrombin was required for cleavage of PAR4-P46A which was reflected in the 35-fold decrease in the kcat compared to PAR4-wt. Further, the kcat/Km for each PAR4 mutant exodomain was confirmed at low substrate concentration where equation 2 is valid as described above for PAR1 (Table 3). Taken together, these data indicated that mutating the P5, P4 or P2 position of PAR4 exodomain did not influence PAR4 binding thrombin as assessed by the Km; however each of these positions were important for efficient cleavage of PAR4 by α-thrombin as reflected in the decrease in kcat.
Figure 2.

Cleavage of PAR4-wt or mutant exodomains with α-thrombin. Reactions with purified exodomains (20-400 μM) in reaction buffer (10 mM Tris-HCl, 150 mM NaCl, pH 8.0) were initiated by the addition of α-thrombin (5.0 nM for A, B and C, or 50 nM for D). Acid quenched reactions were resolved on HPLC and initial rates were determined by calculating the peak areas at early time points where the progress curves were linear and compared to a standard curve generated by completely cleaved exodomain. Curve A is α-thrombin cleavage of PAR4-wt exodomain; curve B, PAR4-L43A; curve C, PAR4-P44A; and curve D, PAR4-P46A. The data were fit to the Henri-Michaelis-Menten equation with nonlinear least squares analysis to determine the kinetic constants. Error bars represent the standard deviation and the apparent absence of error bars indicates small standard deviation.
The ability of PAR1 or PAR4 exodomains to inhibit thrombin hydrolyzing a chromogenic substrate has been used in previous studies to obtain important information regarding the nature of the thrombin-PAR interactions (12,14,29). A separate set of experiments determined the ability of PAR1-wt, PAR4-wt or mutant exodomains to inhibit α-thrombin hydrolysis of a small chromogenic substrate (Sar-Pro-Arg-pNA). Previous studies have demonstrated that PAR1-wt was a noncompetitive inhibitor of α-thrombin due to the high affinity exosite I binding site on the PAR1 exodomain (K51YEPF55) (14). In our studies, PAR1-wt also was a pure noncompetitive inhibitor of thrombin hydrolyzing Sar-Pro-Arg-pNA with a Ki of 26 μM (α = 1) (Figure 3A and Table 4). To confirm the role of exosite I binding region and verify that PAR1 exodomain was a noncompetitive inhibitor, thrombin was pre-incubated with 10 μM hirugen. In the presence of hirugen, PAR1-wt was unable to inhibit hydrolysis of Sar-Pro-Arg-pNA at concentrations up to 100 μM (Table 4). The P4 and P3 mutant PAR1 exodomains also were pure noncompetitive inhibitors with a Ki of 38 μM for PAR1-L38A, 7.1 μM for PAR1-D39A (Figure 3B-C and Table 4). However, the P2 mutant (PAR1-P40A) displayed mixed type of inhibition with a Ki of 26 μM and α of 3.8 (Figure 3D and Table 4). The change from noncompetitive to mixed type of inhibition may be due to a change in the orientation of the exodomain at the active site of thrombin. However, the difference between the two models was small for mixed inhibition versus pure noncompetitive inhibition (R2 = 0.991 versus 0.989 and AIC = -213.8 versus -204.7, respectively). Further, within error, α was equal to 1 which would signify pure noncompetitive inhibition (Table 4). Therefore, one cannot rule out that the PAR1-P40A mutant is also a pure noncompetitive inhibitor (α = 1).
Figure 3.

α-Thrombin hydrolysis of Sar-Pro-Arg-pNA in the absence or presence of PAR1-wt or mutant exodomain. α-Thrombin (0.5 nM) was added to Sar-Pro-Arg-pNA (70-1400 μM) in the absence (●) or presence of 5 (○), 10 (▼), 25 (∇) or 50 (
 ) μM PAR1-wt exodomain (A), PAR1-L38A (B), PAR1-D39A (C) or PAR1-P40A (D). Initial velocities data were fit to 8 models of enzyme inhibition using a global analysis nonlinear least squares regression analysis to determine the Ki and the type of inhibition (see Methods). Double reciprocal plots (insets) are shown only for graphical representation of the model and were not used to determine the Ki or type of inhibition.
) μM PAR1-wt exodomain (A), PAR1-L38A (B), PAR1-D39A (C) or PAR1-P40A (D). Initial velocities data were fit to 8 models of enzyme inhibition using a global analysis nonlinear least squares regression analysis to determine the Ki and the type of inhibition (see Methods). Double reciprocal plots (insets) are shown only for graphical representation of the model and were not used to determine the Ki or type of inhibition.
Table 4.
Inhibtion of α-thrombin hydrolysis of Sar-Pro-Arg-pNA by PAR exodomain.
| Ki (μM)a | Inhibition typeb | R2c | |
|---|---|---|---|
| PAR1-wt | 26 ± 7.7 | NC | 0.97 |
| PAR1-L38A | 38 ± 9.9 | NC | 0.98 |
| PAR1-D39A | 7.1 ± 1.0d | NC | 0.99 |
| PAR1-P40A | 26 ± 12
α = 3.8 ± 2.8 |
M | 0.99 |
| PAR4-wt | 64 ± 23 | C | 0.99 |
| PAR4-L43A | 56 ± 16 | C | 0.98 |
| PAR4-P44A | 41 ± 15 | C | 0.98 |
| PAR4-P46A | 68 ± 25 | C | 0.99 |
| PAR1-wte | (100)f | NI | - |
| PAR4-wte | 110 ± 15 | C | 0.98 |
10 mM Tris·HCl, 150 mM NaCl, pH 8.0, 25°C
± 95% confidence interval
NC (Noncompetitive), C (Competitive), (M) Mixed, (NI) No Inhibition
goodness of fit to inhibition model
p < 0.05 compared to wild type sequence
In the presence of 10 μM hirugen
No inhibition at the concentration in parentheses
Unlike PAR1, PAR4 does not have a hirudin-like sequence. As a result, the primary site of interaction with α-thrombin is at the active site. Therefore, due to the single point of interaction with α-thrombin, one would predict that the PAR4 exodomain would be a competitive inhibitor of α-thrombin. PAR4-wt was a competitive inhibitor of thrombin hydrolyzing Sar-Pro-Arg-pNA with a Ki of 64 μM (Figure 4A and Table 4). PAR4-L43A, PAR4-P44A and PAR4-P46A were also competitive inhibitors, and like the Km, the Ki was unchanged from PAR4-wt 56 (p = 0.62),41 (p = 0.24) and 68 (p = 0.95) μM, respectively (Figure 4B-D and Table 4). In the presence of hirugen, the PAR4-wt exodomain remained a competitive inhibitor, however the Ki shifted from 64 to 110 μM (p = 0.035) (Table 4)
Figure 4.

α-Thrombin hydrolysis of Sar-Pro-Arg-pNA in the absence or presence of PAR4-wt or mutant exodomain. α-Thrombin (0.5 nM) was added to Sar-Pro-Arg-pNA (70-1400 μM) in the absence (●) or presence of 50 (
 ), 100 (▼), 120 (∇) or 200 (○) μM PAR4-wt (A), PAR4-L43A (B), PAR4-P44A (C), or PAR4-P46A (D). Initial velocities data were fit to 8 models of enzyme inhibition using a global analysis nonlinear least squares regression analysis to determine the Ki and the type of inhibition (see Methods). Double reciprocal plots (insets) are shown only for graphical representation of the model and were not used to determine the Ki or type of inhibition.
), 100 (▼), 120 (∇) or 200 (○) μM PAR4-wt (A), PAR4-L43A (B), PAR4-P44A (C), or PAR4-P46A (D). Initial velocities data were fit to 8 models of enzyme inhibition using a global analysis nonlinear least squares regression analysis to determine the Ki and the type of inhibition (see Methods). Double reciprocal plots (insets) are shown only for graphical representation of the model and were not used to determine the Ki or type of inhibition.
Discussion
Using a library of PAR1 and PAR4 exodomains with single amino acid alanine substitutions, we have examined their interaction with α-thrombin. These studies determined the P4 (Leu38) and P2 (Pro40) are important for thrombin to efficiently cleave PAR1 as mutations at these positions decreases the kcat. For PAR4, mutations at P5, P4 or P2 also did not influence the Km of α-thrombin cleaving of PAR4 exodomain. However, alanine substitutions at P5 (Leu43) and P4 (Pro44) reduced the kcat of this reaction to the same extent. The alanine substitution of Pro46 at P2 results in a more dramatic decrease in kcat. Cleary et al. demonstrated that Leu43 at P5 interacts with Pro44 or Pro46 to stabilize secondary structure of a PAR4 peptide (21). Ayala et al. have suggested that Leu43 also interacts with thrombin based on molecular modeling data (13). These investigations show for the first time with full exodomains that Leu at P4 (PAR1) or P5 (PAR4) critically influences the kinetics of α-thrombin cleavage of PAR1 and PAR4 exodomains.
Alanine substitution at P4 and P2 of PAR1 exodomain had no effect on the Km of α-thrombin cleavage, but the kcat was reduced in both cases (Table 3). Each of these PAR1 mutants has an intact hirudin-like sequence (K51YEPF55) that binds α-thrombin’s exosite I. This high affinity interaction allows each of the mutants to bind to α-thrombin with the same affinity. In contrast, the mutations at the P4 or P2 affect how the cleavage site fits into the active site of α-thrombin adversely affecting the rate of the reaction. Interestingly, the Leu at P4 had a more dramatic effect than the Pro at P2. Structural data indicate that the P4 position of L-amino acid polypeptide substrates would point to the hydrophobic binding site adjacent to the S1 pocket termed the aryl binding site (30). Our data indicate that this interaction is critical for the proper orientation of the PAR1 cleavage site into the active site of thrombin for efficient cleavage. In contrast, mutating Asp39 at P3 lowered the Km 3-fold over PAR1-wt exodomain (Table 3). However, the catalytic efficiency was the same as PAR1-wt due to a concomitant decrease in kcat. The simultaneous decrease in Km and kcat indicate nonproductive binding (28). These data argue that Asp at P3 is important for orienting the PAR1 exodomain in the active site of thrombin. The increased binding efficiency of this mutant may interfere with turnover of this reaction. When PAR1 peptides were co-crystallized with thrombin, structures were solved in which the active site of thrombin interacted with L38DPR41 or the exosite I region interacted with K51YEPF55; however none of these structures had the exosite I and the active site simultaneously filled (22). The hirudin-like sequence (K51YEPF55) induces a change of conformation when the Ala190-Gly197 region of thrombin with the Glu192 side chain becomes disordered helping to accommodate the negatively charged Asp at P3 for PAR1. The crystals in which the active site is filled by the L38DPR41 sequence shows that Leu38 occupies the aryl binding pocket formed by Ile174 and Trp215 as predicted by Bode et al (30). The intervening sequence between L38DPR41 and K51YEPF55 of PAR1 (F43LLRNP48) is disordered with no electron density (22). Using the data from the two sets of crystals in Mathews et al. (PDB ID codes 1NRS and 1NRN), Huntington has proposed models for a single PAR1 peptide interacting with thrombin’s active site and exosite I simultaneously (31).
In the cleavage experiments, a single α-thrombin binds to a single PAR1 exodomain. In contrast, for the Ki experiments, α-thrombin binds to a PAR1 exodomain (at exosite I) and Sar-Pro-Arg-pNA (at the active site) simultaneously. As a result, the PAR1-wt, PAR1-L38A and PAR1-D39A exodomains are noncompetitive inhibitors of α-thrombin cleaving Sar-Pro-Arg-pNA. As in the cleavage experiments, the P3 mutant (PAR1-D39A) also binds α-thrombin more tightly in this assay. In contrast, the PAR1-P40A exodomain displayed mixed type of inhibition. These data may indicate that mutating the P2 position alters the fit of the exodomain in the active site of thrombin. However, as indicated in the Results, the difference between the two models was small for mixed inhibition versus pure noncompetitive inhibition. Therefore, one cannot rule out that this interaction is also a pure noncompetitive inhibitor. The importance of the exosite I binding region on PAR1 for thrombin interaction was further confirmed by blocking this interaction with hirugen. In the presence of hirugen, PAR1-wt exodomain was unable to inhibit hydrolysis of the chromogenic substrate. Jacques et al. demonstrated that PAR1 exodomain mutants that have the hirudin-like sequence (K51EYPF55) deleted or mutated to alanine were also unable to inhibit thrombin hydrolyzing a chromogenic substrate further emphasizing the role of this region (29). In sum, these combined data indicate that the exosite I interactions are the primary determinants of PAR1 binding and explain why mutations at the cleavage site do not affect binding but have an influence on the kcat. The influence of exosite interactions have also been described for prothrombin activation by prothrombinase in which point mutations at cleavage sites do not influence overall binding due to exosite interactions, but influence the rate of cleavage (32).
Jacques and Kuliopulos prepared chromogenic substrates based on the PAR1, PAR3 and PAR4 cleavage sites from mouse and human sequences of these receptors and after cleavage studies concluded that α-thrombin interacts with PAR4 through the prolines at P4 and P2 (29). We directly tested the role of these residues interacting with α-thrombin using full exodomains and our data support these conclusions. However, our study also demonstrates that Leu43 at P5 influences the rate of PAR4 cleavage by thrombin to the same extent as Pro44 at P4. NMR studies with the PAR4 peptide S38TPSILPAPR47 demonstrates that Leu43 interacts with Pro44 or Pro46 and has a minor role in interacting with thrombin. Our data with entire exodomains suggest that Leu43 is as important as Pro44 at P4 for efficient cleavage of PAR4 by thrombin. Thus, the hydrophobic properties of Leu43 at P5 may be required for the proper 3-dimensional arrangement of the amino acids at the α-thrombin cleavage site. Jacques et al. also demonstrates that the addition of an acetyl group on the N-terminus lowered the Km from 270 to 61 μM and increases the kcat from 49 to 81 s-1 with a net effect of raising the kcat/Km 7.2-fold (29). These authors suggest that the acetyl group serves as a surrogate for the hydrophobic residue at P5 resulting in the lower Km and higher kcat. Our data support this hypothesis for P5 increasing the kcat. Like PAR4-wt, each of the PAR4 mutants are competitive inhibitors of thrombin hydrolyzing Sar-Pro-Arg-pNA, supporting the hypothesis that PAR4 interacts primarily with the active site of thrombin. The point mutations at P5, P4 or P2 do not influence the ability of PAR4 to bind thrombin; however, the amino acids at the cleavage site likely are coordinated for proper insertion into the active site of thrombin for efficient cleavage because these mutations influence the rate of the reaction. The presence of hirugen shifted the Ki of PAR4-wt from 64 to 110 μM (Table 4). These data are in agreement with a published report in which mutating the proposed region for exosite I interaction on PAR4 to alanine shifted the Ki to 113 μM compared to 35 μM for wild type (29). Further, the Km of thrombin cleavage of these mutants was increased from 56 to 208 μM (29). Therefore, PAR4 also has an extended interaction with thrombin. Our data in the current report indicate that PAR4’s extended interaction with thrombin minimizes the influence of individual point mutations on overall binding. However, as with PAR1, these residues are important for productive binding in order to achieve efficient cleavage.
Finally, it is of interest that the presence of the exosite binding region on PAR1, only lowers the Km of α-thrombin cleavage of PAR1 exodomain 2-fold. In contrast, 10 fold more thrombin is required to activate PAR4 than PAR1 on platelets which express both receptors. However, when PAR4 was expressed in COS-7 cells in the absence of PAR1, the EC50 was > 400 fold higher than for PAR1 (29). Therefore, there is a dramatic difference in the rate of PAR4 activation depending on the context in which it is expressed. A recent report has reconciled the difference of PAR4 activation on COS-7 cells and platelets by demonstrating that PAR1 is a cofactor for PAR4 activation (33). When PAR1 is coexpressed with PAR4, the PAR4 EC50 is lowered 10 fold. This cofactor mechanism is similar to PAR4 activation via PAR3 in mice (34). Thrombin activates PAR4 while its exosite I is bound to the hirudin-like region of PAR1 or PAR3, respectively. However, this mechanism does not completely explain the dramatic difference in PAR1 and PAR4 activation on cells compared to the 2 fold difference in Km of purified exodomain. GPIb has been shown to enhance PAR1 activation on platelets by interacting with exosite II of thrombin (31,35). However, GPIb is specific for platelets and is unlikely expressed on COS7. An alternative possibility is that the length of the PAR4 exodomain is shorter than that of PAR1 (75 versus 62 amino acids). Further, the cleavage site of PAR4 is 32 amino acids from the transmembrane region compared to 60 amino acids for PAR1. Therefore, a potential explanation for the dramatically slower activation of PAR4 on the cell surface is that PAR4 cleavage site is less accessible because it is closer to the plasma membrane. Further kinetic studies are needed on the complete receptors on cells to characterize the complete mechanism(s) of PAR activation.
These studies suggest that compounds directed to the thrombin cleavage site on PAR1 and/or PAR4 may function as selective thrombin receptor activation antagonists. The fact that prolines in the P2 or P4 and leucines in P4 or P5 positions influence thrombin’s interaction with these exodomains suggest that there may be a common 3-dimensional structure that allows for one compound to inhibit thrombin activation of both receptors. Such a finding would explain how peptide RPPGF, the angiotensin converting enzyme breakdown product of bradykinin, binds and blocks the proteolytic cleavage of PAR1 and 4 exodomains and inhibits thrombin-induced platelet activation of both human and mouse platelets (23,36). Based on the results of the current report, the Leu38 of PAR1 and Leu43 are potential targets for the development of thrombin receptor antagonists that act simultaneously on PAR1 and PAR4.
Acknowledgments
The authors would like to thank Dr. Henry I. Mosberg, Dr. Katarzyna Sobczyk-Kojiro and Fernanda Marques (Department of Medicinal Chemistry, College of Pharmacy, University of Michigan) for technical expertise and use of HPLC while at The University of Michigan.
Abbreviations
- PAR
Protease Activated Receptor
Footnotes
This work was supported by Grants HL52779, HL57346, HL61981 (AHS) and NIH Ruth L. Kirschstein National Research Service Award (HL07853) from HHLBI (MTN).
References
- 1.Mann KG, Brummel K, Butenas S. What is all that thrombin for? J Thromb Haemost. 2003;1:1504–1514. doi: 10.1046/j.1538-7836.2003.00298.x. [DOI] [PubMed] [Google Scholar]
- 2.Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407:258–264. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- 3.Swift S, Sheridan PJ, Covic L, Kuliopulos A. PAR1 thrombin receptor-G protein interactions. Separation of binding and coupling determinants in the galpha subunit. J Biol Chem. 2000;275:2627–2635. doi: 10.1074/jbc.275.4.2627. [DOI] [PubMed] [Google Scholar]
- 4.Verrall S, Ishii M, Chen M, Wang L, Tram T, Coughlin SR. The thrombin receptor second cytoplasmic loop confers coupling to Gq-like G proteins in chimeric receptors. Additional evidence for a common transmembrane signaling and G protein coupling mechanism in G protein-coupled receptors. J Biol Chem. 1997;272:6898–6902. doi: 10.1074/jbc.272.11.6898. [DOI] [PubMed] [Google Scholar]
- 5.Steinhoff M, Buddenkotte J, Shpacovitch V, Rattenholl A, Moormann C, Vergnolle N, Luger TA, Hollenberg MD. Proteinase-activated receptors: transducers of proteinase-mediated signaling in inflammation and immune response. Endocr Rev. 2005;26:1–43. doi: 10.1210/er.2003-0025. [DOI] [PubMed] [Google Scholar]
- 6.Riewald M, Petrovan RJ, Donner A, Ruf W. Activated protein C signals through the thrombin receptor PAR1 in endothelial cells. J Endotoxin Res. 2003;9:317–321. doi: 10.1179/096805103225002584. [DOI] [PubMed] [Google Scholar]
- 7.Ruf W, Dorfleutner A, Riewald M. Specificity of coagulation factor signaling. J Thromb Haemost. 2003;1:1495–1503. doi: 10.1046/j.1538-7836.2003.00300.x. [DOI] [PubMed] [Google Scholar]
- 8.Quinton TM, Kim S, Derian CK, Jin J, Kunapuli SP. Plasmin-mediated activation of platelets occurs by cleavage of protease-activated receptor 4. J Biol Chem. 2004;279:18434–18439. doi: 10.1074/jbc.M401431200. [DOI] [PubMed] [Google Scholar]
- 9.Sambrano GR, Huang W, Faruqi T, Mahrus S, Craik C, Coughlin SR. Cathepsin G activates protease-activated receptor-4 in human platelets. J Biol Chem. 2000;275:6819–6823. doi: 10.1074/jbc.275.10.6819. [DOI] [PubMed] [Google Scholar]
- 10.Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, Kuliopulos A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell. 2005;120:303–313. doi: 10.1016/j.cell.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 11.Vu TK, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64:1057–1068. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- 12.Liu LW, Vu TK, Esmon CT, Coughlin SR. The region of the thrombin receptor resembling hirudin binds to thrombin and alters enzyme specificity. J Biol Chem. 1991;266:16977–16980. [PubMed] [Google Scholar]
- 13.Ayala YM, Cantwell AM, Rose T, Bush LA, Arosio D, Di Cera E. Molecular mapping of thrombin-receptor interactions. Proteins. 2001;45:107–116. doi: 10.1002/prot.1130. [DOI] [PubMed] [Google Scholar]
- 14.Jacques SL, LeMasurier M, Sheridan PJ, Seeley SK, Kuliopulos A. Substrate-assisted catalysis of the PAR1 thrombin receptor. Enhancement of macromolecular association and cleavage. J Biol Chem. 2000;275:40671–40678. doi: 10.1074/jbc.M004544200. [DOI] [PubMed] [Google Scholar]
- 15.Myles T, Le Bonniec BF, Stone SR. The dual role of thrombin’s anion-binding exosite-I in the recognition and cleavage of the protease-activated receptor. Eur J Biochem. 2001;1268:70–77. doi: 10.1046/j.1432-1327.2001.01844.x. [DOI] [PubMed] [Google Scholar]
- 16.Vu TK, Wheaton VI, Hung DT, Charo I, Coughlin SR. Domains specifying thrombin-receptor interaction. Nature. 1991;353:674–677. doi: 10.1038/353674a0. [DOI] [PubMed] [Google Scholar]
- 17.Harris JL, Backes BJ, Leonetti F, Mahrus S, Ellman JA, Craik CS. Rapid and general profiling of protease specificity by using combinatorial fluorogenic substrate libraries. Proc Natl Acad Sci U S A. 2000;97:7754–7759. doi: 10.1073/pnas.140132697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le Bonniec BF, Esmon CT. Glu-192----Gln substitution in thrombin mimics the catalytic switch induced by thrombomodulin. Proc Natl Acad Sci U S A. 1991;88:7371–7375. doi: 10.1073/pnas.88.16.7371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ehrlich HJ, Grinnell BW, Jaskunas SR, Esmon CT, Yan SB, Bang NU. Recombinant human protein C derivatives: altered response to calcium resulting in enhanced activation by thrombin. EMBO J. 1990;9:2367–2373. doi: 10.1002/j.1460-2075.1990.tb07411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ishii K, Gerszten R, Zheng YW, Welsh JB, Turck CW, Coughlin SR. Determinants of thrombin receptor cleavage. Receptor domains involved, specificity, and role of the P3 aspartate. J Biol Chem. 1995;270:16435–16440. doi: 10.1074/jbc.270.27.16435. [DOI] [PubMed] [Google Scholar]
- 21.Cleary DB, Trumbo TA, Maurer MC. Protease-activated receptor 4-like peptides bind to thrombin through an optimized interaction with the enzyme active site surface. Arch Biochem Biophys. 2002;403:179–188. doi: 10.1016/s0003-9861(02)00220-5. [DOI] [PubMed] [Google Scholar]
- 22.Mathews II, Padmanabhan KP, Ganesh V, Tulinsky A, Ishii M, Chen J, Turck CW, Coughlin SR, Fenton JW. Crystallographic structures of thrombin complexed with thrombin receptor peptides: existence of expected and novel binding modes. Biochemistry. 1994;33:3266–3279. doi: 10.1021/bi00177a018. [DOI] [PubMed] [Google Scholar]
- 23.Nieman MT, Pagan-Ramos E, Warnock M, Krijanovski Y, Hasan AA, Schmaier AH. Mapping the interaction of bradykinin 1-5 with the exodomain of human protease activated receptor 4. FEBS Lett. 2005;579:25–29. doi: 10.1016/j.febslet.2004.11.041. [DOI] [PubMed] [Google Scholar]
- 24.Lottenberg R, Jackson CM. Solution composition dependent variation in extinction coefficients for p-nitroaniline. Biochim Biophys Acta. 1983;742:558–564. doi: 10.1016/0167-4838(83)90273-x. [DOI] [PubMed] [Google Scholar]
- 25.Johnson ML, Faunt LM. Parameter estimation by least-squares methods. Methods Enzymol. 1992;210:1–37. doi: 10.1016/0076-6879(92)10003-v. [DOI] [PubMed] [Google Scholar]
- 26.Beechem JM. Global analysis of biochemical and biophysical data. Methods Enzymol. 1992;210:37–54. doi: 10.1016/0076-6879(92)10004-w. [DOI] [PubMed] [Google Scholar]
- 27.Myung JI, Pitt MA. Model comparison methods. Methods Enzymol. 2004;383:351–366. doi: 10.1016/S0076-6879(04)83014-3. [DOI] [PubMed] [Google Scholar]
- 28.Fersht A. Enzyme structure and mechanism. W.H. Freeman and Company; San Francisco: 1977. p. 95. [Google Scholar]
- 29.Jacques SL, Kuliopulos A. Protease-activated receptor-4 uses dual prolines and an anionic retention motif for thrombin recognition and cleavage. Biochem J. 2003;376:733–740. doi: 10.1042/BJ20030954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bode W, Turk D, Karshikov A. The refined 1.9-A X-ray crystal structure of D-Phe-Pro-Arg chloromethylketone-inhibited human alpha-thrombin: structure analysis, overall structure, electrostatic properties, detailed active-site geometry, and structure-function relationships. Protein Sci. 1992;1:426–471. doi: 10.1002/pro.5560010402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huntington JA. Molecular recognition mechanisms of thrombin. J Thromb Haemost. 2005;3:1861–1872. doi: 10.1111/j.1538-7836.2005.01363.x. [DOI] [PubMed] [Google Scholar]
- 32.Krishnaswamy S. Exosite-driven substrate specificity and function in coagulation. J Thromb Haemost. 2005;3:54–67. doi: 10.1111/j.1538-7836.2004.01021.x. [DOI] [PubMed] [Google Scholar]
- 33.Leger AJ, Jacques SL, Badar J, Kaneider NC, Derian CK, Andrade-Gordon P, Covic L, Kuliopulos A. Blocking the protease-activated receptor 1-4 heterodimer in platelet-mediated thrombosis. Circulation. 2006;113:1244–1254. doi: 10.1161/CIRCULATIONAHA.105.587758. [DOI] [PubMed] [Google Scholar]
- 34.Nakanishi-Matsui M, Zheng YW, Sulciner DJ, Weiss EJ, Ludeman MJ, Coughlin SR. PAR3 is a cofactor for PAR4 activation by thrombin. Nature. 2000;404:609–613. doi: 10.1038/35007085. [DOI] [PubMed] [Google Scholar]
- 35.DE Candia E, Hall SW, Rutella S, Landolfi R, Andrews RK, DE Cristofaro R. Binding of thrombin to glycoprotein Ib accelerates the hydrolysis of Par-1 on intact platelets. J Biol Chem. 2001;276:4692–4698. doi: 10.1074/jbc.M008160200. [DOI] [PubMed] [Google Scholar]
- 36.Nieman MT, Warnock M, Hasan AA, Mahdi F, Lucchesi BR, Brown NJ, Murphey LJ, Schmaier AH. The preparation and characterization of novel peptide antagonists to thrombin and factor VIIa and activation of protease-activated receptor 1. J Pharmacol Exp Ther. 2004;311:492–501. doi: 10.1124/jpet.104.069229. [DOI] [PubMed] [Google Scholar]