


Abstract
The role of Thr729 in modulating the enzymatic function of human topoisomerase I has been characterized by molecular dynamics (MD) simulation. In detail, the structural–dynamical behaviour of the Thr729Lys and the Thr729Pro mutants have been characterized because of their in vivo and in vitro functional properties evidenced in the accompanying paper. Both mutants can bind to the DNA substrate and are enzymatically active, but while Thr729Lys is resistant even at high concentration of the camptothecin (CPT) anti-cancer drug, Thr729Pro shows only a mild reduction in drug sensitivity and in DNA binding. MD simulations show that the Thr729Lys mutation provokes a structural perturbation of the CPT-binding pocket. On the other hand, the Thr729Pro mutant maintains the wild-type structural scaffold, only increasing its rigidity. The simulations also show the complete abolishment, in the Thr729Lys mutant, of the protein communications between the C-terminal domain (where the active Tyr723 is located) and the linker domain, that plays an essential role in the control of the DNA rotation, thus explaining the distributive mode of action displayed by this mutant.
INTRODUCTION
Topoisomerases are a ubiquitous family of enzymes that are essential for the cellular regulation of DNA supercoiling, generated by processes such as replication, transcription and recombination (1). Human topoisomerase I (hTop1p) is composed of 765 amino acids, arranged in four domains: the NH2-terminal domain (residues 1–214), the core domain (residues 215–635), the linker domain (residues 636–712) and the COOH-terminal domain (residues 713–765) (2,3). Changes in DNA topology are achieved by introducing a transient break in the phosphodiester bond of one strand in the duplex DNA, and the formation of a transient covalent phosphotyrosine bond between the catalytic Tyr723 and the 3′-end of the broken DNA strand (4). Relaxation of supercoiled DNA is obtained by rotation of the scissile DNA strand around the DNA intact one (5). After relaxation, the transient bond between protein and DNA is broken and the DNA strand is religated. The strict control of the rotation step by the enzyme yields to a processive mechanism of catalysis, i.e. the enzyme maintains the binding to a plasmid supercoiled DNA until complete relaxation, without dissociation (5). If key protein regions involved in this step are perturbed, as the linker and nose cone helices, the enzyme carries out a distributive relaxation, i.e. it dissociates from the substrate after the release of only few superhelical turns (6).
HTop1p is of significant medical interest since it is the only target of anti-cancer drugs, such as the camptothecin (CPT) and its water-soluble derivatives. CPT is a plant alkaloid that rapidly blocks both DNA and RNA synthesis, binding specifically and reversibly to the transient covalent enzyme–DNA complex inhibiting the rotation and religation steps of the catalysis. Collision between the stabilized covalent complex intermediate and the replication fork results in double-strand DNA breaks and triggers a cascade of event leading to apoptosis (7).
Several hTop1p mutations, located either near or far from the active site, have been shown to render the enzyme resistant to the CPT family drug (7). X-ray structures of hTop1p in complex with DNA and bound to several anti-cancer agents have explained several aspects of the mechanism of drug resistance (8–10).
The accompanying paper describes the effect of mutation on residue Thr729, located in helix 21 of the C-terminal domain, to Ala, Glu, Lys or Pro (11). The Thr729Ala mutant has been pointed by Kubota and co-authors (12) as resistant to irinotecan in a human lung cancer cell line. The same mutation, when expressed in yeast Saccharomyces cerevisiae, does not show CPT resistance that is achieved only when Thr729 is mutated to Glu, Lys or Pro. Thr729Glu CPT resistance can be explained by the strong inhibition of DNA binding and enzymatic activity, while the structural/dynamic effects on the Thr729Lys mutant are more complex. The plasmid relaxation activity, in fact, is comparable to that of the wild-type enzyme, even though it shows a distributive character, restored to a processive one in excess of substrate, only. Mutation to Pro, finally, shows only a mild reduction in CPT sensitivity and DNA binding, and a relaxation activity comparable to the wild-type system, with a functional processive mechanism.
In this article, we present a molecular dynamics (MD) simulation of the wild type and two of these mutants, Thr729Lys and Thr729Pro, in non-covalent complex with a 22-bp DNA. Our analysis shows the essential role of Thr729 in the protein long range communications functional to the catalytic cycle, being helix 21 in interaction with helices 16–17 in the core domain, helix 20 in the C-terminal domain and helix 19 in the linker domain.
METHODS
The initial configuration of hTop1p was modelled from the 3D structure of the enzyme without the N-terminal domain (topo70), in complex with a linear double helix DNA substrate 22-bp long. In particular, the starting positions for residues 215–633 and 641–765 were obtained from the crystal structure 1a36 (5), and those for residues 203–214 from the crystal structure 1ej9 (13). The seven residues constituting the loop region that connects the linker to the core domain (residues 634–640, which are lacking in the 1a36 PDB structure because of thermal fluctuation) were added to the system by molecular modelling, as already described (14,15), using the SYBYL program (St Louis, MO, USA). The same procedure has been followed to reconstruct the partially missing side chains of the residues not fully detected in the X-ray diffraction structure. The spatial environment of each new residue was checked for close contacts or overlaps with neighbouring residues, and stereochemical regularization of the structures was obtained by the Powell minimization method implemented in the SYBYL program. Residue 729 has been changed from Thr to Lys and Pro to generate the two mutant topo70–DNA complexes.
We used the GROMACS MD package version 3.3.1 (16) to carry out all the simulation, while modelling the systems with the AMBER95 all-atom force field (17). The porting of the AMBER force field in the GROMACS code was implemented by Sorin and Pande (18). The topo70–DNA complexes were placed in a rectangular box (86 × 110 × 130 Å3) filled with TIP3P water molecules (19), and rendered electro neutrals by the addition of 21 Na+ counterions, for the topo70 wild-type and Thr729Pro mutant, or 20 for the Thr729Lys mutant, using the genion program of the GROMACS package, that replaces solvent molecules by ions at the position with the most favourable electrostatic potential. The resulting total systems contained 9415 protein atoms, 1403 DNA atoms, 21 Na+ counterions and 37 235 water molecules for topo70, giving a total of 122 544 atoms; 9415 protein atoms, 1403 DNA atoms, 21 Na+ counterions and 37 302 water molecules for the Thr729Pro mutant, giving a total of 122 745 atoms; and 9423 protein atoms, 1403 DNA atoms, 20 Na+ counterions and 37 302 water molecules for the Thr729Lys mutant, giving a total of 122 752 atoms. Optimization and relaxation of solvent and ions were initially performed keeping the protein and DNA atoms constrained to their initial position with decreasing force constants of 1000 and 300 kJ/(mol nm), for 300 ps. The systems were then simulated for 13 ns in periodic boundary conditions, at a constant temperature of 300 K using the Berendsen's method (20) and at a constant pressure of one bar; the pressure was kept constant (1 bar) using the Rahman-Parrinello barostat (21) and with a 2 fs time step. Pressure and temperature coupling constants were 1 and 0.5 ps, respectively. The electrostatic interactions were taken into account by means of the Particle Mesh Ewald method (22), using a cut-off radius of 0.9 nm in real space, while a cut-off radius of 1.2 nm was chosen for the van der Waals interactions. The lengths of all bonds were kept constant with the Lincs algorithm (23). One nanosecond of simulation of each system required 23 h of wall clock time on a server HP ProLiant DL585 G2, equipped with four AMD Opteron 8220 SE Dual Core for a total of 8 cores at 2.8 GHz each.
The dynamic cross-correlation (DCC) maps of the systems were built with in-house written codes, taking into account only the coordinates of the 563 protein C-alpha atoms since they contain enough information to describe the largest system motions. The elements of the DCC map (Cij) were computed as: 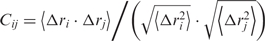 , where Δri is the displacement from the mean position of the i-th atom and the 〈〉 represents the time average over the whole trajectory. Positive Cij values represent a correlated motion between residues i and j (i.e. the residues move in the same direction). Negative values of Cij represent an anti-correlated motion between residues i and j (i.e. they move in opposite directions). Direct hydrogen bonds, root mean square deviations (RMSD) and fluctuations (RMSF) have been carried out using the GROMACS MD package version 3.3.1 (24).
, where Δri is the displacement from the mean position of the i-th atom and the 〈〉 represents the time average over the whole trajectory. Positive Cij values represent a correlated motion between residues i and j (i.e. the residues move in the same direction). Negative values of Cij represent an anti-correlated motion between residues i and j (i.e. they move in opposite directions). Direct hydrogen bonds, root mean square deviations (RMSD) and fluctuations (RMSF) have been carried out using the GROMACS MD package version 3.3.1 (24).
The analyses reported in this article refer to the last 12 ns of the trajectory (i.e. from 1 ns to 13 ns), since the trend of the RMSD indicates that the systems are well stabilized after the first nanosecond. In order to appreciate the statistical convergence of the structural and dynamic results, the average per-residue RMSF and DCC maps analysed after 8 ns of simulation (i.e. from 1 ns to 8 ns) shown in Figures 11–13 in Supplementary data are to be compared with Figures 1, 4 and 6, respectively, analysed in the 1–13 ns time window. The analyses are very similar, indicating a substantial convergence of the simulations. Figures 2, 3, 5 and 10 in Supplementary data were created using the VMD visualization package (http://www.ks.uiuc.edu/Research/vmd/; 25). Graphs have been obtained with the Grace program (http://plasma-gate.weizmann.ac.il/Grace/).
RESULTS
Mutation of residue 729 in lysine or proline has an opposite effect on the fluctuations of human topoisomerase I
The per residue RMSF analysis (Figure 1) indicates that the linker domain of the Thr729Pro mutant is less fluctuating (green line in Figure 1) when compared to the wild protein and the Thr729Lys mutant (black and red lines, respectively). Figure 1 also shows differences for the residues 601–615 fluctuations (in the C-terminal region of the core domain), being very high, intermediate and low for the Thr729Lys mutant, wild-type and Thr729Pro mutant simulations, respectively. Actually, the whole C-terminal region of the core domain, the linker and the C-terminal domain are less mobile in the Thr729Pro system, as compared to the other two systems.
Figure 1.

Average per-residue RMSF represented as a function of the residue number for the wild-type hTop1p protein (black line), Tyr729Lys mutant (red line) and Tyr729Pro mutant (green line).
The average RMSD as a function of simulation time have a similar behaviour among the three systems and their amplitudes are similar to those obtained in other simulations of the topo70–DNA complex (6,14), i.e. up to 3 Å (data not shown). The mean RMSD values vary between 1 Å and 2 Å after elimination of the linker domain from the calculation, confirming a high contribution of this domain on the whole RMSD behaviour.
Interactions between helix 21 and helices 16, 17 and 20
Thr729 is located in the middle of helix 21 (residues 727–735 in the C-terminal domain, Figures 2A and 3A) and its interactions with helices 16 (residues 586–606 in the core domain), 17 (residues 612–629 in core domain) and 20 (713–722 in C-terminal domain) are fundamental for a correct functioning of hTop1p (3). The catalytic residue Tyr723, in fact, is located in the loop between helices 20 (residues 718–722) and 21 and several residues in this region, such as Gly717, Thr718 and Asn722, have been reported to modulate drug sensitivity and enzyme activity, or produce a lethal phenotype in human or yeast topoisomerase I (14,26–28). In the wild-type protein, the only direct helix 21–17 interaction is the hydrogen bond between Thr729 and Tyr619 (Figure 2C), maintained for the whole simulation time. Both the simulations of the two mutated enzymes show the lack of this interaction, but the structural and dynamical effects are different in the two systems. Mutation of residue 729 in Lys, in fact, slightly pulls away helix 17 from the N-terminal portion of helix 21, as observed plotting the distance between the C-alpha atoms of Tyr619 and residue 729 as a function of simulation time (Figure 7 in Supplementary data). However, the most dramatic structural effect caused by this mutation is the occurrence of a new strong interaction between Lys729 and Lys720 that is present for the 80% of the simulation time (Figure 2B). This interaction is never observed in the other two simulations and it induces a conformational perturbation of the drug-binding pocket, as evidenced by the shortening of the Lys729–Asn722 distance when compared to its value in the wild-type and Thr729Pro mutant (Figure 8 in Supplementary data). Note that Asn722, in the crystallographic structure of the ternary topo70–DNA–topotecan complex (8), forms a water-mediated hydrogen bond with the topotecan drug. Moreover, mutation of Ans722 to Ala leads to hTop1p drug resistance (29–31), while mutation to Ser, Asn or His interferes with both enzyme activity and drug sensitivity (27,32).
Figure 2.

(A) Helix 17, in core domain, and helices 20 and 21, in the C-terminal domain, are highlighted in red and light blue colours, respectively. (B–D) Only the helices are shown in representative snapshots of the Tyr729Lys, wild-type and Tyr729Pro simulations, respectively. The side chains of Lys720, in helix 20, of the mutated residue 729, in helix 21 and of Tyr619, in helix 17, are shown in ball and stick. Stable direct hydrogen bonds, when present, are indicated with a white line.
Figure 3.

(A) Helix 16, in core domain, and helix 21, in the C-terminal domain, are highlighted in red and light blue colours, respectively. (B–D) Only the helices are shown in representative snapshots of the Tyr729Lys, wild-type and Tyr729Pro simulations, respectively. The side chains of Thr606 and Trp732, in helixes 16 and 17, respectively, are shown in ball and stick. Stable direct hydrogen bonds, when present, are indicated with a white line.
On the contrary, the Thr729Pro mutation induces a slight reduction of the distance between helix 17 and the N-terminal portion of helix 21 (Figure 7 in Supplementary data), likely correlated with the larger rigidity of the C-terminal portion of the core domain, the linker and the C-terminal domain, as compared to the wild-type and Thr729Lys mutant (Figure 1). Therefore, the Thr729Pro mutation does not perturb the CPT-binding site, being for instance the Pro729–Asp722 distance identical to that in the wild type (Figure 8 in Supplementary data).
Helix 16 takes part to the hTop1p active site with Arg590 and it forms several interactions with the DNA intact strand (3). In the wild-type enzyme, helices 21 and 16 interact through a direct hydrogen bond between Trp732 and Thr606 (Figure 3C). This interaction is maintained in the Thr729Pro mutant (Figure 3D), but not in the Thr729Lys one (Figure 3B). The distance between the C-alpha atoms of Thr606 and Trp732, in the Thr729Pro mutant, is indistinguishable from that of the wild-type protein (Figure 9 in Supplementary data), while it strongly fluctuates in the Thr729Lys system. Thr606 is located in the C-terminal portion of helix 16 (Figure 10 in Supplementary data), in the 601–615 region that strongly fluctuates in the Thr729Lys mutant simulation, but not in the other two systems (Figure 1). Analysis of the secondary structure along with the simulation time shows a partial loss of secondary structure of the C-terminal region of helix 16 in Thr729Lys mutant enzyme (Figure 10B and C in Supplementary data). A similar loss of secondary structure of the C-terminal region of helix 16 is observed in the crystal structure of the topo70–DNA covalent complex in the absence of topotecan (PDB id 1K4S) (8), where the linker domain is not detected for its high flexibility. In contrast, in the crystal structure of the ternary topo70–DNA–TPT complex (PDB id 1K4T) (8), the linker is resolved and the secondary structure of helix 16 is intact, indicating a structural correlation between the drug-binding pocket, the 601–615 region and the linker domain. Our data propose a structural and dynamical interpretation for the key role played by residue 729 in these long-range protein communications.
Long-range effects of residue 729 mutations on the hTop1p dynamics
The global motions of the proteins have been investigated by means of the DCC map that provides an aggregate picture of the correlated motions occurring between protein residues. Highly positive peaks of the Cij elements of the map (red, yellow and green in Figures 4 and 6), in fact, are indicative of strong correlation in the movement of residues i and j, while negative Cij values (dark and light blue in Figures 4 and 6) indicate that the two residues move along opposite direction (anti-correlated motion). Comparison of the DCC maps for the wild-type and Thr729Lys systems (Figure 4, lower right triangle and upper left triangle, respectively) shows a dramatic change in the protein dynamics. The linker domain in the wild-type system, in fact, moves in a highly anti-correlated way with both the C-terminal domain and the C-terminal region of the core domain (Figure 4, rectangles A1 and C1, respectively). Moreover, these two regions have a strong correlated motion, one with the other (Figure 4, rectangle B1). Mutation of residue 729 in Lys completely abolishes these correlated and anti-correlated motions (Figure 4, rectangles A2, B2 and C2). Therefore, although the fluctuations of the linker domain in the Thr729Lys protein are not significantly different from the wild type (Figure 1), its motion is randomic and it is hindered to participate to the protein domain communications, characterizing the enzymatic functionality of hTop1p (9,33), as also shown by the distributive mode of action described in the accompanying paper (see Figure 6 of the accompanying paper). The perturbation of the local interactions around the mutation site (Figure 2A; and Figure 8 in Supplementary data) suggests that the uncoupling of the correlated motions in the linker domain of the Thr729Lys mutant is triggered by the lack of interactions between helices 21–16 and 21–17, respectively (Figures 2 and 3). The linker domain, in fact, interacts with helix 17 (the only protein region that directly contacts the linker domain also in the X-ray structures) in the three simulations by means of several hydrophobic residues: Val703, Ile714 and Leu716 in helix 19, and Pro613 and Leu617 in helix 17 (Figure 5). Therefore, the helix 17 altered motion in the Thr729Lys mutant, that is a consequence of its different interactions with helix 21, is directly transmitted to the linker domain dynamics.
Figure 4.

Dynamic cross correlation map for the topo70 (lower right triangle) and Tyr729Lys mutant (upper left triangle). Correlated values with 0.50 ≤ Cij < 0.65, 0.65 ≤ Cij < 0.80 and 0.80 ≤ Cij < 1 are represented in green, yellow and red, respectively, while the anti-correlation values −0.80 ≤ Cij, <−0.65 and −0.65 ≤ Cij <−0.50 are represented in dark and light blue, respectively. The correlation range 0 ≤ Cij < 0.50 and the anti-correlation range −0.50 < Cij < 0 are represented in a grey scale depending on the absolute value of the correlation. The value 1, along the diagonal (correlation of the residue with itself), is represented in black.
Figure 6.

Dynamic cross correlation map for the topo70 (lower right triangle) and Tyr729Pro mutant (upper left triangle). Correlated values with 0.50 ≤ Cij < 0.65, 0.65 ≤ Cij < 0.80 and 0.80 ≤ Cij < 1 are represented in green, yellow and red, respectively, while the anti-correlation values −0.80 ≤ Cij, <−0.65 and −0.65 ≤ Cij <−0.50 are represented in dark and light blue, respectively. The correlation range 0 ≤ Cij < 0.50 and the anti-correlation range −0.50 < Cij < 0 are represented in a grey scale depending on the absolute value of the correlation. The value 1, along the diagonal (correlation of the residue with itself), is represented in black.
Figure 5.

(A) Helix 17, in core domain, and helix 19, in the linker domain, are highlighted in red and green colours, respectively. (B–D) Only the helices are shown in representative snapshots of the Tyr729Lys, wild-type and Tyr729Pro simulations, respectively. The side chains of Pro613, Leu617, Ala627, in helix 17, and Val703, Ile714, Leu716, in helix 19, are shown in ball and stick.
Comparison of the DCC maps for the wild-type and Thr729Pro systems (Figure 6, lower right and upper left triangles, respectively) shows the vanishing of the correlated and anti-correlated motions already observed for the Thr729Lys mutant (highlighted in Figure 6 by rectangles A1, B1 and C1 in the wild-type system) and the appearance of a new strong correlated motion among the linker domain residues (highlighted in Figure 6 by circle A2). All the regions involved in these interactions (i.e. the C-terminal region of the core domain, the linker domain and the C-terminal domain) show an increased rigidity in the Thr729Pro mutant (Figure 1), likely responsible for the reduction of correlated motions. An increase of anti-correlated motion between the linker domain and the region containing the nose cone helices 5 and 6 (in the core sub-domain II and I, respectively) is also observed (highlighted in Figure 6 by rectangle B2), indicating, in the Thr729Pro mutant, a strong communication between the two protein regions involved in the control of the DNA strand rotation. This is in line with the experimental observation of a more processive mode of relaxation for this mutant (see Figure 6 of the accompanying paper).
DISCUSSION
The experimental characterization of Thr729 mutations in hTop1p, described in the accompanying paper, draws a complex picture of the role of this residue in enzymatic activity and drug sensitivity, since the four studied mutations (Ala, Lys, Pro and Glu) exert different behaviours. Mutation of residue 729 to Ala does not significantly alter the in vivo and in vitro properties of the enzyme, when compared to the wild type. Mutation to Glu shows in vivo drug resistance (Figures 1 and 4 of the accompanying paper), a strong reduction in DNA binding (Figure 7 of the accompanying paper) and thus a significant reduction in DNA-relaxation activity (Figures 2 and 6 of the accompanying paper). Mutation to Lys induces CPT resistance (Figures 1 and 4 of the accompanying paper), a less dramatic reduction in DNA binding (Figure 7 of the accompanying paper) and a DNA-relaxation activity nearly comparable to that of the wild-type enzyme (Figure 2 of the accompanying paper). In this mutant, the relaxation mechanism is altered when tested in excess of DNA substrate (Figure 6A of the accompanying paper), and a mode of relaxation similar to wild type is restored only when the reaction is carried out in excess of enzyme (Figure 6B of the accompanying paper). Mutation to Pro induces an intermediate behaviour between the wild-type and the Thr729Lys mutant, showing a mild reduction in drug sensitivity and in DNA binding (Figures 1, 4 and 7 of the accompanying paper) and a mode of relaxation similar to wild type also in excess of DNA substrate (Figure 6A of the accompanying paper).
We have carried out a structural and dynamical characterization of the two mutants, i.e. Thr729Lys and Thr729Pro that experimentally maintain DNA binding and relaxation activity while showing a large difference in drug resistance.
MD simulation of the non-covalent hTop1p–DNA complex with the single Thr729Lys mutation shows a structural alteration of the drug-binding pocket provoked by the loss of the 729–619 interaction and the occurrence of the Lys729–Lys720 one (Figure 2B; and Figure 8 in Supplementary data). The shortening of the Lys729–Asn722 distance explains the experimentally observed drug resistance of the Thr729Lys mutant, since Asn722 in the wild-type enzyme forms a water-mediated contact with the topotecan anti-cancer drug and its mutation leads to hTop1 drug resistance (8,27,29–32).
Residue 729 plays an essential role in catalysis also. The MD simulation of the wild-type enzyme shows how helix 21, containing the mutated residue, is involved in a network of interactions between helices 21–16, 21–17 and 17–19, that mechanically couples the region controlling the DNA rotation (the linker and the nose cone helices) with the active site region. Mutation of residue 729 to Lys abolishes these protein communications (Figure 4), giving a rationale to the experimentally observed distributive mechanism of DNA relaxation. These long-range effects are, in fact, triggered by the 729 mutation, through perturbation of the helix 21–helix 17 interaction that is transmitted to both the drug-binding pocket (through helix 21–helix 20 interactions) and the linker domain (through helix 17–helix 19 interactions).
On the contrary, the Thr729Pro mutation does not perturb the drug-binding pocket geometry, being the effect of the mutation opposite to that induced by the Thr729Lys mutation, i.e. the geometrical arrangement of helices 21–16–17 is maintained while their dynamic motions are reduced in amplitude, thus suggesting the structural reason for its CPT sensitivity. Moreover, the linker domain and nose cone helices show a strong increase in correlation motion, demonstrating the efficiency of communication in the protein region involved in the control of DNA rotation, again in line with the experimental evidence of the DNA-relaxation processivity.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGMENTS
The authors wish to thank Alessandro Grottesi and Giordano Mancini for helpful discussions and the CASPUR consortium for the computing facilities used in this work. A.D. acknowledges support by the AIRC project ‘Characterization of human topoisomerase I mutants resistant to camptothecin and its derivatives’.
FUNDING
C.L. was supported by an Adriano Buzzati-Traverso fellowship. Funding for open access charge: CASPUR.
REFERENCES
- 1.Wang JC. DNA topoisomerases. Ann. Rev. Biochem. 1996;65:635–692. doi: 10.1146/annurev.bi.65.070196.003223. [DOI] [PubMed] [Google Scholar]
- 2.Stewart L, Ireton GC, Champoux JJ. The domain organization of human topoisomerase I. J. Biol. Chem. 1996;271:7602–7608. doi: 10.1074/jbc.271.13.7602. [DOI] [PubMed] [Google Scholar]
- 3.Redinbo MR, Stewart L, Kuhn P, Champoux JJ, Hol WGJ. Crystal structures of human topoisomerase I in covalent and noncovalent complexes with DNA. Science. 1998;279:1504–1513. doi: 10.1126/science.279.5356.1504. [DOI] [PubMed] [Google Scholar]
- 4.Pommier Y, Pourquier P, Fan Y, Strumberg D. Mechanism of action of eukaryotic DNA topoisomerase I and drugs targeted to the enzyme. Biochim. Biophys. Acta. 1998;1400:83–106. doi: 10.1016/s0167-4781(98)00129-8. [DOI] [PubMed] [Google Scholar]
- 5.Stewart L, Redinbo MR, Qiu X, Hol WGJ, Champoux JJ. A model for the mechanism of human topoisomerase I. Science. 1998;279:1534–1541. doi: 10.1126/science.279.5356.1534. [DOI] [PubMed] [Google Scholar]
- 6.Chillemi G, Redinbo M, Bruselles A, Desideri A. Role of the linker domain and the 203-214 N-terminal residues in the human topoisomerase I DNA complex dynamics. Biophys. J. 2004;87:4087–4097. doi: 10.1529/biophysj.104.044925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat. Rev. Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 8.Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB, Stewart L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc. Natl Acad. Sci. USA. 2002;99:15387–15392. doi: 10.1073/pnas.242259599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chrencik JE, Staker BL, Burgin AB, Pourquier P, Pommier Y, Stewart L, Redinbo MR. Mechanisms of camptothecin resistance by human topoisomerase I mutations. J. Mol. Biol. 2004;339:773–784. doi: 10.1016/j.jmb.2004.03.077. [DOI] [PubMed] [Google Scholar]
- 10.Staker BL, Feese MD, Cushman M, Pommier Y, Zembower D, Stewart L, Burgin AB. Structures of three classes of anticancer agents bound to the human topoisomerase I-DNA covalent complex. J. Med. Chem. 2005;48:2336–2345. doi: 10.1021/jm049146p. [DOI] [PubMed] [Google Scholar]
- 11.Losasso C, Cretaio E, Fiorani P, D'Annessa I, Chillemi G, Benedetti P. A single mutation in the 729 residue modulates human DNA topoisomerase IB DNA binding and drug resistance. Nucleic Acids Res. doi: 10.1093/nar/gkn557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kubota N, Kanazawa F, Nishio K, Takeda Y, Ohmori T, Fujiwara T, Terashiman Y, Saijo N. Detection of topoisomerase I gene point mutation in CPT-11 resistant lung cancer cell line. Biochem. Biophys. Res. Comm. 1992;188:571–577. doi: 10.1016/0006-291x(92)91094-7. [DOI] [PubMed] [Google Scholar]
- 13.Redinbo MR, Champoux JJ, Hol WG. Novel insights into catalytic mechanism from a crystal structure of human topoisomerase I in complex with DNA. Biochemistry. 2000;39:6832–6840. doi: 10.1021/bi992690t. [DOI] [PubMed] [Google Scholar]
- 14.Chillemi G, Fiorani P, Castelli S, Bruselles A, Benedetti P, Desideri A. Effect on DNA relaxation of the single Thr718Ala mutation in human topoisomerase I: a functional and molecular dynamics study. Nucleic Acids Res. 2005;33:3339–3350. doi: 10.1093/nar/gki642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chillemi G, Bruselles A, Fiorani P, Bueno S, Desideri A. The open state of human topoisomerase I as probed by molecular dynamics simulation. Nucleic Acids Res. 2007;35:3032–3038. doi: 10.1093/nar/gkm199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. GROMACS: fast, flexible and free. J. Comp. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 17.Cornell WD, Cieplak P, Bayly CI, Gould IR, Kenneth M, Merz J, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, et al. A second generation force field for the simulation of proteins, nucleic acids and organic molecules. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- 18.Sorin EJ, Pande VS. Exploring the Helix-coil transition via all-atom equilibrium ensemble simulations. Biophys. J. 2005;88:2472–2493. doi: 10.1529/biophysj.104.051938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 20.Berendsen HJC, Postma JPM, van Gusteren WF, Di Nola A, Haak JR. Molecular dynamics with coupling to an external bath. J. Comput. Phys. 1984;81:3684–3690. [Google Scholar]
- 21.Parrinello M, Rahman A. Polymorphic transitions in single-crystals - a new molecular-dynamics method. J. Appl. Phys. 1981;52:7182–7190. [Google Scholar]
- 22.Darden T, York D, Pedersen L. Particle mesh Ewald-an N.log(n) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 23.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. [Google Scholar]
- 24.Lindahl E, Hess B, van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Mod. 2001;7:306–317. [Google Scholar]
- 25.Humphrey W, Dalke A, Shulten K. VMD-visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 26.van der Merwe M, Bjornsti M-A. Mutation of Gly721 alters DNA topoisomerase I active site architecture and sensitivity to camptothecin. J. Biol. Chem. 2008;283:3305–3315. doi: 10.1074/jbc.M705781200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fertala J, Megonigal MD, Bjornsti M-A. Substitutions of Asn-726 in the active site of yeast DNA topoisomerase I define novel mechanisms of stabilizing the covalent-enzyme DNA intermediate. J. Biol. Chem. 2000;275:15246–15253. doi: 10.1074/jbc.275.20.15246. [DOI] [PubMed] [Google Scholar]
- 28.Fiorani P, Amatruda JF, Silvestri A, Butler RH, Bjornsti MA, Benedetti P. Domain interactions affecting human DNA topoisomerase I catalysis and camptothecin sensitivity. Mol. Pharmacol. 1999;56:1105–1115. doi: 10.1124/mol.56.6.1105. [DOI] [PubMed] [Google Scholar]
- 29.Knab AM, Fertala J, Bjornsti M-A. Mechanisms of camptothecin resistance in yeast DNA topoisomerase I mutants. J. Biol. Chem. 1993;268:22322–22330. [PubMed] [Google Scholar]
- 30.Knab AM, Fertala J, Bjornsti M-A. A camptothecin-resistant DNA toposiomerase I mutant exhibits altered sensitivities to other DNA topoisomerase poisons. J. Biol. Chem. 1995;270:6141–6148. doi: 10.1074/jbc.270.11.6141. [DOI] [PubMed] [Google Scholar]
- 31.Hann CL, Carlberg AL, Bjornsti M-A. Intragenic suppressors of mutant DNA topoisomerase I-induced lethality diminish enzyme binding of DNA. J. Biol. Chem. 1998;273:31519–31527. doi: 10.1074/jbc.273.47.31519. [DOI] [PubMed] [Google Scholar]
- 32.Leteurtre F, Fujimori A, Tanizawa A, Chhabra A, Mazumder A, Kohlhagen G, Nakano H, Pommier Y. Saintopin, a dual inhibitor of DNA topoisomerases I and II, as a probe for drug-enzyme interactions. J. Biol. Chem. 1994;269:28702–28707. [PubMed] [Google Scholar]
- 33.Chillemi GFP, Benedetti P, Desideri A. Protein concerted motions in the DNA-human topoisomerase I complex. Nucleic Acids Res. 2003;31:1525–1535. doi: 10.1093/nar/gkg242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.