

Abstract
To improve the efficacy and selectivity of virotherapy for malignant glioma, we designed a strategy to amplify adenoviral replication in conjunction with radiotherapy using a radio-inducible promoter. First, we compared the radiation inducible activity of FLT-1, VEGF, DR5, Cox2, and survivin. We then examined the capacity of the optimal promoter to modulate transgene expression followed by E1A activity in vitro and in vivo in a glioma stem cell model. In the presence of radiation, survivin mRNA activity increased 10-fold. Luciferase transgene expression was dose dependent and optimal at 2Gy. A novel oncolytic adenovirus, CRAd-Survivin-pk7, showed significant toxicity and replication against a panel of passaged and primary CD133+ glioma stem cells. Upon delivery of radiation, the toxicity associated with CRAd-Survivin-pk7 increased by 20–50% (p<0.05). At the same time, the level of E1A activity increased 3-10 fold. In vivo, treatment of U373MG CD133+ stem cells with CRAd-Survivin-pk7 and radiation significantly inhibited tumor growth (p<0.05). At the same time, the level of E1A activity was 100 fold increased vs. CRAd-Survivin-pk7 alone. Selected genes linked to radioinducible promoters whose expression can be regulated by ionizing radiation may improve the therapeutic ratio of virotherapy. In this study, we have identified a new radioinducible promoter, survivin, which greatly enhances the activity of an oncolytic adenovirus in the presence of low dose radiotherapy.
Keywords: Glioblastoma multiforme (GBM), Conditionally replicating adenovirus (CRAd), Radiotherapy, Survivin, CD133, tem cells, Radioinducible promoters
Introduction
Glioblastoma multiforme (GBM) represents one of the most aggressive forms of primary brain cancer. The two-year survival rate is 26.5 percent with radiotherapy plus temozolomide and 10.4 percent with radiotherapy alone (1). These tumors are morphologically diverse, highly invasive, and generally recur close to the original tumor resection secondary to disseminated micrometastases (2, 3). Recently, a fraction of cells resistant to therapy due to activation of the HH-GLI pathway (4, 5) also known as CD133+ cancer stem-cells, have been identified in the context of glial tumors (6). These cells are believed to be responsible for glioma recurrences and decreased survival (5). CD133+ glioma stem cells are resistant to current modes of therapy, including radiation and temozolomide (TMZ) based chemotherapy and are present at a higher fraction in recurrent gliomas (7). These cells also upregulate anti-apoptotic proteins such as Bcl-2, Bcl-XL, cIAP-1 and survivin (6), indicating that they evade normal cellular regulation. Novel therapies are therefore being pursued in order to improve prognosis and increase the length of survival of patients with GBM by selectively targeting these highly resistant stem cells.
One potential therapy involves the use of oncolytic viral vectors. Recent preclinical studies suggest potential efficacy of these viruses and clinical studies in patients have confirmed the safety profile of these agents (8–11). However, a major limitation of these vectors has been poor replication and poor transduction of neighboring tumor cells following intracranial injection. In order to bypass these problems, a new generation of vectors are being developed which selectively target tumor cells and which allow for increased viral replication. This can be achieved by use of tumor specific promoters or modifications of viral capsids to enhance viral transduction. Our group has previously identified the survivin promoter as a tumor specific promoter (12, 13) (TSP) and shown that an adenoviral vector which utilizes the survivin promoter and binds to heparan sulfate proteoglycans (CRAd-Survivin-pk7) can greatly enhance anti-tumor efficacy in an experimental glioma model (12).
In the present study, we sought to (a) evaluate a series of tumor associated promoters with respect to radioinducible properties, (b) examine the role of radiotherapy in conjunction with oncolytic virotherapy, and (c) test the capacity of our novel oncolytic adenovirus, CRAd-S-pk7, to effectively target CD133+ glioma stem cells. The rationale for this work is based on the fact that very few radioinducible promoters have been identified to date and the possibility of enhancing viral replication via radiation induced promoter modulation could significantly enhance the potential efficacy of virotherapy, especially in the context of glioma stem cells. To the best of our knowledge, this is the first study to show that the survivin promoter is a novel radioinducible promoter which can increase viral replication and enhance the cytopathic effect observed with oncolytic vectors in vivo. Moreover, we also show that CRAd-S-pk7 targets CD133+ cells in-vitro and in vivo, which is relevant in light of the fact that these cells confer resistance to chemo- and radiotherapy in glioma patients and are responsible for recurrence of these malignancies.
Materials and Methods
Cell lines
The human glioma U373MG cancer cell line was obtained from American Type Culture Collection (Rockville, MD). U118MG was provided by Dr. Joanne Douglass (University of Alabama Birmingham, AL, USA). These cells were grown in MEM with 10% fetal calf serum, 100 μg/mL penicillin, 100 μg/mL streptomycin and 100X Antibiotic-antimycotic solution (Invitrogen, Carlsbad, CA). Glioblastoma multiforme samples (GBM) were obtained directly from patients undergoing a resection in accordance with a protocol approved by the Institutional Review Board at the University of Chicago. The diagnosis of a grade IV astrocytoma was confirmed by an attending neuropathologist.
Culture of primary glioblastoma cells
Specimens were washed in sterile saline at 4°C and processed immediately after resection by dissociation with mechanical disruption through a 40 μm strainer (Fisher Scientific, USA). Erythrocytes were lysed using NH4Cl. Trypan blue staining confirmed >80% viability after the procedure. Tumor cells were resuspended in Neurobasal media (Cat #10888022, Invitrogen), containing 20 ng/ml of recombinant basic fibroblast growth factor (bFGF, Cat #PH60024, Invitrogen), 20 ng/ml of Epidermal Growth Factor (EGF, Cat #13247051, Invitrogen), N2 supplement (Cat #17502048, Invitrogen), B27 supplement (Cat #17504044, Invitrogen) and 10% heat-inactivated FBS (Atlanta Biochemicals, Atlanta, GA). The cultured cells were maintained for 2 passages.
Magnetic separation of CD133+ glioma stem cells
For magnetic separation of CD133 glioma stem cells, the samples were dissociated and resuspended in PBS containing 0.5% bovine serum albumin. CD133+ cells were isolated using the Miltenyi Biotec CD133 isolation kit (Miltenyi Biotec, Auburn, CA). Positive magnetic cell separation (MACS) was done using several MACS columns in series. The purity of isolated cells was determined by staining with CD133/2-APC (Miltenyi Biotec) or isotype control antibody following analysis on a BD FACSCalibur (BD Biosciences, San Jose, CA). Sterile aliquots of CD133+ and CD133- cells were resuspended in complete media mentioned above and maintained for experiments.
Viruses
The AdCMV-Luc, AdVEGF, AdDR5-Luc and AdSurvivin-Luc are replication-incompetent adenoviruses. AdCMV-Luc and AdSurvivin-Luc have been characterized previously (14, 15). These vectors contain luciferase genes replacing the E1A region under control of human promoters as follows: 0.51 kb of CMV (16), 2.6 kb of VEGF promoter region (17), 1.2 kb of DR5 (18) and 0.26 kb of survivin (19), respectively. The replication deficient adenoviral vectors were constructed based on homologous recombination between pCMV, pVEGF, pDR5, or pSurvivin shuttle vectors and pVK500 adenoviral backbone that contain the entire adenoviral genome with deletion at E1A region. After rescuing in HEK293 cells, viral particles were purified by double CsCl gradient ultracentrifugation followed by titration of viral particles. Physical particle concentration (viral particles [vp]/ml) was determined by measuring absorbance at OD 260 nM.
The competent vector CRAd-S-pk7 was generated by our group and has been described earlier (12). Briefly, the CRAd virus was constructed using a shuttle vector pSc/Survivin/PA, which carries a human survivin promoter and pVK700-based adenoviral wildtype 5 backbone containing polylysine modification in the fiber knob (CRAd-S-pk7) by homologous recombination in E. coli. AdWT replication competent vector was used as a negative control (20). All viruses were selected from single plaque on 911 cells, expanded in A549 cells, and then purified by double CsCl gradient ultracentrifugation (21).
Cellular response to radiation
For optimal dose determination, 5× 104 U373MG were plated in 6-well plates. The cells were then infected with either AdCMV-Luc or AdSurvivin-Luc (1000 vp per cell). After one-hour adsorption, virus-containing media was replaced with fresh growth media. After 24 hours, cells were irradiated (XRT) with doses of 0, 2, 4, 8 and 12 Gy using a γ-irradiator (Philips MG324, Hamburg, Germany). Twenty four hour later cells were analyzed by Luciferase assay. For in vitro experiments, 1× 103 of unsorted or CD133+ cells (U373MG, GBM1 or GBM2) were grown in 96-well flat-bottom plates (Costar, Corning Inc, Corning, NY). Twenty-four hours later, the cells were infected with AdWT, CRAd-S-pk7, or mock infected (1000 vp per cell) as described above. After 24 hrs, the cells were exposed to radiation at 2Gy followed by addition of fresh growth media. Twenty-four hours after XRT, virus-induced toxicity was measured by CytoTox Cytotoxicity Assay kit (Promega, Madison, WI) as per manufacturers’ instruction. A fraction of the cells was subjected to qPCR analysis for E1A expression.
Quantitative RT-PCR and PCR
Total RNA (5μg) was isolated by RNeasy kit (Qiagen, Valencia, CA) and used for cDNA synthesis by oligo (dT) and SuperScript II RNAse H reverse transcriptase (Invitrogen, Carlsbad, CA). Quantitative real-time PCR using SYBR Green PCR master mix (Applied Biosystems, Foster City, CA, USA) was performed with Opticon 2 detection system (Bio-Rad, Hercules, CA) according to manufacturer’s protocol. Primers used in the RT–PCR assays were described before (8, 14, 22). Briefly the following E1A Forward and Reverse primers were used: 5′-AAC CAG TTG CCG TGA GAG TTG-3′ and 5′-CTC GTT AAG CAA GTC CTC GAT ACA T-3′. All the assays were carried out in triplicates and three independent experiments were performed to verify the reproducibility of the results. The results were normalized against human GAPDH expression in each of the samples. For E1A replication experiments, total DNA were extracted from infected/non-infected cells by DNeasy Blood and Tissue kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol. Gene expression was quantified by quantitative PCR using SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) and primers recognizing E1A region. For each experiment, a known amount of AdWT template DNA (106, 105, 104, 103, 102, 101 and 100 copies/μl) was used as a standard curve to quantify the E1A copy numbers of the experimental samples. The total volume of the reaction was 10 μL. The primers used for amplification of E1A have been described above (23). DNA amplification was carried out using Opticon 2 system (Bio-Rad, Hercules, CA). All samples were run in triplicate. The relative quantity of target DNAs was quantified against an internal control.
Detection of luciferase expression
Firefly luciferase enzyme activity was measured using the standard single luciferase assay (Promega, Madison, WI) as per manufacturer’s instruction. To compensate for the differences in cell numbers in different samples, the protein concentration of the lysates was measured using the Bradford protein assay (Bio-Rad, Hercules, CA). Luciferase expression is presented as normalized to cellular protein concentration.
In vivo tumor formation and treatment
For in vivo experiments, 8–10 week old nude mice (n=6/group) (Jackson Laboratories, Bar Harbor, ME) were subcutaneously implanted with 3 × 105 of U373MG CD133+ human glioma cells in the right leg. Treatment was started 8 days later when tumor had been established in all mice (100–150 mm3). Tumor bearing mice were randomly assigned a treatment group (CRAd-S-pk7, CRAd-S-pk7 + XRT, AdWT, AdWT + XRT, XRT alone or mock treatment) and were injected with 1000vp/cell of CRAd-S-pk7, AdWT or sterile PBS for mock controls. Twenty-four hours after virus injection, irradiation of the leg bearing the tumor was done using 2Gy with a γ-irradiator (JL Shepherd Associates Model 143-45). Mice were examined daily for evaluation of tumor growth. Tumors were measured with calipers in three mutually perpendicular diameters (x, y, and z) and the volume (V) was calculated as V = (x * y * z)/2. The data is representative of three independent experiments.
Statistical analysis
Results are expressed as mean ± standard deviation (SD). Student’s t-test was used according to the distribution of experimental values. P-values less than 0.05 were accepted as significant differences between groups. In the animal studies, tumor volumes were expressed as median ± quartile deviation. To test for significant differences in tumor size between treatment groups, one way analysis of variance (ANOVA) was conducted.
Results
Survivin is a radio-inducible promoter
To examine the specific expression of possible candidates for promoter inducible elements, we evaluated the level of mRNA expression before and after radiation exposure. As seen in Figure 1A, the mRNA levels of all these genes were upregulated upon exposure to radiation. However while the levels of expression of FLT1, DR5, VEGF and Cox2 increased two to three-fold, the survivin gene was upregulated to about 10 fold, indicating a strong radiation inducible promoter.
Figure 1. Real-Time PCR detection of promoter activity after radiation exposure.
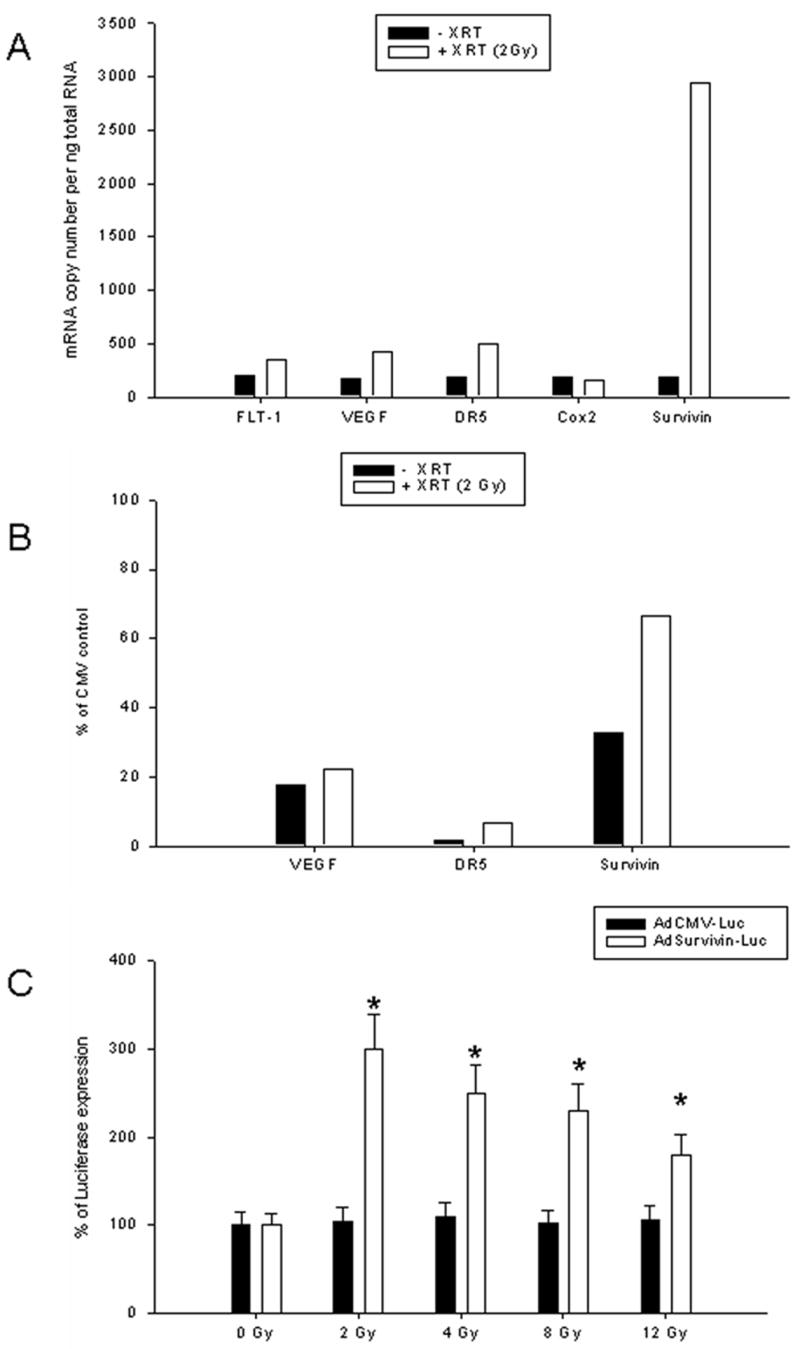
(A) Five different human promoters: FLT-1, DR5, VEGF, Cox2, and Survivin were assessed for expression of mRNA in U118MG cells 2 hours after exposure to 2Gy radiation using quantitative RT-PCR. The levels of mRNA expression in treated and untreated cells was normalized using GAPDH expression and presented as a bar diagram. Experiments were performed three times in triplicates each. (B) Detection of luciferase expression using replication deficient adenovirus before and after radiation. Adenoviral constructs containing DR5, Survivin or VEGF promoter driving luciferase expression were used for infection of U118MG cells. AdVEGF-Luc and AdDR5-Luc demonstrated an increase of 5–7% in luciferase expression in response to radiation. AdSurvivin-Luc, however, showed an increase from 30% to 70% in response to radiation. (C) Dose–dependent transcriptional activity of survivin promoter measured by luciferase assay. U373MG cells were infected with adenoviral constructs (1000vp/cell) containing the luciferase gene either under control of survivin or CMV promoters. After virus infection (2 h), cells were exposed to 2, 4, 6, 8 or 12 Gy radiation and grown for an additional 24h. Total luciferase activity was measured and values were normalized to amounts of total protein. Experiments were performed twice in triplicates. Mean ± SD is presented. p values were < 0.05 compared to non-radiated control (*).
To further investigate the ability of the survivin promoter to drive a luciferase gene in an adenoviral background, we used replication deficient Ad constructs and examined luciferase expression. We infected U118MG cell lines and 24 hrs later irradiated these cells with 2Gy of radiation. Luciferase assay was performed 24 hours later and the results are shown as percentage of CMV promoter induced luciferase expression. As shown in Figure 1B, AdVEGF-Luc and AdDR5-Luc demonstrated an increase of 5–7% in luciferase expression in response to radiation. AdSurvivin-Luc, however, showed an increase from 30% to 70% in response to radiation.
In order to find the optimal radiation dose for the activation of the survivin promoter, we conducted a dose response assay. U373MG cells were infected with AdSurvivin-Luc and irradiated with a single dose of 2, 4, 8 and 12 Gy. Luciferase assay was performed 24 hours later. As seen at Figure 1C, the maximum response to radiation exposure was observed at dose 2Gy (~3-fold). At higher radiation doses, the response diminished to 2 to 1.5 fold. We therefore chose 2Gy for further studies.
An oncolytic vector that utilizes the survivin promoter is effective against glioma
Using primary tumor samples from three GBM patients along with U373MG cell line, we checked whether CRAd-Survivin-pk7 (CRAd-S-pk7) was able to efficiently target these cells and exert its virolytic effect. Figure 2A shows that significant cell death occurred in response to treatment with CRAd-S-pk7 in cells from the primary tumor samples as well as the passaged glioma cell-line. In all cases, the toxicity was increased by about 25–40% compared to AdWT infected cells (p<0.05). To establish that the cytotoxic effect of the adenovirus was due to replication of the viral genome inside the tumors and not due to any extraneous parameter, we looked at the copy number of the viral protein E1A in the cells. Our results show that the virolytic effect exhibited by CRAd-S-pk7 correlates with E1A copies detected in each samples (Figure 2B). This is consistent with toxicity (Fig 2A) as determined by LDH release and shows that human glioma samples preferentially support replication of CRAd-S-pk7 over AdWT (U373MG 6.7×102 vs. 903.41 copies; GBM samples 1.92×102 vs. 254; 2.93×104 vs. 386; and 3.9×103 vs. 212 copies per ng DNA).
Figure 2. Cytotoxicity and replication of CRAd-S-pk7 in unsorted brain tumor samples.
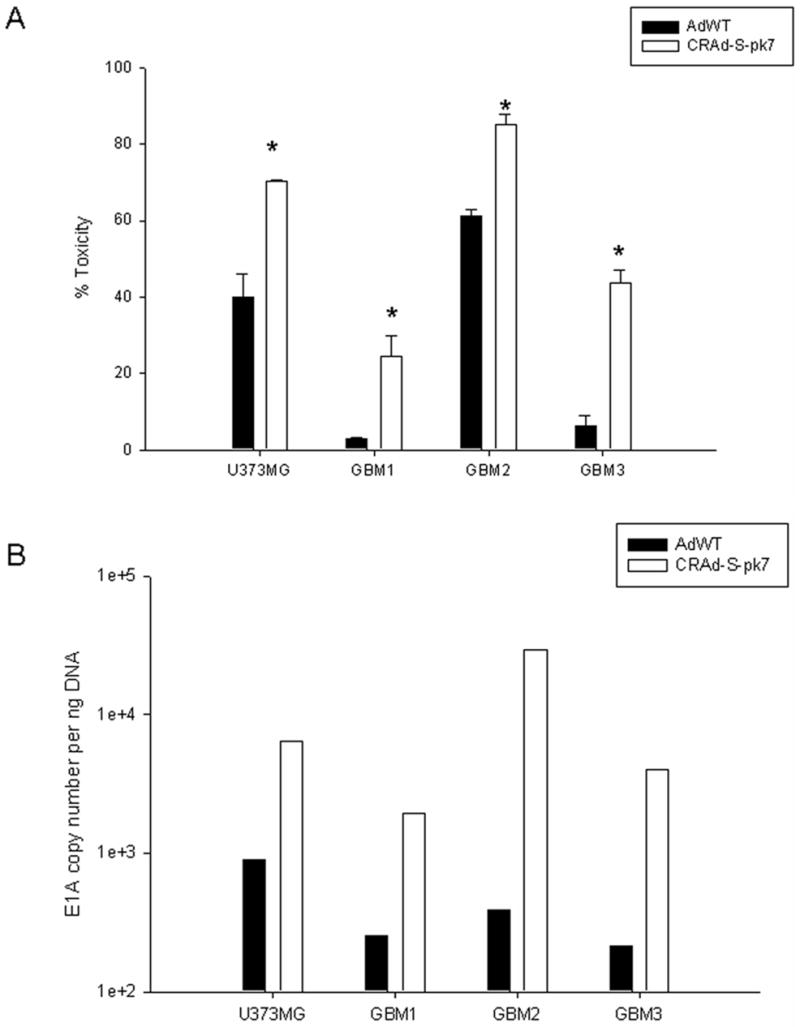
Brain tumor samples were infected with either AdWT or CRAd-S-pk7 and (A) were assayed for cytotoxicity 72 h after infection using CytoTox Cytotoxicity Assay kit or (B) E1A copy numbers were measured by qPCR. In Figure 2A, the results were normalized to untreated cells and are presented as percentage toxicity. Each experiment was performed in 6 replicates. In Figure 2B, E1A copy number is expressed per ng DNA. Measurements were performed in triplicates. Mean ± SD is presented. * p< 0.05.
CRAd-S-pk7 effectively targets CD133+ glioma stem cells
We next examined the response of brain tumor samples enriched for CD133+ cells in response to CRAd-S-pk7 and radiation. First, we analyzed the response of CD133+ glioma stem cells to radiation. As shown in Figure 3, the percentage of CD133+ tumor cells in both primary (GBM1, GBM2) and passaged cell line (U373MG) increased in response to 2Gy radiation (0.6 to 0.7%, 1.7 to 1.9% and 0.75 to 0.9 % respectively), suggesting that stem cells have increased proliferative capacity following radiation therapy. The limiting amounts of cells in the GBM3 sample prevented us from doing this assay in this tumor sample.
Figure 3. Enrichment of CD133-positive cells after radiation in vitro.
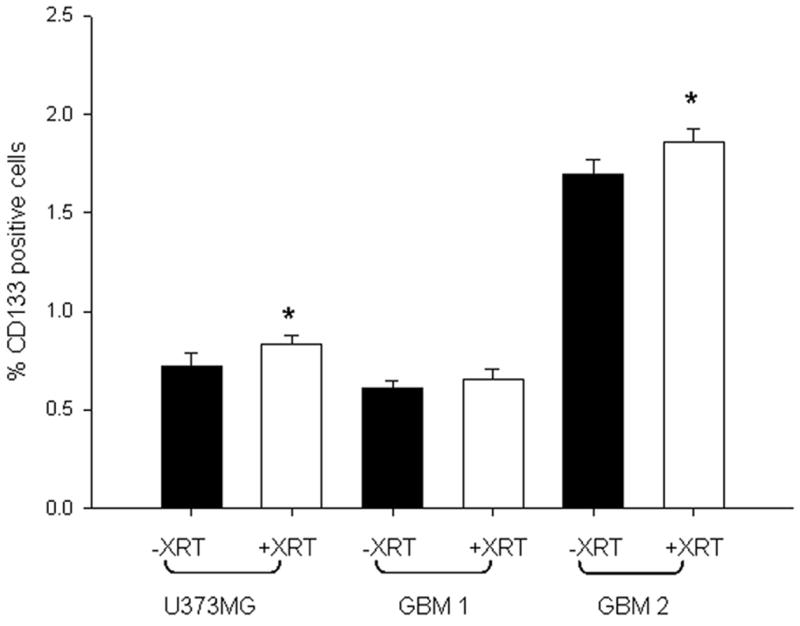
Patient samples GBM1, GBM2 and the cell line U373MG were exposed to 2Gy radiation and grown for 48h along with unexposed controls. CD133+ subpopulation was determined by flow cytometric analysis. Mean ± SD is presented. * p< 0.05.
To further dissect the preferential target of CRAd-S-pk7, we examined its efficacy against CD133- and CD133+ glioma stem cells. First, CD133- cells were infected with wild-type virus or CRAd-S-pk7 ± XRT and then assayed for cytotoxicity and replication efficiency. As shown in Figure 4, we observed an increase in toxicity from 20% to 57% and from 30% to 38% in U373 and GBM1, respectively. There was also a concomitant increase in viral replication of about 5-fold and 2-fold in CRAd-S-pk7 and GBM1, respectively.
Figure 4. Survivin induced killing of CD133- cells in vitro in response to radiation.
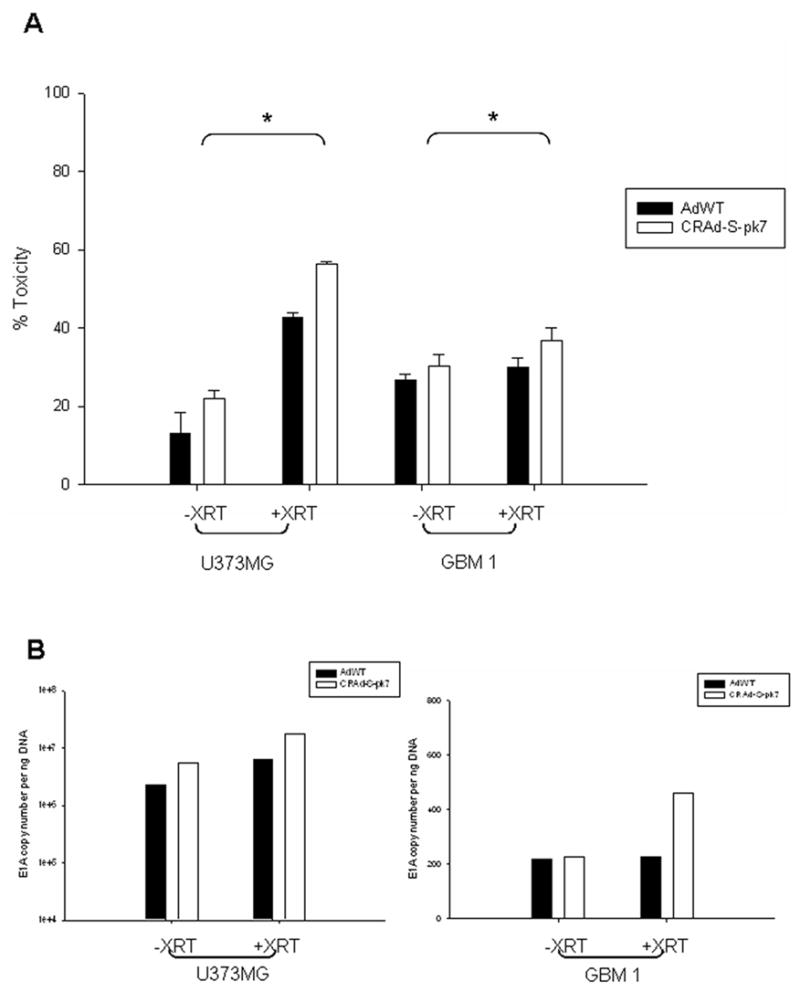
Cells were infected with CRAd-S-pk7 or AdWT and 24h later exposed to 2Gy of radiation. They were then grown for a 24 hr more in complete media. (A) Cytotoxicity was assayed by LDH release and is presented as percent toxicity and normalized to untreated cells (Mean ± SD is presented, * p<0.05). (B) Viral replication was determined by E1A copy number using qPCR and presented as copies per ng total DNA. Experiments were performed twice in triplicates.
Next, to see whether our CRAd could preferentially target CD133+ cells in conjunction with radiation, CD133+ cells from U373MG, GBM1 and GBM2 were infected with wild-type and CRAd-S-pk7 adenovirus. The cells were then assayed for cytotoxicity and replication efficiency. We observed significantly higher toxicity in cells or tumor tissues infected by CRAd-S-pk7 compared to those that were AdWT infected (U373MG 22.09 ± 2.05 vs. 12.9 ± 5.49; GBM1 80.41 ± 2.82 vs. 46.6 ± 7.58; and GBM2 41.9 ± 1.85 vs. 30.98 ± 1.97) (p<0.05) as shown in Figure 5A. The virolytic effect of CRAd-S-pk7 was further enhanced when the cells were exposed to 2Gy radiation. The level of toxicity, as measured by LDH release, increased to 38.99 ± 0.76, 96.82 ± 3.11, and 80.41 ± 12.82 (p<0.05) for U373MG, GBM1 and GBM2, respectively (Figure 5A). The absolute increase in toxicity was therefore greater for CD133+ than CD133- stem cells. This data correlated very well with the increased viral replication in radiated cells particularly those infected with CRAd-S-pk7 (Figure 5B). In fact, radiation exposure induced 1.97, 43.62, and 42.63 fold increase of viral replication for U373MG, GBM1 and GBM2 respectively.
Figure 5. Survivin induced killing of CD133+ cells in vitro in response to radiation.
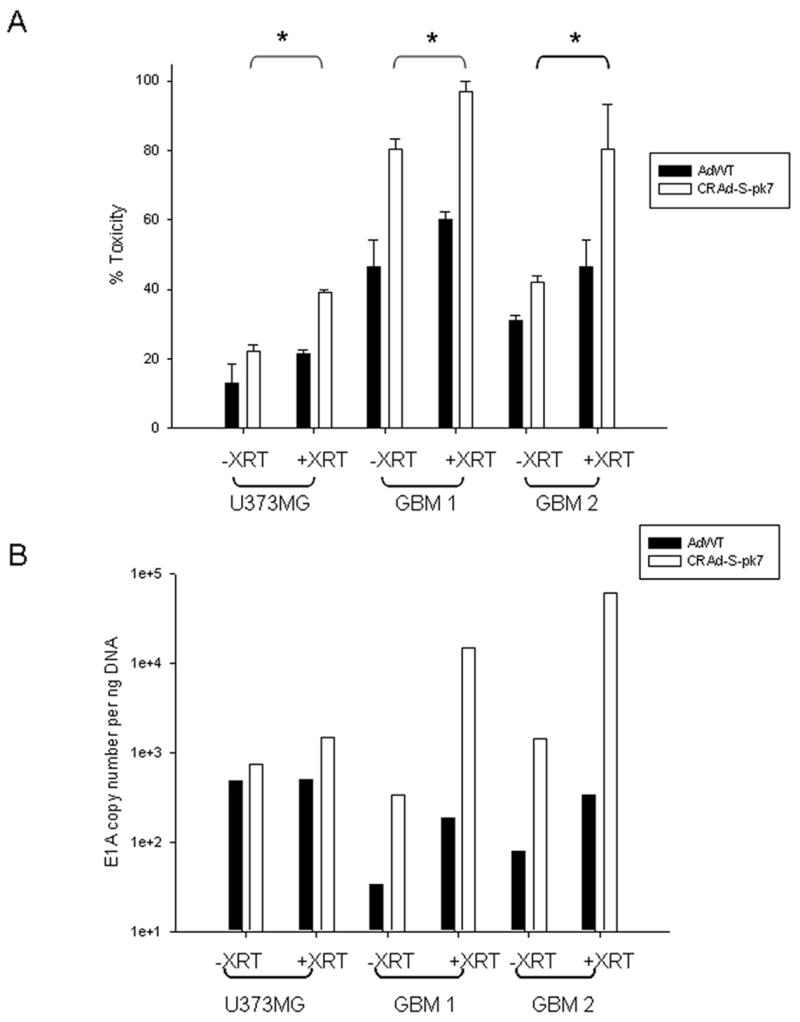
(A) CD133+ enriched tumor cells were infected with either AdWT or CRAd-S-pk7, 24h prior to exposure to radiation (2Gy) followed by 24 hour growth in complete media. Cytotoxicity was measured by LDH assay. (Mean ± SD is presented, * p<0.05) Experiments were performed twice with 6 replicates per condition. (B) Viral replication was determined via E1A copy number measurement using qPCR and presented as copies per ng total DNA. Experiments were performed twice in triplicates.
Tumors formed from CD133+ enriched cells in vivo are targeted by CRAd-S-pk7 and low-dose radiation in a synergistic manner
In order to examine the hypothesis that survivin regulated viral toxicity will be increased in vivo in response to low-dose radiation, we injected nude mice with 3 × 105 of CD133+ U373MG cells. The cells were injected under the skin to facilitate local radiation and allow for precise tumor measurement. After the tumors reached a volume of 100mm3, the mice were randomly divided into six groups and treated as described in the materials and methods. As shown in Figure 6A, median tumor volumes were plotted over a 6-day period. In response to radiation, CRAd-S-pk7 treatment group achieved the most significant tumor growth inhibition (p<0.05). To ascertain the viral replication in response to radiation, three mice from each group were sacrificed at day six after irradiation and the tumors were resected. Viral copy number was ascertained from DNA isolated from these tumors (Figure 6B). The CRAd-S-pk7 + XRT group showed about a 100 fold increase in viral replication compared to CRAd-S-pk7 alone.
Figure 6. In vivo tumor growth rate in nude mice.
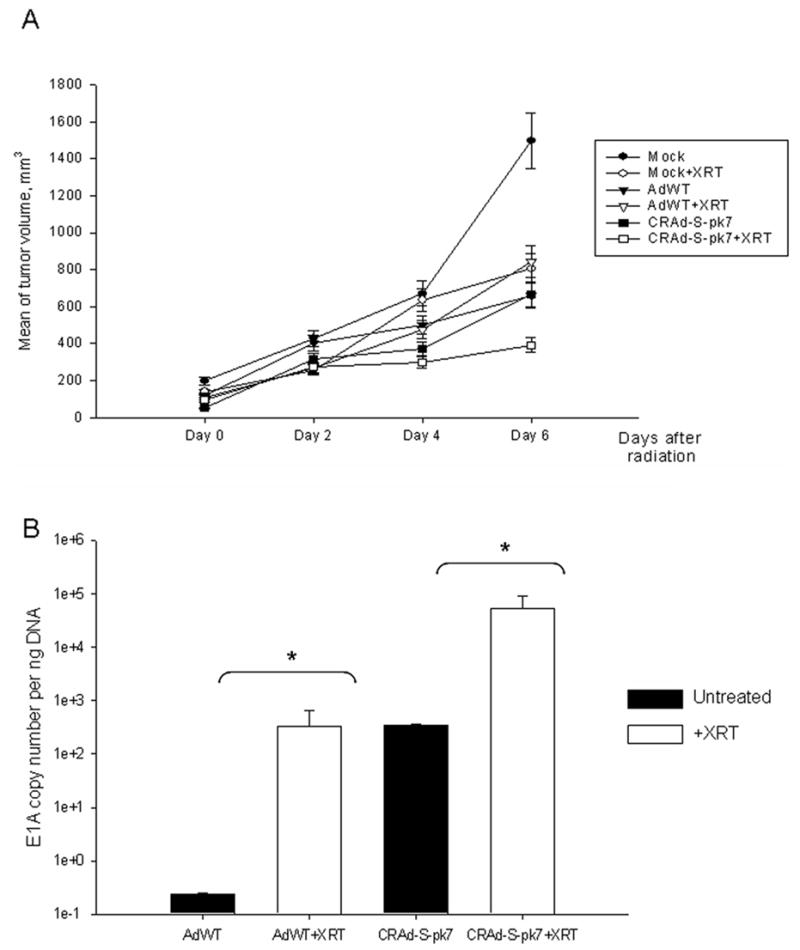
(A) CD133+ enriched U373MG cells were injected s.c. in the right hind leg for tumor formation. After 8 days, when the tumor reached 100mm3, mice were randomly divided in three groups: mock, AdWT or CRAd-S-pk7 treatment and were injected with respective virus at 1000vp/cell or PBS for mock. Twenty four hours later, each group was further divided in two: one group received single dose of radiation of 2Gy, the other group was the non-irradiated control. Tumor volumes were measured for 6 days and the median volumes ± SD were plotted. (B) Viral replication in tumors as measured by E1A qPCR at day six post radiation in each group.
Discussion
A major limitation of oncolytic virotherapy consists of poor in vivo replication and viral spread. This problem, at least in part, is due to the need for attenuation of viral backbone which in turn has been shown to decrease viral propagation and spread. Two examples of this phenomenon include ONYX-015 and G207. The attenuation of E1B in the former and the deletion of the gamma 34.5 (RL1) in the latter, are both associated with decreased viral replication (9). As such, clinical studies of ONYX-015 (Ad virus) or G207 (an HSV virus containing the 34.5 deletion) have failed to show efficacy, partly due to the fact that the virus does not replicate with robust efficiency (8, 10). The ability to enhance viral replication, by means of using tumor selective promoters, is likely to enhance virotherapy. We have previously shown the survivin promoter may represent an optimal TSP and in this study provide compelling data to suggest that radiation, a mainstay of glioma therapy, further enhances survivin mediated adenoviral replication and in vivo efficacy.
To date, very few radioinducible promoters have been described in the literature. A RecA promoter was used as a radiation inducible promoter to increase TNF-alpha production in Clostridium sp (24). The Egr-1 promoter has also been used as a radio-inducible promoter to deliver TNFα to tumor cells (25, 26). The Egr-1 promoter was also studied in the context of radioprotective effect of Flt3 in SCID mice (27) for in-vitro studies on gene activation (28) and for gene expression in context of a hypoxia-inducible promoters (29). However, these genes are neither specifically expressed in tumor cells nor are they upregulated in gliomas. Our current study suggests that survivin is a radioinducible promoter which not only increases transgene expression but also modulates E1A adenoviral activity. Both the in vitro and in vivo studies demonstrate the enhanced activity of a survivin driven oncolytic adenovirus in the presence of low dose radiotherapy, a finding with significant implications for patients who may one day be treated with this virus and subsequently receive radiotherapy.
In addition to showing the radioinducible characteristics of survivin, we also show that a survivin driven CRAd preferentially targets CD133+ glioma stem cells, a novel finding that was not previously reported. Moreover, we show for the first time that in the presence of radiotherapy, there is enhanced viral replication in CD133+ cells as measured by E1A copy number, a finding that translates to improved tumor growth inhibition. CRAd-S-pk7 retains it ability to kill these cells and exposure to low-dose radiation increases the ability of the virus to target these tumors more effectively. In comparison to unsorted cells, the replication of CRAd-S-pk7 increased 10 to 25% in irradiated CD133+ enriched primary GBM tissues compared to the non-irradiated tissue. This indicates specific targeting of the cells by the oncolytic CRAd-S-pk7. The excellent correlation between virus induced cell death and replication of the virus in response to radiation was also observed in a mouse tumor model. We show that mice treated with CRAd-S-pk7 and then exposed to low-dose radiation had significantly smaller tumor size than mice treated with CRAd-S-pk7 alone. While we acknowledge that one potential limitation of our study was the flank glioma model, it nevertheless allowed us the opportunity to precisely monitor tumor growth in response to radiotherapy and virotherapy in vivo. Although studies examining the intracranial model are currently in progress, our current results provide a “proof-of-principle” with regard to the sensitivity of survivin to radiation and enhanced adenoviral replication and killing both in vitro and in vivo.
The exact mechanism responsible for increased E1 activity in response to radiation is unclear. However, in studies involving the Egr-1 gene, the ERK1/2 and the SAP/JNK pathways were found to be activated (30). Moreover the p53 and the NF-κβ transcription factors have been consistently found to be activated in response to clinically relevant levels of radiation (31). While it is possible that the survivin promoter is activated by one of these pathways via the action of these two important transcription factors, further studies are needed to analyze their induction.
In conclusion, we show that exposure to low-dose radiation increases the replication of CRAd-S-pk7 and therefore enhances the cytopathic effect in the context of CD133+ glioma stem cells. CD133+ cells represent the stem-cell fraction of gliomas and the ability of CRAd-S-pk7 to target these cells in the presence of radiotherapy is an important step in improving the prognosis of patients with GBM.
Acknowledgments
We thank Drs. Nikolay Khodarev from the Department of Radiation Oncology for his technical support and Dr. Dingcai Cao for statistical analysis.
Financial support: This work was supported by the National Cancer Institute (R01-CA122930; MSL), National Institute of Neurological Disorders and Stroke (K08-NS046430; MSL), The Alliance for Cancer Gene Therapy Young Investigator Award (MSL), and the American Cancer Society (RSG-07-276-01-MGO; MSL).
References
- 1.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Lesniak MS. Gene therapy for malignant glioma. Expert Rev Neurother. 2006;6:479–88. doi: 10.1586/14737175.6.4.479. [DOI] [PubMed] [Google Scholar]
- 3.Lesniak MS, Brem H. Targeted therapy for brain tumours. Nat Rev Drug Discov. 2004;3:499–508. doi: 10.1038/nrd1414. [DOI] [PubMed] [Google Scholar]
- 4.Stecca B, Ruiz i Altaba A. Brain as a paradigm of organ growth: Hedgehog-Gli signaling in neural stem cells and brain tumors. J Neurobiol. 2005;64:476–90. doi: 10.1002/neu.20160. [DOI] [PubMed] [Google Scholar]
- 5.Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol. 2007;17:165–72. doi: 10.1016/j.cub.2006.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu G, Yuan X, Zeng Z, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–60. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 8.Chiocca EA, Abbed KM, Tatter S, et al. A phase I open-label, dose-escalation, multi-institutional trial of injection with an E1B-Attenuated adenovirus, ONYX-015, into the peritumoral region of recurrent malignant gliomas, in the adjuvant setting. Mol Ther. 2004;10:958–66. doi: 10.1016/j.ymthe.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 9.Yazaki T, Manz HJ, Rabkin SD, Martuza RL. Treatment of human malignant meningiomas by G207, a replication-competent multimutated herpes simplex virus 1. Cancer Res. 1995;55:4752–6. [PubMed] [Google Scholar]
- 10.Markert JM, Medlock MD, Rabkin SD, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000;7:867–74. doi: 10.1038/sj.gt.3301205. [DOI] [PubMed] [Google Scholar]
- 11.Sonabend AM, Ulasov IV, Lesniak MS. Gene therapy trials for the treatment of high-grade gliomas. Gene Ther Mol Biol. 2007;11:79–92. [PMC free article] [PubMed] [Google Scholar]
- 12.Ulasov IV, Zhu ZB, Tyler MA, et al. Survivin-driven and fiber-modified oncolytic adenovirus exhibits potent antitumor activity in established intracranial glioma. Hum Gene Ther. 2007;18:589–602. doi: 10.1089/hum.2007.002. [DOI] [PubMed] [Google Scholar]
- 13.Van Houdt WJ, Haviv YS, Lu B, et al. The human survivin promoter: a novel transcriptional targeting strategy for treatment of glioma. J Neurosurg. 2006;104:583–92. doi: 10.3171/jns.2006.104.4.583. [DOI] [PubMed] [Google Scholar]
- 14.Cooney R, Hynes SO, Duffy AM, Sharif F, O’Brien T. Adenoviral-mediated gene transfer of nitric oxide synthase isoforms and vascular cell proliferation. J Vasc Res. 2006;43:462–72. doi: 10.1159/000095163. [DOI] [PubMed] [Google Scholar]
- 15.Ulasov IV, Rivera AA, Sonabend AM, et al. Comparative Evaluation of Survivin, Midkine, and CXCR4 Promoters for Transcriptional Targeting of Glioma Gene Therapy. Cancer Biol Ther. 2007;6:679–85. doi: 10.4161/cbt.6.5.3957. [DOI] [PubMed] [Google Scholar]
- 16.Zhu ZB, Makhija SK, Lu B, et al. Transcriptional targeting of adenoviral vector through the CXCR4 tumor-specific promoter. Gene Ther. 2004;11:645–8. doi: 10.1038/sj.gt.3302089. [DOI] [PubMed] [Google Scholar]
- 17.Takayama K, Reynolds PN, Adachi Y, et al. Vascular endothelial growth factor promoter-based conditionally replicative adenoviruses for pan-carcinoma application. Cancer Gene Ther. 2007;14:105–16. doi: 10.1038/sj.cgt.7700991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoshida T, Maeda A, Tani N, Sakai T. Promoter structure and transcription initiation sites of the human death receptor 5/TRAIL-R2 gene. FEBS Lett. 2001;507:381–5. doi: 10.1016/s0014-5793(01)02947-7. [DOI] [PubMed] [Google Scholar]
- 19.Zhu ZB, Makhija SK, Lu B, et al. Transcriptional targeting of tumors with a novel tumor-specific survivin promoter. Cancer Gene Ther. 2004;11:256–62. doi: 10.1038/sj.cgt.7700679. [DOI] [PubMed] [Google Scholar]
- 20.Zhu ZB, Makhija SK, Lu B, et al. Targeting mesothelioma using an infectivity enhanced survivin-conditionally replicative adenoviruses. J Thorac Oncol. 2006;1:701–11. doi: 10.1097/01243894-200609000-00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Graham FL. Growth of 293 cells in suspension culture. J Gen Virol. 1987;68(Pt 3):937–40. doi: 10.1099/0022-1317-68-3-937. [DOI] [PubMed] [Google Scholar]
- 22.Immonen A, Vapalahti M, Tyynela K, et al. AdvHSV-tk gene therapy with intravenous ganciclovir improves survival in human malignant glioma: a randomised, controlled study. Mol Ther. 2004;10:967–72. doi: 10.1016/j.ymthe.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 23.Kawakami Y, Li H, Lam JT, Krasnykh V, Curiel DT, Blackwell JL. Substitution of the adenovirus serotype 5 knob with a serotype 3 knob enhances multiple steps in virus replication. Cancer Res. 2003;63:1262–9. [PubMed] [Google Scholar]
- 24.Nuyts S, Van Mellaert L, Theys J, et al. Radio-responsive recA promoter significantly increases TNFalpha production in recombinant clostridia after 2 Gy irradiation. Gene Ther. 2001;8:1197–201. doi: 10.1038/sj.gt.3301499. [DOI] [PubMed] [Google Scholar]
- 25.Weichselbaum RR, Kufe DW, Advani SJ, Roizman B. Molecular targeting of gene therapy and radiotherapy. Acta Oncol. 2001;40:735–8. doi: 10.1080/02841860152619151. [DOI] [PubMed] [Google Scholar]
- 26.Weichselbaum RR, Kufe DW, Hellman S, et al. Radiation-induced tumour necrosis factor-alpha expression: clinical application of transcriptional and physical targeting of gene therapy. Lancet Oncol. 2002;3:665–71. doi: 10.1016/s1470-2045(02)00900-2. [DOI] [PubMed] [Google Scholar]
- 27.Du N, Feng K, Luo C, Li L, Bai C, Pei X. Radioprotective effect of FLT3 ligand expression regulated by Egr-1 regulated element on radiation injury of SCID mice. Exp Hematol. 2003;31:191–6. doi: 10.1016/s0301-472x(02)01082-2. [DOI] [PubMed] [Google Scholar]
- 28.Schmidt M, Heimberger T, Gruensfelder P, Schler G, Hoppe F. Inducible promoters for gene therapy of head and neck cancer: an in vitro study. Eur Arch Otorhinolaryngol. 2004;261:208–15. doi: 10.1007/s00405-003-0621-z. [DOI] [PubMed] [Google Scholar]
- 29.Chadderton N, Cowen RL, Sheppard FC, et al. Dual responsive promoters to target therapeutic gene expression to radiation-resistant hypoxic tumor cells. Int J Radiat Oncol Biol Phys. 2005;62:213–22. doi: 10.1016/j.ijrobp.2005.01.031. [DOI] [PubMed] [Google Scholar]
- 30.Meyer RG, Kupper JH, Kandolf R, Rodemann HP. Early growth response-1 gene (Egr-1) promoter induction by ionizing radiation in U87 malignant glioma cells in vitro. Eur J Biochem. 2002;269:337–46. doi: 10.1046/j.0014-2956.2001.02658.x. [DOI] [PubMed] [Google Scholar]
- 31.Criswell T, Leskov K, Miyamoto S, Luo G, Boothman DA. Transcription factors activated in mammalian cells after clinically relevant doses of ionizing radiation. Oncogene. 2003;22:5813–27. doi: 10.1038/sj.onc.1206680. [DOI] [PubMed] [Google Scholar]