

Abstract
XIAP is a central apoptosis regulator that inhibits apoptosis by binding to and inhibiting the effectors caspase-3/-7 and an initiator caspase-9 through its BIR2 and BIR3 domains, respectively. Smac protein in its dimeric form effectively antagonizes XIAP by concurrently targeting both its BIR2 and BIR3 domains. We report the design, synthesis and characterization of a non-peptide, cell-permeable, bivalent small-molecule (SM-164) which mimics Smac protein for targeting XIAP. Our study shows that SM-164 binds to XIAP containing both BIR domains with an IC50 value of 1.39 nM, being 300 and 7000-times more potent than its monovalent counterparts and the natural Smac AVPI peptide, respectively. SM-164 concurrently interacts with both BIR domains in XIAP and functions as an ultra-potent antagonist of XIAP in both cell-free functional and cell-based assays. SM-164 targets cellular XIAP and effectively induces apoptosis at concentrations as low as 1 nM in leukemia cancer cells, while having a minimal toxicity to normal human primary cells at 10,000 nM. The potency of bivalent SM-164 in binding, functional and cellular assays is 2–3 orders of magnitude higher than its corresponding monovalent Smac mimetics.
Introduction
Apoptosis, or programmed cell death, is a critical cell process in normal development and homeostasis of multicellular organisms. Inappropriate regulation of apoptosis has been implicated in many human diseases, including cancer.1–3 It is now recognized that dysfunction of the apoptosis machinery is a hallmark of cancer.2 Accordingly, targeting critical apoptosis regulators is an attractive approach for the development of new classes of therapies for the treatment of cancer and other human diseases.1–3
The X-linked inhibitor of apoptosis protein (XIAP) is a member of IAP proteins and a central apoptosis regulator, although its role may not be limited to the regulation of apoptosis.5,6 XIAP potently inhibits apoptosis by directly binding to and effectively inhibiting three members of caspases, the two effectors, caspase-3 and -7, and an initiator, caspase-9 (Figure 1A), whose activity is critical for execution of apoptosis. XIAP contains three baculovirus IAP repeat (BIR) domains. The third BIR domain (BIR3) of XIAP selectively targets caspase-9, whereas the BIR2 domain, together with the linker preceding BIR2, inhibits both caspase-3 and caspase-7 (Figure 1B).5 Consistent with its potent apoptosis-suppressing function, XIAP is found to be highly expressed in many human tumor cell lines and tumor samples from patients6 and plays an important role in conferring resistance on cancer cells to a variety of anticancer drugs.7
Figure 1.

(A). A schematic, simplified apoptosis pathway. XIAP inhibits apoptosis by directly binding to and inhibition of caspase-9, caspase-3 and -7. Smac protein binds to XIAP and antagonizes XIAP to promote activation of caspases and apoptosis. (B). Design of bivalent Smac mimetics to target both the BIR2 and BIR3 domains of XIAP by mimicking the binding of dimeric Smac protein.
In cells, the anti-apoptotic function of XIAP is antagonized by Smac/DIABLO (second mitochondria-derived activator of caspases or direct IAP binding protein with low pI) (Figure 1A).8,9 Smac/DIABLO has been identified as a protein released from mitochondria into the cytosol in response to apoptotic stimuli.8,9 It forms an elongated dimer10 and targets both the BIR2 and BIR3 domains in XIAP (Figure 1B).11 It removes the XIAP inhibition of caspase-9 by binding to the BIR3 domain in XIAP through its AVPI tetrapeptide binding motif and directly competing with a similar ATPF tetrapeptide in the processed caspase-9 (Figure 1B).11–15 Conclusive understanding of the mechanism by which Smac removes the inhibition of XIAP to caspase-3/-7 remains elusive.11 Crystal structures16–18 show that the linker preceding BIR2 in XIAP binds to caspase-3/-7 and is the major energetic determinant, while the BIR2 domain itself plays a regulatory role. Modeling suggests that Smac protein also binds to XIAP BIR2 through its AVPI motif and prevents the binding of XIAP to caspase-3/-7.16–19 In this manner, dimeric Smac protein effectively removes the inhibition of XIAP to caspase-9 and to caspase-3/-7 by concurrently targeting both the BIR2 and BIR3 domains in XIAP (Figure 1B).19
Because XIAP blocks apoptosis at the down-stream effector phase, a point where multiple signaling pathways converge, it represents a particularly attractive molecular target for the design of new classes of anticancer drugs aimed at overcoming the apoptosis resistance of cancer cells.7 To date, a number of research laboratories, including ours, have pursued the design of small-molecule antagonists of XIAP.20–28 One approach focuses on the design of small molecules that target the XIAP BIR3 domain, antagonizing the inhibition of XIAP to caspase-9.20–25 These efforts have so far yielded small-molecules that bind to the XIAP BIR3 domain with high affinities. A number of these compounds are effective in inhibition of cell growth and induction of apoptosis in cancer cells.20–25 Another approach is to design small-molecule inhibitors that target the BIR2 domain, blocking the interaction of XIAP with caspase-3/-7.26,27 For example, polyurea compounds that target the caspase-3/XIAP interaction have been shown to achieve broad anticancer activity and are synergistic when used in combination with chemotherapeutic agents.27 Collectively, these studies have provided convincing evidence that targeting XIAP is an effective strategy to overcome the apoptosis resistance of cancer cells.
Since both the BIR2 and BIR3 domains in XIAP effectively suppress apoptosis by targeting caspase-3/-7 and caspase-9 respectively, we reason that bivalent small molecules which concurrently interact with both BIR domains could be particularly efficient and potent XIAP antagonists (Figure 1B). Indeed, a small-molecule mimic containing two Smac AVPI binding motifs was shown to antagonize XIAP with a potency exceeding that of Smac protein in a cell-free functional assay and to enhance apoptosis induction of other therapeutic agents in cancer cells.28 It was proposed28 that its high potency as an XIAP antagonist is attributable to its bivalency, but its precise mode of action in targeting XIAP remains unclear.
In this study, we present the structure-based design, synthesis and detailed characterization of a novel, non-peptidic, bivalent small-molecule (SM-164) using a series of complementary biochemical, functional and cellular assays. We demonstrate that SM-164 concurrently targets the BIR2 and BIR3 domains in the same XIAP molecule, achieves an extremely high binding affinity to XIAP, and functions as an ultra-potent antagonist of XIAP in cell-free functional and cellular assays. It binds potently to cellular XIAP and effectively induces apoptosis in leukemia cancer cells at concentrations as low as 1 nM. Importantly, SM-164 has minimal toxicity to normal human primary cells at concentrations as high as 10,000 nM. Its potency in binding, functional and cellular assays is 2–3 orders of magnitude higher than its corresponding monovalent Smac mimetics.
Results
Design of a cell-permeable monovalent Smac mimetic
Smac protein binds to both the BIR2 and BIR3 in XIAP via its AVPI binding motif.11 It was shown13 that short Smac-based peptides containing the AVPI sequence bind to recombinant XIAP BIR3 and BIR2 proteins with Kd values of 0.4–0.7 μM and 6–9 μM, respectively. The Smac-based AVPI peptide thus provides a suitable template for the design of bivalent small molecules that can concurrently interact with both the BIR2 and BIR3 domains. Unfortunately, Smac-based peptides are not cell-permeable. Thus, for the design of cell-permeable, bivalent Smac mimetics, it was essential to first derive cell-permeable, monovalent Smac mimetics that can bind to both XIAP BIR2 and BIR3 with good affinities.
We have employed a structure-based strategy to design potent, cell-permeable, small molecules (monovalent Smac mimetics) that mimic the Smac AVPI binding motif, although our initial purpose was to target the XIAP BIR3 domain.21–23 Using this approach, we have designed compound 1 (SM-122) as a conformationally constrained, Smac AVPI mimetic containing a [8,5] bicyclic system (Figure 2) and evaluated its binding affinities to XIAP BIR2 and BIR3 proteins. To facilitate the investigation of the molecular mechanism of action for Smac mimetics, we have also designed a biotinylated analogue of SM-122 (2, named BL-SM-122, Figure 2).
Figure 2.
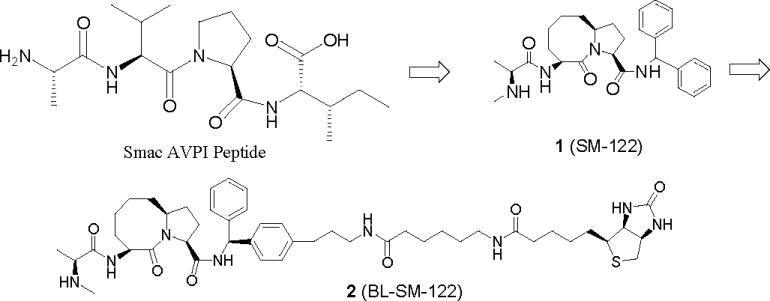
Design of a potent, conformationally constrained, monovalent Smac mimetic SM-122 and its biotinylated analogue.
The binding affinities of compounds 1, 2 and the Smac AVPI peptide to the XIAP BIR3 domain protein were evaluated using a previously established sensitive fluorescence-polarization (FP) assay (Figure 3).29 Compound 1 binds to XIAP BIR3 protein with an IC50 value of 91 nM. Its biotinylated analogue 2 has an IC50 value of 112 nM, very close to that of compound 1, indicating that the biotin label does not have a significant effect on the binding of compound 1 to XIAP BIR3 protein. In comparison, the Smac AVPI peptide has an IC50 value of 1,183 nM and its calculated Ki value is 425 nM, similar to the reported Kd value for this peptide.30 Therefore, compound 1 binds to XIAP BIR3 with an affinity 13-times higher than that of the Smac AVPI peptide.
Figure 3.

Competitive binding of our designed monovalent Smac mimetics 1, 2, 3 and the AVPI peptide to XIAP BIR3 (residues 240–356) as determined using an FP-based assay.
We next evaluated the binding affinity of SM-122 (1) to XIAP BIR2. For this purpose, we attempted to develop an FP assay for XIAP BIR2, similar to that for XIAP BIR3.29 Unfortunately, the fluorescently labeled Smac-based peptide tracer we used previously to establish the competitive binding FP assay for XIAP BIR3 domain29 has a low binding affinity to XIAP BIR2, and is unsuitable as a tracer for the development of an FP assay for XIAP BIR2. To address this limitation, we employed the Biacore surface plasmon resonance (SPR) technique to directly evaluate the binding affinity of biotinylated compound 2 to XIAP BIR2 protein with compound 2 immobilized on the streptavidin chip. Our analysis showed that compound 2 binds to XIAP BIR2 with a Kd value of 1.85 μM (Kon = 1118 1/Ms and Koff = 0.0018 1/s) (Figure 4A). We next evaluated compound 1 for its binding to XIAP BIR2 in a competitive SPR assay with biotinylated compound 2 immobilized on the streptavidin chip and XIAP BIR2 protein and compound 1 in the mobile phase. Our results showed that compound 1 blocks the binding of XIAP BIR2 to compound 2 in a dose-dependent manner and has an IC50 value of 5.6 μM (Figure 4B). Therefore, our SPR experiments showed that both compounds 1 and 2 bind to XIAP BIR2 with a good affinity.
Figure 4.

(A). Analysis of the binding of biotin-labeled compound 2 to XIAP L-BIR2 protein (residues 120–240) using the Biacore surface plasmon resonance (SPR) technique with different concentrations of the protein (from the top: 50, 25, 12.5, 6.25 and 3.125 μM). (B) Competitive binding of compound 1 to XIAP L-BIR2 as determined using the Biacore SPR technique. Biotinylated SM-122-BL was immobilized on the streptavidin chip.
Our FP and SPR assays showed that compound 1 binds to XIAP BIR2 and BIR3 proteins with good affinities. Importantly, as will be demonstrated later in the paper, compound 1 effectively inhibits cell growth and induces apoptosis in cancer cells, indicative of its excellent cell permeability. Hence, compound 1 represents a promising monovalent Smac mimetic template for the design of bivalent Smac mimetics to target concurrently the BIR2 and BIR3 domains.
Design of bivalent Smac mimetics
We next identified suitable sites in compound 1 for chemically tethering two molecules together for the design of bivalent Smac mimetics. For this purpose, we employed computational modeling to predict the binding of 1 to BIR2 and BIR3 domains of XIAP using high-resolution crystal structures of these individual domains.12,18 The predicted binding models (Figure 5) showed that one phenyl group of 1 in the pro-(S) configuration does not insert into the binding pocket of either BIR2 or BIR3 and consequently is exposed, making this phenyl ring suitable as an anchoring site for tethering two molecules of 1 together to produce bivalent Smac mimetics.
Figure 5.

Predicted binding model of Smac mimetic 1 in complex with (A) XIAP BIR2 domain and (B) BIR3 domain.
Modeling also suggested that the pro-(S) phenyl group in 1 may be replaced by other aromatic rings without compromising the binding. Thus, compound 3 was designed and synthesized to test if the pro-(S) phenyl group can be replaced by a [1,2,3]-triazole so that the “click chemistry,” a highly efficient coupling method31, can be readily applied to facilitate the synthesis of bivalent Smac mimetics (Figure 6). Consistent with our modeling prediction, compound 3 was determined to bind to XIAP BIR3 with an IC50 value of 57 nM, essentially the same as that of compound 1 (91 nM) (Figure 3). In addition, compound 3 was also shown to bind to XIAP BIR2 with the same affinity as that of compound 1 in our SPR assay (data not shown). Thus, compounds 1 and 3 have similar binding affinities to either BIR2 or BIR3 domain.
Figure 6.

Design of a new class of bivalent Smac mimetics based upon a conformationally constrained monovalent Smac mimetic.
The experimentally determined three-dimensional structure of XIAP containing both BIR2 and BIR3 domains has not been reported but our modeling analysis showed that the linker region between BIR2 and BIR3 containing approximately 25 residues is quite flexible. Accordingly, we designed a bivalent Smac mimetic SM-164 (4, Figure 6), which contains two monovalent Smac mimetics tethered together through a flexible linker which when extended, provides a distance of approximately 15 Å between the two triazole rings. We reasoned that such a linker would provide sufficient length and flexibility for the two monovalent binding units in 3 to bind concurrently to both the BIR2 and BIR3 domains in the same XIAP molecule. Based upon our predicted binding models for compound 1 (Figure 5), the methyl group in 1 inserts into a small hydrophobic cavity in both BIR2 and BIR3 and the N-methyl amino group participates in a hydrogen-bonding network with protein residues in both models. To confirm our predicted binding models and to determine the specificity of bivalent Smac mimetic 4, compound 5 was designed as an inactive control (Figure 6). In 5, the methyl group in each monovalent binding unit was replaced by a (1H-indolyl)-methyl group, a much larger hydrophobic group, to disrupt the hydrophobic interactions at this site and the methylamino group was replaced by an acetamido group to block the hydrogen bond formation. To test if the stereospecificity is critical for binding, compound 6, a stereoisomer of 4, was designed and synthesized.
Chemistry
The synthesis of compound 1 is shown in Scheme 1. The key intermediate 8 was synthesized from pyroglutamic acid 7 in eight steps according to a reported method.32 Hydrogenation of the double bond in 8 catalyzed by 10% Pd-C, followed by hydrolysis of the methyl ester gave the acid 9. Condensation of 9 with aminodiphenylmethane followed by removal of the Boc protecting group yielded an ammonium salt, condensation of which with Boc-N-methyl-L-alanine furnished an amide. Removal of the Boc protecting group from this amide afforded compound 1 (SM-122).
Scheme 1.

Synthesis of compound 1
Reagents and Conditions: (a) i. 10% Pd-C, H2, MeOH; ii. 2N LiOH, 1,4-dioxane, then 1N HCl, 96% over two steps; (b) i. aminodiphenylmethane, EDC, HOBt, N,N-diisopropylethylamine, CH2Cl2, overnight; ii. 4N HCl in 1,4-dioxane, MeOH; iii. Boc-N-methyl-L-alanine, EDC, HOBt, N,N-diisopropylethylamine, CH2Cl2; iv. 4N HCl in 1,4-dioxane, MeOH, 72% over four steps.
The synthesis of SM-157 (3) is shown in Scheme 2. The chiral amine 11 was prepared according to a reported method from the alcohol 10 in five steps.33 Condensation of acid 9 with amine 11 gave an amide 12. After removal of the Boc protecting group, the resulting ammonium salt was condensed with Boc-N-methyl-L-alanine to afford amide 13. Cycloaddition of 13 with azidomethylbenzene catalyzed by CuSO4-(+)-sodium-L-ascorbate yielded a triazole.31 Removal of the Boc protecting group from this triazole furnished compound 3 (SM-157).
Scheme 2.
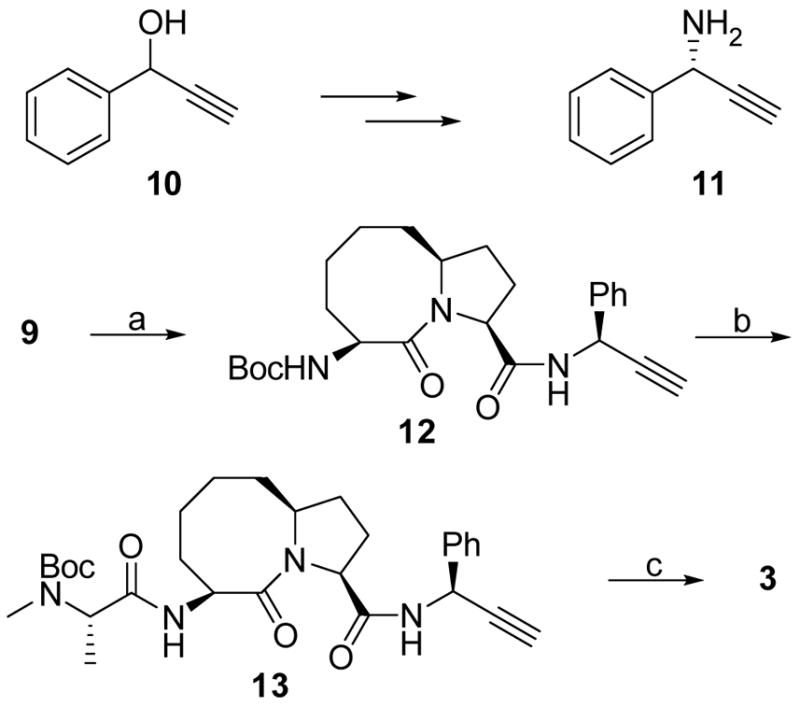
Synthesis of compound 3.
Reagents and conditions: (a) 11, EDC, HOBt, N,N-diisopropylethylamine, CH2Cl2, overnight, 91%; (b) i. 4 N HCl in 1,4-dioxane, MeOH; ii. Boc-N-methyl-L-alanine, EDC, HOBt, N,N-diisopropylethylamine, CH2Cl2, 86% over two steps; (c) i. azidomethylbenzene, CuSO4, (+)-sodium -L-ascorbate, tert-butanol - H2O 2:1; ii. 4 N HCl in 1,4-dioxane, MeOH, 74% over two steps.
The synthesis of the bivalent compounds 4 (SM-164), 5 (SM-173) and 6 (SM-206) is shown in Scheme 3. The chiral amine 14 was prepared according to a reported method.33 Compound 15 was synthesized with the same method as that used for compound 13. Removal of the Boc protecting group in 12 followed by condensation of the resulting ammonium salt with L-Nα-Boc-Trp yielded an amide. Removal of the two Boc protecting group in this amide followed by condensation of the resulting salt with acetic anhydride provided compound 16. Cycloaddition of 2 eq of compound 13 or 15 with 1 eq of 1,4-bis-(4-azidobutyl)-benzene respectively under the catalysis of CuSO4-(+)-sodium-L-scorbate gave two bis-triazoles.31 Removal of the two Boc protecting groups in these bis-triazoles yielded compounds 4 and 6, respectively. Cycloaddition of 2 eq of compound 16 with 1 eq of 1,4-bis-(4-azido-butyl)-benzene catalyzed by CuSO4-(+)-sodium-L-ascorbate gave compound 5.31
Scheme 3.
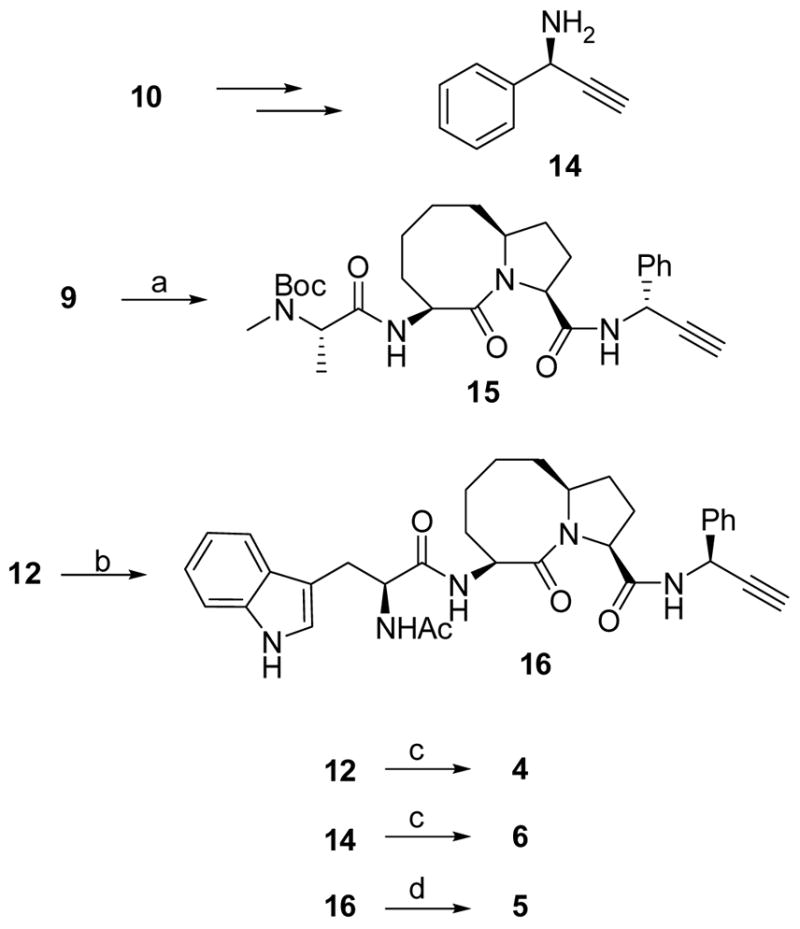
Synthesis of compounds 4, 5 and 6.
Reagents and conditions: (a) i. 14, EDC, HOBt, N,N-diisopropylethylamine, CH2Cl2, overnight; ii. 4 N HCl in 1,4-dioxane, MeOH; iii. Boc-N-methyl-L-alanine, EDC, HOBt, N,N-diisopropylethylamine, CH2Cl2, 73% over three steps; (b) i. 4 N HCl in 1,4-dioxane; ii. N-Boc-L-Trp, EDC, HOBt, N,N-diisopropylethylamine, CH2Cl2; iii. 4 N HCl in 1,4-dioxane, MeOH, iv. acetic anhydride, N,N-diisopropylethylamine, CH2Cl2, 67% over four steps; (c) i. 1,4-bis-(4-azido-butyl)-benzene, CuSO4, (+)-sodium -L-ascorbate, tert-butanol - H2O 2:1; ii. 4 N HCl in 1,4-dioxane, MeOH, 68% for 4 and 66% for 6; (d) 1,4-bis-(4-azido-butyl)-benzene, CuSO4, (+)-sodium -L-ascorbate, ter t-butanol - H2O 2:1, 76%.
The synthesis of biotinylated compound 2 (BL-SM-122) is shown in Scheme 4. The chiral amine 18 was synthesized according to our reported method.34 Condensation of acid 9 with 18 followed by removal of the Boc protecting group and condensation of the resulting ammonium salt with L-N-Boc-N-methyl-alanine yielded an amide. Cleavage of the Cbz protecting group in this amide by hydrogenation followed by condensation of the resulting amine with Cbz-6-aminohexanoic acid provided amide 19. Removal of the Cbz protecting group in this amide (19) followed by condensation with (+)-biotin N-hydroxy-succinimide ester furnished a biotinylated amide. Removal of the Boc protecting group in the amide gave our designed biotinylated compound 2 (BL-SM-122).
Scheme 4.

Synthesis of compound 2 (BL-SM-122)
Reagents and conditions: (a) i. 18, EDC, HOBt, N,N-diisopropylethylamine, CH2Cl2, overnight; ii. 4 N HCl in 1,4-dioxane, MeOH; iii. Boc-N-methyl-L-alanine, EDC, HOBt, N,N-diisopropylethylamine, CH2Cl2; iv. H2, 10% Pd-C, MeOH; v. Cbz-6-aminohexanoic acid, EDC, HOBt, N,N-diisopropylethylamine, CH2Cl2, 42% over five steps; (b). i. H2, 10% Pd-C, MeOH; ii. (+)-biotin N-hydroxy-succinimide ester, N,N-diisopropylethylamine, CH2Cl2; iii. 4 N HCl in 1,4-dioxane, MeOH, 74% over three steps.
Design of a fluorescently tagged, bivalent Smac peptide and development of a competitive binding assay to XIAP containing BIR2 and BIR3 domains
To evaluate accurately the binding affinities of our designed bivalent Smac mimetics, it was critical to develop a sensitive and reliable binding assay for XIAP containing both BIR2 and BIR3 domains. For this purpose, we designed and synthesized a fluorescently tagged Smac-based peptide, Smac-1F (Figure 7), which contains two basic binding units (AKPF) chemically tethered through two lysines by a flexible linker. This design was based upon a biochemical study,30 which showed that the second residue (valine) in the Smac AVPI peptide can be replaced by a lysine and the fourth residue by a phenylalanine to yield new peptides with a better binding affinity than the original Smac AVPI peptide to XIAP BIR3. Saturation experiments determined that Smac-1F binds to L-BIR2-BIR3 (residues 120–356) with a Kd value of 2.3 nM (Figure 7). We further determined that the linker preceding BIR2 does not contribute to the binding to Smac-1F.35 Our extensive investigations conclusively showed that Smac-1F indeed interacts with both BIR2 and BIR3 domains simutaneously in XIAP.35 Using Smac-1F and XIAP L-BIR2-BIR3 protein, we have established a competitive fluorescence-polarization (FP) assay to determine quantitatively the binding affinities of our designed Smac mimetics to XIAP containing both BIR2 and BIR3 domains.
Figure 7.

(A). Chemical structure of Smac-1F, a bivalent Smac-based peptide labeled with a fluorescence probe. (B). Saturation curve of XIAP (residues 120–356) containing both BIR2 and BIR3 domains to Smac-1F.
Quantitative determination of the binding affinities of designed Smac mimetics to XIAP protein
The binding affinities of compounds 1, 3, 4, 5, 6 and the Smac AVPI peptide to XIAP L-BIR2-BIR3 protein were determined in a competitive FP assay using Smac-1F as the tracer (Figure 8). Compounds 1, 3, 4, 6 and the Smac AVPI peptide bind to XIAP with IC50 values of 438, 376, 1.39, 71.5 and 10,396 nM, respectively, whereas the designed inactive control 5 shows no appreciable binding at 100 μM. Hence, the bivalent Smac mimetic 4 is 271–351 times more potent than the monovalent compounds 1 and 3 and >7,000 times more potent than the Smac AVPI peptide, respectively. The stereoisomer 6 is 51-times less potent than compound 4, confirming the importance of the stereospecificity. There was no significant difference with binding affinities for each of these Smac mimetics when either XIAP L-BIR2-BIR3 or BIR2-BIR3 protein (residues 156–356) was used in the binding assays (data not shown), indicating that the linker preceding BIR2 domain does not interact with these Smac mimetics. Our binding data demonstrate that the designed bivalent Smac mimetic 4 achieves an extremely high binding affinity to XIAP proteins containing both BIR2 and BIR3 domains and is much more potent than monovalent Smac mimetics 1 and 2 and the natural Smac AVPI peptide.
Figure 8.

Competitive binding of Smac mimetics to XIAP protein (residues 120–356) containing BIR2 and BIR3 domains determined in a competitive FP-based assay.
Probing the binding modes of Smac mimetics to XIAP proteins by analytical gel filtration
Although compound 4 was designed to target both the BIR2 and BIR3 domains in XIAP simultaneously, it was unclear if this is indeed happening. To probe the binding mode of 1, 3 and 4 to XIAP, we performed analytical gel filtration experiments using recombinant XIAP BIR3 (residues 241–356) and BIR2-BIR3 (residues 156–356) proteins (Figure 9). The BIR2-BIR3 protein without the linker preceding the BIR2 domain was used because this protein has a better solubility than the L-BIR2-BIR3 protein and compound 4 binds to these two proteins with the same affinities in our FP binding assay.
Figure 9.

Analytical gel filtration of (A) XIAP BIR3-only protein or (B) XIAP BIR2-BIR3 protein alone or in the presence of monovalent Smac mimetics 1 and 3 and bivalent Smac mimetic 4. The apparent molecular weights are shown in parentheses. The calculated molecular weight (MW) for BIR3 and BIR2-BIR3 proteins in monomeric form are 15.7 and 28.0 kDalton, respectively.
When the recombinant XIAP protein containing only the BIR3 domain was used in the gel filtration experiment, compound 4 induced dimerization of the protein (Figure 9A), indicating that each of the two monovalent binding units in 4 binds to one XIAP BIR3-only protein and forms a 1:2 complex between 4 and the XIAP BIR3-only protein. In sharp contrast, when the recombinant XIAP protein containing both BIR2 and BIR3 domains was used in the gel filtration experiment, compound 4 did not induce dimerization of the protein (Figure 9B). Instead, compound 4 appeared to make the protein become more compact, judging by the apparent molecular weight derived from the gel filtration data. Furthermore, monovalent compounds 1 and 3 failed to cause dimerization of either BIR3 or BIR2-BIR3 protein. These data suggest that when 4 interacts with the BIR3-only protein, one molecule of compound 4 binds to two molecules of XIAP BIR3 protein and induces dimerization of the protein. But when presented with XIAP containing both BIR2 and BIR3 domains, it binds simultaneously to both the BIR2 and BIR3 domains in the same XIAP molecule.
Although our binding data and gel filtration experiments using wild-type XIAP proteins strongly suggested that bivalent Smac mimetic 4 interacts simultaneously with both the BIR2 and BIR3 domains in the same XIAP molecule, these data alone do not conclusively show that either BIR2 or BIR3 is directly involved in the binding. To confirm that both BIR2 and BIR3 domains are directly involved in the binding of XIAP BIR2-BIR3 protein to the bivalent Smac mimetic 4, we generated mutations in each of the BIR domains. Based upon our modeling, E219 in the BIR2 domain is involved in a charge-charge interaction with compound 1 (Figure 5A), suggesting an important role of E219 in the interaction between 1 and the BIR2 domain. Our modeled structure also showed that the E314 residue in the BIR3 domain has a strong charge-charge interaction with 1 and W323 has hydrophobic contacts with the 8-membered ring in 1, suggesting that these two residues may play a key role in the binding of 1 to XIAP BIR3. Accordingly, we have prepared two mutated proteins, BIR2(E219R)-BIR3 and BIR2-BIR3(E314S,W323E), and used them to probe the involvement of these two individual domains in the binding of compound 4 to wild-type XIAP BIR2-BIR3. The gel filtration experiments using wild-type and mutated XIAP proteins containing both BIR2 and BIR3 domains showed that in the absence of 4, all these proteins behave as a monomer (Figure 10A). In the presence of compound 4, complete dimerization of the XIAP BIR2(E219R)-BIR3 mutated protein is observed, which is in sharp contrast to the absence of dimerization when the wild-type XIAP BIR2-BIR3 protein was used (Figure 10B). These data clearly showed that BIR2 is directly involved in the interaction of XIAP BIR2-BIR3 with compound 4. The gel filtration experiments further showed that compound 4 induces partial dimerization of the XIAP BIR2-BIR3(E314S,W323E) mutated protein, presumably via binding to two native BIR2 domains (Figure 10B). The incomplete dimerization of the XIAP BIR2-BIR3(E314S,W323E) protein may be due to the relatively weak binding of its monovalent binding unit in 4 to the BIR2 domain but nevertheless clearly demonstrates that BIR3 is also directly involved in the binding of XIAP BIR2-BIR3 with compound 4. These gel filtration experiments using both native and mutated proteins, together with the binding data, provide clear and convincing evidence that the bivalent Smac mimetic 4 interacts simultaneously with both the BIR2 and BIR3 domains in XIAP, achieving an extremely tight binding to the protein.
Figure 10.

Analytical gel filtration of (A) wild-type XIAP BIR2-BIR3, BIR2(E219R)BIR3 (BIR2M-BIR3), BIR2-BIR3(E314S,W323E) (BIR2-BIR3M) proteins alone (B) or in the presence of bivalent Smac mimetic 4. The apparent molecular weights (App. MW) are shown in parentheses.
Analysis of the binding of Smac mimetics to XIAP BIR2-BIR3 protein by HSQC NMR spectroscopy
Heteronuclear single quantum correlation (HSQC) NMR spectroscopy was employed to further investigate if a single molecule of bivalent compound 4 concurrently interacts with the BIR2 and BIR3 domains in XIAP and if monovalent compound 1 and bivalent compound 4 have different binding modes to XIAP containing both BIR domains. The BIR2-BIR3 protein (residues 156–356) was also used in our NMR experiments for its better solubility than the L-BIR2-BIR3 protein.
It was found that most of the resonances within the BIR3 domain in XIAP BIR2-BIR3 protein appear at identical positions as those from the BIR3-only protein (residues 241–356), indicating that the BIR3 domain structure is well preserved in XIAP BIR2-BIR3 protein (data not shown). By comparing the spectra of BIR2-only, BIR3-only and BIR2-BIR3 proteins, we were able to assign most of the peaks for the BIR3 domain and to identify peaks associated with the BIR2 domain in the XIAP BIR2-BIR3 protein.
When compound 4 was added to uniformly 15N labeled BIR2-BIR3 protein, 15N HSQC spectra showed that many residues in the protein are affected by the compound (Supporting Information). Line width analysis of the residues within the core structural domains indicated that XIAP BIR2-BIR3 protein, with or without compound 4, has approximately the same molecular size, confirming that compound 4 does not cause dimerization of the protein, consistent with our gel filtration results.
There are striking differences between the 15N HSQC spectra of XIAP BIR2-BIR3 protein with compounds 1 and 4 (Supporting Information). Overall, more of the protein residues are affected by compound 4 than by compound 1, and the changes are apparent at the lowest concentration (10 μM) of compound 4 tested, which is at 1:10 ratio of the protein concentration used in the NMR experiments. In comparison, higher concentrations of compound 1 are required for similar changes in chemical shifts, indicating that compound 1 has a lower binding affinity to XIAP than compound 4, consistent with FP binding data.
Analysis of the residues in BIR3 domain of BIR2-BIR3 protein showed that compounds 1 and 4 affected the same BIR3 residues (Supporting Information), indicating that they bind to the same site in the BIR3 domain. However, compound 4 at the same concentrations has a greater effect on these BIR3 residues than compound 1. For example, both compounds 4 and 1 induce significant chemical shifts on valine 298 (V298) (Figure 11(a)–(c)) and a number of other residues (Supporting Information) located in the BIR3 domain at the lowest concentration tested, but compound 4 at the same concentrations has a greater effect than compound 1. These data showed that although both compounds bind to BIR3 strongly, compound 4 binds to BIR3 with an even higher affinity than compound 1. Of note, V298 directly contacts with compound 1 in our predicted binding model (Figure 5) and with the isoleucine residue in the Smac AVPI peptide in both crystal and NMR solution structures.12,13
Figure 11.

Small regions of overplayed 15N HSQC NMR spectra of 15N labeled XIAP BIR2-BIR3 with various concentrations of compounds 1 and 4 (blue: 0 μM, orange: 10 μM, red: 30 μM, and green: 150 μM (for panels e and f only). (a)-(c) Residue valine 298 (V298) in BIR3 domain of BIR2-BIR3 protein with different concentrations of compounds 4 and 1, respectively; (d)-(f) a residue in BIR2 domain of BIR2-BIR3 proteins with different concentrations of compounds 4 and 1, respectively.
The BIR2 residues affected with different concentrations of compound 4 follow the same trends as seen with the BIR3 residues and there are also significant changes in chemical shifts for BIR2 residues when compound 4 was tested at 1:10 ratio of the protein concentration. In sharp contrast, even at the highest concentration (150 μM) of compound 1 tested, which is 50% in excess of the protein concentration, BIR2 residues are affected much more weekly than the BIR3 residues (Figure 11 and Supporting Information). These data showed that compound 1 binds to BIR2 with a much weaker affinity than to BIR3 but compound 4 binds to both BIR domains with high affinities.
Taken together, our NMR data provide additional and strong evidence that bivalent compound 4 interacts concurrently with both BIR2 and BIR3 domains in the same XIAP molecule and monovalent compound 1 binds to the BIR3 domain with a higher affinity than to the BIR2 domain. Furthermore, compound 4 binds to XIAP with a higher affinity than compound 1. These NMR data are entirely consistent with binding data and the results from gel filtration experiments using native and mutated XIAP proteins.
Antagonism by Smac mimetics of XIAP activation of caspase-9 and –3/-7 in cell free functional assays
Since XIAP functions as a potent inhibitor of caspase-9, -3 and -7, we evaluated 1, 4, 5 and the AVPI peptide for their ability to antagonize XIAP in cell-free caspase functional assays (Figure 12). In these assays, XIAP L-BIR2-BIR3 (residues 120–356) was found to dose-dependently inhibit the activity of caspase-9 and caspase-3/-7 and achieve complete inhibition at 50 nM (Figure 12). Both compounds 1 and 4 antagonize XIAP in a dose-dependent manner and are capable of restoring the activity of caspase-9, as well as that of caspase-3/-7. Consistent with their binding affinities to XIAP, compound 4 is 100 times more potent than 1, and significantly, at an equal molar concentration of XIAP, it completely overcomes the inhibition of XIAP and fully restores the activity of caspase-9 and -3/-7, indicating its extremely high potency to antagonize XIAP. In comparison, 100 μM of the Smac AVPI peptide, (2,000 times of the concentration of XIAP protein) is needed to completely restore the activity of caspase-9 and caspase-3/-7 and the inactive control 5 has a minimal effect at 100 μM. These functional data show that bivalent Smac mimetic 4 functions as an ultrapotent antagonist of XIAP and is 100- and 2000-times more potent than the corresponding monovalent Smac mimetic 1 and the Smac AVPI peptide, respectively, entirely consistent with their binding affinities to XIAP.
Figure 12.

Functional antagonism of Smac mimetics to XIAP protein (residues 120–356) in cell-free (A) caspase-9 and (B) caspase-3/-7 activity assays.
Cellular antagonism to XIAP in isogenic cell lines transfected with XIAP or vector control
Our cell-free functional assays demonstrate that our designed bivalent Smac mimetic 4 functions as an ultrapotent antagonist of XIAP and is much more potent than its monovalent counterpart 1. We next determined if compounds 1 and 4 are capable of antagonizing XIAP in cells and if 4 is also much more potent than 1. For this purpose, we employed a pair of isogenic Jurkat cell lines transfected with either XIAP (XIAP-JK) or vector control (Vec-JK).36 Western blot analysis showed that XIAP-JK cells have much higher levels of XIAP than Vec-JK cells and transfection of XIAP did not alter the levels of other IAP proteins, including cIAP-1 and cIAP-2 (Figure 13A). XIAP-JK cells become highly resistant to TRAIL (tumor necrosis factor-related apoptosis-inducing ligand) as compared to Vec-JK cells (Figure 13B). Both 1 and 4 can antagonize XIAP to cell death induction by TRAIL, but 4 is 100-times more potent than 1 in overcoming the resistance of XIAP to cell death induction by TRAIL. Of note, both 1 and 4 have no significant effect on their own in these two Jurkat cell lines. These data clearly showed that 4 is a highly effective, cell-permeable antagonist of XIAP and is 100-times more potent than its monovalent counterpart 1.
Figure 13.

(A). Western blot analysis of expression levels of IAP proteins in Jurkat cells transfected with vector control (Vec-JK), or full length of XIAP (XIAP-JK). (B). Induction of cell death by TRAIL alone or in combination with compounds 1 and 4. Cells were treated with TRAIL alone or in combination for 24 hours and cell viability was assessed by trypan blue exclusion assay.
Inhibition by Smac mimetics of cell growth and induction of apoptosis in leukemia cells
Consistent with its role as a potent inhibitor of apoptosis, XIAP is highly expressed in many cancer cell lines and tumor samples from patients.6 We hypothesize that in some, but not all, cancer cell lines, XIAP may function as the final defensive mechanism to protect cancer cells from apoptosis induction and a potent Smac mimetic may be capable of inducing apoptosis and inhibiting cell growth as a single agent without using another apoptotic stimulus by efficiently antagonizing the XIAP mediated protection.
Indeed, we found that compounds 1, 3, 4 and 6 effectively inhibit cell growth in the HL-60 human leukemia cell line (Figure 14A) and a number of other cancer cell lines with diverse tumor types. Compound 4 achieves an IC50 value of 1 nM in inhibition of cell growth in the HL-60 cell line. In comparison, monovalent compounds 1 and 3 have IC50 values of 1400 and 2000 nM, respectively, the stereoisomer 6 has an IC50 value of 90 nM and the inactive control 5 has an IC50 value >3000 nM. Therefore, the bivalent Smac mimetic 4 is 1000-times more potent than the monovalent compounds 1 and 3 in inhibition of cell growth and their cell growth activities correlate well with their binding affinities to XIAP.
Figure 14.

(A). Inhibition of cell growth by Smac mimetics in the HL-60 leukemia cancer cell line. HL-60 cells were treated with these Smac mimetics for 4 days and cell growth was analyzed by the WST-8 assay. (B). Induction of apoptosis by Smac mimetics 1, 3, 4 and 5 in the HL-60 leukemia cell line. HL-60 cells were treated with compounds 1, 3, 4 and 5 for 24 hours. Cells were harvested, stained with annexin-V-FITC and Propidium Iodide (PI) double staining and analyzed by flow cytometer. (C). Western blot analysis of cleavage of PARP (CL-PARP), caspase-3 (CL-C3) and -9 (CL-C9) in the HL-60 cell line induced by compounds 1, 3, 4 and 5. HL-60 cells were treated with Smac mimetics for 24 hours. Whole cell lysates were analyzed with Western blotting using specific antibodies. β-actin was used as the loading control.
Because cell growth inhibition is a combination of cell growth arrest and cell death induction, we examined if compounds 1, 3, 4 and 5 can induce apoptosis in the HL-60 cell line by flow cytometry (Figure 14B). Our data showed that compound 4 potently and effectively induces apoptosis in the HL-60 cell line in a dose-dependent manner (Figure 14B). Compound 4 at 10 and 100 nM for 24-hours induces 57% and 69% of HL-60 cells to undergo apoptosis, respectively. In fact, compound 1 at concentrations as low as 1 nM for 24-hours induces 17% of HL-60 cells to undergo apoptosis, higher than the untreated control (9%). Monovalent Smac mimetics 1 and 3 at 1000 nM induce 25% of HL-60 cells to undergo apoptosis but are at least 100-times less effective than compound 4. Importantly, the inactive control compound 5 at 1000 nM has no significant effect, indicating the specific effect by compound 4.
Since compound 4 effectively antagonizes XIAP and promotes activation of caspase-9 and -3 in cell-free systems, we evaluated if 4 also activates caspase-9 and -3 in the HL-60 leukemia cells. In addition, we investigated the ability of compound 4 to induce cleavage of PARP (poly(ADP-ribose)- polymerase), a substrate of caspase-3 and a biochemical marker of apoptosis. The results are shown in Figure 14C. As shown, within 24 hours compound 4, at concentrations as low as 10 nM, induces robust activation of caspase-9 and -3 and cleavage of PARP and is 100-times more effective than the monovalent Smac mimetics 1 and 3. The inactive control compound 5 has no effect on caspase activation and PARP cleavage at 1000 nM.
Taken together, our data showed that these Smac mimetics can effectively inhibit cell growth and induce apoptosis as single agents and the bivalent Smac mimetic 4 is 100–1000 times more potent than the corresponding monovalent Smac mimetics 1 and 3. Compound 4 achieves an IC50 value of 1 nM in the HL-60 leukemia cell line in the cell growth assay and induces significant apoptosis at concentrations as low as 1 nM in a 24-hour treatment. The activity for compound 4 is highly specific since the inactive control compound 5 has no or minimal effect in all these assays at concentrations as high as 1000 nM.
Intracellular targets of designed Smac mimetics; co-immunoprecipitation assay
Our data show that compound 4 potently induces apoptosis in the HL-60 leukemia cell line at low nanomolar concentrations. To provide direct evidence that Smac mimetics 1, 3 and 4 target XIAP in cells, we have performed co-immunoprecipitation assays using biotinylated BL-SM-122 in the HL-60 cell lysates (Figure 15).
Figure 15.
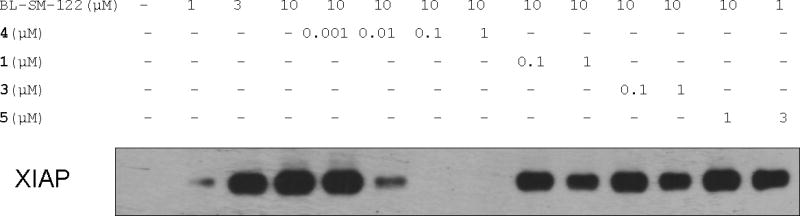
Probing the interaction of Smac mimetics to cellular XIAP in the HL-60 leukemia cell line by a competitive, co-immunoprecipitation pull-down assay using biotinylated SM-122 (BL-SM-122). HL-60 whole cell lysates were incubated with BL-SM-122 alone, or pre-incubated with compounds 1, 3, 4 and 5, followed by co-incubation with BL-SM-122. Complexes formed between BL-SM-122 and its targeted proteins were recovered by incubation with Streptavidin-argarose beads. XIAP protein associated with beads was eluted by heating and detected by western blotting using a monoclonal XIAP antibody.
These studies showed that BL-SM-122 pulls down XIAP protein in a dose-dependent manner in the HL-60 cell lysates. Compound 4 effectively competes off the binding of BL-SM-122 to XIAP dose-dependently. Compound 4 at 10 nM competes off more than 50% of XIAP bound to BL-SM-122 at 10 μM, and at 100 nM completely blocks the binding of XIAP to BL-SM-122. Monovalent Smac mimetics 1 and 3 at 1 μM reduce the amount of XIAP protein bound to BL-SM-122 only slightly, whereas the inactive control compound 5 has no effect at 1 or 3 μM. These data clearly indicate that bivalent Smac mimetic 4 binds to cellular XIAP with a much higher binding affinity than BL-SM-122 and the unlabeled monovalent Smac mimetics 1 and 3.
Summary
We have designed compound 4 (SM-164) as a cell-permeable, bivalent Smac mimetic to mimic the mode of binding of Smac protein to XIAP. Our study showed that SM-164 binds to XIAP protein containing both the BIR2 and BIR3 domains with an IC50 value of 1.39 nM and functions as an extremely potent antagonist of XIAP in our cell-free functional assays and in cells. Our gel filtration experiments using wild-type and mutated XIAP proteins, together with our NMR experiments, established that SM-164 achieves an extremely high affinity to XIAP by concurrently interacting with both the BIR2 and BIR3 domains. It potently binds to cellular XIAP protein, promotes activation of caspases and effectively induces apoptosis in leukemia cancer cells at concentrations as low as 1 nM. Significantly, SM-164 has a minimal toxicity to normal human primary cells at concentrations as high as 10,000 nM, displaying an outstanding selectivity. Our study provides convincing evidence that concurrent targeting of the BIR2 and BIR3 domains in XIAP using a non-peptide, bivalent small-molecule is a highly effective strategy to antagonize XIAP, promoting apoptosis in cancer cells. SM-164 is a powerful tool for further elucidation of the cellular functions of XIAP and a very promising lead compound in the development of a new class of anticancer therapy aimed at overcoming apoptosis resistance of cancer cells.
Experimental Section
I. Chemistry
General Methods
1H NMR spectra were acquired at 300 MHz and 13C spectra at 75 MHz. 1H chemical shifts are reported with CDCl3 (7.27 ppm) or HDO (4.70 ppm) as internal standards. 13C chemical shifts are reported relative to CDCl3 (77.00 ppm) or 1,4-dioxane (67.16 ppm) as internal standards. The final products were purified by C18 reverse phase semi-preparative HPLC column with solvent A (0.1% of TFA in H2O) and solvent B (0.1% of TFA in CH3CN) as eluents.
(3S,6S,10aS)-6-(tert-Butoxycarbonylamino)-5-oxodecahydropyrrolo[1,2-a]azocine-3-carboxylic acid (9)
To a solution of compound 8 (1.35 g, 4 mmol) in MeOH (20 mL) was added 10% Pd-C (0.2 g). The mixture was stirred under H2 for 8 h then filtered through celite and the filtrate was evaporated to give an ester. To a solution of this ester in 1,4-dioxane (5 mL) was added 2N LiOH (6 mL). The mixture was stirred at room temperature for 2 h and then neutralized with 1N HCl until the pH was 4. After extraction of the mixture with ethyl acetate (3×30 mL), the combined organic layers were dried over Na2SO4 and removal of the solvent gave the acid 9 (1.25 g. 96% over two steps). This acid can be used in the following steps without purification or can be purified by recrystallization from ethyl acetate/hexane (1:1). 1H NMR (CDCl3) δ 5.62 (brs, 1H), 4.63 (m, 1H), 4.50 (m, 1H), 4.25 (m, 1H), 2.41-2.09 (m, 3H), 2.07-1.50 (m, 9H), 1.45 (brs, 9H); 13C NMR (75 MHz, CDCl3) δ 174.77, 172.50, 155.24, 79.72, 59.96, 59.59, 50.97, 36.39, 32.21, 28.36, 26.56, 25.24, 22.65; ESI MS: m/z 349.2 (M+Na)+; HR ESI MS for C16H26N2O5Na required: 349.1739, found: 349.1727.
(3S,6S,10aS)-N-benzhydryl-6-((S)-2-(methylamino)propanamido)-5-oxodecahydropyrrolo[1,2-a]azocine-3-carboxamide (1)
To a solution of the acid 9 (160 mg, 0.5 mmol) in CH2Cl2 (10 mL) was added aminodiphenylmethane (140 mg, 0.77 mmol), EDC (150 mg, 0.77 mmol), HOBt (105 mg, 0.78 mmol) and N,N-diisopropylethyl amine (0.5 mL) at 0°C with stirring. The mixture was stirred at room temperature for eight hours and then condensed. The residue was purified by chromatography to give an amide. To a solution of this amide in MeOH (5 mL) was added HCl (4N in 1,4-dioxane, 3 mL). The solution was stirred at room temperature overnight and then evaporated to give an ammonium salt. To a mixture of this salt in CH2Cl2 (10 mL) was added Boc-N-methyl-L-alanine (150 mg, 0.74 mmol), EDC (154 mg, 0.8 mmol), HOBt (103 mg, 0.76 mmol) and N,N-diisopropylethyl amine (0.7 mL) at 0°C with stirring. The mixture was stirred at room temperature overnight and then condensed. The residue was purified by chromatography to give an amide. To a solution of this amide in MeOH (5 mL) was added HCl solution (4N in 1,4-dioxane, 3 mL). The solution was stirred at room temperature overnight and then condensed to give crude compound 1 as a salt with HCl (184 mg, 72% over four steps). The crude product was purified by C18 reversed-phase semi-preparative HPLC with a gradient from 70% of solvent A and 30% of solvent B to 50% of solvent A and 50% of solvent B in 40 min. 1H NMR (300 M Hz, D2O) δ 7.30-7.18 (m, 10H), 5.95 (s, 1H), 4.74 (m, 1H), 4.34 (m, 1H), 4.24 (m, 1H), 3.83 (m, 1H), 2.57 (s, 3H), 2.21-1.50 (m, 11H), 1.40 (d, J = 7.0 Hz, 3H), 1.38 (m, 1H); 13 173.77, C NMR (D2O) δ 172.62, 169.95, 141.56, 141.28, 129.54, 129.44, 128.40, 128.35, 128.01, 127.82, 62.50, 61.40, 58.19, 57.49, 51.42, 36.16, 33.30, 32.64, 31.61, 28.07, 25.36, 22.14, 15.95; ESI MS: m/z 477.3 (M+H)+; HR ESI MS for C28H37N4O3 required: 477.2866, found: 477.2849.
tert-Butyl-(3S,6S,10aS)-5-oxo-3-((R)-1-phenylprop-2-ynylcarbamoyl)decahydropyrrolo[1,2-a]azo-cin-6-ylcarbamate (12)
To a solution of acid 9 (650 mg, 2 mmol) in CH2Cl2 (20 mL) was added the chiral amine 11 (380 mg, 2.9 mmol), EDC (580 mg, 3 mmol), HOBt (405 mg, 3 mmol) and N,N-diisopropylethyl amine (1.5 mL) at 0 °C with stirring. The mixture was stirred at room temperature for a further eight hours and then condensed. The residue was purified by chromatography to give amide 12 (798 mg, 91%). 1H NMR (CDCl3): δ 7.80 (brd, J = 8.6 Hz, 1H), 7.50-7.40 (m, 2H), 7.33-7.20 (m, 3H), 5.91 (dd, J = 8.6, 2.4 Hz, 1H), 5.45 (brd, J = 7.9 Hz, 1H), 4.64 (dd, J = 8.3, 6.3 Hz, 1H), 4.52 (m, 1H), 4.10 (m, 1H), 2.60 (m, 1H), 2.45 (d, J = 2.4 Hz, 1H), 2.20-1.60 (m, 5H), 1.40-1.22 (m, 14H), 1.05 (m, 1H); 13C NMR (CDCl3): δ 172.25, 169.30, 154.95, 138.50, 128.55, 128.04, 127.04, 81.71, 79.53, 72.75, 59.50, 59.09, 50.95, 44.70, 36.48, 36.28, 31.92, 28.29, 24.89, 23.87, 22.94; ESI MS: m/z 439.3 (M+Na)+; HR ESI MS for C25H33N3O4Na required: 462.2369, found: 462.2354.
tert-Butylmethyl((S)-1-oxo-1-((3S,6S,10aS)-5-oxo-3-((R)-1-phenylprop-2-ynylcarbamoyl)deca-hydropyrrolo[1,2-a]azocin-6-ylamino)propan-2-yl)carbamate (13)
To a solution of compound 12 (440 mg, 1 mmol) in MeOH (5 mL) was added HCl solution (4N in 1,4-dioxane, 5 mL). The solution was stirred at room temperature overnight and then condensed to give an ammonium salt. To a mixture of this salt and CH2Cl2 (10 mL) was added Boc-N-methyl-L-alanine (303 mg, 1.5 mmol), EDC (287 mg, 1.5 mmol), HOBt (204 mg, 1.5 mmol) and N,N-diisopropylethyl amine (1.2 mL) at 0°C with stirring. The mixture was stirred at room temperature overnight then condensed. The residue was purified by chromatography to give amide 13 (450 mg, 86% over two steps). 1H NMR (CDCl3): δ 7.75 (brd, J = 8.5 Hz, 1H), 7.50-7.40 (m, 2H), 7.33-7.20 (m, 3H), 5.91 (dd, J = 8.5, 2.3 Hz, 1H), 6.88 (brs, 1H), 4.80 (m, 1H), 4.60 (dd, J = 8.4, 6.3 Hz, 1H), 4.58 (brm, 1H), 4.10 (m, 1H), 2.80 (s, 3H), 2.60 (m, 1H), 2.49 (d, J = 2.3 Hz, 1H), 2.20-1.60 (m, 5H), 1.50-1.05 (m, 18H); 13C NMR (CDCl3): δ 171.55, 170.51, 169.29, 154.95, 138.52, 128.59, 128.11, 127.09, 81.69, 80.57, 72.81, 59.59, 59.03, 49.88, 44.77, 36.50, 35.86, 31.92, 30.04, 28.36, 24.85, 23.98, 23.01, 13.72; ESI MS: m/z 547.3 (M+Na)+; HR ESI MS for C29H40N4O5Na required: 547.2896, found: 547.2897.
(3S,6S,10aS)-N-((S)-(1-Benzyl-1H-1,2,3-triazol-4-yl)(phenyl)methyl)-6-((S)-2-(methylamino)-propanamido)-5-oxodecahydropyrrolo[1,2-a]azocine-3-carboxamide (3)
To a solution of compound 13 (105 mg, 0.2 mmol) and azidomethylbenzene (201 mg, 1.5 mmol) in tert-butanol (10 mL) was added a mixture of CuSO4 (20 mg, 0.13 mmol) and (+)-sodium-L-ascorbate (60 mg) in H2O (5 mL). The mixture was stirred at room temperature overnight and then extracted with CH2Cl2 (3×15 mL). The combined organic layer was washed with brine, dried over Na2SO4 and condensed. The residue was purified by chromatography to give a triazole. To a solution of this triazole in MeOH (5 mL) was added HCl solution (4N in 1,4-dioxane, 2 mL). The solution was stirred at room temperature overnight and then condensed to give crude compound 3 as a salt with HCl (88 mg, 74% over two steps). This crude product was purified by C18 reversed phase semi-preparative HPLC and the purity was determined by analytical HPLC to be over 98%. The gradient ran from 70% of solvent A and 30% of solvent B to 50% of solvent A and 50% of solvent B in 40 min. 1H NMR (D2O) δ 7.66 (s, 1H), 7.35-7.10 (m, 10H), 6.04 (m, 1H), 5.36 (s, 2H), 4.70 (m, 1H), 4.28 (m, 1H), 4.19 (m, 1H), 3.80 (m, 1H), 2.56 (s, 3H), 2.20-1.50 (m, 11H), 1.40 (d, J = 7.1 Hz, 3H), 1.38 (m, 1H); 13C NMR (D2O) δ 173.14, 172.21, 169.50, 148.55, 139.09, 134.99, 129.40, 129.27, 129.04, 128.50, 128.31, 127.38, 124.02, 61.97, 60.97, 57.20, 54.22, 51.08, 50.36, 35.91, 33.05, 32.28, 31.32, 27.65, 25.06, 21.92, 15.63; ESI MS: m/z 580.3 (M+Na)+; HR ESI MS for C31H39N7O3Na required: 580.3012, found: 580.3017.
(S,3S,3′S,6S,6′S,10aS,10a′S)-N,N′-((1S,1′S)-(1,1′-(4,4′-(1,4-Phenylene)bis(butane-4,1-diyl))bis(1H-1,2,3-triazole-4,1-diyl))bis(phenylmethylene))bis(6-((S)-2-(methylamino)propanamido)-5-oxodecahydropyrrolo[1,2-a]azocine-3-carboxamide) (4)
To a solution of compound 13 (280 mg, 0.53 mmol) and 1,4-bis-(4-azidobutyl)-benzene (71 mg, 0.26 mmol) in tert-butanol (10 mL) was added a mixture of CuSO4 (20 mg, 0.13 mmol) and (+)-sodium-L-ascorbate (60 mg) in H2O (5 mL). The mixture was stirred at room temperature overnight and then extracted with CH2Cl2 (3×30 mL). The combined organic layer was washed with brine, dried over Na2SO4 and evaporated to give a residue which was purified by chromatography to give a bis-triazole. To a solution of this bis-triazole in MeOH (5 mL) was added HCl solution (4N in 1,4-dioxane, 5 mL). The solution was stirred at room temperature overnight and then condensed to give crude compound 4 as a salt with HCl (215 mg, 68% over two steps). This crude product was purified by C18 reversed phase semi-preparative HPLC and the purity checked by analytical HPLC to be over 98%. 1H NMR (D2O): δ 7.40 (s, 2H), 7.20-7.05 (m, 10H), 6.60 (s, 4H), 6.05 (s, 2H), 4.70 (m, 2H), 4.28 (m, 2H), 4.15 (m, 2H), 4.06 (m, 4H), 3.80 (m, 2H), 2.55 (s, 6H), 2.28-1.04 (m, 38H); 13C NMR (D2O): δ 172.75, 172.09, 169.49, 148.33, 139.53, 129.24, 128.54, 127.41, 123.44, 61.89, 60.89, 57.18, 51.02, 50.30, 35.95, 34.28, 33.09, 32.35, 31.34, 29.22, 27.86, 25.11, 21.92, 15.69; ESI MS: m/z 1121.6 (M+H)+; HR ESI MS for C62H85N14O6 required: 1121.6777, found: 1121.6777.
(3S,6S,10aS)-6-((S)-2-Acetamido-3-(1H-indol-3-yl)propanamido)-5-oxo-N-((R)-1-phenylprop-2-ynyl)decahydropyrrolo[1,2-a]azocine-3-carboxamide (16)
To a solution of compound 12 (434 mg, 1 mmol) in MeOH (5 mL) was added HCl solution (4N in 1,4-dioxane, 5 mL). The solution was stirred at room temperature overnight and then condensed to give an ammonium salt. To a mixture of this salt in CH2Cl2 (10 mL) was added Nα-Boc-L-Trp (360 mg, 1.2 mmol), EDC (290 mg, 1.5 mmol), HOBt (202 mg, 1.5 mmol) and N,N-diisopropylethyl amine (1.2 mL) at 0°C with stirring. The mixture was stirred at room temperature overnight and then condensed. The residue was purified by chromatography to give an amide. To a solution of this amide in MeOH (5 mL) was added HCl solution (4N in 1,4-dioxane, 5 mL). The solution was stirred at room temperature overnight and then evaporated to give an ammonium salt. To a mixture of this salt was added acetic anhydride (0.3 mL) and N,N-diisopropylethyl amine (0.8 mL) at 0°C with stirring. The mixture was stirred at room temperature overnight then condensed and the residue was purified by chromatography to give the amide 16 (379 mg, 67% over four steps). 1H NMR (CDCl3): δ 8.71 (brs, 1H), 8.13 (d, J = 1.6 Hz, 1H), 8.01 (d, J = 8.5 Hz, 1H), 7.45-7.15 (m, 7H), 7.10-6.92 (m, 2H), 6.77 (d, J = 1.6 Hz, 1H), 6.39 (d, J = 8.5 Hz, 1H), 5.91 (dd, J = 8.5, 2.4 Hz, 1H), 5.20 (m, 1H), 5.05 (m, 1H), 4.34 (t, J = 8.2 Hz, 1H), 4.12 (m, 1H), 3.20 (m, 1H), 2.90 (m, 1H), 2.45 (d, J = 2.4 Hz, 1H), 2.20-1.45 (m, 15H); 13C NMR (CDCl3): δ 170.79, 170.72, 170.66, 170.11, 137.60, 135.81, 128.61, 127.83, 127.59, 127.06, 122.57, 121.69, 118.94, 118.30, 111.13, 110.55, 81.66, 72.60, 60.45, 60.18, 53.37, 49.38, 44.47, 36.59, 35.96, 31.99, 30.05, 26.44, 25.56, 22.98, 22.34, 20.63; ESI MS: m/z 590.3 (M+Na)+; HR ESI MS for C33H37N5O4Na required: 590.2743, found: 590.2748.
(S,3S,3′S,6S,6′S,10aS,10a′S)-N,N′-((1S,1′S)-(1,1′-(4,4′-(1,4-Phenylene)bis(butane-4,1-diyl))bis(1H-1,2,3-triazole-4,1-diyl))bis(phenylmethylene))bis(6-((S)-2-acetamido-3-(1H-indol-3-yl)propan-amido)-5-oxodecahydropyrrolo[1,2-a]azocine-3-carboxamide) (5)
To a solution of compound 16 (190 mg, 0.34 mmol) and 1,4-bis-(4-azidobutyl)-benzene (45 mg, 0.17 mmol) in tert-butanol (10 mL) was added a mixture of CuSO4 (20 mg, 0.13 mmol) and (+)-sodium-L-ascorbate (60 mg) in H2O (5 mL). The mixture was stirred at room temperature overnight then extracted with CH2Cl2 (3×30 mL). The combined organic layer was washed with brine, dried over Na2SO4 and evaporated to give a residue which was purified by chromatography to give compound 5 (282 mg, 76%). 1H NMR (CDCl3): δ 8.70 (s, 2H), 8.32 (d, J = 8.0 Hz, 2H), 7.60 (d, J = 7.4 Hz, 2H), 7.40-6.80 (m, 26 H), 6.47 (d, J = 7.9 Hz, 2H), 6.25 (d, J = 8.0 Hz, 2H), 4.81 (m, 2H), 4.70 (m, 2H), 4.50 (m, 2H), 4.27 (t, J = 7.1 Hz, 4H), 3.95 (m, 2H), 3.30-3.08 (m, 4H), 2.60 (t, J = 2.61 Hz, 4H), 2.45-2.25 (m, 2H), 2.12-1.20 (m, 36H); 13C NMR (CDCl3): δ 170.78, 170.41, 170.14, 169.88, 148.00, 140.45, 138.98, 136.19, 128.53, 128.44, 127.68, 127.56, 127.28, 123.70, 121.67, 121.55, 119.13, 118.57, 111.29, 110.25, 59.98, 59.06, 54.01, 50.40, 50.20, 50.01, 47.76, 35.93, 34.61, 31.74, 29.53, 29.09, 28.08, 24.83, 23.30, 22.95, 20.82; ESI MS: m/z 1429.7 (M+Na)+; HR ESI MS for C80H94N16O8Na required: 1429.7338, found: 1429.7341.
tert-Butylmethyl((S)-1-oxo-1-((3S,6S,10aS)-5-oxo-3-((S)-1-phenylprop-2-ynylcarbamoyl)deca-hydropyrrolo[1,2-a]azocin-6-ylamino)propan-2-yl)carbamate (15)
To a solution of the acid 9 (480 mg, 1.47 mmol) in CH2Cl2 (15 mL) was added chiral amine 14 (290 mg, 2.21 mmol), EDC (430 mg, 2.25 mmol), HOBt (297 mg, 2.2 mmol) and N,N-diisoopropylethyl amine (1.5 mL) at 0 °C with stirring. The mixture was stirred at room temperature for eight hours and then condensed. The residue was purified by chromatography to give an amide. To a solution of this amide in MeOH (5 mL) was added HCl solution (4N in 1,4-dioxane, 5 mL). The solution was stirred at room temperature overnight and then condensed to give an ammonium salt. To a mixture of this salt in CH2Cl2 (10 mL) was added Boc-N-methyl-L-alanine (404 mg, 2.0 mmol), EDC (390 mg, 2.0 mmol), HOBt (272 mg, 2.0 mmol) and N,N-diisopropylethyl amine (2.0 mL) at 0°C with stirring. The mixture was stirred at room temperature overnight and then concentrated. The residue was purified by chromatography to give the amide 15 (563 mg, 73% over three steps). 1H NMR (CDCl3): δ 7.85 (brd, J = 8.5 Hz, 1H), 7.50-7.42 (m, 2H), 7.35-7.20 (m, 3H), 6.98 (brd, 1H), 4.86 (m, 1H), 5.90 (dd, J = 8.5, 2.4 Hz, 1H), 4.73 (brm, 1H), 4.62 (m, 1H), 4.21 (m, 1H), 2.81 (s, 3H), 2.55 (m, 1H), 2.42 (d, J = 2.4 Hz, 1H), 2.13 (m, 1H), 2.08-1.55 (m, 10H), 1.38 (brs, 9H), 1.32 (d, J = 7.1 Hz, 3H); 13C NMR (CDCl3): δ 172.23, 171.10, 170.27, 138.61, 129.13, 128.55, 127.35, 82.06, 73.18, 59.89, 59.54, 50.37, 44.93, 37.22, 36.60, 32.29, 30.53, 28.78, 25.66, 24.38, 23.92, 14.29; ESI MS: m/z 547.3 (M+Na)+; HR ESI MS for C29H40N4O5Na required: 547.2896, found: 547.2906.
(S,3S,3′S,6S,6′S,10aS,10a′S)-N,N′-((1R,1′R)-(1,1′-(4,4′-(1,4-Phenylene)bis(butane-4,1-diyl))bis(1H-1,2,3-triazole-4,1-diyl))bis(phenylmethylene))bis(6-((S)-2-(methylamino)propanamido)-5-oxodeca-hydropyrrolo[1,2-a]azocine-3-carboxamide) (6)
To a solution of compound 14 (240 mg, 0.46 mmol) and 1,4-bis-(4-azidobutyl)-benzene (60 mg, 0.22 mmol) in tert-butanol (10 mL) was added a mixture of CuSO4 (20 mg, 0.13 mmol) and (+)-sodium-L-ascorbate (60 mg) in H2O (5 mL). The mixture was stirred at room temperature overnight and then extracted with CH2Cl2 (3×30 mL). The combined organic layer was washed with brine, dried over Na2SO4 and condensed. The residue was purified by chromatography to give a bis-triazole. To a solution of this bis-triazole in MeOH (5 mL) was added HCl solution (4N in 1,4-dioxane, 5 mL). The solution was stirred at room temperature overnight and then condensed to give compound 6 as a salt with HCl (180 mg, 66% over two steps). The product was purified by C18 reversed phase semi-preparative HPLC and the purity determined by analytical HPLC to be over 98%. 1H NMR (D2O): δ 7.44 (s, 2H), 7.30-6.80 (m, 10H), 6.49 (s, 4H), 5.99 (s, 2H), 4.63 (m, 2H), 4.28 (m, 2H), 4.06 (m, 2H), 3.92 (m, 2H), 3.80 (m, 4H), 2.55 (s, 6H), 2.28-0.95 (m, 42H); 13C NMR (D2O): δ 175.04, 174.33, 171.97, 150.65, 143.08, 141.97, 131.63, 131.00, 130.59, 129.60, 126.25, 64.31, 63.33, 59.74, 53.47, 52.92, 38.47, 36.87, 35.85, 34.86, 33.87, 31.85, 30.42, 27.65, 24.52, 18.26. ESI MS: m/z 1143.7 (M+Na)+; HR ESI MS for C62H85N14O6Na required: 1143.6596, found: 1143.6616.
(3S,6S,10aS)-6-[2-(tert-Butoxycarbonyl-methyl-amino)-propionylamino]-5-oxo-decahydro-pyrrolo[1,2-a]azocine-3-carbonic acid (S)-1-(4-(3-(4-benzoxycarbonylamino-butyrylamino)-prop-yl)-phenyl)-1′-phenyl amide (18)
To a solution of acid 9 (220 mg, 0.67 mmol) in CH2Cl2 (10 mL) was added amine 17 (250 mg, 0.67 mmol), EDC (190 mg, 1.0 mmol), HOBt (135 mg, 1.1 mmol) and N,N-diisopropylethyl amine (1 mL) at 0°C with stirring. The mixture was stirred at room temperature for eight hours and then evaporated. The residue was purified by chromatography to give an amide. To a solution of this amide in MeOH (5 mL) was added HCl (4N in 1,4-dioxane, 3 mL). The solution was stirred at room temperature overnight and then condensed to give an ammonium salt. To a mixture of this salt in CH2Cl2 (10 mL) was added Boc-N-methyl-L-alanine (160 mg, 0.79 mmol), EDC (191 mg, 1.0 mmol), HOBt (140 mg, 1.04 mmol) and N,N-diisopropylethyl amine (1 mL) at 0°C with stirring. The mixture was stirred at room temperature overnight and then condensed. The residue was purified by chromatography to give an amide. To a solution of this amide in MeOH (10 mL) was added 10% Pd-C (100 mg). The solution was stirred under 1 atm of H2 at room temperature overnight before being filtered through celite and condensed. The residue was dissolved in CH2Cl2 (10 mL) and Cbz-6-amino-hexanoic acid (212 mg, 0.8 mmol), EDC (193 mg, 1 mmol), HOBt (134 mg, 1 mmol) and N,N-diisopropylethyl amine (1 mL) were added at 0°C with stirring. The mixture was stirred at room temperature overnight and then concentrated to give a residue which was purified by chromatography to give compound 19 (245 mg, 42% over five steps). 1H NMR (CDCl3) δ 7.95 (brd, J = 8.5 Hz, 1H), 7.40-7.05 (m, 14H), 6.95 (brm, 1H), 6.19 (brd, J = 8.6 Hz, 1H), 5.49 (m, 1H), 5.10 (brs, 2H), 4.95-4.76 (m, 2H), 4.70 (m, 1H), 4.60 (brm, 1H), 4.14 (m, 1H), 3.33-3.12 (m, 4H), 2.86 (s, 3H), 2.68-2.49 (m, 3H), 2.20-1.15 (m, 33H); 13C NMR (CDCl3) δ172.71, 171.65, 170.58, 169.54, 156.41, 141.80, 140.50, 139.28, 136.59, 128.57, 128.47, 128.05, 127.45, 127.26, 127.14, 80.61, 66.54, 59.90, 59.16, 56.88, 50.00, 40.74, 39.11, 36.47, 35.91, 32.88, 31.94, 31.12, 30.04, 29.61, 28.37, 26.20, 25.14, 24.89, 24.12, 23.17, 13.77; ESI MS: m/z 903.5 (M+Na)+; HR ESI MS for C50H68N6O8 required: 903.4996, found: 903.5009.
(3S,6S,10aS)-6-((S)-2-(Methylamino)propanamido)-5-oxo-N-((S)-(4-(3-(6-(5-((3aS,4S,6aR)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanamido)hexanamido)propyl)phenyl)(phenyl)-methyl)decahydropyrrolo[1,2-a]azocine-3-carboxamide (2, BL-SH-122)
To a solution of compound 19 (110 mg, 0.13 mmol) in MeOH (10 mL) was added 10% Pd-C (50 mg). The solution was stirred under 1 atm of H2 at room temperature overnight before filtering through celite and concentration. The residue was dissolved in CH2Cl2 (10 mL) and (+)-biotin N-hydroxy-succinimide ester (48 mg, 0.15 mmol) and N,N-diisoopropylethylamine (0.5 mL) were added to the solution. The mixture was stirred overnight and then condensed. The residue was purified by chromatography to yield a biotinylated amide. To a solution of the amide in MeOH was added a solution of HCl in 1,4-dioxane. The solution was stirred at room temperature overnight and then the solvent was evaporated to give a crude product which was purified by C18 reversed phase semi-preparative HPLC to give compound 19 (84 mg, 74% over three steps). The purity was checked by analytical HPLC to be over 98%. The gradient ran from 70% of solvent A and 30% of solvent B to 50% of solvent A and 50% of solvent B in 40 min. 1H NMR (D2O): δ 7.22-7.04 (m, 5H), 7.02-6.90 (m, 4H), 5.87 (s, 1H), 4.65 (m, 1H), 4.32 (m, 1H), 4.22 (m, 1H), 4.12 (m, 1H), 4.01 (m, 1H), 3.80 (m, 1H), 3.05-2.82 (m, 5H), 2.62 (m, 1H), 2.52 (s, 3H), 2.46 (m, 1H), 2.45-2.30 (m, 2H), 2.18-1.05 (m, 33H); 13C NMR (D2O): δ 176.33, 176.30, 172.70, 172.07, 169.46, 165.37, 141.42, 139.27, 129.10, 127.94, 127.63, 62.31, 62.03, 60.99, 60.46, 57.43, 57.19, 55.76, 51.00, 40.08, 39.40, 39.24, 35.98, 35.83, 32.59, 31.31, 30.56, 28.49, 28.37, 28.08, 25.99, 25.66, 25.47, 15.66; ESI MS: m/z 895.5 (M+Na)+; HR ESI MS for C47H68N8O6S required: 895.4880, found: 895.4878.
II. Molecular modeling
The crystal structure of XIAP BIR3 in a complex with the Smac protein12 (PDB code:1G73) was used to predict the binding models of XIAP BIR3 bound to designed compounds. The XIAP BIR2 structure from the X-ray structure18 of the complex of XIAP BIR2 and caspase-3 (PDB code: 1I3O) was used to predict the binding models of designed compounds to XIAP BIR2.
A previous study37 has demonstrated that the N-terminus of the small subunit of caspase 3 (sequence: SGVDD) interacts with XIAP BIR2 in the same binding groove as is involved in the XIAP BIR2-Smac peptide interaction. This groove can be seen clearly in the asymmetric crystallographic unit between XIAP BIR2 and caspase 3. The Swiss PDB viewer program was used to generate the asymmetric crystallographic unit in 1I3O while retaining the structure of XIAP BIR2 and the SGVDDDM peptide from caspase 3 for further molecular dynamics simulations. Our initial experiments docking compound 1 with XIAP BIR2 using the X-ray structure of XIAP BIR2 from 1I3O failed to yield satisfactory results. Examination of the crystal structure of XIAP BIR2 and comparison with that of XIAP BIR3 showed that the first aspartic acid residue, D in the SGVDDDM peptide interacts with the side chain of Q197 of XIAP BIR2. Thus, the binding pocket in the crystal structure of XIAP BIR2, which interacts with this aspartate residue, may be forced to adopt a conformation that is incompatible with its interaction with a hydrophobic residue in the fourth position of the Smac AVPI peptide. To resolve this problem, we performed an extensive molecular dynamics simulation of the XIAP BIR2 structure. In order to investigate if a hydrophobic residue in the fourth position can induce a conformational change of the XIAP BIR2 and achieve favorable interactions, we used Sybyl38 to mutate this residue, converting SGVDDDM to SGVFDDM and added an N-methyl group to the C-terminus of the peptide. A molecular dynamics simulation of XIAP BIR2 in a complex with this mutated peptide was then performed to refine the structure.
Prior to the MD simulation, counter ions were added to neutralize the peptide-protein complex before solvating the system with a 10Å cube of explicit waters. The four residues bonded with the Zn2+ ion were constrained by moderate harmonic forces to prevent unfolding of the protein. The protocol for the MD simulation was as follows: A 1000-step minimization of the solvated system was performed and followed by 6 ps of MD simulation to gradually heat the system from 0K to 298K. The system was then equilibrated and refined by a further 94 ps simulation at 298 K. During the simulation, the four residues covalently bonded with the zinc ion and the backbone atoms of the remaining residues in XIAP BIR2 are constrained by harmonic forces with force constants equal to 15 kcal/mol. Based on this protocol, only the side chain atoms of XIAP BIR2 are refined and adapted to the mutated peptide (SGVFDDM) as compared to the SGVDDM determined in the crystal structure. This refined structure of XIAP BIR2 was then used to predict the models of our designed compounds binding to XIAP BIR2.
We used the Amber program suite39 (version 7) to perform all molecular dynamics (MD) simulations. A recent version of the Amber force field (ff96) 40 was used for the natural amino acids in the complex and the TIP3P model41 was used for water molecules. There is one Zn2+ ion covalently bound to C200, C203, H220, and C227 in the XIAP BIR2 domain and to C300, C303, H320, and C327 in the XIAP BIR3 domain. This Zn2+ ion, while important for structural integrity, has no direct interaction with the ligands. We used parameters developed by Ryde42 for the Zn2+ ion and its coordination with the neighboring four residues to model this chelating structure in our simulation. All the MD simulations were carried out at NTP. The SHAKE algorithm43 was used to fix the bonds involving hydrogen. The PME method44 was used to account for long range electrostatic interactions and the non-bonded cutoff distance was set at 10Å. The time step was 2 fs, and the neighboring pairs list was updated after every 20 steps. For the refinement of the structure between SM-122 and XIAP BIR2 and BIR3, the protocol is as follows: A 500-step minimization of the solvated system was performed followed by 6 ps of MD simulation to gradually heat the system from 0K to 298K. The system was then equilibrated by another 34 ps simulation at 298K. Finally, the 1 ns production simulation was run and the snapshots of conformations (typically 2000), evenly spaced in time, were collected for structural analysis.
All the binding poses of designed ligands with XIAP BIR2 and BIR3 were developed with the GOLD program 45 (version 2.2). The centers of the binding sites for XIAP BIR2 and BIR3 were set at T208 and T308 respectively and the radius of the binding site was defined as 13 Å, large enough to cover the binding pockets. For each genetic algorithm (GA) run, a maximum number of 200,000 operations were performed on a population of 5 islands of 100 individuals. Operator weights for crossover, mutation and migration were set to 95, 95 and 10 respectively. The docking simulations were terminated after 20 runs for each ligand. GoldScore implemented in Gold 2.2 was used as the fitness function to evaluate the docked conformations. The 20 conformations ranked highest by each fitness function were saved for analysis of the predicted docking modes. For the docking poses reported, these were the highest ranked conformations from the docking simulations.
III. Protein expression and purification, binding assays to different constructs of XIAP, cell-free functional assays for the activity of caspase-9 and caspase-3/-7, and analytical gel filtration assays
Protein expression and purification
Different constructs of human XIAP proteins, including linker-BIR2-BIR3 (residues 120–356), BIR3 (residues 241–356) and linker-BIR2 (residues 120–240) were cloned into a pET28 vector (Novagen) containing an N-terminal 6xHis tag. BIR2-BIR3 (residues 156–356), without the linker preceding BIR2, was cloned into a modified HIS-TEV vector with an N-terminal 8xHis tag. The mutated proteins, BIR2(E219R)-BIR3 and BIR2-BIR3(E314S,W323E) were created using QuikChange mutagensis (Stratagene) on a BIR2-BIR3 (residues 156–356) template. Proteins were produced in E. coli BL21(DE3) cells grown at 37°C in 2xYT containing kanamycin to an OD600 of 0.6. Protein expression was induced by 0.4 mM IPTG at 20°C for 20 hours (linker-BIR2-BIR3), 20°C for 4 hours (BIR2-BIR3), 37°C for 3 hours (linker-BIR2), or 27°C for 4 hours (BIR3). Cells were lysed by sonication in buffer containing Tris pH 7.5 (50 mM), NaCl (200 mM), ZnAc (50 μM), 0.1% βME and Leupectin/Aprotin protease inhibitors. Proteins were purified from the soluble fraction using Ni-NTA resin (QIAGEN) followed by gel filtration on a Superdex 75 column in Tris pH 7.5 (20 mM), NaCl (200 mM), ZnAc (50 μM), and dithiothretal (DTT, 1 mM). After purification, DTT was added to a final concentration of 10 mM.
Fluorescence-polarization-based binding for XIAP BIR3 protein
A sensitive in vitro binding assay using the fluorescence polarization (FP)-based method29 was used to determine the binding affinity of Smac mimetics to XIAP BIR3 protein. In this assay, 5-carboxyfluorescein was coupled to the lysine side chain of a mutated Smac peptide with the sequence (AbuRPFK-Fam). This fluorescently tagged peptide, named SM5F, was used as the fluorescent tracer in the FP-based binding assay of different compounds to the XIAP BIR3 protein. The recombinant XIAP BIR3 protein of human XIAP (residues 241–356) fused to His-tag was stable and soluble, and was used for the FP-based binding assay. The Kd value of SM5F peptide to XIAP BIR3 protein was determined to be 17.9 nM.29
Dose-dependent binding experiments were carried out with serial dilutions of the tested compounds. An aliquot of the samples and preincubated XIAP BIR3 protein (0.030 μM) and SM5F peptide (5 nM) in the assay buffer (100 mM potassium phosphate, pH 7.5; 100 μg/ml bovine gamma globulin; 0.02% sodium azide, purchased from Invitrogen Life Technology), were added to Dynex 96-well, black, round-bottom plates (Fisher Scientific). For each assay, the controls included XIAP BIR3 protein and SM5F (equivalent to 0% inhibition), and SM5F only (equivalent to 100% inhibition). The polarization values were measured after 3 hours of incubation using an ULTRA READER (Tecan U.S. Inc., Research Triangle Park, NC). IC50 values, the inhibitor concentration at which 50% of the bound tracer is displaced, were determined from a plot using nonlinear least-squares analysis. Curve fitting was performed using GRAPHPAD PRISM software (GraphPad Software, Inc., San Diego, CA).
Surface Plasmon Resonance (SPR) affinity measurements and SPR competitive binding assays to XIAP BIR2 and BIR3 domains
SPR experiments were performed at room temperature on a Biacore 3000 biosensor with HBS-P [HEPES, (pH 7.4, 10 mM), NaCl (150 mM), and 0.005% Tween 20] as the running buffer. Biotin labeled Smac mimetic, BL-SM-122, was immobilized onto streptavidin (SA) sensorchips by using streptavidin-biotin coupling chemistry. BL-SM-122 was immobilized on two chips with different densities, 272 and 105 response units on the sensor chip surface.
To collect kinetic binding data of BIR2 XIAP protein for BL-SM-122 directly, the BIR2 protein solution with different concentrations in running buffer was injected over the ligand and reference flow cells at a constant flow rate of 20 μl/min. During each injection, the ligand/protein complex was allowed to associate/dissociate for 240 and 300 seconds, respectively. Residual bound protein was desorbed with NaOH (50 mM), followed by two washes with the running buffer. Binding kinetics were derived from sensorgrams after subtraction of baseline responses and the data were fit globally to a 1:1 interaction model (A + B = AB) by using the BIA evaluation software.
For competitive binding assay, a fixed concentration of XIAP BIR2-only protein (1 μM) was incubated with different concentrations of the tested compound in HBS-P buffer for ~2 hr at room temperature. The mixtures were then injected over a sensorchip containing a channel with BL-SM-122 immobilized on the chip and a control without BL-SM-122. The baseline response (control) was subtracted to obtain the specific binding response. Taking the response for the protein alone as the maximal response (100%), the relative residual binding (%) in the presence of different concentrations of the tested compound at a given injection time point (225 s) was then calculated. The relative residual responses were plotted against initial concentrations of the tested compound and fitted using a nonlinear least-squares equation using GRAPHPAD PRISM software (GraphPad Software, Inc., San Diego, CA).
Fluorescence-polarization-based binding for XIAP L-BIR2-BIR3 protein
A FP-based competitive binding assay was established for quantitative determination of the binding affinities of our designed Smac mimetics to XIAP containing both BIR2 and BIR3 domains. In the competitive binding experiments, the tested compound was incubated with XIAP protein (residues 120–356, 3 nM) and Smac-1F (0.5 nM) in the assay buffer [potassium phosphate, pH 7.5 (100 mM); bovine gamma globulin (100 μg/ml); 0.02% sodium azide] in Dynex 96-well, black, round-bottom plates (Fisher Scientific). In each experiment, the controls included XIAP and Smac-1F peptide (equivalent to 0% inhibition), and Smac-1F only (100% of inhibition). Polarization values were measured after 2 hours incubation, using the Ultra plate reader. The IC50 value, the inhibitor concentration at which 50% of bound tracer was displaced, was determined from the plot using nonlinear least-squares analysis For each assay, Smac AVPI peptide was used as the control. Curve fitting was performed using GRAPHPAD PRISM software (GraphPad Software, Inc., San Diego, CA).
Cell-free caspase functional assays
MDA-MB-231 cell lysates were prepared by solubilizing cells in ice cold buffer containing KCl (50 mM), EGTA (5 mM), MgCl2 (2 mM) DTT (1 mM), 0.2% CHAPS and HEPES, (pH 7.5 50 mM), containing cocktail protease inhibitors, incubating on ice for 10 minutes, then freezing in liquid nitrogen. Cytochrome c and dATP were added to the cell lysates, which were then incubated at 30°C in a water bath for 60 minutes to activate caspase-9 and -3/-7. Addition of recombinant XIAP L-BIR2-BIR3 protein (50 nM) in the cell lysates completely suppressed the activity of caspase-9, and caspase-3/-7. Different concentrations of a tested Smac mimetic (1 nM - 100 μM) were added to determine the restoration of the activity of these caspases.
For determination of caspase activity, 25 μM of either caspase-9 substrate (Z-LEHD-AFC), or caspase-3/-7 substrate (Z-DEVD-AFC) (BioVision Inc.) was added. Fluorescence detection of substrate cleavage by caspase-9 or -3/-7 for their specific substrate was carried out on an ULTRA READER using an excitation wavelength of 400 nm and an emission wavelength of 505 nm. The reaction was monitored for 1–2 hours.
Analytical gel filtration experiments
To probe the binding mode of the designed Smac mimetics to XIAP proteins, we performed analytical gel filtration experiments with BIR3 (residues 241–356) protein, BIR2-BIR3 (residues 156–356) protein, BIR2(E219R)-BIR3 (residues 156–356) protein, and BIR2-BIR3(E314S,W323E) protein. Analytical gel filtration experiments were performed on a Superdex 75 column (GE Healthcare) attached to an AKTA Purifier-10 system in Tris-HCl, (pH 7.5, 20 mM), NaCl (200 mM), zinc acetate (50 μM), and DTT (1 mM). Recombinant protein was run on the column at a concentration of 1 mg/ml alone or after incubation with a 1:1 molar ratio of Smac mimetic. Molecular weight standards from Amersham-Pharmacia were used to calibrate the column.
IV. NMR HSQC Experiments
Expression and purification of 15N labeled proteins
The BIR3 domain (residues 241–356) of human XIAP fused to His-tag (pET28b, Novagen), the BIR2 domain (residues 156–260) and the BIR2-BIR3 protein (residues 156–356) of human XIAP in pHis-TEV vector were used to express respective proteins from BL21(DE3) cells (Nogaven) in M9 medium containing 15NH4Cl to label the protein uniformly with 15N. Most of the proteins were found in the soluble fraction and they were purified using TALON (Clontech) affinity chromatography, followed by Q-XL ion exchange and G75 size-exclusion chromatography (GE Healthcare) with AKTA purifier (GE Healthcare).
NMR HSQC experiments
15N HSQC NMR spectra were recorded on a Bruker AVANCE 500MHz NMR spectrometer with samples containing 100 μM of the 15N labeled proteins in 50 mM Tris (pH 7.2), 50 μM ZnCl2, 1 mM DTT at 25°C with or without test compound at a final concentration between 10–150 μM. The spectra were then compared in order to identify residues affected by the interaction with the test compound. Within the BIR3 domain, the known chemical shift assignments of the backbone atoms,13,46 were used to identify the residues corresponding to the affected peaks. The backbone atom resonance assignments of the BIR3 domain were also confirmed by 3D NMR triple resonance experiments (HNCA, HNCACB, HN(CO)CBCA, HNCO, TOCSY-HSQC, C(CO)NH). All NMR data were processed and analyzed using the nmrPipe/nmrDraw package (Dr. Frank Delaglio, NIDDK, NIH).47
V. Assays for cell growth, cell viability, apoptosis, Western blotting and biotin-streptavidin pull-down
Cell lines
Human HL-60 leukemia cell line was purchased from the American Type Culture Collection (Manassas, Virginia, USA), maintained in RPMI 1640 (Invitrogen), supplemented with 10% fetal bovine serum (Invitrogen) and 1% penicillin-streptomycin, at 37°C in 5% CO2. Human Jurkat T leukemic cell lines stably transfected with the pEBB-HA (VEC-JK), pEBB-HA-XIAP (XIAP-JK) were a kind gift of Dr. Colin Duckett (Departments of Pathology and Internal Medicine, University of Michigan). Jurkat cell lines were cultured in RPMI 1640 supplemented with 10% fetal bovine serum (Invitrogen) and 1% penicillin-streptomycin in the presence of puromycin from EMD Biosciences (2 μg/ml).
Cell growth assay
The effect of Smac mimetics on HL-60 cell growth was evaluated by a WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt assay (Dojindo Molecular Technologies, Inc). Cells (3000–4000 cells in each well) were cultured in 96-well tissue culture plates in medium (200 μl) containing various concentrations of Smac mimetics for indicated time. At the end of incubation, WST-8 dye (20 μl) was added to each well and incubated for an additional 1–3 h, then the absorbance was measured in a microplate reader (Molecular Devices) at 450 nm. Cell growth inhibition was evaluated as the ratio of the absorbance of the sample to that of the control.
Cell viability assay
Cell viability was quantitated by microscopic examination in a trypan blue exclusion assay. Cells were treated in triplicate for 24 hours, harvested and stained with an equal volume of 0.04% trypan blue. Both blue cells and morphologically shrunk cells were scored as non-viable cells. At least 100 cells from each treatment, performed in triplicate, were counted.
Apoptosis assay
Apoptosis assays were performed with an annexin-V/propidium iodide (PI) apoptosis detection kit (Roche) according to manufacturer’s instructions. Briefly, cells were harvested, washed with ice-cold PBS and then stained with annexin-V-FITC and PI for 15 minutes at room temperature in the dark. Stained cells were analyzed in a FACS calibur flow cytometer. Annexin-V (+) cells were measured as apoptotic cells, annexin V (−) and PI (+) cells were measured as death cells.
Western blotting
Cells were harvested and washed with cold PBS. Cell pellets were lysed in double lysis buffer (DLB; 50 mmol/L Tris, 150 mmol/L sodium chloride, (1 mmol)/L EDTA, 0.1% SDS and 1% NP-40) in the presence of L PMSF (1 mmol) and protease inhibitor cocktail (Roche) for 10 min on ice, then centrifuged at 13,000 rpm at 4°C for 10 min. Protein concentrations were determined using a Bio-Rad protein assay kit (Bio-Rad Laboratories).
Proteins were electrophoresed onto either 4% or 20% gradient SDS-PAGE (Invitrogen) then transferred to PVDF membranes. Following blocking in 5% milk, membranes were incubated with a specific primary antibody, washed, and incubated with horseradish peroxidase–linked secondary antibody (Amersham). The signals were visualized with the Chemiluminescent HRP antibody detection reagent (Denville Scientific). When indicated, the blots were stripped and reprobed with a different antibody. Primary antibodies against caspase-9, cleaved caspase 3 and β-actin were purchased from Cell Signaling Technology; primary antibody against cleaved PARP was purchased from Epitomics.
Biotin-avidin pull-down assay
The interaction between compounds 1, 2, 3 and 4 and cellular XIAP was investigated using a biotin-streptavidin pull-down assay. Cells were lysed in lysis buffer [20 mmol/L Tris, NaCl (150 mM/L) and 1% NP40] for 20 min. Cell lysates were precleared with streptavidin-agarose beads, incubated with biotinylated SM-122, named BL-SM-122, alone for pull-down assay, or pre-incubated with compound 1, 2, 3 and 4 for 5 min followed by co-incubation with BL-SM-122 for competitive experiments. Complexes formed between the Smac mimetics and the associated proteins were recovered by incubation with streptavidin-agarose beads (100 μl) on a shaker for 2 h at 4°C then centrifuged at 1000 × g for 1 min. The complexes were then washed 3 times with lysis buffer at 4°C and eluted from the beads by boiling in SDS loading buffer (100 μl). The eluted proteins were detected by Western blotting.
Supplementary Material
Complete references of 24, 27 and 39, NMR HSQC spectra of XIAP BIR2-BIR3 protein with different concentrations of compounds 1 and 4 and enlarged view for several BIR3 residues available. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We are grateful for the financial support from the Breast Cancer Research Foundation, the Prostate Cancer Foundation, the Department of Defense Prostate Cancer Program (W81XWH-04-1-0213), Ascenta Therapeutics, and the National Cancer Institute, NIH (R01CA109025). Jurkat cells were kind gifts of Dr. Colin Duckett from the University of Michigan. The authors thank Dr. Dennis Torchia at the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health for helpful discussions of the NMR experiments, Dr. Jason Gestwicki at the Life Sciences Institute and Department of Pathology at the University of Michigan for assistance and discussions of the SPR experiments, and Dr. G.W.A. Milne for critical reading of the manuscript.
References
- 1.Ponder BA. Nature. 2001;411:336–341. doi: 10.1038/35077207. [DOI] [PubMed] [Google Scholar]
- 2.Lowe SW, Lin AW. Carcinogenesis. 2000;21:485–495. doi: 10.1093/carcin/21.3.485. [DOI] [PubMed] [Google Scholar]
- 3.Nicholson DW. Nature. 2000;407:810–816. doi: 10.1038/35037747. [DOI] [PubMed] [Google Scholar]
- 4.Deveraux QL, Reed JC. Genes Dev. 13:239–252. doi: 10.1101/gad.13.3.239. [DOI] [PubMed] [Google Scholar]
- 5.Salvesen GS, Duckett CS. Nat Rev Mol Cell Biol. 2002;3:401–410. doi: 10.1038/nrm830. [DOI] [PubMed] [Google Scholar]
- 6.Tamm I, Kornblau SM, Segall H, Krajewski S, Welsh K, Kitada S, Scudiero DA, Tudor G, Qui YH, Monks A, Andreeff M, Reed JC. Clin Cancer Res. 2000;6:1796–1803. [PubMed] [Google Scholar]
- 7.Holcik M, Gibson H, Korneluk RG. Apoptosis. 2001;6:253–261. doi: 10.1023/a:1011379307472. [DOI] [PubMed] [Google Scholar]
- 8.Du C, Fang M, Li Y, Wang X. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 9.Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 10.Chai J, Du C, Wu JW, Kyin S, Wang X, Shi Y. Nature. 2000;406:855–62. doi: 10.1038/35022514. [DOI] [PubMed] [Google Scholar]
- 11.Shiozaki EN, Shi Y. Trends Biochem Sci. 2004;29:486–494. doi: 10.1016/j.tibs.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 12.Wu G, Chai J, Suber TL, Wu JW, Du C, Wang X, Shi Y. Nature. 2000;408:1008–1012. doi: 10.1038/35050012. [DOI] [PubMed] [Google Scholar]
- 13.Liu Z, Sun C, Olejniczak ET, Meadows RP, Betz SF, Oost T, Herrmann J, Wu JC, Fesik SW. Nature. 2000;408:1004–1008. doi: 10.1038/35050006. [DOI] [PubMed] [Google Scholar]
- 14.Srinivasula S, Hegde R, Saleh A, Datta P, Shiozaki E, Chai J, Lee RA, Robbins PD, Fernandes-Alnemri T, Shi Y, Alnemri ES. Nature. 2001;410:112–116. doi: 10.1038/35065125. [DOI] [PubMed] [Google Scholar]
- 15.Shiozaki EN, Chai J, Rigotti DJ, Riedl SJ, Li P, Srinivasula SM, Alnemri ES, Fairman R, Shi Y. Mol Cell. 2003;11:519–527. doi: 10.1016/s1097-2765(03)00054-6. [DOI] [PubMed] [Google Scholar]
- 16.Chai J, Shiozaki E, Srinivasula SM, Wu Q, Dataa P, Alnemri ES, Shi Y. Cell. 2001;104:769–780. doi: 10.1016/s0092-8674(01)00272-0. [DOI] [PubMed] [Google Scholar]
- 17.Huang Y, Park YC, Rich RL, Segal D, Myszka DG, Wu H. Cell. 2001;104:781–790. [PubMed] [Google Scholar]
- 18.Riedl SJ, Renatus M, Schwarzenbacher R, Zhou Q, Sun C, Fesik SW, Liddington RC, Salvesen GS. Cell. 2001;104:791–800. doi: 10.1016/s0092-8674(01)00274-4. [DOI] [PubMed] [Google Scholar]
- 19.Huang Y, Rich RL, Myszka DG, Wu H. J Biol Chem. 2003;278:49517–49522. doi: 10.1074/jbc.M310061200. [DOI] [PubMed] [Google Scholar]
- 20.Fulda S, Wick W, Weller M, Debatin KM. Nature Med. 2002;8:808–815. doi: 10.1038/nm735. [DOI] [PubMed] [Google Scholar]
- 21.Sun H, Nikolovska-Coleska Z, Yang CY, Xu L, Liu M, Tomita Y, Pan H, Yoshioka Y, Krajewski K, Roller PP, Wang S. J Am Chem Soc. 2004;126:16686–16687. doi: 10.1021/ja047438+. [DOI] [PubMed] [Google Scholar]
- 22.Sun H, Nikolovska-Coleska Z, Yang CY, Xu L, Tomita Y, Krajewski K, Roller PP, Wang S. J Med Chem. 2004;47:4147–4150. doi: 10.1021/jm0499108. [DOI] [PubMed] [Google Scholar]
- 23.Sun H, Nikolovska-Coleska Z, Lu J, Qiu S, Yang CY, Gao W, Meagher J, Stuckey J, Wang S. J Med Chem. 2006;49:7916–7920. doi: 10.1021/jm061108d. [DOI] [PubMed] [Google Scholar]
- 24.Oost TK, et al. J Med Chem. 2004;47:4417–4426. doi: 10.1021/jm040037k. [DOI] [PubMed] [Google Scholar]
- 25.Zobel K, Wang L, Varfolomeev E, Franklin MC, Elliott LO, Wallweber HJ, Okawa DC, Flygare JA, Vucic D, Fairbrother WJ, Deshayes K. ACS Chem Biol. 2006;1:525–533. doi: 10.1021/cb600276q. [DOI] [PubMed] [Google Scholar]
- 26.Wu TY, Wagner KW, Bursulaya B, Schultz PG, Deveraux QL. Chem Biol. 2003;10:759–67. doi: 10.1016/s1074-5521(03)00157-1. [DOI] [PubMed] [Google Scholar]
- 27.Schimmer AD, et al. Cancer Cell. 2004;5:25–35. doi: 10.1016/s1535-6108(03)00332-5. [DOI] [PubMed] [Google Scholar]
- 28.Li L, Thomas RM, Suzuki H, De Brabander JK, Wang X, Harran PG. Science. 2004;305:1471–4. doi: 10.1126/science.1098231. [DOI] [PubMed] [Google Scholar]
- 29.Nikolovska-Coleska Z, Wang R, Fang X, Pan H, Tomita Y, Li P, Roller PP, Krajewski K, Saito NG, Stuckeys JA, Wang S. Anal Biochem. 2004;332:261–73. doi: 10.1016/j.ab.2004.05.055. [DOI] [PubMed] [Google Scholar]
- 30.Kipp RA, Case MA, Wist AD, Cresson CM, Carrell M, Griner E, Wiita A, Albiniak PA, Chai J, Shi Y, Semmelhack MF, McLendon GL. Biochemistry. 2002;41:7344–7349. doi: 10.1021/bi0121454. [DOI] [PubMed] [Google Scholar]
- 31.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 32.Duggan HME, Hitchcock PB, Young DW. Org Biomol Chem. 2005;3:2287–2295. doi: 10.1039/b503014e. [DOI] [PubMed] [Google Scholar]
- 33.Messina F, Botta M, Corelli F, Schneider MP, Fazio F. J Org Chem. 1999;64:3767–3769. doi: 10.1021/jo982513i. [DOI] [PubMed] [Google Scholar]
- 34.Sun H, Nikolovska CZ, Yang CY, Wang S. Tetrahedron Letters. 2005;46:7015–7018. [Google Scholar]
- 35.Nikolovska-Coleska Z, Meagher JL, Jiang S, Kawamoto SA, Gao W, Yi H, Qin D, Roller PR, Stuckey JA, Wang S. Analytical Biochemistry. doi: 10.1016/j.ab.2007.10.032. (submitted) [DOI] [PubMed] [Google Scholar]
- 36.Wilkinson JC, Cepero E, Boise LH, Duckett CS. Mol Cell Biol. 2004;24:7003–7014. doi: 10.1128/MCB.24.16.7003-7014.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scott FL, Denault JB, Riedl SJ, Shin H, Renatus M, Salvesen GS. EMBO J. 2005;24:645–655. doi: 10.1038/sj.emboj.7600544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sybyl, a molecular modeling system, is supplied by Tripos, Inc., St. Louis, MO 63144.
- 39.Case DA, et al. AMBER. Vol. 7. University of California; San Francisco: 2002. [Google Scholar]
- 40.Kollman PA, Dixon R, Cornell W, Fox T, Chipot C, Pohorille A. In: Computer Simulation of Biomolecular Systems. Wilkinson A, Weiner P, van Gunsteren WF, editors. Vol. 3. Elsevier; 1997. p. 83. [Google Scholar]
- 41.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 42.Ryde U. Proteins: Struct Funct Genet. 1995;21:41–56. [Google Scholar]
- 43.Rychaert JP, Ciccotti G, Berendsen HJC. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 44.Darden TA, York DM, Pedersen L. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 45.Jones G, Willett P, Glen RC, Leach AR, Taylor R. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 46.Sun C, Cai M, Meadows RP, Xu N, Gunasekera AH, Herrmann J, Wu JC, Fesik SW. J Biol Chem. 2000;275:33777–33781. doi: 10.1074/jbc.M006226200. [DOI] [PubMed] [Google Scholar]
- 47.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Complete references of 24, 27 and 39, NMR HSQC spectra of XIAP BIR2-BIR3 protein with different concentrations of compounds 1 and 4 and enlarged view for several BIR3 residues available. This material is available free of charge via the Internet at http://pubs.acs.org.