

Abstract
Glycogen storage disease type Ia (GSD-Ia) patients deficient in glucose-6-phosphatase-α manifest a disturbed glucose homeostasis. We hypothesized that disturbed glucose homeostasis might affect myeloid functions. Here we show that GSD-Ia mice exhibit normal neutrophil activities but have elevated myeloid progenitor cells in the bone marrow and spleen. Interestingly, GSD-Ia mice exhibit a persistent increase in peripheral blood neutrophil counts along with elevated serum levels of granulocyte colony stimulating factor and cytokine-induced neutrophil chemoattractant. Taken together, our results suggest that a loss of glucose homeostasis can compromise the immune system, resulting in neutrophilia. This may explain some of the unexpected clinical manifestations seen in GSD-Ia.
Keywords: glycogen storage disease type I, glucose-6-phosphatase-alpha, neutrophilia, granulocyte colony stimulating factor, cytokine-induced neutrophil chemoattractant, myeloid functions
1. Introduction
Glycogen storage disease type I (GSD-I) is caused by a deficiency in the glucose-6-phosphatase-α (G6Pase-α) complex that consists of a glucose-6-phosphate transporter (G6PT) that translocates glucose-6-phosphate (G6P) from the cytoplasm into the lumen of the endoplasmic reticulum, and a G6Pase-α catalytic subunit (also known as G6PC), that hydrolyses endoluminal G6P to glucose and phosphate (1). Together, they work to maintain blood glucose homeostasis between meals. A deficiency of G6Pase-α causes GSD type Ia (GSD-Ia, MIM232200) and a deficiency in G6PT causes GSD type Ib (GSD-Ib, MIM232220) (1). While G6Pase-α is expressed primarily in the liver, kidney and intestine (2), G6PT is expressed ubiquitously (3). G6Pase-α and G6PT activities are functionally linked (4, 5) and a detrimental mutation in either protein prevents the other from functioning efficiently and leads to the same metabolic phenotype, characterized by fasting hypoglycemia, hepatomegaly, nephromegaly, hyperlipidemia, hyperuricemia, lactic acidemia, and growth retardation (1). While the metabolic abnormalities of GSD-Ia and GSD-Ib are almost identical (1), GSD-Ib patients exhibit neutropenia and myeloid dysfunctions (1, 6, 7). There are no clinical reports of neutrophil dysfunctions in GSD-Ia patients, and it has been assumed that the GSD-Ib defects arise from alternative activities of G6PT in myeloid tissues. We suggest an alternative hypothesis, namely that the loss of glucose homeostasis does underlie the neutrophil defects, but that the dysfunctions in GSD-Ia do not rise to the level of clinical observation or significance seen in GSD-Ib. In this report, we report evidence to support our hypothesis.
To study the biology and pathophysiology of GSD-I, we have generated G6Pase-α-deficient (4) and G6PT-deficient (8) mouse strains that mimic the human disorders. Both manifest the metabolic abnormalities characteristics of a disturbed glucose homeostasis and the GSD-Ib mice also suffer from neutropenia and neutrophil dysfunctions characteristic of GSD-Ib. As with the human disease, serum levels of granulocyte colony stimulating factor (G-CSF) and cytokine-induced neutrophil chemoattractant (KC) are markedly increased in GSD-Ib mice (8). We also showed that G6PT expression in bone marrow and neutrophils is required for normal myeloid functions (9). However, our study also suggests that some of the myeloid abnormalities observed in GSD-Ib are caused by the disturbed glucose homeostasis (9).
The similarity in metabolic abnormalities between GSD-Ia and GSD-Ib prompted us to examine myeloid functions in GSD-Ia mice. We show that the GSD-Ia neutrophils have a normal respiratory burst, chemotaxis, and calcium flux activities. Like GSD-Ib mice, bone and spleen of GSD-Ia mice are developmentally delayed and both organs exhibit hypocellularity and moderate increases in myeloid progenitor cell numbers. Interestingly, serum levels of G-CSF and KC are also elevated in GSD-Ia mice. In contrast to neutropenia manifested by GSD-Ib patients and mice, peripheral blood neutrophil counts are consistently elevated in GSD-Ia mice.
2. Materials and methods
2.1. Respiratory Burst, Chemotaxis, and [Ca2+]i Measurements
All animal studies were conducted under an animal protocol approved by the NICHD Animal Care and Use Committee. A glucose therapy was administered to the GSD-Ia mice as described previously (4) and mice that survived weaning were given unrestricted access to Mouse Chow (Zeigler Bros., Inc., Gardners, PA). GSD-Ia is an autosomal recessive disorder (1). Since the phenotypes of wild-type and heterozygous mice are identical, with both exhibiting normal development, metabolic functions and myeloid functions (4), we will use the designation “unaffected” or “control” mice to refer to either wild-type or heterozygous mice.
Neutrophils were isolated from thioglycollate-stimulated peritonea of 6-7 week-old mice as described previously (8). Neutrophils were activated with 200 ng/ml of phorbol myristate acetate (PMA; Sigma Chemical Co.) and neutrophil respiratory burst was monitored by luminal-amplified chemiluminescence using the LumiMax Superoxide Anion Detection kit (Stratagene, La Jolla, CA) as described previously (8).
Neutrophil chemotaxis in response to f-Met-Leu-Phe (fMLP, Sigma Chemical Co.), KC, or macrophage inflammatory protein-2 (MIP-2) (PeproTech Inc., Rocky Hill, NJ) was performed in 48-well chambers (NeuroProbe, Gaithersburg, MD) using polyvinylpyrrolidone-free polycarbonate membranes with 3 μm pores as described previously (8). Cells were counted in six randomly selected fields at 400-fold magnification and reported as the mean average.
Intracellular calcium ion concentration measurements ([Ca2+]i) in neutrophils were performed in poly-L-lysine coated 96-well plates (Greiner, Longwood, FL). The neutrophils (106 cells) were suspended in 100 μl of 1X HBSS containing 10 mM HEPES and followed by 100 μl of FLIPER calcium 3 assay kit component A (Molecular Devices, Sunnyvale, CA) dissolved in HEPES-HBSS Buffer. The plate was then loaded into a Flexstation Fluorimeter (Molecular Devices) set at 37 °C, the appropriate ligands added to the plate robotically, and cells were excited at 485 nm, and the fluorescence intensities detected and recorded every 0.5 sec at 515 nm.
2.2. Hematological and phenotype analyses
For the differential leukocyte counts, blood samples were collected from the tail vein of mice using EDTA-containing CAPIJECT tubes (TerumoMedical Co., Elkton, MD). Manual 200-cell leukocyte differential counts of peripheral blood cells were performed on Hema 3 (Fisher Scientific, Pittsburgh, PA.) stained smears as described previously (8). The cytokines, G-CSF and KC were quantified using Quantikine ELISA kits (R&D Systems Inc., Minneapolis, MN). For hematoxylin and eosin (H&E) staining, tissues were preserved in 10% neutral buffered formalin, embedded in paraffin, and sectioned at 4-6 micron thickness.
2.3. Hematopoietic progenitor cell assays
Progenitor cells were assayed in semisolid agar cultures by plating 2 × 104 bone marrow or 105 spleen cells in 1 ml of methylcellulose media (MethoCult M3231, Stem Cell Technologies, Vancouver, Canada) supplemented with G-CSF (10 ng/ml), granulocyte macrophage-CSF (GM-CSF) (10 ng/ml), or macrophage-CSF (M-CSF) (2.5 ng/ml). The number of colonies containing more than 50 cells was counted on days 7 to 9.
2.4. Statistical analysis
The unpaired t test was performed using the GraphPad Prism Program, version 4 (GraphPad Software, San Diego, CA). Values were considered statistically significant at p < 0.05.
3. Results
3.1. The GSD-Ia mice do not manifest neutrophil dysfunctions characteristics of GSD-Ib
Neutrophils from GSD-Ib patients (6, 7) and mice (8) exhibit impaired respiratory burst, chemotaxis, and calcium flux activities. We therefore examined neutrophil activities in thioglycollate-recruited peritoneal neutrophils from GSD-Ia and their unaffected littermates. Results in Fig. 1A show that after exposure to PMA, superoxide production was markedly increased in both control and GSD-Ia neutrophils and both activities remained elevated for more than 16 minutes. Moreover, neutrophils from control and GSD-Ia mice exhibited nearly identical dose-dependent chemotactic responses to fMLP, KC, and MIP-2 (Fig. 1B). In a similar fashion the thioglycollate-elicited peritoneal neutrophils from control and GSD-Ia mice showed similar increases in Ca2+ mobilization in response to fMLP, KC, and MIP-2 (Fig. 1C).
Fig. 1.
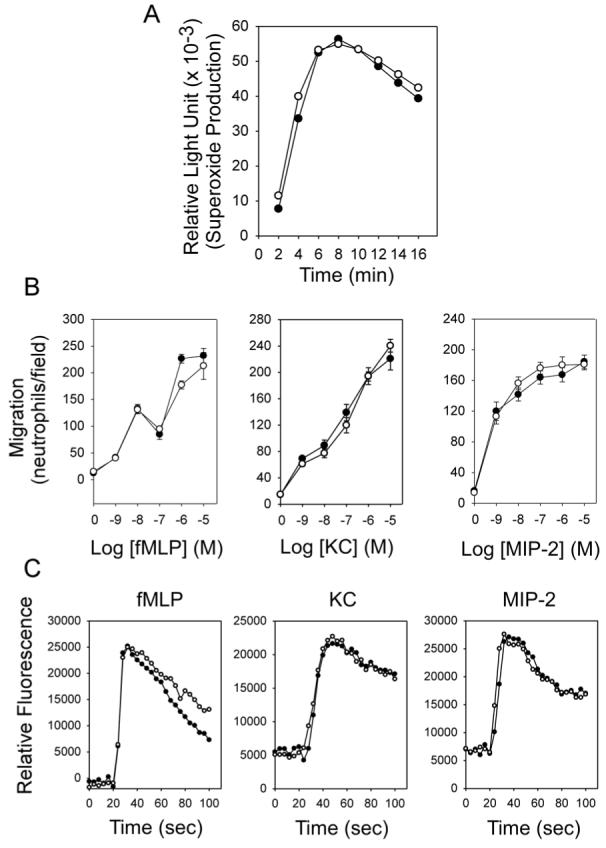
GSD-Ia neutrophils exhibit no defect in respiratory burst, chemotaxis, and calcium flux. Neutrophils were isolated from thioglycollate-induced peritoneum of 6-7 week-old unaffected (○) and GSD-Ia (●) mice. (A) Neutrophil respiratory burst activity. Representative experiments are shown. (B) Concentration-dependent chemotaxis in response to fMLP, KC or MIP-2. Values represent mean ± SEM of quadruplet determinations. (C) Ca2+ flux in response to 10-6 M of fMLP, KC or MIP-2. Representative experiments are shown.
3.2. Altered hematopoiesis in the bone marrow and spleen of GSD-Ia mice
After birth, bone marrow is the primary site of maturation and development of hematopoietic cells (10). In neonatal mice, the spleen is also a hematopoietic organ (11). In GSD-Ib mice, both the bone and spleen are developmentally delayed (8) and the same is observed in the GSD-Ia mice (Fig. 2). In the unaffected mice the epiphyses and growth plate of femoral and tibia bones were well formed at 2 weeks of age (Fig. 2A), but in GSD-Ia mice the epiphyses and growth plate were not evident at 2 weeks of age becoming well formed only at 4-5 weeks of age (Fig. 2A). Similarly the white pulps in the spleen of the unaffected mice are evident at 1 week of age and well formed by 2 weeks of age (Fig. 2B), but in the GSD-Ia mice are not evident until 3 weeks of age, becoming well formed only at 5 weeks of age (Fig. 2B).
Fig. 2.
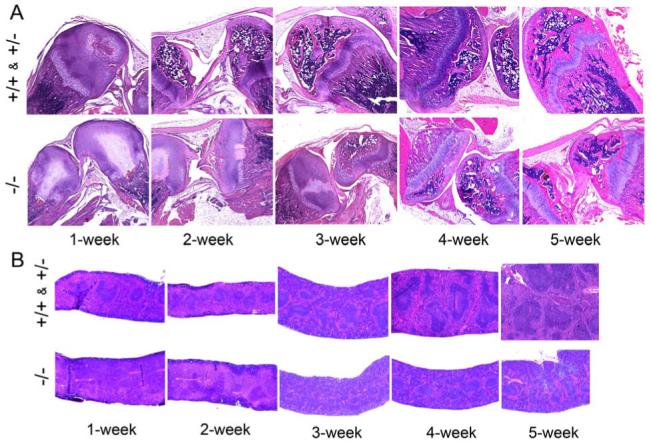
Histological analyses of bone and spleen. The unaffected (+/+ & +/-) and GSD-Ia (-/-) mice between 1 and 5 weeks of age were examined. (A) H&E stained bone sections at magnifications of ×50. (B) H&E stained spleen sections at magnifications of ×50.
We then examined colony-forming progenitor cells in the bone marrows and spleens of 3-week-old GSD-Ia and control littermates. In bone marrow aspirates combined from the femur and tibia, the total numbers of cells in the unaffected mice are 3.3-fold higher than those in the GSD-Ia mice (Fig. 3A), consistent with the delay in postnatal development. The colony forming units (CFU) in bone marrow aspirates after in vitro stimulation with G-CSF, GM-CSF, or M-CSF revealed 1.3-fold more CFU-G, 1.7-fold more CFU-GM, and 1.4-fold more CFU-M in GSD-Ia mice compared to the control littermates (Fig. 3A). In 3-week-old GSD-Ib mice, the numbers of bone marrow CFU-G, CFU-GM, and CFU-M were 3.7-, 10.5-, and 3.9-fold higher, respectively than age-matched control mice (8). Therefore, the elevations in GSD-Ia mice are reminiscent of, though not as highly elevated as the numbers observed in GSD-Ib mice (8).
Fig.3.
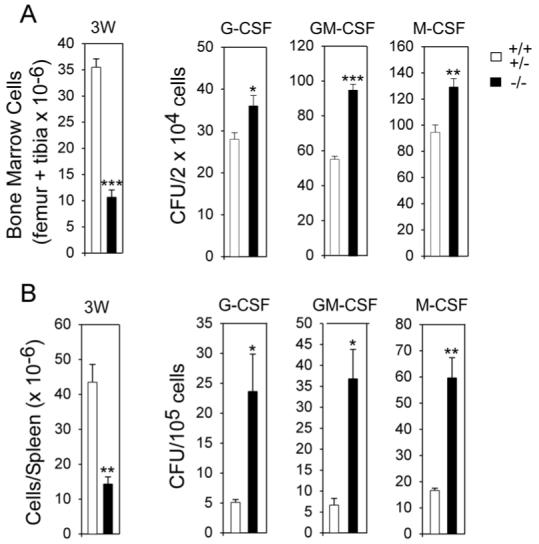
Total cell counts and myeloid progenitor cells in the femur plus tibia and spleen of GSD-Ia mice. The myeloid functions were examined in 3-week-old unaffected (+/+ & +/-) and GSD-Ia (-/-) mice. CFU were determined following stimulation bone marrow or spleen cells with G-CSF, GM-CSF, or M-CSF. Results are the mean ± SEM from four separate experiments in which each mouse was assessed individually. (A) Bone marrow. (B) Spleen. *p < 0.05; **p < 0.005; ***p < 0.0001.
The total numbers of cells in the spleen in 3-week-old control mice were 3-fold higher than those in the GSD-Ia littermates and the numbers of splenic CFU-G, CFU-GM, and CFU-M in GSD-Ia mice were 4.7-, 5.5-, and 3.6-fold higher, respectively than age-matched control mice (Fig. 3B). In 3-week-old GSD-Ib mice, the numbers of splenic CFU-G, CFU-GM, and CFU-M were similarly elevated, being 3.7-, 10.5-, and 3.9-fold higher, respectively than age-matched control mice (8).
3.3. GSD-Ia mice exhibit neutrophilia along with increased serum levels of G-CSF and KC
In GSD-Ib mice, serum levels of G-CSF and KC were abnormally increased and G-CSF and KC in 3-week-old GSD-Ib mice were 5.5- and 6.1-fold higher, respectively, than the unaffected littermates (8). In 3-week-old GSD-Ia mice, serum G-CSF and KC values were also increased but only to 2- and 3-fold higher, respectively, than the control animals (Fig. 4A). Serum G-CSF and KC remained elevated being 1.7- and 3-fold higher, respectively, than the unaffected littermates at age 6 weeks (Fig. 4A).
Fig. 4.
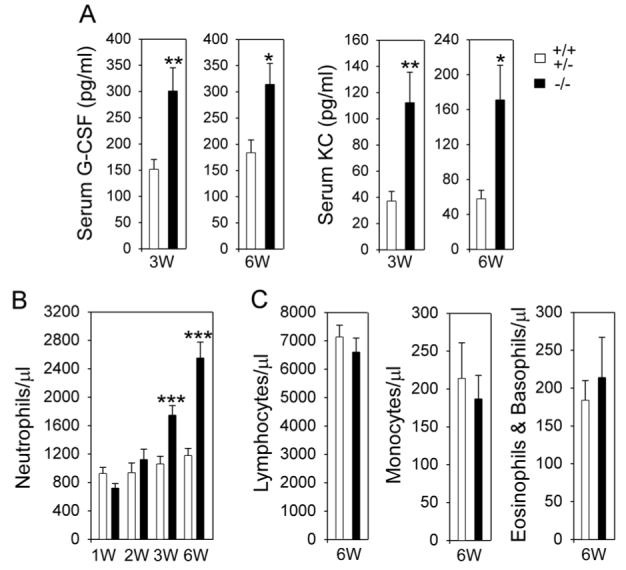
Analysis of serum levels of G-CSF and KC and peripheral blood leukocyte counts. The unaffected (+/+ & +/-) and GSD-Ia (-/-) mice between 1 and 6 weeks of age were examined. (A) Serum cytokines in mice at 3 weeks (n = 37 for +/+ & +/- and -/-) and 6 weeks (n = 12 for +/+ & +/- and n = 20 for -/-) of age. (B) Blood neutrophil counts in mice during post-natal development. *p < 0.05; **p < 0.005; ***p < 0.001. The differences in neutrophil counts between control and GSD-Ia mice at 1 and 2 weeks of age are not statistically significant. (C) Blood lymphocyte, monocyte, and eosinophil/basophil counts in 6-week-old mice. The differences between control and GSD-Ia mice are not statistically significant.
Differential peripheral blood leukocyte counts revealed that neutrophil counts in 1-week-old GSD-Ia mice averaged 78% of the counts in the control mice (Fig. 4B). While the absolute neutrophil counts in the unaffected mice increased slightly during postnatal development, neutrophil counts in GSD-Ia mice increased markedly with age (Fig. 4B). As a result, neutrophil counts in 2-, 3-, and 6-week-old GSD-Ia mice averaged 1.2-, 1.7-, and 2.2-fold higher, respectively, than the counts in the control littermates. On the other hand, blood lymphocyte, monocyte, and eosinophil/basophil counts were similar between 6-week-old GSD-Ia and control mice (Fig. 4C). The neutrophilia seen in GSD-Ia mice was a marked contrast to the neutropenia manifested by GSD-Ib patients (6, 7) and mice (8) despite a higher increase in serum levels of G-CSF and KC or interleukin-8 (IL-8) in GSD-Ib (8).
4. Discussion
GSD-Ia and GSD-Ib share similar metabolic abnormalities caused by the loss of interprandial glucose homeostasis (1). However GSD-Ib patients and mice manifest additional symptoms of neutrophil dysfunctions. Since the G6PT gene has a ubiquitous expression profile (3) compared to the liver/kidney/intestine restricted profile of the G6Pase-α gene (2), it has been assumed that the neutrophil dysfunctions reflect a secondary activity of the G6PT protein in myeloid tissues. While we have shown that there is good evidence for a secondary activity for G6PT, we have also hypothesized that disruption of glucose homeostasis could also contribute, in part, to the myeloid dysfunction (9). If this is true, GSD-Ia neutrophil activity should also differ from the controls. Consistent with our hypothesis, the GSD-Ia mice exhibit an elevated myeloid progenitor cell frequency in the bone marrow and spleen, and have increased serum concentrations of G-CSF and KC, reminiscent of GSD-Ib (8). Notably, these changes are significantly less pronounced in GSD-Ia than GSD-Ib mice. More interestingly, while GSD-Ib patients and mice exhibit neutropenia, the GSD-Ia mice exhibit neutrophilia. Functionally, the GSD-Ia mice exhibit no defects in neutrophil respiratory burst, chemotaxis, or calcium flux, indicating that G6Pase-α expression is not required for normal neutrophil functions. This is not unexpected because neutrophils do not express the G6Pase-α gene (2). Taken together, our study suggests that myeloid functions in GSD-Ia are also abnormal, if not clinically overt, and arise from the disturbance of glucose homeostasis.
The neutrophil number in the blood is tightly regulated and represents a balance between the production, release, and clearance of neutrophils from the circulation. A key component in this is G-CSF, a hematopoietic growth factor that regulates granulopoiesis (12, 13). KC, a member of the CXC chemokine family, also modulates the neutrophil number, by recruiting neutrophils to local sites of inflammation (14, 15). In this study, the serum levels of G-CSF and KC in GSD-Ia mice were 2- and 3-fold higher, respectively than the levels in the unaffected littermates, indicating an underlying challenge to the immune system (14-16). Consistent with studies that correlate overexpression of G-CSF and IL-8, the human homolog of KC, with neutrophilia (17, 18); we also observed neutrophilia in GSD-Ia mice. The cause of the increase in cytokines in GSD-Ia mice is unclear; however, since there is a long-term damage to both the liver and kidney in GSD-Ia, despite dietary controls, it is reasonable to suggest there is a progressive low level of immune challenge reflected by these findings. In this regard it is interesting that the increased secretion of chemokines has been detected in a wide variety of diseases, including glomerulonephritis, ischemia-reperfusion, alcoholic hepatitis, and hepatocellular carcinoma (14, 15, 19-21).
GSD-Ia (4) and GSD-Ib (8) mice are both growth retarded. Likewise, bone and spleen are developmentally delayed in both GSD-Ia (this study) and GSD-Ib (8) mice, and both are accompanied by reduced cellularity and increased myeloid progenitor cell frequencies. When compared, the decrease in cellularity and the increase in myeloid progenitor cells are more pronounced in GSD-Ib mice, than the GSD-Ia mice, which correlates with the serum levels of G-CSF and KC being elevated more in GSD-Ib than GSD-Ia mice. Our current findings therefore suggest that myeloid dysfunction in GSD-Ib could be caused by the intrinsic G6PT deficiency in combination with the disturbed glucose homeostasis.
The question remains why despite the marked increase in serum G-CSF and IL-8 or KC in GSD-Ib patients and mice (8) they manifest neutropenia. The explanation may lie in the enhanced neutrophil apoptosis observed in GSD-Ib (22) which is sufficient to counter the neutrophilia. G-CSF therapy has successfully treated neutropenia and the severity of infections manifested by GSD-Ib patients (23). However, G-CSF therapy does not rescue neutrophil apoptosis in GSD-Ib patients (22). Also, a normal number of functional neutrophils were required for hematopoietic stem cell mobilization while neutropenic phase impairs such mobilization (24). This suggests that enhanced neutrophil apoptosis which cannot be rescued by G-CSF therapy and the impairment in neutrophil-mediated hematopoietic stem cell mobilization contribute to neutropenia seen in GSD-Ib.
In summary, we have shown that a loss of glucose homeostasis in GSD-I challenges the immune system leading to an increase in serum G-CSF and KC. In GSD-Ia this leads to neutrophilia. Our findings highlight the critical role blood glucose homeostasis can play in normal myeloid function.
Acknowledgements
This research was supported in part by the Intramural Research Program of the NICHD, NIH.
The abbreviations used are
- GSD-Ia
glycogen storage disease type Ia
- G6Pase-α
glucose-6-phosphatase-α
- GCSF
granulocyte colony stimulating factor
- KC
cytokine-induced neutrophil chemoattractant
References
- 1.Chou JY, Matern D, Mansfield BC, Chen Y-T. Type I glycogen storage diseases: disorders of the glucose-6-phosphatase complex. Curr. Mol. Med. 2002;2:121–143. doi: 10.2174/1566524024605798. [DOI] [PubMed] [Google Scholar]
- 2.Pan C-J, Lei K-J, Chen H, Ward JM, Chou JY. Ontogeny of the murine glucose-6-phosphatase system. Arch. Biochem. Biophys. 1998;358:17–24. doi: 10.1006/abbi.1998.0849. [DOI] [PubMed] [Google Scholar]
- 3.Lin B, Annabi B, Hiraiwa H, Pan C-J, Chou JY. Cloning and characterization of cDNAs encoding a candidate glycogen storage disease type 1b protein in rodents. J. Biol. Chem. 1998;273:31656–31670. doi: 10.1074/jbc.273.48.31656. [DOI] [PubMed] [Google Scholar]
- 4.Lei K-J, Chen H, Pan C-J, Ward JM, Mosinger B, Lee EJ, Westphal H, Chou JY. Glucose-6-phosphatase dependent substrate transport in the glycogen storage disease type 1a mouse. Nat. Genet. 1996;13:203–209. doi: 10.1038/ng0696-203. [DOI] [PubMed] [Google Scholar]
- 5.Hiraiwa H, Pan C-J, Lin B, Moses SW, Chou JY. Inactivation of the glucose-6-phosphate transporter causes glycogen storage disease type 1b. J. Biol. Chem. 1999;274:5532–5536. doi: 10.1074/jbc.274.9.5532. [DOI] [PubMed] [Google Scholar]
- 6.Beaudet AL, Anderson DC, Michels VV, Arion WJ, Lange AJ. Neutropenia and impaired neutrophil migration in type 1B glycogen storage disease. J. Pediatr. 1980;97:906–910. doi: 10.1016/s0022-3476(80)80418-5. [DOI] [PubMed] [Google Scholar]
- 7.Chou JY, Mansfield BC. Glucose-6-phosphate transporter: the key to glycogen storage disease type Ib. In: Broer S, Wagner CA, editors. Membrane Transporter Diseases. Springer; New York: 2003. pp. 191–205. [Google Scholar]
- 8.Chen L-Y, Shieh J-J, Lin B, Pan C-J, Gao J-L, Murphy PM, Roe TF, Moses S, Ward JM, Westphal H, Lee EJ, Mansfield BC, Chou JY. Impaired glucose homeostasis, neutrophil trafficking and function in mice lacking the glucose-6-phosphate transporter. Hum. Mol. Genet. 2003;12:2547–2558. doi: 10.1093/hmg/ddg263. [DOI] [PubMed] [Google Scholar]
- 9.Kim SY, Nguyen AD, Gao J-L, Murphy PM, Mansfield BC, Chou JY. Bone-Marrow Derived Cells Require a Functional Glucose-6-Phosphate Transporter for Normal Myeloid Functions. J. Biol. Chem. 2006;281:28794–28801. doi: 10.1074/jbc.M604964200. [DOI] [PubMed] [Google Scholar]
- 10.Sieff CA, Nathan DG, Clark SC. The anatomy and physiology of hematopoiesis. In: Nathon DG, Orkin SH, editors. Hematology of Infancy and Childhood. 5th ed. Vol. 1. W.B. Saunders; Philadelphia: 1998. pp. 161–236. [Google Scholar]
- 11.Wolber FM, Leonard E, Michael S, Orschell-Traycoff CM, Yoder MC, Srour EF. Roles of spleen and liver in development of the murine hematopoietic system. Exp. Hematol. 2002;30:1010–1019. doi: 10.1016/s0301-472x(02)00881-0. [DOI] [PubMed] [Google Scholar]
- 12.Barreda DR, Hanington PC, Belosevic M. Regulation of myeloid development and function by colony stimulating factors. Dev. Comp. Immunol. 2004;28:509–554. doi: 10.1016/j.dci.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 13.Christopher MJ, Link DC. Regulation of neutrophil homeostasis. Curr. Opin. Hematol. 2007;14:3–8. doi: 10.1097/00062752-200701000-00003. [DOI] [PubMed] [Google Scholar]
- 14.Luster AD. Chemokines-chemotactic cytokines that mediate inflammation. N. Engl. J. Med. 1998;338:436–445. doi: 10.1056/NEJM199802123380706. [DOI] [PubMed] [Google Scholar]
- 15.Murdoch C, Finn A. Chemokine receptors and their role in inflammation and infectious diseases. Blood. 2000;95:3032–3043. [PubMed] [Google Scholar]
- 16.Kawakami M, Tsutsumi H, Kumakawa T, Abe H, Hirai M, Kurosawa S, Mori M, Fukushima M. Levels of serum granulocyte colony-stimulating factor in patients with infections. Blood. 1990;76:1962–1964. [PubMed] [Google Scholar]
- 17.Serizawa I, Amano K, Ishii H, Ichikawa T, Kusaka M, Taguchi T, Kiyokawa N, Fujimoto J. Long-term overexpression of human granulocyte colony-stimulating factor in transgenic mice: persistent neutrophilia with no increased mortality for more than one year. Cytokine. 2000;12:630–635. doi: 10.1006/cyto.2000.0669. [DOI] [PubMed] [Google Scholar]
- 18.Aggarwal A, Baker CS, Evans TW, Haslam PL. G-CSF and IL-8 but not GM-CSF correlate with severity of pulmonary neutrophilia in acute respiratory distress syndrome. Eur. Respir. J. 2000;15:895–901. doi: 10.1034/j.1399-3003.2000.15e14.x. [DOI] [PubMed] [Google Scholar]
- 19.Ren Y, Poon RT, Tsui HT, Chen WH, Li Z, Lau C, Yu WC, Fan ST. Interleukin-8 serum levels in patients with hepatocellular carcinoma: correlations with clinicopathological features and prognosis. Clin. Cancer Res. 2003;9:5996–6001. [PubMed] [Google Scholar]
- 20.Leemans JC, Stokman G, Claessen N, Rouschop KM, Teske GJ, Kirschning CJ, Akira S, van der Poll T, Weening JJ, Florquin S. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J. Clin. Invest. 2005;115:2894–2903. doi: 10.1172/JCI22832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kobayashi Y. Neutrophil infiltration and chemokines. Crit. Rev. Immunol. 2006;26:307–331. doi: 10.1615/critrevimmunol.v26.i4.20. [DOI] [PubMed] [Google Scholar]
- 22.Kuijpers TW, Maianski NA, Tool AT, Smit GP, Rake JP, Roos D, Visser G. Apoptotic neutrophils in the circulation of patients with glycogen storage disease type 1b (GSD1b) Blood. 2003;101:5021–5024. doi: 10.1182/blood-2002-10-3128. [DOI] [PubMed] [Google Scholar]
- 23.Calderwood S, Kilpatrick L, Douglas SD, Freedman M, Smith-Whitley K, Rolland M, Kurtzberg J. Recombinant human granulocyte colony-stimulating factor therapy for patients with neutropenia and/or neutrophil dysfunction secondary to glycogen storage disease type 1b. Blood. 2001;97:376–382. doi: 10.1182/blood.v97.2.376. [DOI] [PubMed] [Google Scholar]
- 24.Pruijt JF, Verzaal P, van Os R, de Kruijf EJ, van Schie ML, Mantovani A, Vecchi A, Lindley IJ, Willemze R, Starckx S, Opdenakker G, Fibbe WE. Neutrophils are indispensable for hematopoietic stem cell mobilization induced by interleukin-8 in mice. Proc. Natl. Acad. Sci. USA. 2002;99:6228–6233. doi: 10.1073/pnas.092112999. [DOI] [PMC free article] [PubMed] [Google Scholar]