

Abstract
The objective of this study was to identify and characterize a self-renewing subpopulation of human ovarian tumor cells (ovarian cancer-initiating cells, OCICs) fully capable of serial propagation of their original tumor phenotype in animals. Ovarian serous adenocarcinomas were disaggregated and subjected to growth conditions selective for self-renewing, nonadherent spheroids previously shown to derive from tissue stem cells.To affirm the existence of OCICs, xenoengraftment of as few as 100 dissociated spheroid cells allowed full recapitulation of the original tumor (grade 2/grade 3 serous adenocarcinoma), whereas >105 unselected cells remained nontumorigenic. Stemness properties of OCICs (under stem cell—selective conditions) were further established by cell proliferation assays and reverse transcription—PCR, demonstrating enhanced chemoresistance to the ovarian cancer chemotherapeutics cisplatin or paclitaxel and up-regulation of stem cell markers (Bmi-1, stem cell factor, Notch-1, Nanog, nestin, ABCG2, and Oct-4) compared with parental tumor cells or OCICs under differentiating conditions.To identify an OCIC cell surface phenotype, spheroid immunostaining showed significant up-regulation of the hyaluronate receptor CD44 and stem cell factor receptor CD117 (c-kit), a tyrosine kinase oncoprotein. Similar to sphere-forming OCICs, injection of only 100 CD44+CD117+ cells could also serially propagate their original tumors, whereas 105 CD44−]CD117− cells remained nontumorigenic. Based on these findings, we assert that epithelial ovarian cancers derive from a subpopulation of CD44+CD117+ cells, thus representing a possible therapeutic target for this devastating disease.
Introduction
Ovarian cancer, the most lethal malignancy of the female reproductive system, results annually in over 14,000 U.S. and 114,000 worldwide deaths (1). Greater than 90% of ovarian cancers arise from the surface epithelium (2), and tumorigenesis has been associated with ovulation-associated wound repair and/or inflammation, possibly leading to abnormal stem cell expansion (2, 3).
Formation of ascites, a pathologic accumulation of peritoneal fluid containing inflammatory and disseminated tumor cells, is common in advanced disease (4). Whereas standard therapy, cytoreductive surgery followed by platinum/taxane, results in complete response in 70% of patients (5), most will relapse within 18 months with chemoresistant disease (5). Thus, improved targeted therapies and chemosensitization strategies are essential for reducing the mortality of this devastating malignancy.
One emerging model for the development of drug-resistant tumors invokes a pool of self-renewing malignant progenitors known as cancer-initiating cells (CICs). In that scenario, CICs generate a caricature (i.e., an abnormal organ) of the tissue from which they derive, with a hierarchy of cell types at distinct stages of differentiation (6). Consequently, they relapse after remission is likely due to failure to eradicate CICs, which, despite bulk tumor shrinkage, can subsequently reproduce the entire malignant phenotype (7). Indeed, normal stem cells ( from which CICs may originate; ref. 8) possess several characteristics that might confer chemoresistance (expression of membrane efflux transporters, enhanced DNA repair, low mitotic index; refs. 7, 8).
CICs were first identified in acute myeloid leukemia as possessing the cell surface antigenic phenotype CD34+CD38− and the capacity to reproduce the complete leukemic hierarchy upon xenoengraftment (9). Similar to the hematopoietic system, epithelial linings of most tissue surfaces undergo continuous turnover and are organized according to a stem cell hierarchy (6, 10). In 2003, CD24−CD44+ cells were isolated from human breast tumors that could serially propagate in animals and recapitulate their original phenotype (11). CICs have since been identified for numerous other epithelial malignancies (melanoma, lung, head/ neck, pancreas, prostate, and colon cancers; refs. 12-17). A consensus of five defining criteria has been established to affirm the existence of CICs: (a) self-renewal, (b) restriction to a small minority of the total tumor population, (c) reproducible tumor phenotype, (d) multipotent differentiation into nontumorigenic cells, and (e) expression of distinctive cell surface markers, permitting consistent isolation (6, 18).
In ovarian cancer, Bapat and colleagues (19) isolated two clones from patient ascites that could organize anchorage-independent, spherical structures (spheroids) in culture, similar to those naturally found in ascites (20). These clones were capable of forming xenografts in nude mice, with a histopathology similar to parental human tumors, serial propagation in animals (19), and expressed the stem cell factor receptor CD117 (c-kit), a well-known proto-oncoprotein (21). Another ovarian cancer study identified a subpopulation of dye-excluding side population—cultured murine cells, representing membrane transporter-expressing putative stem cells, that were highly tumorigenic in mice compared with dye-staining (i.e., noneffluxing) cells (22). To satisfy the CIC criterion of distinctive cell surface markers (6), expression of CD117 (similar to the ascites study) and hyaluronate receptor CD44 (also a marker for CICs from several other solid tumors; refs. 12, 13, 16, 17, 23) was shown in those murine cells.
In the current study, using primary human ovarian tumors, we isolated and characterized ovarian CICs (OCICs) fully capable of reestablishing their original tumor hierarchy in vivo. OCICs organized self-renewing, anchorage-independent spheres and were reproducibly isolatable using antibodies against both CD44 and CD117. Moreover, OCICs were also capable of intraperitoneal tumorigenesis (demonstrating activity in their native microenvironment) and could serially propagate tumors in animals. Consequently, OCICs fulfill all currently accepted criteria for the existence of a subpopulation of tumor-initiating cells (6, 18), and their specific detection and targeting could be highly valuable for therapy of recurrent, chemoresistant disease.
Materials and Methods
Collection and culture of dissociated human tumor cells
All studies were approved by Institutional Review Boards of Indiana University and Ohio State University. Tumors were obtained at diagnostic radical surgeries of ovarian cancer patients; the five tumors used in this study (designated T1—T5) were categorized as malignant Fesddration Internationale des Gynaecologistes et Obstetristes (FIGO) stage III serous adenocarcinomas. Fresh tumors were minced, suspended in DMEM/F12 medium (Invitrogen), and mixed with 300 units/mL of both collagenase (Invitrogen) and hyaluronidase (Calbiochem), followed by overnight incubation (37°C, 5% CO2). Enzymatically disaggregated suspensions were filtered (40-μm cell strainer) and washed twice with PBS, and RBCs were removed by Histopaque-1077 (Sigma). The resulting single tumor cells were placed under stem cell conditions (24) by resuspension in serum-free DMEM/F12 supplemented with 5 μg/mL insulin (Sigma), 20 ng/mL human recombinant epidermal growth factor (EGF; Invitrogen), 10 ng/mL basic fibroblast growth factor (bFGF; Invitrogen), and 0.4% bovine serum albumin (BSA; Sigma), followed by culturing in Ultra Low Attachment plates (Corning) and subsequent organization into spheres.
Assessments of spheroid differentiation
To examine ovarian tumor-like epithelial differentiation of anchorage-independent cells, spheres were dissociated by trypsin and single cells were plated on collagen-coated dishes (Corning) under standard differentiating conditions [DMEM/F12 supplemented with 10% fetal bovine serum (FBS) without growth factors]. Cell morphology was assessed 11 d after plating using a Zeiss Axiovert 40 inverted microscope with Axio-Vision software (Carl Zeiss MicroImaging). Further (immunofluorescent) examination of differentiation into ovarian tumor epithelium used monoclonal antibodies against cytokeratin-7 (CK-7) or cancer antigen-125 (CA-125; 1:500 each; Santa Cruz Biotechnology), followed by incubation with a FITC-labeled goat anti-mouse IgG secondary antibody (Santa Cruz; 1:400). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Santa Cruz). Microscopy was performed using a Nikon E800 fluorescence microscope.
Chemotherapy sensitivity assays
To assess chemosensitivity of sphere-forming cells to cisplatin and paclitaxel under stem cell conditions, spheres were dissociated (trypsinization and filtering through 40-μm cell strainers) and seeded at 3,000 cells per well (96-well Ultra Low plates; Corning) in 200 μL serum-free DMEM/F12 medium supplemented with growth factors. In parallel, sphere-forming cells were plated, allowed to differentiate for 11 d, trypsinized, and replated at 3,000 cells per well (standard, coated 96-well culture plates; Corning). After 24 h, both undifferentiated and differentiated cells were treated for 72 h with 0.1 to 100 μmol/L cisplatin (BD Biosciences) or 0.1 nmol/L to 10 μmol/L paclitaxel (Sigma; n = 5 per drug dose). Relative cell numbers were determined by standard 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays (described previously; ref. 25). Dose-response experiments were performed in duplicate. Doses required for 50% growth inhibition (IC50 values) were determined using Prism 4 (GraphPad Software) as means ± SD. Spheroid cell drug cytotoxicities, under both stem cell and differentiating conditions, were compared after 3-h treatment with cisplatin (30 μmol/L) and/or paclitaxel (2 μmol/L), representing the near Cmax values clinically achievable for a 3-h infusion (26). After drug treatments, cells were incubated for 72 h in drug-free medium and MTT assays were performed with relative cell survival defined as the percentage of drug-treated cells divided by untreated control. Statistical comparisons were performed by Student's t test.
Reverse transcription—PCR analysis of stem cell marker genes
Total RNA was extracted from spheroid cells, differentiated spheroid cells, or parental bulk tumors using an RNA Mini Kit (Qiagen), reverse-transcribed into cDNA, amplified for 30 cycles in 25-μL reactions with 10 pmol primers (gene-specific primer sequences in Supplementary Table S1), and PCR products were electrophoresed on 1% agarose gels using β-actin as a loading control.
Spheroid immunofluorescence studies
Spheroids were deposited by cytospin onto glass slides, fixed in ice-cold 4% paraformaldehyde (4°C, 10 min), and blocked (30 min with normal serum). An indirect immunofluorescent labeling technique was used to identify CD44-expressing and CD117-expressing cells using mouse anti-CD44 1:200 and rat anti-CD117 1:200 monoclonal antibodies (Santa Cruz) in PBS with 2% normal serum (1 h at room temperature). Slides were washed (PBS, 5 min) and incubated in the dark at room temperature for 30 min with phycoerythrin-conjugated goat anti-mouse IgG (against anti-CD44) and FITC-conjugated chicken anti-rat IgG (against anti-CD117; Santa Cruz).
To examine expression of other stem cell markers, adherent sphere-forming cells were grown on coverslips for 14 d and incubated with monoclonal anti-nestin or polyclonal anti-Nanog/anti—Oct-4 antibodies (Abcam; 1:150 dilution each), washed, and stained with FITC-labeled goat anti-mouse IgG secondary antibody (1:400). Positive control cells were stained, in parallel, for each antibody, and negative controls were performed by substituting primary antibodies with mouse nonspecific IgG. Nuclei were counterstained with DAPI. Fluorescence microscopy was performed (Nikon E800 fluorescent microscope fitted with FITC and PE filters), and images were acquired digitally using MagnaFire Software (Optronics) and processed in Adobe Photoshop.
Fluorescence-activated cell sorting analysis
For fluorescence-activated cell sorting (FACS), small pieces of tumors (primary ovarian T3, xenograft T1, T2) were dissociated into single cells, washed, and RBCs removed (described above). Cells were suspended in 2% BSA/PBS and labeled with anti-CD44, anti-CD117, and (phycoerythrin-labeled and FITC-labeled) secondary antibodies. For FACS of xenograft tumors, possible contaminating mouse cells were eliminated by discarding H2K+ (mouse histocompatibility class I) cells (mouse anti-mouse H-2Kd monoclonal, Santa Cruz); nonviable (i.e., membrane-permeable) cells were excluded by DAPI staining. Isolation of CD44+, CD117+, or CD44+CD117+ cells was performed using a FACSAria flow cytometer (BD Biosciences) and analyzed by WinMDI (Scripps Research Institute). For tumors T4 and T5, disaggregated tumor cells were first propagated as spheroids for 2 mo ( for OCIC enrichment), dissociated, and subjected to FACS isolation of CD44+CD117+ cells for subsequent mouse engraftment. Cells were routinely sorted twice to assess purity (typically >99%).
In vivo xenograft experiments
All animal studies adhered to protocols approved by the Institutional Animal Care and Use Committee of Indiana University. To assess tumorigenicity of sphere-forming, CD44+, CD117+,or CD44+CD117+ cells, dissociated spheroid or tumor cells were counted, resuspended in 40 μL 1:1 PBS/Matrigel (BD Biosciences), and injected s.c. into the left flanks of 3- to 4-wk-old female nude athymic mice (BALB/ c-nu/nu; Harlan). Engrafted mice were inspected biweekly for tumor appearance by visual observation and palpation, and tumor latencies (23) were then determined. Mice were sacrificed by cervical dislocation at a tumor diameter of 1 cm or at 6 mo posttransplantation. Xenograft tumors were resected, fixed in 10% neutral, buffered formalin, and embedded in paraffin for sectioning (5 μm) on a rotary microtome, followed by slide mounting, H&E staining, and histologic assessment by a pathologist for tumor type, grade, and stage. To determine xenograft recapitulation of the parental tumor phenotype, the same process was performed on human tumors. CA-125 immunodetection in xenograft tumors was performed using an ImmunoPure ABC Staining kit (Santa Cruz) and imaged (described above). Negative controls contained no primary antibody. To evaluate formation of ovarian tumors in their native environment, nude mice were injected i.p. with 5,000 spheroid-derived cells, monitored biweekly for weight loss and ascites formation, and euthanized upon excessive abdominal distention or palpable tumor growth.
Sequential tumorigenicity of putative OCICs was assessed using three convergent approaches. First, xenograft tumors were minced (1-mm pieces), implanted s.c. into a new host mouse, and allowed to grow to 1 cm (entire procedure repeated up to five consecutive times). Second, to determine reproducible OCIC isolation using the cell surface markers CD44 and CD117, FACS sorting and engraftment were performed (described above), and the resulting tumor was resorted for injection into another mouse (total of three serially injected animals). Third, graft tumors were digested and plated as single tumor cells (stem cell culture conditions). After reformation and dissociation of spheroids, ∼100 single cells were reinjected into mice and, similar to the tumor mincing studies, the entire process was repeated five times. To eliminate tumor-infiltrating mouse cells before injection, anti-H2K cells were discarded by FACS (described above).
Results
Anchorage-independent, self-renewing sphere formation by a subpopulation of human ovarian tumor cells
Previous studies have shown normal and cancer stem cells to organize anchorage-independent, autonomous, three-dimensional spherical structures (spheroids; refs. 14, 19, 27-30). In ovarian cancer, similar structures are observed in patient ascites (20), which contain a small subpopulation of tumor-propagating cells capable of organizing spheroids (19). Based on that prior ascites study, we attempted to isolate a self-renewing stem cell population from solid ovarian tumors, using a method for anchorage-independent (i.e., stem cell-selective) culturing of breast CICs (26). Primary tumor specimens obtained from five different patients (denoted as T1—T5, FIGO stage III, grade 2/grade 3 primary serous adenocarcinomas) were dissociated and inoculated on uncoated culture plates in serum-free medium with EGF, bFGF, and insulin (24). One week after plating, nonadherent spherical clusters of cells were observable (Fig. 1A, top left). Those floating spheres were enzymatically dissociated weekly, with the resulting single cells generating secondary spheres. After ∼10 passages, ∼1% of the spheres remained (Fig. 1A, top right), appearing as distinct prototypical spheroids (Fig. 1A, bottom left), similar to those found in patient ascites (21, 22) and spheroids highly enriched for tissue stem cells in mammary and neural cultures (30-33). Single spheroids were collected and dissociated, yielding 100 to 500 cells for replating and generation of ∼10 secondary spheroids after 2 months (n = 3 independent experiments). Using this approach, we obtained sustainable spheroids from all patient tumors (T1—T5) under stem cell conditions, with ∼103-fold increase after 6 months, establishing the ability of these anchorage-independent structures to self-renew.
Figure 1.
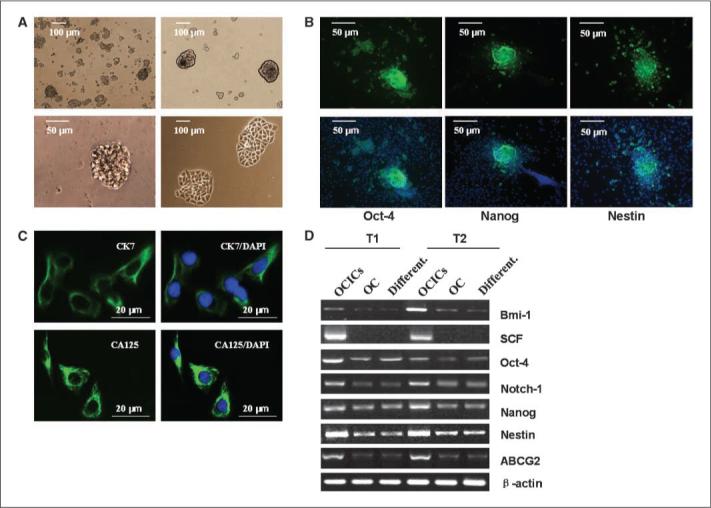
A subpopulation of human ovarian tumor cells form self-renewing, anchorage-independent spheroids under stem cell—selective conditions and are capable of epithelial differentiation. A, cell suspensions form small, nonadherent clusters 1 wk after plating (top left). Magnification, 100×. After ∼10 passages, a minor (1%) fraction of spheres persist as larger, symmetric, prototypical spheroids (top right). Magnification, 100×. Typical spheroids contained ∼100 viable cells and could be serially passaged for >6 mo (bottom left). Magnification, ×320. Under differentiating conditions for 11 to 14 d, dissociated sphere-forming cells adhere to plates and form symmetric holoclones (bottom right). Magnification, 100×. B, immunofluorescence of undifferentiated (top) or differentiated (bottom) spheroids or single cells under differentiating conditions, using antibodies against the stem cell markers Oct-4 (left), Nanog (center), and nestin (right). Nuclei were stained with DAPI. Magnification, 20×. C, under differentiating conditions, sphere-forming cells express the epithelial markers CK-7 and ovarian CA-125, as shown by fluorescence microscopy. Nuclei were stained with DAPI. D, as shown by RT-PCR, sphere-forming cells (OCICs), under stem cell—selective conditions, overexpress several stem cell marker genes compared with parental bulk tumor population cells (OC) and OCICs under differentiating conditions (Different.). Lanes 1 to 3 correspond to tumor T1 gene expression under the three conditions: lane 1, stem cell–selective (OCICs); lane 2, bulk tumor (OC); lane 3, differentiating (Different.). Lanes 4 to 6 similarly denote tumor T2 sphere-forming cell gene expression under the same conditions (β-actin used as a control).
When single cells from spheroids were cultured under differentiating conditions (i.e., after withdrawal of growth factors and addition of 10% FBS) for 11 to 14 days, floating cells could adhere, acquire an epithelial morphology, form symmetric colonies known as holoclones (ref. 31; Fig. 1A, bottom right), and survive subsequent passages. Secondly, we examined these spheroid-derived cells, under both stem cell—selective and differentiating conditions, for stem cell marker expression (Oct-4, Nanog, nestin), essential proteins for embryogeneis, neurogenesis, or hematopoiesis (32-34). Expression of these proteins was lost or greatly reduced after 14 days under differentiating conditions (Fig. 1B, bottom versus top, undifferentiated spheroids). To further support possible differentiation of these sphere-forming cells, expression of epithelial markers CK-7 and CA-125 (35) was examined, demonstrating positivity for both (Fig. 1C). Taken together, these data indicate that a subpopulation of spheroids from human ovarian tumors self-renew under stem cell—selective conditions and, under differentiation conditions, assume an epithelial tumor phenotype.
Ovarian tumor spheroid cells overexpress stem cell genes
After demonstration of tumor-derived cells to organize self-renewing spheroids, expression of genes specific to tissue and/or embryonic stem cells was examined in spheroids. RNA from spheroid or unselected bulk tumor cells was analyzed by reverse transcription—PCR(RT-PCR) for Oct-4, nestin, and Nanog (protein expression shown in Fig. 1B). Each of these genes, in addition to SCF, Notch-1, and Bmi-1, are essential for developmental processes (embryogeneis, neurogenesis, stem cell expansion, and hematopoiesis; refs. 32-34), whereas Notch-1 is also overexpressed in ovarian cancer and Bmi-1 is a well-known oncogene (36, 37). We also assessed expression of ABCG2, encoding a membrane efflux transporter expressed in hematopoietic stem cells and also associated with chemotherapy resistance (38). Sphere-forming cells overexpressed each stem cell marker (versus bulk tumor or differentiated cells; Fig. 1D), providing further evidence for their undifferentiated phenotype (6, 39).
Sphere-forming cells are resistant to conventional chemo-therapies
To examine whether self-renewing spheroid cells (having higher expression of ABCG2; Fig. 1D) possess a hypothesized cancer stem cell chemoresistant phenotype, we assessed the sensitivity of T1 and T2 sphere-forming cells to cisplatin and paclitaxel under stem cell—selective versus differentiating conditions. Compared with T1 and T2 spheroid cells under differentiating conditions, cisplatin IC50 values were greater (P < 0.05) under stem cell conditions (3.3-fold and 16.1-fold higher, respectively), whereas paclitaxel IC50 values also increased (P < 0.05) by 5.1-fold and 3.1-fold, respectively (Supplementary Table S2). Similarly, cell survival assays showed greater resistance of T1-derived (Fig. 2A; P < 0.05) and T2-derived (Fig. 2B; P < 0.01) spheroid cells to both agents (singly and in combination) under stem cell versus differentiating conditions. These results support a role for these stem-like cells in ovarian cancer chemoresistance (i.e., failure to eradicate progenitors resulting in tumor regrowth).
Figure 2.
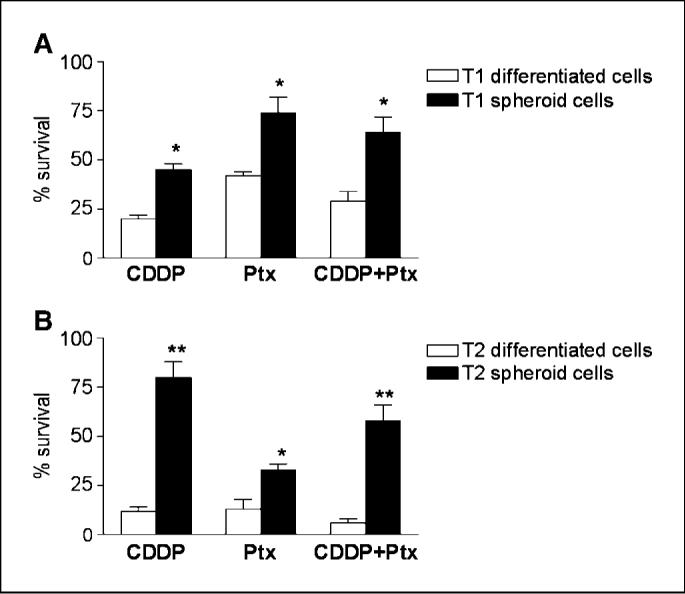
OCICs, under stem cell conditions, are highly resistant to conventional chemotherapies. A, sphere-forming OCICs from tumor T1, under stem cell—selective (black columns) or differentiating (white columns) conditions, were treated with cisplatin (CDDP;30 μmol/L; left), paclitaxel (Ptx;2 μmol/L; center), or cisplatin (30 μmol/L) and paclitaxel (2 μmol/L) for 3 h (CDDP + Ptx; right); cell survival was determined by MTT assays. B, same treatments as in A for OCICs derived from tumor T2, with white columns denoting differentiating conditions and black columns indicating stem cell conditions; *, P < 0.05; **, P < 0.01.
Sphere-forming cells serially propagate tumors of identical phenotypes
To investigate the tumorigenicity of sphere-forming cells, we examined whether exponentially smaller numbers (compared with unselected bulk tumor cells) were capable of tumorigenesis, as previously shown for other epithelial cancer CICs (11-13, 16, 23). T1 or T2 sphere-forming cells (primary tumor samples) or their differentiated or corresponding parental bulk tumor cells were injected s.c. into flanks of nude mice. With injections of only 100 cells per mouse, both T1 and T2 spheroid cells were tumorigenic in two of two athymic nude mice (Fig. 3A; Table 1), with tumor latencies of 73 to 102 days, similar to or less than CICs of other malignancies (11, 13, 16). Correspondingly, injections of 103 and 104 T1 and T2 spheroid cells were also tumorigenic in two of two mice with shorter tumor latencies (Table 1). Without nonadherent spheroid selection, T1 and T2 bulk tumor cells failed to form tumors even at 106 cells per engraftment, whereas one of two mice injected with 106 (but not 105) T1 and T2 differentiated cells were tumorigenic, albeit with extended latency (84−92 days; Table 1). All subcutaneous xenograft tumors derived from T1 and T2 spheroid cells were categorized as serous adenocarcinomas of moderate/poor differentiation (grade 2/ grade 3), similar to the parental primary patient tumors (H&E-stained sections; Fig. 3B, top, left versus right); these also expressed CA-125, an ovarian adenocarcinoma marker (Fig. 3B, bottom left; ref. 40). In all cases, no architectural/cytologic differences were observed between primary and graft tumors. Based on this enhanced, reproducible tumorigenicity, we designated these sphere-forming cells ”OCICs”, in accord with previously accepted terminology (41).
Figure 3.
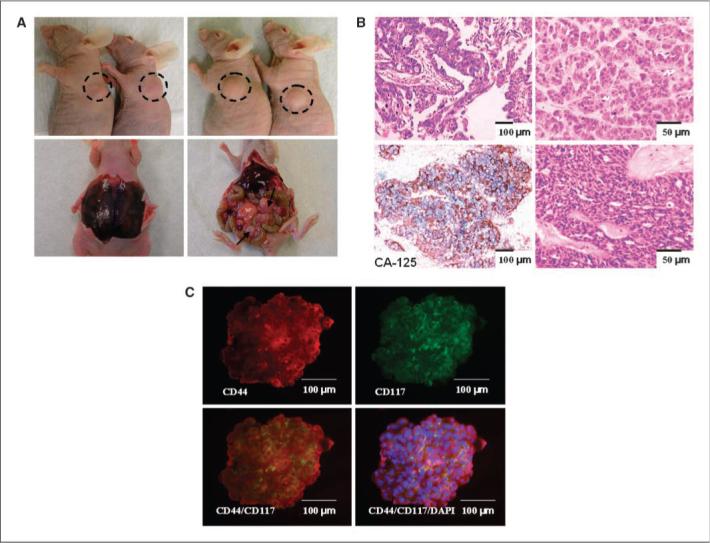
Robust in vivo propagation of human ovarian tumors (with reproducible histologic phenotypes) in nude mice by sphere-forming OCICs. A, xenograft tumor formed after injection of sphere-forming OCICs derived from patient tumors T1 and T2. Injection of ∼100 OCICs per mouse from T1 (left top)or T2(right top) dissociated spheroids generated tumors with 2/2 efficiency. I.p. injection of T2 OCICs gave rise to bloody ascites (left bottom) and peritoneal metastasic lesions (right bottom); black arrows denote metastases on the colon. B, representative H&E staining sections of T2 primary tumor (top left; magnification, 100×) and subcutaneous graft tumor from T2-derived spheroids (top right; magnification, 200×). Both tumors were classified as advanced grade (2/3) serous adenocarcinomas. Expression of the epithelial tumor marker CA125 in human xenograft tumor derived from T2 spheroids, as determined by immunohistochemistry; specific peroxidase staining is indicated by the brown color, and nuclei (blue) were counterstained with hematoxylin magnification at 100× (bottom left). H&E staining of intraperitoneal tumor derived from T2 spheroids (bottom right; magnification, 200×). C, representative double staining for CD44 and CD117 in T1 spheroids by immunofluorescence; similar results were obtained for tumors T2—T5. Immunofluorescence staining of anti-CD44 monoclonal antibodies (PE-conjugated secondary antibody, red) in ovarian tumor spheroid (top left); immunofluorescence staining of anti-CD117 monoclonal antibodies (FITC-conjugated secondary antibody, green) in ovarian tumor sphere cells (top right); CD44+ sphere cells colocalize with CD117+ cells (orange overlay, bottom left) or overlaid and additionally stained with DAPI (blue; bottom right). Magnification, 200×.
Table 1.
In vivo tumorigenicity of ovarian tumor sphere-forming cells
| Sample/cell type | Cell dose* | Injection site | Tumor formation† | Latency (d)‡ | Serial transplantation rate |
|---|---|---|---|---|---|
| T1 | |||||
| Sphere-forming | 100 | S.c. | 2/2 | 86, 102 | 5/5 |
| 1,000 | S.c. | 2/2 | 74, 81 | — | |
| 10,000 | S.c. | 2/2 | 60, 77 | — | |
| Bulk tumor | 10,000 | S.c. | 0/2 | — | — |
| 100,000 | S.c. | 0/2 | — | — | |
| 1,000,000 | S.c. | 0/2 | — | — | |
| Differentiated | 10,000 | S.c. | 0/2 | — | — |
| 100,000 | S.c. | 0/2 | — | — | |
| 1,000,000 | S.c. | 1/2 | 84 | — | |
| T2 | |||||
| Sphere-forming | 100 | S.c. | 2/2 | 73, 96 | 5/5 |
| 1,000 | S.c. | 2/2 | 65, 75 | — | |
| 10,000 | S.c. | 2/2 | 62, 69 | — | |
| 5,000 | I.p. | 1/1 | 75 | 3/3 | |
| Bulk tumor | 10,000 | S.c. | 0/2 | — | — |
| 100,000 | S.c. | 0/2 | — | — | |
| 1,000,000 | S.c. | 0/2 | — | — | |
| 50,000 | I.p. | 0/1 | — | — | |
| 500,000 | I.p. | 0/1 | — | — | |
| Differentiated | 10,000 | S.c. | 0/2 | — | — |
| 100,000 | S.c. | 0/2 | — | — | |
| 1,000,000 | S.c. | 1/2 | 92 | — | |
| 50,000 | I.p. | 0/1 | — | — | |
| 500,000 | I.p. | 0/1 | — | — |
Number of cells per injection.
Number of tumors formed per number of injection.
The time from injection to the first appearance of a palpable tumor.
One reservation regarding studies of CICs is engraftment into nonnative microenvironments (39, 42). To establish that OCICs faithfully duplicate the well-established progression of ovarian cancer in its native setting, i.p. injection of T2 sphere-forming cells resulted in development of bloody ascites and peritoneal metastasis to the omentum, liver, colon, stomach, and kidney (Fig. 3A, bottom), and intraperitoneal tumor histology similar to both subcutaneous xenograft and primary patient tumors (Fig. 3B, bottom right). In comparison to OCICs, i.p. injection of up to 5 × 105 T2 unselected bulk tumor and differentiated cancer cells failed to produce tumors and bloody ascites (Table 1).
Another essential criterion for CICs is their ability to serially propagate tumors in consecutively engrafted animals (18). To examine this definitive stemness characteristic, serial engraftments of T1 and T2 xenografts were performed by s.c. transplantation of 1-mm tumor pieces into nude mice. Generally, tumors developed ∼3 weeks after transplantation, with a total of five such successful serial transplantations (data not shown). Secondly, dissociated cells from both T1 and T2 xenograft tumors could reform spheroids under stem cell—selective conditions. To eliminate any possible contamination by tumor-infiltrating mouse cells, dissociated xenograft cells were stained with an antibody against mouse-H2K and stained cells were discarded by FACS before culturing under stem cell conditions (Supplementary Fig. S1). Reinjection (s.c.) of 100 such secondary sphere-forming cells resulted in tumors, in two of two animals, with a latency slightly shorter than the parental patient tumor sphere-forming cells (78 and 83 days for T1, passage 2 xenografts; 65 and 80 days for T2, passage 2 xenografts). Also, a total of five such consecutive 100-cell engraftments were performed successfully (Table 1). Furthermore, tumors collected after i.p. injection and recultured under stem cell conditions were (similar to s.c. engraftment) also capable of serial transplantation, in three of three mice (Table 1), demonstrating reproducible tumor formation within their native abdominal environment. These results indicate that sphere-forming OCICs are at least 104 more malignantly potent than their parental tumor cells, demonstrating that a highly tumorigenic subpopulation of cells resides within ovarian neoplasms.
Sphere-forming OCICs express cell surface proteins CD117 and CD44
Previous studies of ovarian cancer tumor progenitors from patient ascites and mouse cultures showed expression of stem cell factor receptor CD117 (c-kit) and hyaluronate receptor CD44 (19, 22). Consequently, we examined expression of these proteins in our sphere-forming, highly tumorigenic OCICs, under stem cell—selective conditions. The vast majority of T1 to T5 (n =5 for each tumor) spheroid cells stained for both CD44 and CD117 (representative T1 spheroids; Fig. 3C); in contrast, T1 and T2 patient parental tumors possessed a greatly reduced number of CD44+-stained and CD117+-stained cells, limited to a region of high cellular density (Supplementary Fig. S2). These results show both CD44 and CD117 to be candidate cell surface markers for ovarian tumor progenitors.
CD44+CD117+ cells are highly tumorigenic and can serially propagate their original tumor phenotype
Based on both our in vivo tumor experiments showing sphere-forming cells to be substantially more tumorigenic than bulk tumor cells and our spheroid immunofluorescence studies, we examined whether CD44 and CD117 could be used to isolate CICs from whole tumors. First, we FACS-purified cells singly positive and negative for each marker from first and second passage T1 and T2 xenografts. Those preliminary studies showed that xenograft-passaged CD44+CD117+ cells were 50-fold and 20-fold more tumorigenic than CD44−CD117− cells, respectively, and could also form polyclonal, heterogeneous tumors (data not shown).
We next determined the tumorigenicity of CD44+CD117+ cells by FACS sorting a total of five human ovarian tumors, including two xenografts (derived from T1 and T2 sphere-forming cells), a patient primary tumor (T3, grade 2/grade 3 serous adenocarcinoma; Fig. 4A, top), and two patient tumors (T4, T5) propagated in culture as spheroids. As described above, RBCs were removed and contaminating mouse cells were eliminated during FACS. For the T1 xenograft, 0.14% of the sorted cells were CD44+CD117+, similar to the T2 xenograft (0.16%) and T3 primary tumors (0.2%; Fig. 4A, top). For tumors T4 and T5, disaggregated tumor cells were first propagated for 2 months as spheroids and then dissociated and subjected to CD44+CD117+ FACS. Of the T4 and T5 sphere-forming cells, 78.5% and 82.2%, respectively, coexpressed both markers (Fig. 4A, bottom left and center), consistent with all tumor (T1—T5)— derived spheroids (Fig. 3C, representative spheroids, bottom). The purity of all isolated cell populations was >99%, as assessed by post—sort flow cytometry (Fig. 4A, representative plot, bottom right). Purified CD44+CD117+ cells and CD44−CD117− cells from all five tumors were then injected s.c. into nude mice and tumor frequencies (defined in Table 1) determined over 6 months. Whereas 5 × 105 CD44−CD117− cells purified from T1 and T2 first passage xenografts were tumorigenic after >3 months, injection of as few as 100 CD44+CD117+ cells resulted in tumor formation, with shorter latencies (52—93 days; Table 2), similar to or less than latencies of our sphere-forming cells and CICs of other malignancies (11, 13, 16). For T3 primary tumor and sphere-forming cells derived from T4 and T5 primary tumors, only CD44+CD117+ cells were tumorigenic; no tumors resulted from injection of 105 CD44−CD117− cells (Table 2).
Figure 4.
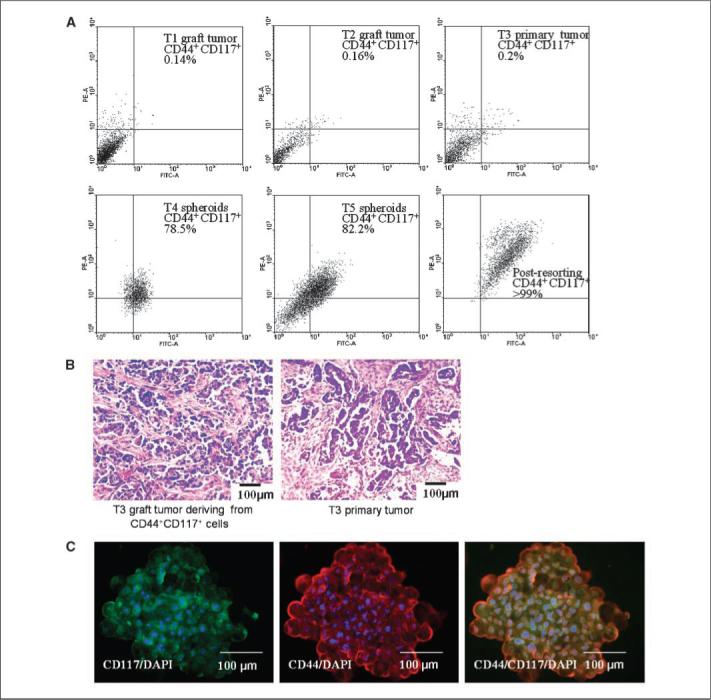
Tumor-derived spheroids stably coexpress CD44 and CD117, and those markers can be used to isolate highly malignant progenitors from whole tumors that reproduce their original phenotype. A, isolation of CD44+CD117+ cells by FACS. Scatter plots represent typical examples of patterns of CD44+CD117+ expression in a panel of human ovarian tumors T1—T3 (top) or spheroids generated from tumors T4 and T5 (bottom left and center). FACS experiments were repeated in duplicate, and the purity of CD44+CD117+ population was >99%, as revealed by postresorting FACS analysis (bottom right). B, H&E staining of the T3 xenograft tumors (left) generated from CD44+CD117+ cells is histologically identical to the corresponding T3 patient primary tumor (right). Both tumors were classified as poorly differentiated (G3) serous adenocarcinoma; magnification, 100×. C, after 30 d in culture, CD44+CD117+-generated spheroids from the T3 primary tumor retained CD44+CD117+ (orange overlay) expression. Nuclei were stained with DAPI (blue). Magnification, 200×.
Table 2.
In vivo tumorigenicity of CD44+CD117+ ovarian tumor cells
| Tumor/cell type | Cell doses and tumor formation and latency (d) |
Serial transplantation rate | ||||||
|---|---|---|---|---|---|---|---|---|
| 100 | 500 | 1,000 | 5,000 | 10,000 | 100,000 | 500,000 | ||
| T1 xenograft | ||||||||
| CD44+CD117+ | 2/2 (77, 93) | — | 2/2 (65, 78) | — | 2/2 (62, 70) | — | — | 3/3 |
| CD44−CD117− | — | 0/2 | 0/2 | — | 0/2 | — | 1/2 (132) | — |
| T2 xenograft | ||||||||
| CD44+CD117+ | 2/2 (52, 72) | — | 2/2 (59, 80) | — | 2/2 (43, 65) | — | — | 3/3 |
| CD44−CD117− | — | 0/2 | 0/2 | — | 0/2 | — | 2/2 (91, 103) | — |
| T3 primary | ||||||||
| CD44+CD117+ | 2/2 (63, 75) | — | 2/2 (50, 74) | 2/2 (65, 89) | — | — | — | 3/3 |
| CD44−CD117− | 0/2 | — | 0/2 | — | 0/2 | 0/2 | — | — |
| T4 spheroids | ||||||||
| CD44+CD117+ | 1/2 (55) | — | 2/2 (42, 60) | — | 2/2 (60, 69) | — | — | N/A |
| CD44−CD117− | — | — | — | 0/2 | 0/2 | 0/2 | 0/2 | — |
| T5 spheroids | ||||||||
| CD44+CD117+ | 2/2 (50, 65) | — | 1/2 (52) | — | 2/2 (45, 62) | — | — | N/A |
| CD44−CD117− | — | — | — | 0/2 | 0/2 | 0/2 | 0/2 | — |
NOTE: All in vivo tumorigenicity experiments were done in the left flank of athymic mice by s.c. injection.
Abbreviation: N/A, not applicable.
In addition to high tumorigenicity, xenograft tumors derived from sorted T1 to T3 CD44+CD117+ cells also histologically reproduced their original tumors (Fig. 4B, representative T3 xenograft-to-tumor comparison, left versus right), demonstrating the additional stemness characteristic of reproducible phenotypes (6, 11, 16, 18). Moreover, CD44+CD117+ cells from T1 and T2 graft tumors were serially transplantable in three consecutively injected mice (Table 2), with serial grafts having similar CD44+CD117+ percentages with mostly CD44−CD117− cells (Supplementary Table S3), thus demonstrating multipotent differentiation of the doubly positive progenitors (8). In addition to the xenograft assays, sorted CD44+CD117+ cells from T3 primary tumor were likewise capable of forming spheroids, which, after 1 month, remained positive for both markers (Fig. 4C), establishing their ability to self-renew under stem cell—selective conditions.
Discussion
In this report, we describe the isolation and characterization of a highly tumorigenic subpopulation of cells from human ovarian adenocarincomas, which we have designated (in accord with previously accepted terminology; ref. 41) OCICs. Whereas others have obtained tumorigenic cells from ovarian cancer patient ascites and mouse cultures (19, 22), we believe this is the first described isolation of malignant progenitors from human ovarian primary tumor tissues. Over the past 5 years, several such CICs have been identified for other epithelial malignancies, including melanoma and cancers of the breast, head/neck, lung, pancreas, colon, and prostate (11-17). Five separate criteria have been established for CICs, including (a) self-renewal, (b) small minority of the total tumor population, (c) reproducible tumor phenotype, (d) multipotent differentiation into nontumorigenic cells, and (e) distinct cell surface antigenic phenotype, permitting consistent isolation (6, 18). Two recent commentaries regarding previously identified CICs, however, questioned their self-renewal and multi-potency (39, 43), whereas another report suggested possible misinterpretations resulting from engraftment of hematologic CICs into nonnative (e.g., subcutaneous) growth environments (42).
Initially, to identify candidate OCICs, we cultured disaggregated tumor cells under stem cell–selective conditions. A small minority of cells could survive and form anchorage-independent clusters that subsequently coalesced into larger, self-renewing spheroids (14, 19, 27, 28) morphologically similar to spheroids isolated from patient ascites (20). Interestingly, while patient spheroids were found to bind hyaluronate, that binding was not inhibitable by anti-CD44 antibodies; however, CD44 expression was not examined in those spheroids (20). In addition to anchorage independence, our tumor-derived spheroids expressed numerous stem cell markers and were sustainable indefinitely under stem cell—selective conditions (thus fulfilling CIC criteria a and b). As xenografts, sphere-forming cells were >104 more tumorigenic than unselected parental tumor cells, and in addition to cultivation from primary human tumors, serially tumorigenic, sphere-forming OCICs were re-isolatable from graft tumors, demonstrating self-renewal in vivo (criterion a). Moreover, OCIC injection resulted in graft tumors histologically identical (grade 2/grade 3 serous adenocarcinomas) to the original primary tumor (Fig. 3B), a characteristic of progenitors of other epithelial malignancies (11, 13, 16), fulfilling the established CIC requirement of reproducible tumor phenotypes (criterion c).
To address concerns regarding engraftment into nonnative microenvironments (42), we characterized disease progression after i.p. injection of OCICs. Introduction of OCICs into their normal abdominal setting resulted in a pathology essentially identical to the human malignancy, with formation of bloody ascites and extensive peritoneal dissemination (Fig. 3A, bottom and B, bottom right). While we acknowledge that the nude mouse remains a less than ideal host for such studies, we believe these results show typical malignancy progression of OCICs within their natural anatomic surrounding.
As mentioned above, another now-required characteristic of CICs is reproducible isolation using distinct cell surface antigens (criterion 5; refs. 6, 18). Previous studies of ovarian tumorigenic cells from ascites and mouse cultured ovarian cancer cells suggested those progenitors to express the hyaluronate acid receptor CD44 and the oncoprotein c-kit (CD117; refs. 19, 22). Both CD44 and CD117 are overexpressed in advanced ovarian malignancies (44, 45), with cell surface CD44 believed to contribute to hyaluronate binding, to the abdominal mesothelial lining, by exfoliated tumor cells (i.e., peritoneal seeding; ref. 46). Indeed, based on its likely roles in cancer stemness and metastasis, CD44 is now emerging as a possible therapeutic target for highly aggressive malignancies, including ovarian cancer (47). Consequently, after demonstration of tumorigenicity of our sphere-forming OCICs, cultured spheroids were examined for expression of both markers, showing >80% of cells having coexpression (Fig. 3C; Table 2). Furthermore, by FACS, we showed CD44+CD117+ cells to constitute <0.2% of the total tumor cell population while fully able to recapitulate their original phenotype upon engraftment (Fig. 4B; CIC criterion 3). Moreover, that doubly positive fraction remained fairly constant through two xenograft passages, with the vast remainder (>95%) consisting of nontumorigenic CD44−CD117− cells (Fig. 4A; Supplementary Table S3), demonstrating both reproducible isolation and a multipotent capacity to form heterogeneous tumors ( fulfilling CIC criteria 4 and 5).
Whereas 90% of ovarian malignancies arise from the ovarian surface epithelium (OSE), primarily within inclusion cysts but also on the tissue exterior (48), at present, we can only speculate on the precise origin of OCICs. Three major scenarios have been put forth for sporadic ovarian carcinogeneis: (a) incessant ovulation hypothesis, in which repeated, uninterrupted follicular rupture and repair leads to proliferation-induced mutations during wound healing; (b) ovulation-induced inflammatory responses, similarly leading to enhanced proliferation/mutation; (c) up-regulated gonadotropin expression (as occurs after menopause), likewise increasing OSE proliferation (48). It is now commonly accepted that these three scenarios are not mutually exclusive and ovarian tumor initiation likely results from a cumulative effect of each or all three (49); thus, OCICs could arise by any of these homeostatic disruptions of the OSE.
To further suggest an OSE origin for OCICs, we note that the OSE, embryonically derived from the coelomic lining, remains relatively less differentiated than other tissue epithelia (2, 48). Moreover, the OSE also retains a capacity to undergo epithelial-to-mesenchymal transition, believed to contribute to postovulatory repair (48). Consequently, unlike most solid tumors, ovarian cancers become increasingly epithelial during tumor progression, as reflected by acquisition of phenotypes of Müllerian duct—derived endometrium, oviduct, and endocervix (48). Based on such epithelial differentiation, a second hypothesis puts forth that ovarian cancer actually originates from these secondary Müllerian tissues (based on its histology and characteristic gene expression; refs. 50, 51); it would be interesting to examine those tissues for expression of OCIC markers. Similar to human patient ovarian tumor progression, we likewise observed epithelial differentiation of OCICs (evidenced by expression of CA-125, CK-7) both in vitro and in vivo (Figs. 1C and 3B). Recent reports of the efficacy of Müllerian inhibiting substance (a transforming growth factor-β family hormone mediating male developmental regression of female precursor organs) for growth inhibition of ovarian tumors and stem-like side population mouse cultured ovarian cancer cells (22, 52) further support an embryo-genesis-like progression of this malignancy.
With specific regard to the ovarian carcinogenesis models of ovulation-mediated, inflammation-mediated, and gonadotropin-mediated transformation, an autocrine SCF/CD117 cascade has been hypothesized as contributory to OSE proliferation during early tumor initiation/progression (53), supporting a possible role in OSE transformation. Also consistent with a possible origin for OCICs, normal OSE cells have been shown to express both CD44 and CD117, both on the ovarian surface and in the epithelium surrounding inclusion cysts (53, 54).
Whereas advanced ovarian cancer is generally initially responsive to standard chemotherapies (cisplatin and paclitaxel), that response is almost inevitably followed by development of a drug-resistant phenotype (1, 4). One increasingly accepted hypothesis of chemoresistance posits that standard therapies fail to target tumor progenitors, which are believed to express normal stem cell phenotypes, such as a low mitotic index, enhanced DNA repair, and expression of membrane efflux transporters (e.g., ABCG2; refs. 6-8). In accord with that hypothesis, we showed that OCICs, under stem cell—selective conditions, overexpress ABCG2 (Fig. 1D) and are more resistant to cisplatin and paclitaxel (Fig. 2), suggesting a possible role for these cells in ovarian cancer chemoresistance.
In summary, we have identified a subpopulation of highly neo-plastic progenitors from solid human ovarian tumors. We strongly assert that these ovarian cancer-initiating cells fulfill all currently accepted requirements for solid tumor progenitors (6, 18). As our laboratory has previously investigated epigenetic markers of ovarian cancer (55), we are now initiating comprehensive studies of distinct chromatin and DNA methylation alterations in these tumor-propagating cells. Further characterization of such progenitors will likely lead to a greater understanding of early events leading to the genesis of this highly elusive disease, in addition to providing new therapeutic targets aimed at the cells directly responsible for its propagation.
Supplementary Material
Acknowledgments
Grant support: NIH grants CA085289 and CA113001, Walther Cancer Institute (Indianapolis), Ovar'coming Together (Indianapolis), NHRI-EX97-9717NC (Taiwan), and Phi Beta Psi Sorority (Brownsburg, IN).
We thank Sue Childress, Dr. Mark Braun, Christiane Hassel, Dr. Frederick Stehman, and Indiana Molecular Biology Institute Microscopy Facility (Jim Powers, Barry Stein).
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest No potential conflicts of interest were disclosed.
References
- 1.Pecorelli S, Favalli G, Zigliani L, Odicino F. Cancer in women. Int J Gynaecol Obstet. 2003;82:369–79. doi: 10.1016/s0020-7292(03)00225-x. [DOI] [PubMed] [Google Scholar]
- 2.Godwin AK, Testa JR, Hamilton TC. The biology of ovarian cancer development. Cancer. 1993;71:530–6. doi: 10.1002/cncr.2820710207. [DOI] [PubMed] [Google Scholar]
- 3.Ness RB, Cottreau C. Possible role of ovarian epithelial inflammation in ovarian cancer. J Natl Cancer Inst. 1999;91:1459–67. doi: 10.1093/jnci/91.17.1459. [DOI] [PubMed] [Google Scholar]
- 4.Cannistra SA. Cancer of the ovary. N Engl J Med. 2004;351:2519–29. doi: 10.1056/NEJMra041842. [DOI] [PubMed] [Google Scholar]
- 5.Ozols RF. Treatment goals in ovarian cancer. Int J Gynecol Cancer. 2005;15(Suppl 1):3–11. doi: 10.1111/j.1525-1438.2005.15351.x. [DOI] [PubMed] [Google Scholar]
- 6.Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annu Rev Med. 2007;58:267–84. doi: 10.1146/annurev.med.58.062105.204854. [DOI] [PubMed] [Google Scholar]
- 7.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–84. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 8.Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea-a paradigm shift. Cancer Res. 2006;66:1883–90. doi: 10.1158/0008-5472.CAN-05-3153. [DOI] [PubMed] [Google Scholar]
- 9.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–7. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 10.Miller SJ, Lavker RM, Sun TT. Interpreting epithelial cancer biology in the context of stem cells: tumor properties and therapeutic implications. Biochim Biophys Acta. 2005;1756:25–52. doi: 10.1016/j.bbcan.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 11.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65:10946–51. doi: 10.1158/0008-5472.CAN-05-2018. [DOI] [PubMed] [Google Scholar]
- 13.Dalerba P, Dylla SJ, Park IK, et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci U S A. 2007;104:10158–63. doi: 10.1073/pnas.0703478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fang D, Nguyen TK, Leishear K, et al. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005;65:9328–37. doi: 10.1158/0008-5472.CAN-05-1343. [DOI] [PubMed] [Google Scholar]
- 15.Kim CF, Jackson EL, Woolfenden AE, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121:823–35. doi: 10.1016/j.cell.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 16.Li C, Heidt DG, Dalerba P, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–7. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 17.Prince ME, Sivanandan R, Kaczorowski A, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci U S A. 2007;104:973–8. doi: 10.1073/pnas.0610117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clarke MF, Dick JE, Dirks PB, et al. Cancer stem cells-perspectives on current status and future directions: AACR workshop on cancer stem cells. Cancer Res. 2006;66:9339–44. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 19.Bapat SA, Mali AM, Koppikar CB, Kurrey NK. Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res. 2005;65:3025–9. doi: 10.1158/0008-5472.CAN-04-3931. [DOI] [PubMed] [Google Scholar]
- 20.Burleson KM, Casey RC, Skubitz KM, et al. Ovarian carcinoma ascites spheroids adhere to extracellular matrix components and mesothelial cell monolayers. Gynecol Oncol. 2004;93:170–81. doi: 10.1016/j.ygyno.2003.12.034. [DOI] [PubMed] [Google Scholar]
- 21.Natali PG, Nicotra MR, Sures I, et al. Expression of c-kit receptor in normal and transformed human nonlymphoid tissues. Cancer Res. 1992;52:6139–43. [PubMed] [Google Scholar]
- 22.Szotek PP, Pieretti-Vanmarcke R, Masiakos PT, et al. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian inhibiting substance responsiveness. Proc Natl Acad Sci U S A. 2006;103:11154–9. doi: 10.1073/pnas.0603672103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patrawala L, Calhoun T, Schneider-Broussard R, et al. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene. 2006;25:1696–708. doi: 10.1038/sj.onc.1209327. [DOI] [PubMed] [Google Scholar]
- 24.Ponti D, Costa A, Zaffaroni N, et al. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65:5506–11. doi: 10.1158/0008-5472.CAN-05-0626. [DOI] [PubMed] [Google Scholar]
- 25.Balch C, Yan P, Craft T, et al. Antimitogenic and chemosensitizing effects of the methylation inhibitor zebularine in ovarian cancer. Mol Cancer Ther. 2005;4:1505–14. doi: 10.1158/1535-7163.MCT-05-0216. [DOI] [PubMed] [Google Scholar]
- 26.Judson PL, Watson JM, Gehrig PA, Fowler WC, Jr., Haskill JS. Cisplatin inhibits paclitaxel-induced apoptosis in cisplatin-resistant ovarian cancer cell lines: possible explanation for failure of combination therapy. Cancer Res. 1999;59:2425–32. [PubMed] [Google Scholar]
- 27.Bez A, Corsini E, Curti D, et al. Neurosphere and neurosphere-forming cells: morphological and ultrastructural characterization. Brain Res. 2003;993:18–29. doi: 10.1016/j.brainres.2003.08.061. [DOI] [PubMed] [Google Scholar]
- 28.Dontu G, Abdallah WM, Foley JM, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17:1253–70. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reynolds BA, Weiss S. Clonal and population analyses demonstrate that an EGF-responsive mammalian embryonic CNS precursor is a stem cell. Dev Biol. 1996;175:1–13. doi: 10.1006/dbio.1996.0090. [DOI] [PubMed] [Google Scholar]
- 30.Vescovi AL, Reynolds BA, Fraser DD, Weiss S. bFGF regulates the proliferative fate of unipotent (neuronal) and bipotent (neuronal/astroglial) EGF-generated CNS progenitor cells. Neuron. 1993;11:951–66. doi: 10.1016/0896-6273(93)90124-a. [DOI] [PubMed] [Google Scholar]
- 31.Locke M, Heywood M, Fawell S, Mackenzie IC. Retention of intrinsic stem cell hierarchies in carcinoma-derived cell lines. Cancer Res. 2005;65:8944–50. doi: 10.1158/0008-5472.CAN-05-0931. [DOI] [PubMed] [Google Scholar]
- 32.Loh YH, Wu Q, Chew JL, et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet. 2006;38:431–40. doi: 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- 33.Mohle R, Kanz L. Hematopoietic growth factors for hematopoietic stem cell mobilization and expansion. Semin Hematol. 2007;44:193–202. doi: 10.1053/j.seminhematol.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 34.Wiese C, Rolletschek A, Kania G, et al. Nestin expression-a property of multi-lineage progenitor cells? Cell Mol Life Sci. 2004;61:2510–22. doi: 10.1007/s00018-004-4144-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCluggage WG. Recent advances in immunohistochemistry in the diagnosis of ovarian neoplasms. J Clin Pathol. 2000;53:327–34. doi: 10.1136/jcp.53.5.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hopfer O, Zwahlen D, Fey MF, Aebi S. The Notch pathway in ovarian carcinomas and adenomas. Br J Cancer. 2005;93:709–18. doi: 10.1038/sj.bjc.6602719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Valk-Lingbeek ME, Bruggeman SW, van Lohuizen M. Stem cells and cancer; the Polycomb connection. Cell. 2004;118:409–18. doi: 10.1016/j.cell.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 38.Sharom FJ. ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics. 2008;9:105–27. doi: 10.2217/14622416.9.1.105. [DOI] [PubMed] [Google Scholar]
- 39.Hill RP, Perris R. “Destemming” cancer stem cells. J Natl Cancer Inst. 2007;99:1435–40. doi: 10.1093/jnci/djm136. [DOI] [PubMed] [Google Scholar]
- 40.Berek JS, Bast RC., Jr Ovarian cancer screening. The use of serial complementary tumor markers to improve sensitivity and specificity for early detection. Cancer. 1995;76:2092–6. doi: 10.1002/1097-0142(19951115)76:10+<2092::aid-cncr2820761331>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 41.Smith A. A glossary for stem-cell biology. Nature. 2006;441:1060. [Google Scholar]
- 42.Kelly PN, Dakic A, Adams JM, Nutt SL, Strasser A. Tumor growth need not be driven by rare cancer stem cells. Science. 2007;317:337. doi: 10.1126/science.1142596. [DOI] [PubMed] [Google Scholar]
- 43.Hill RP. Identifying cancer stem cells in solid tumors: case not proven. Cancer Res. 2006;66:1891–5. doi: 10.1158/0008-5472.CAN-05-3450. [DOI] [PubMed] [Google Scholar]
- 44.Raspollini MR, Amunni G, Villanucci A, et al. c-KIT expression and correlation with chemotherapy resistance in ovarian carcinoma: an immunocytochemical study. Ann Oncol. 2004;15:594–7. doi: 10.1093/annonc/mdh139. [DOI] [PubMed] [Google Scholar]
- 45.Saegusa M, Machida D, Hashimura M, Okayasu I. CD44 expression in benign, premalignant, and malignant ovarian neoplasms: relation to tumour development and progression. J Pathol. 1999;189:326–37. doi: 10.1002/(SICI)1096-9896(199911)189:3<326::AID-PATH425>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 46.Cannistra SA, Kansas GS, Niloff J, et al. Binding of ovarian cancer cells to peritoneal mesothelium in vitro is partly mediated by CD44H. Cancer Res. 1993;53:3830–8. [PubMed] [Google Scholar]
- 47.Auzenne E, Ghosh SC, Khodadadian M, et al. Hyaluronic acid-paclitaxel: antitumor efficacy against CD44(+) human ovarian carcinoma xenografts. Neoplasia. 2007;9:479–86. doi: 10.1593/neo.07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Auersperg N, Wong AS, Choi KC, Kang SK, Leung PC. Ovarian surface epithelium: biology, endocrinology, and pathology. Endocr Rev. 2001;22:255–88. doi: 10.1210/edrv.22.2.0422. [DOI] [PubMed] [Google Scholar]
- 49.Ozols RF, Bookman MA, Connolly DC, et al. Focus on epithelial ovarian cancer. Cancer Cell. 2004;5:19–24. doi: 10.1016/s1535-6108(04)00002-9. [DOI] [PubMed] [Google Scholar]
- 50.Cheng W, Liu J, Yoshida H, Rosen D, Naora H. Lineage infidelity of epithelial ovarian cancers is controlled by Hox genes that specify regional identity in the reproductive tract. Nat Med. 2005;11:531–7. doi: 10.1038/nm1230. [DOI] [PubMed] [Google Scholar]
- 51.Dubeau L. The cell of origin of ovarian epithelial tumors and the ovarian surface epithelium dogma: does the emperor have no clothes? Gynecol Oncol. 1999;72:437–42. doi: 10.1006/gyno.1998.5275. [DOI] [PubMed] [Google Scholar]
- 52.Stephen AE, Pearsall LA, Christian BP, et al. Highly purified Mullerian inhibiting substance inhibits human ovarian cancer in vivo. Clin Cancer Res. 2002;8:2640–6. [PubMed] [Google Scholar]
- 53.Parrott JA, Kim G, Skinner MK. Expression and action of kit ligand/stem cell factor in normal human and bovine ovarian surface epithelium and ovarian cancer. Biol Reprod. 2000;62:1600–9. doi: 10.1095/biolreprod62.6.1600. [DOI] [PubMed] [Google Scholar]
- 54.Cannistra SA, Abu-Jawdeh G, Niloff J, et al. CD44 variant expression is a common feature of epithelial ovarian cancer: lack of association with standard prognostic factors. J Clin Oncol. 1995;13:1912–21. doi: 10.1200/JCO.1995.13.8.1912. [DOI] [PubMed] [Google Scholar]
- 55.Wei SH, Balch C, Paik HH, et al. Prognostic DNA methylation biomarkers in ovarian cancer. Clin Cancer Res. 2006;12:2788–94. doi: 10.1158/1078-0432.CCR-05-1551. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.