

Abstract
Brh2 is the Ustilago maydis ortholog of the BRCA2 tumor suppressor. It functions in repair of DNA by homologous recombination through controlling the action of Rad51. A critical aspect in the control appears to be the recruitment of Rad51 to single-stranded DNA regions exposed as lesions after damage or following a disturbance in DNA synthesis. In previous experimentation Brh2 was shown to nucleate formation of the Rad51 nucleoprotein filament that becomes the active element in promoting homologous pairing and DNA strand exchange. Nucleation was found to initiate at junctions of double-stranded and single-stranded DNA. Here we investigated the DNA binding specificity of Brh2 in more detail using oligonucleotide substrates. We observed that Brh2 prefers partially duplex structures with single-stranded branches, flaps, or D-loops. We found also that Brh2 has an inherent ability to promote DNA annealing and strand exchange reactions on free as well as RPA-coated substrates. Unlike Rad51, Brh2 was able to promote DNA strand exchange when preincubated with double-strand DNA. These findings raise the notion that Brh2 may have roles in homologous recombination beyond the previously established Rad51-mediator activity.
Individuals with mutation in the gene encoding BRCA2 are predisposed to breast, ovarian, and other cancers. The function of BRCA2 is in DNA repair by homologous recombination where it mediates interactions of Rad51, a protein needed for proficiency in homologous pairing and DNA strand exchange (1, 2). BRCA2 associates with Rad51 through a BRC-repeat domain located medially plus a second unrelated domain at the extreme C-terminus (3), and interacts with DNA through a domain comprised of a helix rich region and tandem OB folds1 closely related to those present in RPA for association with DNA (4). In studies using the conserved but smaller homolog (Brh2) from Ustilago maydis as a model system for understanding BRCA2 interaction with Rad51, it was found that Brh2 directs Rad51 to junctions of single-stranded (ss) and double-stranded (ds) DNA to initiate filament formation (5). Assembly of Rad51 on ssDNA to form an extended presynaptic filament is a necessary prerequisite for efficient homologous pairing and strand exchange (6, 7). The overall reaction is dependent on ATP, but joint molecule formation and strand exchange over significant lengths do not depend on ATP hydrolysis, only that it is present as a cofactor (8). As dsDNA is readily bound by Rad51 (7, 9, 10), pairing reactions can be inhibited by excess dsDNA since this competes with unbound ssDNA for the available free Rad51(11). Such inhibition can be circumvented by pre-coating ssDNA with Rad51 before addition of dsDNA (7).
Discovery of mediator activity associated with Brh2 followed in the tracks of the earlier pioneering studies demonstrating mediator function of Rad52 protein in promoting Rad51-catalyzed strand exchange (12–15). Yeast lacks a protein orthologous to BRCA2 but relies instead on Rad52 for all aspects of homology-directed repair and recombination events (16). In studies performed in vitro Rad52 was found to stimulate Rad51-promoted strand exchange by enabling Rad51 to displace RPA and access DNA, thus facilitating assembly into a filament (17, 18). This mediator activity stems from Rad52’s highly specific ssDNA binding activity and from its ability to interact physically with Rad51 and RPA. Besides its mediator function, Rad52 has the capacity to promote homologous pairing as measured by D-loop formation and oligonucleotide DNA strand exchange. It greatly accelerates annealing of complementary single-strands (19–21) and carries out the homologous pairing reactions in the absence of ATP in contrast to Rad51 (22, 23). Rad52’s innate capacity to promote recombination reactions as well as to mediate loading of Rad51 on DNA have been interpreted as evidence for multifunctionality underlining its pivotal role in homologous recombination in yeast.
BRCA2 and Rad52 are unrelated at the sequence level and their DNA binding domains bear no relationship to each other. The three OB folds within the DNA binding domain of BRCA2 are arranged in a tandem linear order(4), while with Rad52, an ssDNA-binding groove is thought to form around the outside of an eleven-subunit ring-like complex (24, 25). The Rad51-interacting region in Rad52 has been localized by deletion mapping (26). There is no sequence motif reminiscent of BRC, but the interaction region is not yet defined at the structural level. Nevertheless, there is an interesting functional parallel. In studies with Brh2 and with a truncated form of BRCA2, it was found that these proteins recruit Rad51 to RPA-coated single-stranded DNA and facilitate the nucleation of the filament with concomitant displacement of RPA (5, 27). Thus, there seems to be a measure of functional conformity between Brh2 and Rad52 in the Rad51-loading or mediator function. Here, using oligonucleotide substrates, we examined the DNA binding specificity of Brh2 and tested its ability to promote DNA transactions involving complementary strands in comparison with both Rad51 and Rad52.
EXPERIMENTAL PROCEDURES
Protein expression and purification
All proteins used in this study were recombinant proteins that were purified after being overproduced in Escherichia coli (Fig. 1). As previously described Brh2 was purified as a complex with Dss1(5) with the exception that here Brh2 was purified as an MBP-fusion protein in a complex with His-tagged Dss1 protein. The Brh2/Dss1 complex (Fig. 1A) was used in the experimentation described below and is referred to as Brh2 for simplicity. The Brh2 cDNA was cloned into pMAL-C2 (New England Biolabs) such that the fusion begins at Brh2 residue 106. The Dss1 cDNA was cloned into pACYC Duet vector (Novagen) for expression with an N-terminal hexahistidine affinity tag. The Brh2 and Dss1 fusion constructs are completely functional in complementing the radiation sensitivity of the U. maydis brh2 and dss1 mutants, respectively. The plasmids were sequentially transformed into E. coli BL21 (DE3) cells and cultures grown in LB medium containing ampicillin and chloramphenicol. Cell cultures (3 liters) were grown at 37° to A600 of 0.6, induced by addition of IPTG to 0.1 mM, but maintained at 18° after induction for 10 hrs. After harvesting, cells were resuspended in 50 ml BA buffer (25mM Tris-HCl pH7.5, 200mM KCl, 1mM DTT, 10% glycerol, 0.1% NP40) + 20mM imidazole, crushed by two passes through a French pressure cell at 20,000 p.s.i., and centrifuged at 16,000 rpm in a Sorvall SS34 rotor for 30 min. The supernatant was recovered and centrifuged at 27,000 rpm for 1 hr in a Beckman SW28 rotor. Subsequent procedures were all performed at 0–4°. The supernatant was passed onto a column (6 ml bed volume) of NTA-agarose (Novagen) charged with Ni2+ and equilibrated in the same buffer. The column was washed with 40 ml of buffer and protein eluted with 25 ml BA buffer + 200 mM imidazole. The eluate was loaded on a 5 ml column of crosslinked amylose which was washed with 40 ml of BA buffer followed by 25 ml BA buffer + 10 mM maltose. The fractions containing Brh2 protein were pooled, diluted to 100 mM KCl, and applied to a 1 ml MonoQ column equilibrated in buffer BB (25mM Tris-HCl pH 7.5, 100mM KCl, 1mM DTT, 10% glycerol). After washing with 5 ml of buffer the protein was eluted with a linear gradient (20 ml) from 0.1 to 0.6 M KCl. Peak fractions were pooled and dialyzed against buffer BB, and stored frozen at −80°.
FIGURE 1. Proteins in the analysis.
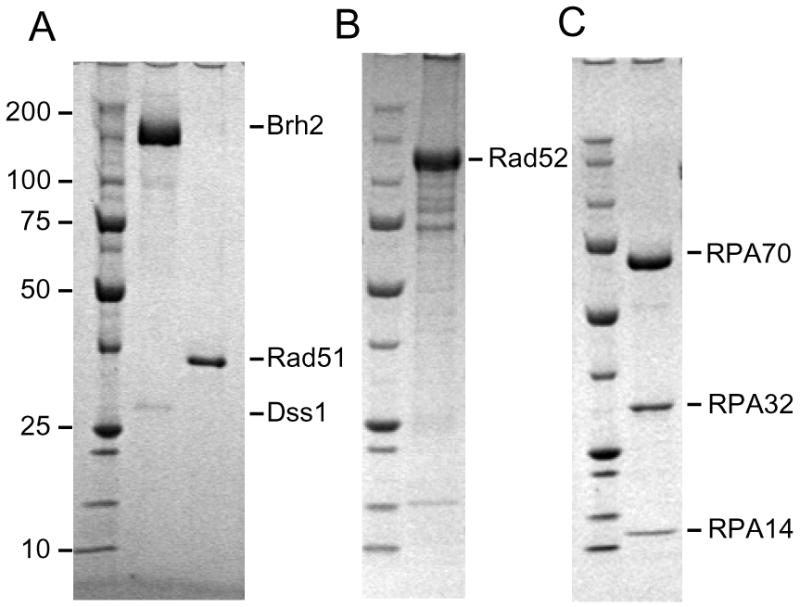
SDS polyacrylamide gels with approximately 15 pmol each of the indicated proteins run along side a ladder of Precision Protein standards (BioRad Laboratories) were stained with Comassie blue. (A) MBP-Brh2/His-Dss1 and Rad51 (B) MBP-Rad52 (C) RPA heterotrimer.
For overproduction of Rad51 in E. coli BL21(DE3) the U. maydis cDNA encoding Rad51 was cloned in a pET vector derivative (Novagen) under control of the T7 promoter so as not to include any affinity tags. Cultures (6 liters) were grown at 37°, induced as above and cells harvested 5 hrs later after continued incubation at 37°. Cells were resuspended in ice cold 50 ml buffer A (25mM Tris-HCl, pH 7.5, 1 mM DTT, 1 mM EDTA, 10% glycerol, and 0.1% NP40) + 0.5 M KCl, crushed and centrifuged as above. Subsequent procedures were all performed at 0–4°. The high speed supernatant was diluted with buffer RA (25mM Tris-HCl, pH 7.5, 1 mM DTT, 1 mM EDTA, 10% glycerol), to a conductivity equivalent to 100 mM KCl, mixed with 200 ml gravity-settled DEAE-cellulose slurry (Whatman DE52), stirred for 45 min, then passed onto a Buchner funnel with sintered glass filter. The resin was washed batchwise on the funnel under gentle vacuum with 2 liters buffer RA + 100 mM KCl, followed by a wash of 200 ml with buffer A + 2M KCl. The 2M eluate was diluted 4-fold then loaded onto a column (20 ml bed volume) of Affi-Gel Blue (BioRad Laboratories) equilibrated in buffer RA + 0.2 M KCl. Following the method described for yeast Rad51 (10), the column was eluted with a linear salt gradient (200 ml) from 0.2 to 1.5 M KCl, peak fractions pooled after identification of those containing Rad51 by SDS-gel electrophoresis and passed over a column (20 ml bed volume) of hydroxyapatite equilibrated with buffer RB (25 mM potassium phosphate pH 7.5, 500 mM KCl, 1 mM DTT, 10% glycerol) which was then eluted with a linear gradient (200 ml) from 25 mM to 0.8 M of potassium phosphate. Peak fractions were pooled and dialyzed against buffer RC (25 mM Tris-HCl, pH 7.5, 100 mM KCl, 1mM DTT, 10 % glycerol, then applied to a 1 ml Mono Q column (Amersham Biosciences), washed with 10 ml of RC buffer and eluted with a linear gradient (20 ml) from 0.1 to 1 M KCl. Peak fractions that appeared >95% homogeneous were pooled (Fig. 1A) and dialyzed against buffer RD, 25 mM Tris-HCl, pH 7.5, 0.5M KCl, 1mM DTT, 0.1 mM EDTA, 20% glycerol, snap frozen in liquid nitrogen and stored at −80°.
For overproduction of Rad52 in E. coli BL21(DE3) the U. maydis cDNA encoding the Rad52 ortholog (GenBank accession number XP761136) was amplified by PCR and cloned in pMAL-C2 (New England Biolabs) as an MBP-fusion protein. Cultures (6 liters) were induced at 23° and processed as above for Rad51 protein. Extracts in buffer A + 0.5 M KCl were passed through a column (100 ml) of DEAE-cellulose. The flow-through fraction was collected and passed onto a 5 ml column of crosslinked amylose (New England Biolabs). The column was washed with 10 ml buffer A + 0.5 M KCl, then eluted with the same buffer + 10 mM maltose. The eluate was passed onto successive columns of Affi-Gel Blue and hydroxyapatite and fractionated by salt and phosphate gradient chromatography, respectively, as above with Rad51. Rad52 prepared in this way was very sensitive to contaminating protease activity and the purified protein preparations quickly accumulated a significant amount of proteolytic fragments deduced by their amylose binding capacity and mobility on SDS gel electrophoresis (Fig. 1B) to be deleted of approximately 300 residues from the C-terminus. We estimate that the most purified fractions contained 70–80% of the full-length protein. However, the U. maydis Rad52 at 722 residues is much larger than the yeast (504 residues) and human (418 residues) orthologs with an unusually long extension at the C-terminal end, a feature not uncommon among U. maydis proteins (28). The conserved DNA binding domain is located within the first 300 residues of the N-terminus so it seems likely that even in the most severely truncated proteolytic products the DNA binding domain was still intact. To date we have been unsuccessful in ridding the preparations of the trace protease activity that appears responsible for generating Rad52 truncated fragments.
For overproduction of RPA in E. coli BL21(DE3) the cDNAs for RPA70 was cloned into pET28a (Novagen) to fuse a hexahistidine tag to the N-terminus and the cDNAs for RPA32 and RPA14 were cloned into pACYC Duet (Novagen). The plasmids were transformed sequentially into BL21(DE3). Cultures grown with shaking in a water bath (New Brunswick G76) were induced as above but in a decreasing temperature gradient from 32° to 23° over the course of 5 hr effected by switching off the water bath’s heating element. Extracts from 3 liters of culture were prepared using BA buffer and protein was loaded on an NTA-agarose column and eluted as above. The eluate was diluted to a conductivity equal to that of 0.1 M KCl loaded onto a 1 ml MonoQ column and eluted with a linear gradient (30 ml) from 0.1 to 0.6 M KCl. Fraction eluting at 0.3 M KCl containing RPA subunits were pooled, diluted to a conductivity equal to that of 0.1 M KCl, loaded onto a heparin Sepharose column (1 ml HiTrap Heparin HP, Amersham Biosciences), and eluted with a linear gradient (20 ml) from 0.1 to 0.4 M KCl. Fractions eluting at 0.2 M KCl appeared to contain equimolar amounts of all three RPA subunits (Fig. 1C) and were pooled and stored frozen at −80°.
DNA substrates
Unless indicated otherwise all DNA concentrations are expressed as moles of DNA molecules. Oligonucleotide sequences are in general based on pBluescript II DNA SK+ (Stratagene) and were synthesized and gel purified by Integrated DNA Technologies. The 100mer (and complementary strand) corresponds to pBluescript II SK+ nucleotides 2–101. After labeling 30 pmol of 100mer (+) or (−) strand at the 5′-end with 32P using T4 polynucleotide kinase and 60 μCi of [γ-32P]ATP, it was annealed with appropriate complementary strands in the presence of 20 mM KCl by heating to 80° then slowly cooling to 22°. Substrates were prepared by annealing the following combination of oligomers: ds --A/B; ds nicked--C/E/B; gap ds-- F/G/B; 3′ overhang-- A/D; 5′ overhang-- C/B; ss - B; Holliday junction-- L/M/N/O; fork-- H/B; ds fork-- H/I/C/B; 3′ flap-- H/I/B; 5′ flap-- H/C/B; bubble-- J/B; D-loop-- J/K/B; 5′ flap D-loop-- J/B/I. Gel purification was performed when required by electrophoresis of annealed products in 12% polyacrylamide gels. The following oligonucleotides designated A-O were utilized: A. 100mer (−): 5′-TAA ATT GTA AGC GTT AAT ATT TTG TTA AAA TTC GCG TTA AAT TTT TGT TAA ATC AGC TCA TTT TTT AAC CAA TAG GCC GAA ATC GGC AAA ATC CCT TAT A-3′; B.100mer (+): 5′-TAT AAG GGA TTT TGC CGA TTT CGG CCT ATT GGT TAA AAA ATG AGC TGA TTT AAC AAA AAT TTA ACG CGA ATT TTA ACA AAA TAT TAA CGC TTA CAA TTT A-3′; C. 49mer (−): 5′-AA ATT GTA AGC GTT AAT ATT TTG TTA AAA TTC GCG TTA AAT TTT TGT TA-3′; D. 49mer (+): 5′-TAA CAA AAA TTT AAC GCG AAT TTT AAC AAA ATA TTA ACG CTT ACA ATT T-3′; E. pBSII (52–101) (−): 5′-AA ATC AGC TCA TTT TTT AAC CAA TAG GCC GAA ATC GGC AAA ATC CCT TAT A-3′; F. pBSII (2–42) (−): 5′-TAA ATT GTA AGC GTT AAT ATT TTG TTA AAA TTC GCG TTA AA-3′; G. pBSII (62–101) (−): 5′-TTT TTT AAC CAA TAG GCC GAA ATC GGC AAA ATC CCT TAT A-3′; H. A HJ-pBSII 51–101 (−): 5′ –GAC GCT GCC GAA TTC TGG CTT GCT AGG ACA TCT TTG CCC ACG TTG ACC CGA ATC AGC TCA TTT TTT AAC CAA TAG GCC GAA ATC GGC AAA ATC CCT TAT A-3′; I. 1A HJ rev comp: 5′-CG GGT CAA CGT GGG CAA AGA TGT CCT AGC AAG CCA GAA TTC GGC AGC GTC -3′; J. pBSII (2–42)- 1A HJ(2–21)- (62–101): 5′-TAA ATT GTA AGC GTT AAT ATT TTG TTA AAA TTC GCG TTA AAC GCT GCC GAA TTC TGG CTT TTT TTT AAC CAA TAG GCC GAA ATC GGC AAA ATC CCT TAT A-3′; K. PBS II (44–59) (−): 5′-T TTT TGT TAA ATC AGC TCA-3′; L.1A HJ: 5′-GAC GCT GCC GAA TTC TGG CTT GCT AGG ACA TCT TTG CCC ACG TTG ACC CG-3′; M. 2A HJ: 5′-CGG GTC AAC GTG GGC AAA GAT GTC CTA GCA ATG TAA TCG TCT ATG ACG TC-3′; N. 3A HJ: 5′-GAC GTC ATA GAC GAT TAC ATT GCT AGG ACA TGC TGT CTA GAG ACT ATC GC-3′; O. 4A HJ: 5′-GCG ATA GTC TCT AGA CAG CAT GTC CTA GCA AGC CAG AAT TCG GCA GCG TC-3′. Heterologous 80mer derived from pUC19: 5′-TTA ACT ATG CGG CAT CAG AGC AGA TTG TAC TGA GAG TGC ACC ATA TGC GGT GTG AAA TAC CGC ACA GAT GCG TAA GGA GA-3′.
Gel mobility shift
Oligonucleotide DNA substrate at 2 nM was incubated with protein for 10 minutes at 37° in reactions (15 μl) with buffer containing 25 mM HEPES, pH 7.5, and 1 mM DTT. Glutaraldehyde was then added to a final concentration of 0.2%, incubation continued for 10 min to crosslink protein-DNA complexes, and Tris-HCl, pH 8.0 added to a final concentration of 120 mM to quench the crosslinking reaction. Products were resolved by electrophoresis on 1% agarose gels run at 4°. Gels were dried onto DE81 paper and radiolabeled bands identified using a Molecular Dynamics phosphorimager. Relative amounts of substrate and shifted product bands were determined using Image Quant software.
Single-strand annealing
Annealing of complementary 100mer oligonucleotides was monitored by a fluorimetric method based on the procedure described by the Kowalczykowski laboratory (29) but modified to utilize PicoGreen (Molecular Probes) instead of DAPI to signal formation of duplex DNA as the former has a higher fluorescence yield (30). Measurements were performed in microtiter plates in 100 μl reactions containing 25 mM Tris-HCl, pH 7.5, 25 mM KCl, 1 mM DTT, and 12 nM each A (−) strand and B (+) strand 100mer oligonucleotides at 37° in the presence of 0.15 μM PicoGreen unless otherwise indicated. Reactions were set up with A strand ss 100mer without or with RPA at a ratio of nucleotide to protein of 25 to 1. The RPA-DNA mixture was deposited on one side of a 96-well white microtiter dish chamber as a small microdroplet (25 μl) and the Brh2 or Rad52 solution as a separate microdroplet (25 μl) on the opposite side of the chamber with care taken to avoid mixing. Reactions were started by addition of a solution (50 μl) with the complementary B strand 100mer plus PicoGreen using a multi-channel pipettor such that mixing of the microdroplets was rapid. Fluorescence measurements were taken at 10 to 30 sec intervals with excitation and emission wavelengths set at 485 nm and 538 nm respectively, using a SpectraMax Gemini fluorimetric plate reader (Molecular Devices). Due to the manual handling several seconds elapsed before the first readings could be recorded. Therefore, the initial time points were not zero. Percent annealing was normalized to the fluorescence intensity of standards consisting of 12 nM 100mer dsDNA made by thermal annealing (100%) and 12 nM 100 ssDNA (0%).
DNA strand exchange
For strand exchange monitored by FRET reactions were performed in two different formats depending on the instrumentation that was available for detection. In one using oligonucleotides modified with a Cy5 fluorophore donor and Iowa Black RQ dark quencher (Integrated DNA Technologies), reactions contained 10 nM ss 100mer (oligonucleotide B) and 10 nM ds 49mer (oligonucleotides C & D) in which the strand complementary to the ss 100mer (oligonucleotide C) was 5′-end labeled with Cy5 and the strand with identity to the ss 100mer (oligonucleotide D) was 3′-end labeled with the fluorescence quencher Iowa Black RQ. Reactions (100 μl) were performed in 96-well white flat-bottom microtiter plates (Costar) and were started by addition of ds 49mer to solutions containing ss 100mer plus proteins. Fluorescence measurements were monitored using the SpectraMax Gemini fluorimetric plate reader as above with excitation and emission wavelengths set at 643 nm and 667 nm, respectively. Percent strand exchange was normalized to the fluorescence intensity of standards consisting of 10 mM ds 5′-Cy5-49mer/3′-Iowa Black-49mer (0%) and 10 nM ss 5′-Cy5-49mer (100%). The second format utilized a FAM fluorophore donor and Black Hole Quencher 2 (Integrated DNA Technologies) in which oligonucleotide C was 5′-end labeled with FAM and oligonucleotide D was 3′-end labeled with Black Hole Quencher 2. Reactions (15 μl) containing 25 mM Tris-HCl, pH 7.5, 25 mM KCl, 1 mM DTT and 30 nM ds 5′-FAM/3′-BH2 49mer were run at 37 ° in MicroAmp optical 96-well PCR plates (Applied Biosystems) and monitored in a TaqMan 7500 Real Time PCR System (Applied Biosystems) at 38 sec intervals with excitation set at 488 nm and emission monitored with Filter A in place. Percent release from quenching, a metric interpreted as equivalent to strand exchange, was normalized to the fluorescence intensity of standards consisting of 30 nM ds 5′-FAM/3′-BH2 49mer with 200 nM Brh2 (0%) and 30 nM ss 5′-FAM-49mer with 200 nM Brh2(100%). E. coli exonuclease III (Exo III, New England Biolabs) was used as a control with units of activity specified by the manufacturer.
For more conventional strand exchange reactions with radiolabeled substrates 60 nM ss100mer (oligonucleotide B) was paired with 6 nM ds 49mer (oligonucleotides C & D) in which the strand complementary to the ss 100mer (oligonucleotide C) was 5′-end labeled with 32P using T4 polynucleotide kinase and [γ-32P]ATP. Preincubations were performed with the first DNA (60 nM ssDNA or 6 nM dsDNA, plus the protein as appropriate for the experiment), then reactions were started by addition of the second DNA substrate (60 nM ssDNA or 6 nM dsDNA, respectively). In the standard reaction with Rad51, 60 nM ss100mer was preincubated with Rad51 protein at 37° in a 15 μl reaction in a buffer containing 25 mM Tris-HCl, pH 7.5, 20 mM KCl, 1 mM DTT, 4 mM CaCl2, 2 mM ATP. After 15 min reactions were started by addition of 6 nM 32P-labeled ds 49mer DNA and incubation continued for additional time. In the standard Brh2 reaction, conditions were the same except that CaCl2 and ATP were omitted and preincubation with ss100mer was not performed unless otherwise indicated. Reactions were stopped by addition of SDS and proteinase K to 1.2 % and 1.7 mg/ml respectively for 30 min. EDTA (20 mM) and loading dye were added and DNA components were resolved by electrophoresis on 10% polyacrylamide gels in 90 mM Tris-borate, pH 7.9, 1mM EDTA buffer. Dried gels were exposed to PhosphorImager plates and developed images were quantified using ImageQuant software as above. The percentage of product was calculated as the radiolabeled product divided by the total amount of radiolabeled DNA in each lane.
RESULTS
Brh2 binds preferentially to DNA structures with D-loops or branches
The Brh2/Dss1 complex (Fig. 1A) was used in the experimentation described below and is referred to as Brh2 for simplicity. In previous studies it was established that Brh2 can recruit Rad51 to DNA to facilitate filament nucleation at a junction between dsDNA and ssDNA (5). The specificity for 3′ protruding single-strand regions implied Brh2 has an inherent ability to recognize junctions with the polarity of a processed DNA double-strand break. Support for this notion was obtained from measurement of the affinity for binding 3′-tailed versus 5′-tailed oligonucleotides using a gel mobility shift assay in which a 2.5 fold preference for 3′ tailed molecules was determined. We were interested in exploring additional DNA structural features that might constitute determinants contributing to Brh2 binding and used a similar methodology to measure specificity for an expanded series of DNA substrates with a variety of different conformations. The set was framed around a 100-mer oligonucleotide derived from a natural sequence stretch within pBluescript II plasmid DNA. By annealing oligomers with various runs of non-complementary and complementary sequences to the 100-mer, we prepared the various substrates shown schematically in Fig. 2. The substrates end-labeled with 32P were incubated with increasing concentrations of Brh2 and protein-bound complexes were then detected by shifts in the mobility of substrates after gel electrophoresis. Tailed molecules with 3′ or 5′ overhangs, forked molecules, and flap molecules were designed to be 100 residues long and consist of half the length as based-paired duplex and half as tails, flaps, etc. 100mer molecules of gapped dsDNA, bubble and D-loop structures contained 20 internal residues that were unpaired.
FIGURE 2. Specificity of Brh2 in DNA binding.
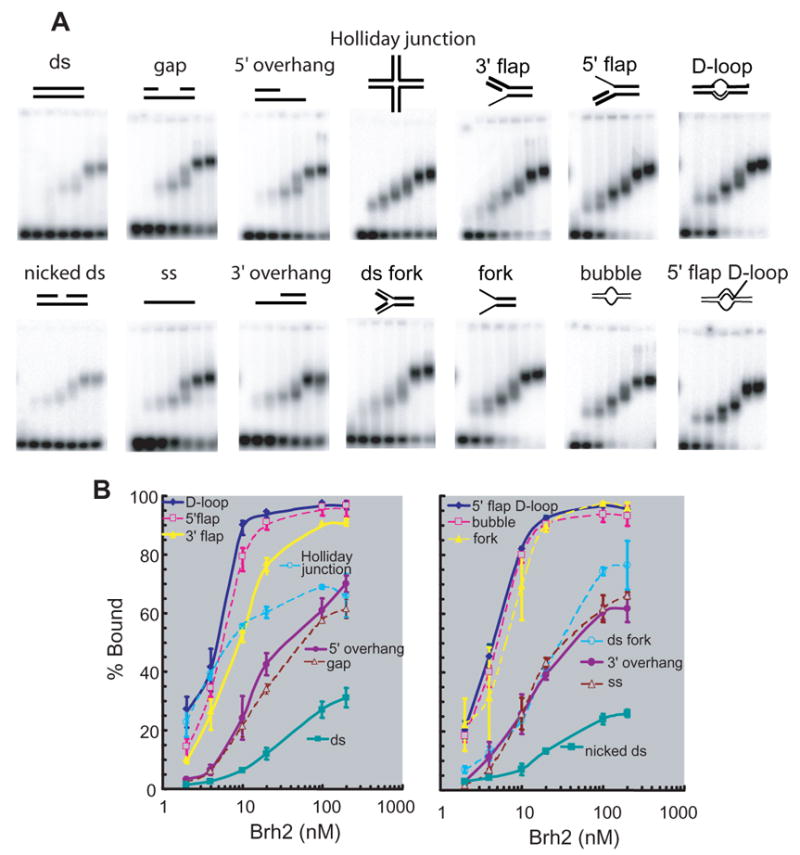
(A) Reactions containing oligonucleotide substrates shown schematically and increasing concentrations of Brh2 (samples from left to right in each frame: 0, 2, 4, 10, 20. 100. 200 nM) were incubated for 10 min, then analyzed by gel electrophoresis as described in EXPERIMENTAL PROCEDURES. Samples were run in triplicate but a single representative is shown. The sample with the 3′ flap substrate ran crooked and the image was straightened using Photoshop software. (B) The level of substrate shifted relative to the total signal in the lane was determined and is shown graphically. For clarity the substrates are listed in order according to their binding affinity and are divided arbitrarily into two groups arranged so that all curves are visible. The left-hand figure contains the data represented in the upper panel of mobility shift assays (A), while the right-hand figure contains the data represented in the lower panel.
Upon testing with Brh2 the substrates appeared to fall into thee general classes according to binding preference. Linear dsDNA molecules that were completely base paired were bound the weakest, linear ssDNA and linear molecules with single-stranded regions were bound substantially better, and dsDNA molecules with forked, branched, or bubble structures were the best. Binding preference did not strictly correlate with branches as the 4-armed Holliday junction appeared somewhat anomalous in its binding. By a slight margin the D-loop substrate appeared to be the top candidate with half of the input DNA shifted at a Brh2 concentration of about 4 nM under these conditions. It was notable that a ladder of up to perhaps four distinctly migrating complexes became evident upon increasing protein concentration using several of the branched DNA substrates. Formation of each more slowly migrating complex appeared dependent on accumulation of the previous complex suggesting the higher order complexes resulted from protein-protein interaction.
Brh2 anneals ssDNA complexed with RPA
In the course of examining the association of Brh2 with various combinations of oligonucleotides, we noticed that oligomers with complementary stretches annealed quickly when mixed in the presence Brh2. To investigate this annealing activity in more detail we used a fluorimetric assay to measure reaction in real time rather than tracking with radiolabeled oligomers by gel electrophoresis. Since spontaneous annealing of complementary oligonucleotides can be fast, monitoring reaction in real time has the advantage of eliminating high background and artifacts that could arise in electrophoretic gel-based assays, in which end points are determined after manipulations that might promote annealing. The fluorimetric assay is based on a method developed to measure annealing by yeast Rad52 protein, which monitors the increase in DAPI fluorescence upon binding annealed dsDNA (18), except that we substituted PicoGreen for DAPI to enhance sensitivity. Picogreen has been developed as a selective reagent for determining dsDNA concentrations.(30).
A 100-mer oligonucleotide and its complement were used as substrates in annealing reactions. Since Rad52 has been reported to promote annealing of complementary ssDNA oligonucleotides (19), we used the U. maydis Rad52 protein as a positive control for annealing. To reduce the rate spontaneous annealing of the oligonucleotides so that the effects of added protein could be measured, we manipulated DNA concentration and temperature of reaction. Under conditions where the spontaneous reaction was low, there was a substantial rate enhancement with added Brh2, although by comparison Brh2 was less active on a molar basis than Rad52 (Fig. 3A & B). Annealing was delayed by 2 to 3 minutes when 4 mM MgCl2 was added to reactions (Fig. 3C), but then proceeded with almost identical kinetics.
FIGURE 3. Brh2 anneals complementary single strands.
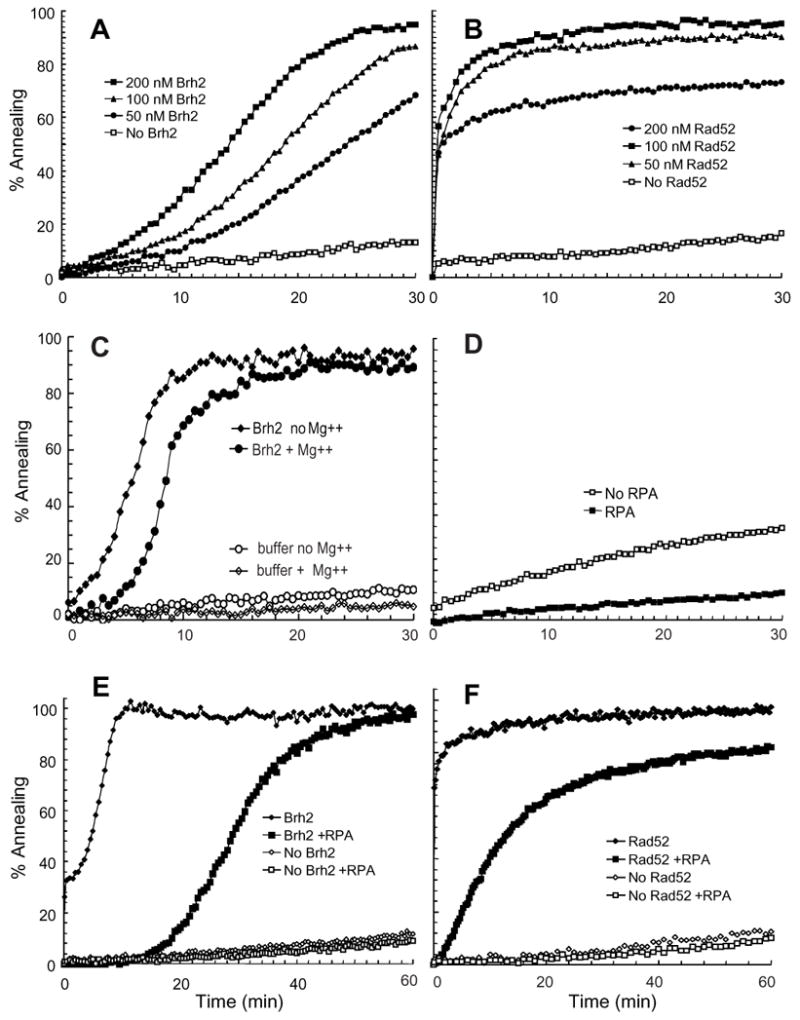
100mer oligonucleotide (+) and (−) strands at 12 nM each were incubated at 30° with the indicated levels of (A) Brh2 or (B) Rad52 in the presence of 0.15 μM Picogreen as described in EXPERIMENTAL PROCEDURES. (C) Reactions as in A with or without 4 mM MgCl2 were incubated at 37°. (D) Reactions containing 100mer (+) and (−) strands at 40 nM each were incubated at 37° plus or minus RPA at a ratio of nucleotide to protein of 25 to 1. Reactions containing 100mer (+) and (−) strands at 12 nM each with (E) Brh2 at 200 nM or (F) Rad52 at 100 nM plus or minus RPA at a nucleotide to protein of 25 to 1 were incubated at 37°.
Because RPA binds tightly to ssDNA it creates a barrier to Rad51 in DNA strand exchange. Rad52 and Brh2 have been demonstrated to serve as mediators in strand exchange by enabling Rad51 to gain access to ssDNA coated with RPA thus facilitating displacement of RPA (5, 12, 14, 15). We tested whether Brh2 could promote annealing of the oligonucleotides when precoated with RPA. Spontaneous annealing was strongly inhibited when the ss 100mers were precoated with RPA at a saturating level of 25 nucleotides per RPA (Fig. 3D). However, Brh2, like Rad52 (29, 31), was still able to promote annealing, (Fig. 3E & F). In the case of Brh2 there was a marked lag before annealing initiated followed by an increasing rate indicative of overcoming a kinetic barrier in reaction. These results reinforce the previous observations that Brh2 has the capacity for specific interaction with RPA-coated ssDNA (5), but also suggest it is capable of enabling displacement of the RPA. Evidence for annealing of RPA-coated oligonucleotides by CeBRC-2, the BRCA2-related protein from Caenorhabditis elegans, was also presented recently but with the use of a gel assay measuring end points (32).
DNA strand exchange monitored by FRET
To test whether Brh2 could promote a pairing transactions more complex than complementary strand annealing, we investigated its activity in reaction involving three DNA strands, namely oligonucleotide strand exchange. Here the substrates were based on the ss 100mer used above and a ds 49mer homologous to the distal end of the ss 100mer. In an approach similar to that taken above to enable monitoring reaction in real time, we developed a fluorimetric assay based on FRET to follow DNA strand exchange. The basis of the assay was the release of emission by a fluorescent reporter from FRET-mediated dark quenching upon strand exchange. The 49mer (−) strand complementary to the ss 100mer (+) strand was labeled at the 5′-end with the fluorophore Cy5 and the 49mer (+) strand was labeled at the 3′-end with the fluorescence quencher Iowa Black (IB). When the 49mer strands were paired the fluorescent signal of the Cy5 donor was quenched by FRET with the Iowa Black energy dissipater due to the close proximity. However, when the Cy5-49mer strand was exchanged with the complementary 100mer, the FRET quenching was abrogated and fluorescence emission of Cy5 revealed. Thus, release from the quencher under these conditions is a measure of DNA strand exchange.
Under the same conditions as those used in the annealing reactions with equimolar concentrations of substrates, there was a substantial increase in fluorescence emission upon addition of Brh2 indicative of Cy5 fluorescence emission released from quenching to a level approaching 20% of the ss Cy5-49mer (Fig. 4A). The increased fluorescence was dependent on addition of homologous ss 100-mer although it made no difference whether (+) strand 100mer or (−) strand 100mer was used. Furthermore, no increase was observed when a heterologous ss 80mer was used in place of the ss 100mer or when ss DNA was omitted altogether (Fig. 4B). On the other hand addition of E. coli exonuclease III resulted in an immediate increase in fluorescence as would be expected upon removal of the 3′-IB quencher given Exo III’s 3′ to 5′ polarity on ds DNA. These controls imply that the increase in fluorescence in the three DNA strand reaction was due to strand transfer of the 5′-Cy5-49mer from the 5′-Cy5/3′-IB ds 49mer to the ss 100mer and not to nucleolytic removal of the quencher from the 5′-Cy5/3′-IB ds 49mer or to ssDNA dependent unwinding activity.
FIGURE 4. FRET assay for DNA strand exchange.
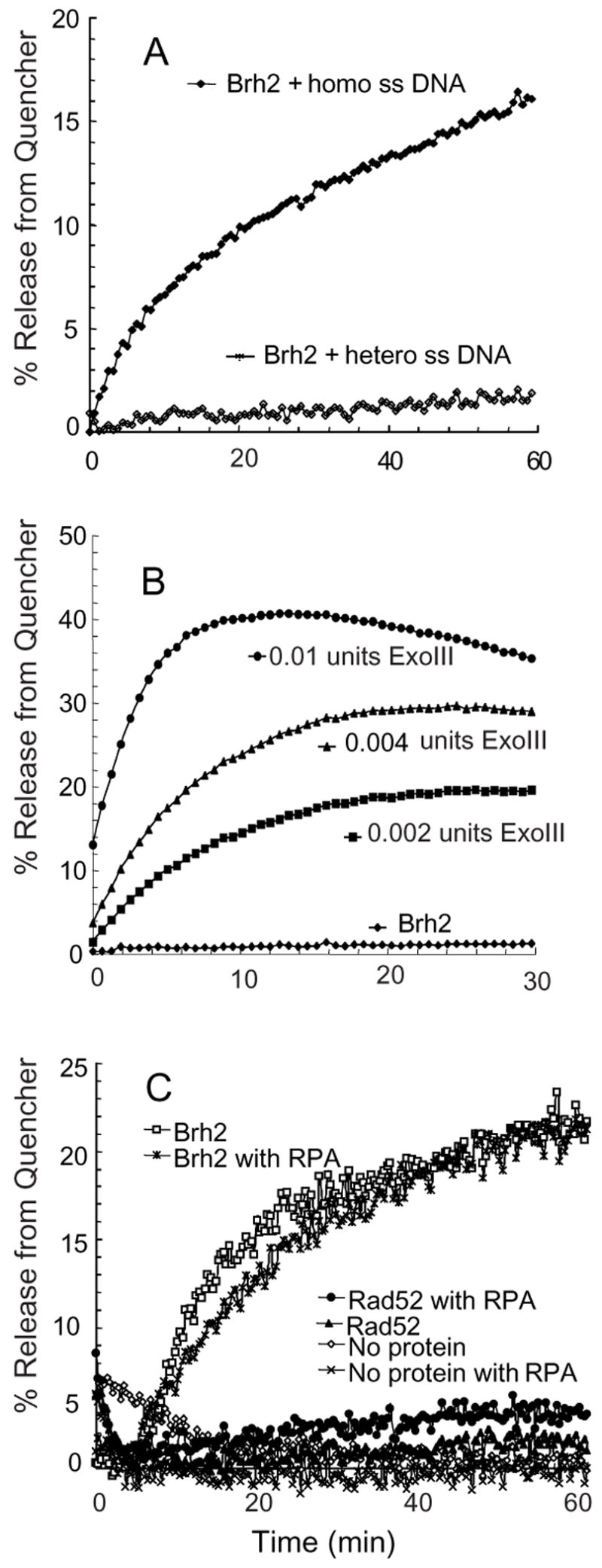
(A) Reactions containing 30 nM ds 5′-FAM-49mer/3′-BHQ2-49mer, 120 nM homologous ss100mer or heterologous ss 80mer were started by addition of 200 nM Brh2 and the increase in fluorescence was monitored. (B) Reactions were the same as in A but containing 30 nM ds 5′-FAM-49mer/3′-BHQ2-49mer only, 4 mM MgCl2, and the indicated amounts of Exo III. (C) Reaction mixtures containing 10 nM ss 100mer were preloaded with 80 nM RPA for 10 min where indicated followed by 200 nM Brh2 or 100 nM Rad52. Reactions were started by addition of ds 5′-Cy5-49mer/3′-IB-49mer.
When the ss DNA was precoated with a saturating amount of RPA as above (25 nucleotide residues per RPA heterotrimer) the reaction proceeded with almost the same rate and to nearly the same extent as in the absence of RPA (Fig. 4C). However, when the analysis was performed with Rad52 under conditions optimal for its annealing, only a low level of strand exchange activity was detectable. This modest activity was slightly elevated when the ss DNA was precoated with RPA. Thus, Brh2 is active in pairing a ss DNA with a complementary strand that is free or duplexed when RPA is present. Moreover, while Brh2 is less potent in the two-stand annealing reaction compared with Rad52, it is more active in the three-strand reaction.
Brh2 strand exchange is insensitive to the ordering of components
Oligonucleotide DNA strand exchange could potentially result from several different mechanisms. In one mode exemplified by Rad51/RecA it is thought that the protein induces an alteration in DNA structure upon binding to enable base pair switching between dsDNA and ssDNA facilitating homologous pairing and strand exchange (33). A hallmark of the Rad51 class of mechanism is the requirement for formation of the nucleoprotein filament on ssDNA. Thus, DNA strand exchange is stimulated when Rad51 is preincubated with ssDNA, but inhibited when preincubated with dsDNA. An alternative mode suggested from studies on the paralog Rad51C is that duplex oligomers are melted by action of the protein and the separated strands can change partners by spontaneously annealing with other strands of complementary sequence that are present (34). We performed experiments to investigate the mechanism of DNA strand exchange observed with Brh2. Here we used the ss 100mer and ds 49mer substrates above but with conventional methodology following the transfer of 32P-labeled (−) strand to the ss 100mer (+) strand by gel electrophoresis. Strand exchange is evident by the shift in label from the faster running ds 49mer to the slower 32P-49mer/100mer heteroduplex. Strand exchange promoted by Brh2 (Fig. 5A) was dose dependent and evident regardless of whether Brh2 was preincubated with ss 100mer or ds 49mer (Fig. 5B), unlike the reaction promoted by Rad51, which proceeded only when Rad51 was preincubated with ss 100mer. Thus, in contrast to Rad51, Brh2 strand exchange was insensitive to the order of addition of components. We considered the possibility that the observed strand exchange might be an artifact arising from trace exonuclease contamination. Here, exposure of a single-stranded tract in dsDNA in the presence of complementary strand and an annealing activity could lead to an apparent strand exchange (35). However, when we examined Brh2 directly for exonuclease activity by incubating it with the 32P-labeled ds 49mer plus Mg2+, a cofactor required for many E. coli deoxyribonuclease, then analyzing the products on a DNA sequencing gel in which release of a single nucleotide could be detected, there was no indication of exonuclease contamination. By comparison controls in which Exo III was added exhibited a ladder of bands indicative of exonucleolytic degradation.
FIGURE 5. Oligonucleotide strand exchange.
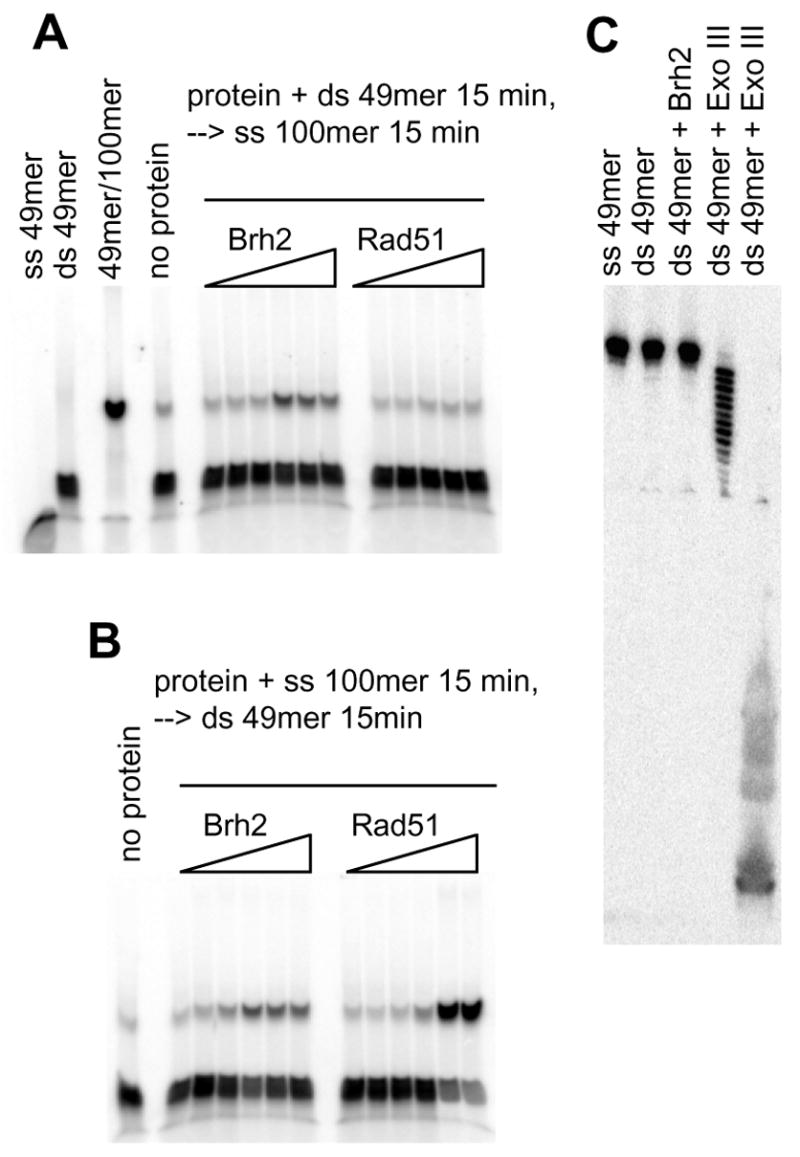
(A) Brh2 or Rad51 was preincubated with 6 nM 32P-labeled ds49mer (label on minus strand) for 15 min (4 mM CaCl2 and 2 mM ATP added for Rad51 reactions), then 60 nM ss100mer was added and incubation continued for 15 min. (B) Brh2 or Rad51 was preincubated with 60 nM ss100mer. After 15 min 6 nM 32P-labeled ds49mer was added and incubation continued for an additional 15 min. Concentrations for the samples of Brh2 in A and B were 0.05, 0.1, 0.25, 0.5. 1, and 2 μM, respectively. For Rad51, concentrations were 0.06, 0.15, 0.6, 1.5. and 3 μM in A and 0.06, 0.15, 0.3, 0.6, 1.5. and 3 μM in B. (C) Brh2 (1 μM) was incubated with 6 nM 32P-labeled ds49mer (label on minus strand) for 30 min in 25 mM Tris-HCl, pH 7.5, 20 mM KCl, 4 mM MgCl2, 1 mM DTT, 2 mM ATP. Similarly, reactions were run containing 0.004 units or 1 unit Exo III, respectively. Samples were mixed with tracking dye stop solution containing formamide and EDTA to yield final concentrations of 50% and 10 mM, respectively, heated at 50° for 5 min, then loaded on an 8% polyacrylamide gel containing 7 M urea that had been prerun for 30 min to warm the gel.
To test whether the mechanism of Brh2 strand exchange involved melting of the 49-mer duplex followed by spontaneous annealing with the ss100-mer, we followed the procedure used in analysis of Rad51C by Kowalczykowski’s group (34). Reactions mixtures containing Brh2 and the ds 49mer were incubated for a set time then mixed with a 10-fold excess of ss 100mer and either deproteinized immediately or else incubated further and then deproteinized. If separated strands of the 49-mer were generated by incubation with Brh2, then the labeled ss 49mer should anneal when the reaction was quenched upon addition of an excess of complementary ss 100mer thus forming the strand exchange product with different mobility. No further increase in strand exchange product would be expected upon continued incubation with ss 100mer. However, no strand exchange above background level was apparent after the initial incubation period of ds 49mer with Brh2 followed by the quench that included addition of excess ss 100mer and immediate deproteinization (Fig. 6A, left panel). Strand exchange only became apparent when there was continued incubation with the added ss 100mer. These results imply that the Brh2 strand exchange reaction occurs without the generation of free ssDNA from the dsDNA as an intermediate. No strand exchange took place when the same procedure was used with Rad51 (Fig. 6A, right panel) as expected. In this case strand exchange with Rad51 became efficient only with extended preincubation time with the ss 100mer (Fig. 6A, middle panel). Conversely, the extent of Brh2-promoted strand exchange was undiminished regardless of the time of incubation of Brh2 with ds49-mer as long as this was followed by a second incubation with ss100-mer, in contrast to the Rad51 reaction (Fig. 6B & 6C). Thus, oligonucleotide strand exchange by Brh2 proceeds by a mechanism that does not involve duplex melting and does not require prior formation of a complex with ssDNA.
FIGURE 6. Order of addition in strand exchange.

Brh2 or Rad51 was preincubated with one DNA substrate. Strand exchange reactions were then started by addition of the second DNA substrate. In A–C proteins were mixed with the first DNA substrate as indicated and preincubated for 15 min. Reactions were started by addition of the second DNA substrate and terminated at 0, 0.5, 1, 1.5, 2, 3, 5, 7.5, 10, 15, 20, and 30 min, respectively. These 12 time points are represented in order from left to right by the 12 lanes in each frame (A) Brh2 (0.5 μM) was preincubated with 6 nM 32P-labeled ds 49mer for 15 min, followed by 60 nM ss100mer. Samples were then taken at the times indicated as above and deproteinized. Control lanes at the far left include 32P-labeled ss 49mer or ds-49mer with no protein. These are followed by control mixtures of 32P-labeled ds 49mer plus ss100mer with no protein but incubated for the indicated times before loading. A low level of slower moving DNA, which is heteroduplex formed by spontaneous annealing of some residual free 32P-labeled ss 49mer with ss100mer, is apparent in all three lanes. (B) 1.5 μM Rad51 was preincubated with 60 nM ss100mer for 15 min. Reactions were initiated by addition of 6 nM 32P-labeled ds49mer and samples taken at the indicated times. (C) 1.5 μM Rad51 was preincubated with 6 nM 32P-labeled ds 49mer for 15 min. Reactions were initiated by addition of 60 nM ss 100mer and samples taken at the times indicated as above. In (D–E) proteins were mixed with the first DNA substrate as indicated and preincubated for variable lengths of time as follows: 0, 0.5, 1, 1.5, 2, 3, 5, 7.5, 10, 15, 20, and 30 min, respectively. Reactions were started by addition of the second DNA substrate and then terminated after 15 min. (D) Brh2 (0.5 μM) or (E) Rad51 (1.5 μM) was preincubated with 6 nM 32P-labeled ds 49mer for variable time as above, followed by addition of 60 nM ss100mer for 15 min. Control lanes at the far left include mixtures of 32P-labeled ds 49mer plus ss100mer with no protein but incubated for the indicated times before loading. (F) Data from the above figures is summarized with percentage strand exchange plotted for Rad51 and Brh2 reactions preincubated with ss or dsDNA.
Given that ds DNA is less stable in the absence of divalent cation, we considered the possibility that the apparent strand exchange might be explained as a protein-enhanced reaction initiated in response to increased spontaneous breathing of the duplex. If so, there should be no reaction when a physiological level of Mg2+ is present. Therefore, we performed reactions in the presence of 4 mM Mg2+, the concentration optimal for the Rad51-promoted strand exchange activity2. Under these conditions there was robust strand exchange promoted by Brh2 and the activity was slightly stimulated by addition of RPA (Fig. 7, lanes c, k). These findings argue against the notion that the observed Brh2-promoted strand exchange results from spontaneous breathing of the ds DNA, but to the contrary, support the idea that the strand exchange activity of Brh2, regardless of the molecular basis, could represent a physiologically significant aspect of its biological function.
FIGURE 7. Multiple components in strand exchange.
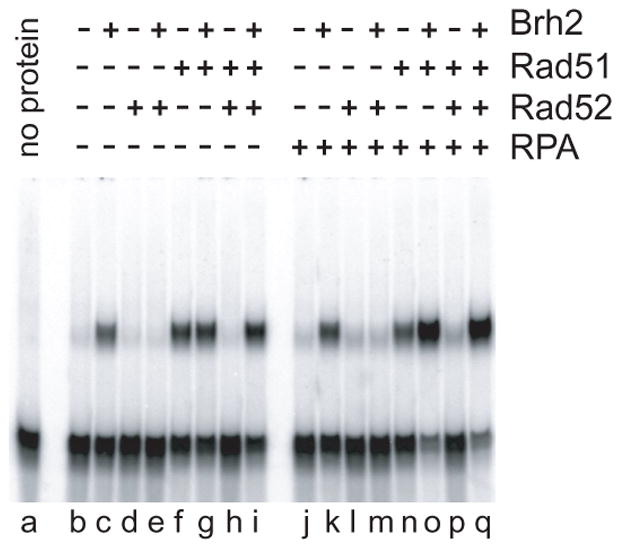
All reactions were carried out in the presence of 4 mM MgCl2 and 2 mM ATP. Lanes b-i: Protein components (Brh2 -500 nM; Rad51 - 330 nM; Rad52 - 330 nM) were mixed together as indicated with 60 nM ss100mer. After 15 min 6 nM 32P-labeled ds49mer was added and incubation continued for an additional 15 min. Lanes j-q: 60 nM ss100mer was preincubated with 160 nM RPA for 10 min. Other proteins were added to the mix as indicated for an additional 15 min. Then 6 nM 32P-labeled ds49mer was added, incubation continued for a further 15 min.
In the yeast system it has been established that Rad52 forms a complex with Rad51and can stimulate Rad51-catalyzed strand exchange. However, the ratio of Rad52 to Rad51 is critical. When the level of Rad52 protomers exceeds Rad51 by 1:3 strand exchange activity is strongly inhibited (17, 36). In agreement we found that when Rad51 was at sub-optimal levels for strand exchange (1 protomer per 18 nucleotides ssDNA) there was almost total inhibition of activity upon addition of Rad52 at a ratio of 1:1 regardless of the presence or absence of RPA (Fig. 7 lanes f, h & lanes n, p). Rad52 also inhibited the Brh2 strand exchange reaction (Fig. 7, lanes e, m) Inhibition of Rad51 by Rad52 was completely relieved when Brh2 was also present Fig. 7, lanes i, q). These results suggest that precise control of the interplay among Brh2, Rad51, and Rad52 is essential for well-ordered DNA strand exchange.
DISCUSSION
Previously Brh2 was shown able to nucleate Rad51 filament formation on RPA-coated DNA at a dsDNA/ssDNA junction (5) thus demonstrating its activity as a mediator. Here we followed up on an aspect of that work by focusing more closely on the interplay of Brh2 with DNA using an oligonucleotide-based model system. Three principal conclusions can be drawn from these studies. First, Brh2 prefers binding to partially dsDNA oligonucleotide structures that contain ssDNA regions as forks, branches, or D-loops. Second, Brh2 can promote annealing of complementary single strand oligonucleotides coated with RPA. Third, Brh2 can promote a type of DNA strand exchange using oligonucleotide substrates without regard for the order of addition of components. These findings raise the possibility that Brh2 might contribute to the homologous recombination system by means not previously considered in the studies on its mediator activity.
A paradoxical feature of Rad51 revealed by in vitro analysis is its sensitivity to dsDNA during the presynaptic stage. Efficient Rad51-promoted strand exchange requires a preloading step on ssDNA to circumvent the powerful inhibitory effects of dsDNA on strand exchange. How then, can the function of Rad51 in vivo be reconciled in view of the largely duplex conformation of DNA within the nuclear environment? In the preceding work establishing that Brh2 could assist Rad51 in forming nucleoprotein filaments on a resected DNA molecule (5), the evidence suggested that Brh2 recognized ds/ssDNA junctions and nucleated Rad51 filament formation in the appropriate polarity for subsequent strand exchange. That Brh2 can promote an oligonucleotide strand exchange reaction, albeit over a limited stretch of residues, with dsDNA and RPA-coated ssDNA present raises the idea that this pairing activity might contribute to initiating Rad51-catalyzed pairing reactions. Is it possible that Brh2 sets up recombination events by establishing an initial synaptic step involving a limited stretch of residues and then recruits Rad51 to potentiate and extend pairing thus forming a stable heteroduplex joint? The binding preference of Brh2 for D-loops and branched structures might be a clue to the mechanism underlying the oligonucleotide strand exchange. Perhaps the Brh2 strand exchange reaction is driven by stabilization of a branched or D-loop intermediate. Alternatively or in addition, perhaps the preferential binding of Brh2 to Holliday junctions suggests a role in stabilizing the cruciform or “chickenfoot” junctions formed at a stalled replication fork possibly enabling lesion bypass through template switching.
More experimentation will be required to establish the mechanism underlying the Brh2 promoted strand exchange, but it seems likely that it will involve some degree of duplex deformation, opening, or unwinding and concerted pairing of the third strand. With its intrinsic ability to promote single-strand annealing, and DNA strand exchange, Brh2 joins a set of proteins found in eukaryotes as well as prokaryotes providing similar functions central to homologous recombination. In prokayrotes these include RecA family members, RecO (37), and RecT (38), and in eukaryotes Rad51 family members and paralogs such as Dmc1(39, 40) and Xrcc3-Rad51C(34, 41), Hop2 (42), and Rad52. An emerging paradigm linking these proteins with a common mechanism is the formation of a presynaptic complex with ssDNA that enables switching of base pairs (33, 43). Brh2 seems to stand apart in being able to initiate DNA strand exchange in the presence of dsDNA, but that feature by no means rules out the possibility that its mechanism for DNA strand invasion proceeds through formation of a nucleoprotein structure favorable for base pair switching. As the U. maydis system offers the powerful advantage of genetic manipulation, it should be possible to generate Brh2 alleles defective in DNA annealing and strand exchange. Analysis of the corresponding mutant phenotypes would be decisive in elucidating the biological significance of these activities.
It seems to be a remarkable example of evolutionary convergence in function that Brh2 in U. maydis and Rad52 in Saccharomyces cerevisiae perform similar mediator reactions in vitro and provide the pivotal genetic control governing homologous recombination in vivo, yet they bear no structural resemblance. That a conserved Rad52 is present in U. maydis, yet relegated to a relatively minor role in recombination3, while a Brh2/BRCA2 ortholog is absent in S. cerevisiae raises the interesting questions of how these proteins evolved to similarly prominent positions in the homologous recombination systems of their respective organisms and what role is played by the U. maydis Rad52?
Acknowledgments
This work greatly benefited from critical comments offered by Steve Kowalczykowski, UC Davis, and Lorraine Symington, Columbia University.
Footnotes
This work was supported in part by a grant from the National Institutes of Health.
Abbreviations used: BH2, Black Hole 2 dark quencher; bp, base pair; ds DNA, double-strand DNA; DTT, dithiothreitol; FAM, 6-carbonylfluorescein; FRET, fluorescence resonance energy transfer; HEPES, N-(hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid); His, hexahistidine affinity tag; IB, Iowa Black RQ dark quencher; IPTG, isopropyl-β-D-thiogalactoside; MBP, maltose binding protein;-mer, oligomer with n residues; NTA, nitrilotriacetate; OB, oligonucleotide/oligosaccharide binding; RPA, replication protein A; ss DNA, single-strand DNA
N. Mazloum and W.K. Holloman, unpublished results
M. Kojic, Q. Zhou, and W. K. Holloman, unpublished results
References
- 1.Pellegrini L, Venkitaraman A. Emerging functions of BRCA2 in DNA recombination. Trends Biochem Sci. 2004;29:310–6. doi: 10.1016/j.tibs.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 2.West SC. Molecular view of recombination proteins and their control. Nat Rev Mol Cell Biol. 2003;4:1–11. doi: 10.1038/nrm1127. [DOI] [PubMed] [Google Scholar]
- 3.Esashi F, Christ N, Gannon J, Liu Y, Hunt T, Jasin M, West SC. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature. 2005;434:598–604. doi: 10.1038/nature03404. [DOI] [PubMed] [Google Scholar]
- 4.Yang H, Jeffrey PD, Miller J, Kinnucan E, Sun Y, Thoma NH, Zheng N, Chen PL, Lee WH, Pavletich NP. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science. 2002;297:1837–48. doi: 10.1126/science.297.5588.1837. [DOI] [PubMed] [Google Scholar]
- 5.Yang H, Li Q, Fan J, Holloman WK, Pavletich NP. The BRCA2 homologue Brh2 nucleates RAD51 filament formation at a dsDNA-ssDNA junction. Nature. 2005:653–7. doi: 10.1038/nature03234. [DOI] [PubMed] [Google Scholar]
- 6.Benson FE, Stasiak A, West SC. Purification and characterization of the human Rad51 protein, an analogue of E. coli RecA. EMBO J. 1994;13:5764–71. doi: 10.1002/j.1460-2075.1994.tb06914.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sung P, Robberson DL. DNA strand exchange mediated by a RAD51-ssDNA nucleoprotein filament with polarity opposite to that of RecA. Cell. 1995;82:453–61. doi: 10.1016/0092-8674(95)90434-4. [DOI] [PubMed] [Google Scholar]
- 8.Chi P, Van Komen S, Sehorn MG, Sigurdsson S, Sung P. Roles of ATP binding and ATP hydrolysis in human Rad51 recombinase function. DNA Repair. 2006;5:381–91. doi: 10.1016/j.dnarep.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 9.Baumann P, Benson FE, West SC. Human Rad51 protein promotes ATP-dependent homologous pairing and strand transfer reactions in vitro. Cell. 1996;87:757–66. doi: 10.1016/s0092-8674(00)81394-x. [DOI] [PubMed] [Google Scholar]
- 10.Zaitseva EM, Zaitsev EN, Kowalczykowski SC. The DNA binding properties of Saccharomyces cerevisiae Rad51 protein. J Biol Chem. 1999;274:2907–15. doi: 10.1074/jbc.274.5.2907. [DOI] [PubMed] [Google Scholar]
- 11.Sigurdsson S, Trujillo K, Song B, Stratton S, Sung P. Basis for avid homologous DNA strand exchange by human Rad51 and RPA. J Biol Chem. 2001;276:8798–806. doi: 10.1074/jbc.M010011200. [DOI] [PubMed] [Google Scholar]
- 12.Sung P. Function of yeast Rad52 protein as a mediator between replication protein A and the Rad51 recombinase. J Biol Chem. 1997;272:28194–7. doi: 10.1074/jbc.272.45.28194. [DOI] [PubMed] [Google Scholar]
- 13.Benson FE, Baumann P, West SC. Synergistic actions of Rad51 and Rad52 in recombination and DNA repair. Nature. 1998;391:401–4. doi: 10.1038/34937. [DOI] [PubMed] [Google Scholar]
- 14.Shinohara A, Ogawa T. Stimulation by Rad52 of yeast Rad51-mediated recombination. Nature. 1998;391:404–7. doi: 10.1038/34943. [DOI] [PubMed] [Google Scholar]
- 15.New JH, Sugiyama T, Zaitseva E, Kowalczykowski SC. Rad52 protein stimulates DNA strand exchange by Rad51 and replication protein A. Nature. 1998;391:407–10. doi: 10.1038/34950. [DOI] [PubMed] [Google Scholar]
- 16.Symington LS. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol Mol Biol Rev. 2002;66:630–70. doi: 10.1128/MMBR.66.4.630-670.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song B, Sung P. Functional interactions among yeast Rad51 recombinase, Rad52 mediator, and replication protein A in DNA strand exchange. J Biol Chem. 2000;275:15895–904. doi: 10.1074/jbc.M910244199. [DOI] [PubMed] [Google Scholar]
- 18.Sugiyama T, Kowalczykowski SC. Rad52 protein associates with replication protein A (RPA)-single-stranded DNA to accelerate Rad51-mediated displacement of RPA and presynaptic complex formation. J Biol Chem. 2002;277:31663–72. doi: 10.1074/jbc.M203494200. [DOI] [PubMed] [Google Scholar]
- 19.Mortensen UH, Bendixen C, Sunjevaric I, Rothstein R. DNA strand annealing is promoted by the yeast Rad52 protein. Proc Natl Acad Sci USA. 1996;93:10729–34. doi: 10.1073/pnas.93.20.10729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Dyck E, Stasiak AZ, Stasiak A, West SC. Visualization of recombination intermediates produced by RAD52-mediated single-strand annealing. EMBO Rep. 2001;2:905–909. doi: 10.1093/embo-reports/kve201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu Y, Sugiyama T, Kowalczykowski SC. DNA annealing mediated by Rad52 and Rad59 proteins. J Biol Chem. 2006;281:15441–15449. doi: 10.1074/jbc.M601827200. [DOI] [PubMed] [Google Scholar]
- 22.Kagawa W, Kurumizaka H, Ikawa S, Yokoyama S, Shibata T. Homologous pairing promoted by the human Rad52 protein. J Biol Chem. 2001;276:35201–8. doi: 10.1074/jbc.M104938200. [DOI] [PubMed] [Google Scholar]
- 23.Bi B, Rybalchenko N, Golub EI, Radding CM. Human and yeast Rad52 proteins promote DNA strand exchange. Proc Natl Acad Sci USA. 2004;101:9568–72. doi: 10.1073/pnas.0403205101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kagawa W, Kurumizaka H, Ishitani R, Fukai S, Nureki O, Shibata T, Yokoyama S. Crystal structure of the homologous-pairing domain from the human Rad52 recombinase in the undecameric form. Mol Cell. 2002;10:359–71. doi: 10.1016/s1097-2765(02)00587-7. [DOI] [PubMed] [Google Scholar]
- 25.Singleton MR, Wentzell LM, Liu Y, West SC, Wigley DB. Structure of the single-strand annealing domain of human. Proc Natl Acad Sci USA. 2002;99:13492–7. doi: 10.1073/pnas.212449899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krejci L, Song B, Bussen W, Rothstein R, Mortensen UH, Sung P. Interaction with Rad51 is indispensable for recombination mediator function of Rad52. J Biol Chem. 2002;277:40132–41. doi: 10.1074/jbc.M206511200. [DOI] [PubMed] [Google Scholar]
- 27.San Filippo J, Chi P, Sehorn MG, Etchin J, Krejci L, Sung P. Recombination mediator and Rad51 targeting activities of a human BRCA2 polypeptide. J Biol Chem. 2006;281:11649–57. doi: 10.1074/jbc.M601249200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kamper J, Kahmann R, Bolke M, Ma LJ, Brefort T, Saville BJ, Banuett F, Kronstad JW, Gold SE, Muller O, Perlin MH, Wosten HA, de Vries R, Ruiz-Herrera J, Reynaga-Pena CG, Snetselaar K, McCann M, Perez-Martin J, Feldbrugge M, Basse CW, Steinberg G, Ibeas JI, Holloman WK, Guzman P, Farman M, Stajich JE, Sentandreu R, Gonzalez-Prieto JM, Kennell JC, Molina L, Schirawski J, Mendoza-Mendoza A, Greilinger D, Munch K, Rossel N, Scherer M, Vranes M, Ladendorf O, Vincon V, Fuchs U, Sandrock B, Meng S, Ho EC, Cahill MJ, Boyce KJ, Klose J, Klosterman SJ, Deelstra HJ, Ortiz-Castellanos L, Li W, Sanchez-Alonso P, Schreier PH, Hauser-Hahn I, Vaupel M, Koopmann E, Friedrich G, Voss H, Schluter T, Margolis J, Platt D, Swimmer C, Gnirke A, Chen F, Vysotskaia V, Mannhaupt G, Guldener U, Munsterkotter M, Haase D, Oesterheld M, Mewes HW, Mauceli EW, DeCaprio D, Wade CM, Butler J, Young S, Jaffe DB, Calvo S, Nusbaum C, Galagan J, Birren BW. Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature. 2006;444:97–101. doi: 10.1038/nature05248. [DOI] [PubMed] [Google Scholar]
- 29.Sugiyama T, New JH, Kowalczykowski SC. DNA annealing by RAD52 protein is stimulated by specific interaction with the complex of replication protein A and single-stranded DNA. Proc Natl Acad Sci USA. 1998;95:6049–54. doi: 10.1073/pnas.95.11.6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singer VL, Jones LJ, Yue ST, Haugland RP. Characterization of PicoGreen reagent and development of a fluorescence-based solution assay for double-stranded DNA quantitation. Anal Biochem. 1997;249:228–38. doi: 10.1006/abio.1997.2177. [DOI] [PubMed] [Google Scholar]
- 31.Shinohara A, Shinohara M, Ohta T, Matsuda S, Ogawa T. Rad52 forms ring structures and cooperates with RPA in single-strand DNA annealing. Genes Cells. 1998;3:145–156. doi: 10.1046/j.1365-2443.1998.00176.x. [DOI] [PubMed] [Google Scholar]
- 32.Petalcorin MI, Sandall J, Wigley DB, Boulton SJ. Rad52 forms ring structures and cooperates with RPA in single-strand DNA annealing. J Mol Biol. 2006;361:231–42. doi: 10.1016/j.jmb.2006.06.020. [DOI] [PubMed] [Google Scholar]
- 33.Nishinaka T, Shinohara A, Ito Y, Yokoyama S, Shibata T. Base pair switching by interconversion of sugar puckers in DNA extended by proteins of RecA-family: a model for homology search in homologous genetic recombination. Proc Natl Acad Sci USA. 1998;95:11071–6. doi: 10.1073/pnas.95.19.11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lio YC, Mazin AV, Kowalczykowski SC, Chen DJ. Complex formation by the human Rad51B and Rad51C DNA repair proteins and their activities in vitro. J Biol Chem. 2003;278:2469–78. doi: 10.1074/jbc.M211038200. [DOI] [PubMed] [Google Scholar]
- 35.Kmiec EB, Holloman WK. Holloman, DNA strand exchange in the absence of homologous pairing. J Biol Chem. 1994;269:10163–8. [PubMed] [Google Scholar]
- 36.Arai N, Ito D, Inoue T, Shibata T, Takahashi H. Heteroduplex joint formation by a stoichiometric complex of Rad51 and Rad52 of Saccharomyces cerevisiae. J Biol Chem. 2005;280:32218–32229. doi: 10.1074/jbc.M507521200. [DOI] [PubMed] [Google Scholar]
- 37.Luisi-DeLuca C, Kolodner R. Purification and characterization of the Escherichia coli RecO protein. Renaturation of complementary single-stranded DNA molecules catalyzed by the RecO protein. J Mol Biol. 1994;236:124–38. doi: 10.1006/jmbi.1994.1123. [DOI] [PubMed] [Google Scholar]
- 38.Noirot P, Gupta RC, Radding CM, Kolodner RD. Hallmarks of homology recognition by RecA-like recombinases are exhibited by the unrelated Escherichia coli RecT protein. EMBO J. 2003;22:324–34. doi: 10.1093/emboj/cdg027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hong EL, Shinohara A, Bishop DK. Saccharomyces cerevisiae Dmc1 protein promotes renaturation of single-strand DNA (ssDNA) and assimilation of ssDNA into homologous super-coiled duplex DNA. J Biol Chem. 2001;276:41906–12. doi: 10.1074/jbc.M105563200. [DOI] [PubMed] [Google Scholar]
- 40.Sehorn MG, Sigurdsson S, Bussen W, Unger VM, Sung P. Human meiotic recombinase Dmc1 promotes ATP-dependent homologous DNA strand exchange. Nature. 2004;429:433–7. doi: 10.1038/nature02563. [DOI] [PubMed] [Google Scholar]
- 41.Kurumizaka H, Ikawa S, Nakada M, Eda K, Kagawa W, Takata M, Takeda S, Yokoyama S, Shibata T. Homologous-pairing activity of the human DNA-repair proteins Xrcc3.Rad51C. Proc Natl Acad Sci USA. 2001;98:5538–43. doi: 10.1073/pnas.091603098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Petukhova GV, Pezza RJ, Vanevski F, Ploquin M, Masson JY, Camerini-Otero RD. The Hop2 and Mnd1 proteins act in concert with Rad51 and Dmc1 in meiotic recombination. Nature Struct Mol Biol. 2005;12:449–53. doi: 10.1038/nsmb923. [DOI] [PubMed] [Google Scholar]
- 43.Gupta RC, Folta-Stogniew E, O’Malley S, Takahashi M, Radding CM. Rapid exchange of A:T base pairs is essential for recognition of DNA homology by human Rad51 recombination protein. Mol Cell. 1999;4:705–714. doi: 10.1016/s1097-2765(00)80381-0. [DOI] [PubMed] [Google Scholar]