

Abstract
Single nucleotide polymorphisms (SNPs) are DNA sequence variations that can affect the expression or function of genes. As a result, they may lead to phenotypic differences between individuals, such as susceptibility to disease, response to medications, and disease progression. Millions of SNPs have been mapped within the human genome providing a rich resource for genetic variation studies. Adenosine-to-inosine RNA editing also leads to the production of RNA and protein sequence variants, but it acts on the level of primary gene transcripts. Sequence variations due to RNA editing may be misannotated as SNPs when relying solely on expressed sequence data instead of genomic material. In this study, we screened the human SNP database for potential cases of A-to-I RNA editing that cause amino acid changes in the encoded protein. Our search strategy applies five molecular features to score candidate sites. It identifies all previously known cases of editing present in the SNP database and successfully uncovers novel, bona fide targets of adenosine deamination editing. Our approach sets the stage for effective and comprehensive genome-wide screens for A-to-I editing targets.
Keywords: RNA editing, single nucleotide polymorphism, adenosine deamination, inosine
INTRODUCTION
Currently the total number of single nucleotide polymorphisms (SNPs) reported in public databases exceeds 9 million (Sherry et al. 2001), making SNPs the most frequently occurring genetic variations in the human genome. They are important molecular markers that link sequence variations to phenotypic changes. Therefore, the characterization of these SNPs advances the understanding of human physiology and the molecular bases of diseases (Taylor et al. 2001). In particular, SNPs that involve an amino acid change (recoding SNP) are of interest for clinicians and researchers, since they often strongly influence the function of the resulting gene product.
It is important to distinguish DNA-based single nucleotide variations (true SNPs) from sequence alterations in gene products (RNA or protein) that originate from recoding events on the level of the RNA transcripts. In particular, the post-transcriptional processing of pre-mRNAs by A-to-I modification has been recognized as an important mechanism for generating RNA and protein diversity (for review, see Bass 2002; Hoopengardner 2006; Maas et al. 2006; Nishikura 2006; Gommans et al. 2008). It is mediated by adenosine deaminases acting on RNAs (ADARs) that specifically bind to and deaminate their partially double-stranded RNA targets (Bass 2002; Gommans et al. 2008). When A-to-I RNA editing occurs within mRNA coding sequences it can result in amino acid substitutions, since inosine is interpreted as guanosine by the translational machinery. Several mammalian genes have been described where the substitution of a single amino acid due to RNA editing leads to a significant alteration in protein function (for review, see Gommans et al. 2008). Especially, neurotransmitter receptors and other brain-specific transcripts are among the previously characterized recoding targets for editing. Generally, A-to-I edited and nonedited gene products are produced side by side within the same cell, thereby increasing the number of protein variants available for cellular functions.
Generally, SNPs are annotated based on sequence analysis of chromosomal DNA from many individuals and subsequent determination of the ratio of the alleles within the population for each site. However, among the millions of validated genomic SNPs, some polymorphisms have been annotated solely based on the analysis of expressed sequences derived from mRNA (Buetow et al. 1999; Irizarry et al. 2000). Therefore, absent of additional genomic confirmation, it is possible that such sequence variations may not represent true SNPs, but instead result from RNA editing events.
Indeed, a few previously annotated SNPs, which are located within noncoding sequences were recently shown to in fact be single nucleotide sequence variations caused by RNA editing (Eisenberg et al. 2005). They were identified, since they coincide with the location of Alu repeat elements that were previously known to be subject to RNA editing at other positions (Athanasiadis et al. 2004; Kim et al. 2004; Levanon et al. 2004).
Apart from the relatively small number of currently known RNA editing events that lead to amino acid substitutions (Gommans et al. 2008), thousands of human genes undergo A-to-I editing within noncoding regions of mRNAs and introns (Athanasiadis et al. 2004; Blow et al. 2004; Kim et al. 2004; Levanon et al. 2004). These cases of editing, which are extrapolated to involve at least >85% of all primary RNA transcripts (Athanasiadis et al. 2004), are due to intramolecular foldback structures formed by oppositely oriented pairs of transposon-derived repeat elements.
The functional consequences of the high frequency of RNA editing in noncoding sequences have not been extensively studied, but in a few instances it was shown that intronic editing can alter splice consensus sites, leading to (or predicting) changes in pre-mRNA splicing patterns (Athanasiadis et al. 2004; Lev-Maor et al. 2007). Furthermore, extensive editing in UTRs can lead to nuclear retention, which in one case has recently been shown to regulate the expression of a mouse calcium transporter (Prasanth et al. 2005).
Key features that characterize the range of known editing cases are summarized in Figure 1. One end of the spectrum is represented by repeat-element mediated editing, which is associated with low site selectivity, a high number of editing sites per gene, and a high inosine content per RNA molecule. At the other end of the spectrum reside the recoding events including glutamate receptor and other brain-specific transcripts. These are characterized by high site specificity and low inosine content per molecule. Tens of thousands of editing sites located in noncoding Alu elements have been identified in humans (Athanasiadis et al. 2004; Blow et al. 2004; Kim et al. 2004; Levanon et al. 2004). In contrast, only a small number of site-selective recoding events are known (Gommans et al. 2008). Most of the latter were identified serendipitously. A few additional cases of recoding in mammals due to RNA editing were recently found through screening approaches (Clutterbuck et al. 2005; Levanon et al. 2005a; Ohlson et al. 2007). However, a major limitation of systematic searches for edited genes in mammals has been a low signal-to-noise ratio (Morse et al. 2002; Morse 2004; Clutterbuck et al. 2005; Levanon et al. 2005b; Ohlson et al. 2005; Gommans et al. 2008).
FIGURE 1.

Frequency of coding RNA editing targets versus noncoding targets. Schematic representation of the range of mRNA molecules that are known to undergo A-to-I RNA editing in humans. A large number of editing events (>30,000) occur in noncoding sequences that are characterized by RNAs with high double-stranded character, which results in a high number of adenosine deamination events with low site selectivity. In contrast, <30 cases of A-to-I editing are known where an mRNA is deaminated at only one or a few positions with high site selectivity in molecules that form a partially double-stranded structure with limited base pairing.
Here we report a combined bioinformatics and experimental strategy to systematically identify A-to-I editing events that lead to amino acid substitutions. In this study, we specifically asked if it is possible to identify novel RNA editing events within the SNP database that lead to nonsynonymous codon changes.
We show that our screening protocol selects all of the previously known editing targets with SNP annotations as high scoring candidates. Furthermore, we experimentally prove the in vivo occurrence of recoding RNA editing in human brain tissue for two additional genes that are among the highest scoring candidates from our screen. Overall, the experimental analysis of 64 predicted sites from four scoring groups revealed a high accuracy of predicting bona fide editing sites, as in our highest scoring group four out of seven sites (57%) are real editing substrates.
RESULTS
Bioinformatics screen for A-to-I RNA editing candidates in the human SNP database
The dbSNP database build 125 contains a total of > 5 ×106 mapped SNPs. From these annotations we extracted all those that are based solely on expressed sequence data using the UCSC genome table browser (Kuhn et al. 2007). This yielded ∼30,000 sites. Figure 2 depicts the subsequent filtering steps that were performed to narrow down the list of SNPs to only those that may represent recoding RNA editing sites within known genes. First, all variations other than A/G or G/A were removed. Those other types of base differences may result from different RNA modification events, but cannot be due to A-to-I editing. Subsequently, only the entries where adenosine (A) is present in the genomic sequence at the putative SNP position were retained, whereas those with G in the genomic sequence were removed. Next, we filtered the sites located within the known coding sequence of genes from sites in noncoding exons, since we wanted to focus on recoding events. This step eliminates potential A-to-I RNA editing sites in small regulatory RNAs such as miRNAs and editing events affecting 5′- and 3′-untranslated regions of mRNAs (for review, see Nishikura 2006; Gommans et al. 2008). In the next step we removed the sites that produce synonymous changes, i.e., the codon change caused by RNA editing leaves the protein sequence unaltered. This narrowed the number of potential editing sites down to 984. Finally, we selected among these 984 positions the ones located within known genes, thus removing entries with “hypothetical” and “unknown protein” annotations. The resulting list of 863 sites constituted the starting point for our bioinformatics analysis to rank the entries in order to identify the ones that have a high probability of representing bona fide RNA editing sites.
FIGURE 2.

Filtering steps of SNP database. Flowchart summarizing the retrieval and filtering of single-nucleotide polymorphisms from the human SNP database (dbSNP). See Results section for a step-by-step description. About 30,000 annotated SNPs are based on expressed sequence data only, therefore representing base discrepancies that may be the result of post-transcriptional modification events. Eight hundred sixty-three of these SNPs constitute A/G mRNA/gDNA discrepancies that are located in the open reading frame of a protein-coding gene and predict a nonsynonymous change within a known gene. In 554 of them the 5′-neighbor nucleotide is either a C, A, or T, and therefore more likely to be edited than with a G as a 5′-neighbor.
The molecular features used to rank and filter each of the 863 potential editing/SNP sites are derived from known properties of previously characterized mammalian RNA editing sites.
First, we downloaded and evaluated the preceding and following base (−1, +1 positions) of all 863 sites in order to score the entries according to the main 5′- and 3′-base preferences of ADARs (Bass 2002; Athanasiadis et al. 2004). Since G has been shown to be selected against preceding an editing site, we removed all entries with a G at (−)1 from the list before the ranking step. For the remainder, the assigned values for the (−)1 position are: 1 for A or T, and 2 for C. Second, for the (+)1 position the values are 1 for G and 2 for either A, T or C.
Third, we manually assigned a value for cross-species conservation. It captures how strongly the potentially edited nucleotide itself as well as the sequence surrounding the modified site is conserved (including mouse, rat, chicken, and zebrafish). Please see the Materials and Methods section for detailed description of the binning process using the PhastCons program (Siepel et al. 2005). As a result of this analysis, the entries were grouped into the bins: H=highly conserved, HM=medium/high, M=medium, ML=medium/low, and L=low. To receive a value of H (highly conserved) the nucleotide at the candidate site for modification must be an adenosine in all examined species and the exonic sequence surrounding this nucleotide must be strongly conserved (> 95%) across these species according to the PhastCons annotation in the UCSC genome browser.
For the remaining 293 sites we performed an in silico editing analysis using BLASTN (see Materials and Methods). Two hundred four candidate sites showed an in silico editing level of 1% or higher and were therefore chosen for analysis of potential RNA secondary structures using the M-fold algorithm (Zuker et al. 1999). Up to 2.5 kb of genomic sequence in both directions from the putative editing site were inspected for RNA foldback structures. Structural scores (STR) were determined for each structure as described in detail in the Materials and Methods section. Candidate structures were then grouped into bins 1–5 based on their STR value (see Materials and Methods). Value 5 indicates that no discernable folding above random could be detected, and a value of 1 is given to structures that show highly base-paired folds with high ratio of G/C base pairs and small intervening sequence between base-paired areas. For example, the known fold-back structure for the serotonin receptor 5-HT2C that includes the editing sites A to E was assigned a value of 1 (STR score=2899), whereas the glutamate receptor GluR-6 I/V-editing site structure obtained a value of 3 (STR score=362).
For each of the described features individual scores were computed using a LODs scoring method and combined to yield an overall score (S) as outlined under Materials and Methods. Table 1, screen A, lists the top candidates that arise when the 15 well-characterized mammalian A-to-I RNA editing sites are used as a reference set of sequences. These include several glutamate receptors, serotonin receptor 5HT-2C, Gabra-3 and potassium channel Kv1.1 (see Supplemental Table S1). For each feature the values of the reference set (positive control regions) are compared to the values of the sample set (all A/G discrepancies) to rank the sample set.
TABLE 1.
High scoring candidates of RNA editing


Interestingly, three out of four recently validated cases of A-to-I RNA editing (Clutterbuck et al. 2005; Levanon et al. 2005b) affecting two genes (bladder cancer-associated protein BC10 and filamin A alpha) and previously annotated as SNPs, were ranked very high (Table 1, screen A, position ranks 3,6,7). The fourth editing site located in the CYFIP coding sequence was ranked at position 41. These results clearly indicate that our search strategy is selecting for real editing targets. Furthermore, no known editing site is missed in our screen since there is no other previously reported recoding editing site among the ∼30,000 entries that formed the starting point for our search. Therefore, none of the real editing targets that have previously been characterized were missed or ranked lower than position 41 among the total of 589 entries of recoding, nonsynomymous SNPs.
Identification of novel sites of A-to-I RNA editing among high scoring candidates
Next we moved the four known cases of A-to-I editing (two in BC10 and one each in FLNA and CYFIP) that were contained in our candidate list into the reference set (now containing 19 sites) and compared the ranking of the resulting high score list (Table 1, screen B) with the previous one. Interestingly, the top 18 entries remained in unchanged order. Apart from the sites within BC10, filaminA, and CYFIP that we removed by adding them to the reference set, only minor changes with respect to the order of entries occur further down in the listing (see Supplemental Table S2).
We subsequently proceeded with the experimental validation analysis of predicted targets using gene-specific RT-PCR and sequencing of cDNAs that were derived from human brain total RNA. In those cases where candidate gene transcripts are tissue specifically expressed outside of the brain, tissue-specific cDNA, and gDNA pairs from other human tissues were analyzed. Genomic DNA from the same tissue specimen was analyzed in parallel to ensure that the presence of a polymorphism at the candidate site could be excluded.
Four groups of genes that span the entire spectrum of the ranked candidates (score ranks I–XXIX) were selected in order to estimate the signal-to-noise ratios across the whole range of the sample set. At least 10 individual genes from each of the four groups were experimentally analyzed yielding a total of 64 analyzed genes (see Table 2).
TABLE 2.
Statistical analysis of experimental validation

No editing was detected in any of the gene candidates from the lower three groups (Table 2, group 2: score ranks III–X [18 of 47 analyzed]; group 3: score ranks XI–XV [31 out of 83 analyzed]; group 4: score ranks XVI–XXIX [12 out of 52 analyzed]) by our RT-PCR and sequencing screening method (see Supplemental Table S2).
It is important to note that this does not prove that editing cannot or does not occur at those positions. Rather, it shows that editing at these positions is not detectable using the RT-PCR screening method in a specific tissue sample isolated at a single time point from a single individual. It cannot be ruled out that editing occurs at a very low rate that is below the detection threshold of this method, or that editing occurs in another specific cell type, or in a temporally restricted fashion.
When we analyzed the top four highest scoring sites that constitute group 1 (score ranks I+II), we clearly detected RNA editing in human brain at three of the four sites. These were located within two genes; the splicing factor SRp25 isoform 3 and insulin-like growth factor binding protein 7 (IGFBP7). This means that within this highest scoring group (summary score of > 2.5) three out of the four (75%) sites turn out to be real positives (see Table 1, screen B). Table 2 summarizes the validation data and the statistical evaluation of expected versus observed outcomes.
Since the apparent editing level for the SRp25 gene based on the RT-PCR sequencing assay was low (between 5% and 10%; Fig. 3A), the PCR amplicon was subcloned and a total of 100 individual clones were sequenced. This revealed that 7(±1)% of cDNAs carried a G instead of an A at the predicted position (see also Supplemental Fig. 1). In addition to the main editing site there may be additional minor editing sites within the same exon. Some of these residues are located within the same predicted RNA secondary structure (Fig. 3C) as the major site and are therefore more likely to represent real base modification events. However, for only one of them (see Fig. 3C) more than one template out of the analyzed 100 displayed a G at this position. Therefore, it cannot be ruled out at this point that these minor sites reflect base changes due to errors during reverse transcription, PCR amplification or sequencing. Figure 3 depicts the SR gene sequence and computer-predicted secondary structure of the pre-mRNA. The main editing site predicts a lysine-to-arginine change within a basic region of the protein. Interestingly, the entire, computer-predicted RNA fold-back structure is made up of exonic RNA sequences.
FIGURE 3.

RNA editing of SR-protein SRp25 in the human brain. (A) Sequence analysis of paired SRp25 genomic DNA (control) and brain cDNA of the same human brain specimen. Electropherograms show the antisense sequence of exon 3 in SRp25 around the predicted editing site. A mixed peak of T and C in the cDNA sample but not in the genomic counterpart indicates the presence of two populations of molecules. Subcloning of the same PCR amplicon used for sequencing revealed an editing level of 7%. (B) Amino acid change caused by RNA editing modification. A section of the open reading frame within exon 3 of SRp25 is shown with the K-to-R codon alteration indicated. (C) Computer-predicted RNA secondary structure of the SRp25 pre-mRNA sequence. A partially double-stranded RNA hairpin consisting entirely of exon 3 sequences is shown with the main editing site (highlighted in red and underlined) as well as potential additional sites (underlined) indicated.
The adenosines in IGFBP7 for which we prove the occurrence of editing are two of three predicted positions (A, B, C; see Fig. 4; Table 1, screen B) within the same exon of this gene, all of which are ranked among the top seven candidates. For two of these sites (A and B) in IGFBP7 it had previously been suggested that they might be subject to A-to-I modification based on database evidence and cDNA sequencing (Eisenberg et al. 2005), however, without experimental proof of an RNA-based mechanism. Our results from analysis of matched cDNA and genomic DNA from the same tissue specimen prove that the adenosine at site B is subject to high level RNA editing in human brain (31[±3]%) and not a genomic polymorphism. The resulting lysine-to-arginine amino acid substitution affects the IGFBP7 protein sequence within a region that represents a heparin binding site and also is close to a protease cleavage site (Sato et al. 1999; Ahmed et al. 2003). Site A is also subject to RNA editing with a level of modification around 55(±6)% according to our analysis of a human brain cDNA tissue sample where again the genomic DNA counterpart shows a signal for A (T in the reverse sequence) only (see Fig. 4).
FIGURE 4.

RNA editing of IGF-binding protein 7. (A) Sequence traces obtained from amplified genomic DNA and matching cDNA samples encompassing the predicted RNA editing sites A and B within the IGFBP7 exon 1 sequence (reverse complement sequence of mRNA is shown). A double-peak of T and C indicates a mixed population of mRNAs in the cDNA samples due to post-transcriptional A-to-I RNA modification in the sense strand. (B) Schematic representation of the IGFBP7 open reading frame indicating functional domains. The positions of the two editing sites with ensuing amino acid change are depicted. SS=signal sequence consisting of amino acid residues 1–26; HBS=heparin binding site encompassing amino acids 89–97; cleavage=protease processing site at amino acid position 97. (C) Predicted (Mfold) RNA fold encompassing the IGFBP7 editing sites A and B (underlined and highlighted in red) within exon 1. Also labeled is a third potential minor editing site (C) that did not show evidence of editing in vivo according to our screening analysis and might represent a true genomic SNP.
An inspection of the human EST database further suggests that position A may represent an editing site with 36 out of 302 human ESTs that carry a G at this site. The predicted amino acid change is an arginine-to-glycine substitution. For position B the EST analysis yields 132 of 302 (=43.7%) in silico editing and for position C eight of 302 ESTs (=2.6%) covering that region carry a G at this site. Candidate position C in IGFBP7 did not show evidence of editing in our sequencing analysis. It may therefore represent a genomic polymorphism, or RNA editing is restricted to specific cell types or occurs at a very low level.
Interestingly, as in SRp25, the entire computer predicted RNA fold-back structure in IGFBP7 is formed by exonic sequences only.
Inspection of mouse and rat mRNA and EST databases suggests that RNA editing at positions A and B also occurs in rodents. In mouse, 48 out of 85 (=56.5%) carry a G at position A and a similar number carry a G at position B (=57.7%). In rat, 75.4% carry a G at position A and 80% at position B.
Within score ranks I–VI, none of the sites lacking evidence for editing turned out to be a genomic SNP based on the analysis of the matched genomic DNA. They might therefore represent genomic SNPs with low penetrance in the population or they could be RNA editing sites with below background editing levels or with editing restricted to certain cell types or at specific times during development. The analysis of genes from groups VII, and VIII revealed three cases that we confirm as genomic SNPs (UBE3, IP5PA, and AK021577) (see Supplemental Table S2). The presence of a genomic SNP does not rule out for the same position to undergo A-to-I editing in transcripts derived from an adenosine-bearing allele, but in the absence of evidence for editing upon experimental validation we assume that editing does not occur at the site.
DISCUSSION
Toward the long-term goal of comprehensively analyzing the prevalence of A-to-I RNA editing in the human transcriptome, we developed a combined bioinformatics and experimental strategy. A critical component of such a strategy is to define selective criteria that capture as many of the true targets as possible while eliminating sequences that are not modified by ADARs in vivo. Although each individual selection feature does not strongly select for a bona fide editing target over background, the combination of scores from five distinct molecular features into a single weighted score has a much stronger predictive value.
Within the prefiltered sample set of 554 human SNPs all known editing sites previously annotated as SNPs that have been identified using various approaches were recaptured in our screen as high-scoring candidates. In fact, when including the novel sites identified and validated in this study, 75% of candidates within the highest scoring groups (I–IV) are known RNA editing targets, whereas only a single known editing site (CYFIP; group VIII) appears within all other tested medium and low scoring groups. For any of the candidates that did not show detectable editing activity in human brain the occurrence of editing in brain or other tissues cannot be ruled out. It is in the nature of the experimental screening method applied here that editing events with levels below ∼5% may be missed. Furthermore, for the testing of larger numbers of candidates only one adult human tissue was analyzed. RNA editing events that are specific for certain cell types or developmental stages may also escape this initial screening.
Splicing factor SRp25 (also known as ADP-ribosylation-like factor 6 interacting protein 4) is an ubiquitously expressed protein (Sasahara et al. 2000) of uncharacterized function. Because of its homology with SR-splicing factors it is believed to be a nuclear protein with a role in splicing regulation (Sasahara et al. 2000). The amino acid substitution due to RNA editing in the SRp25 affects a basic region in the protein that has not been ascribed a specific function. Based on its sequence characteristic it may represent a nuclear localization sequence or a domain that interacts with the nucleic acid backbone. The lysine-to-arginine change does not alter the overall charge of the molecule, and represents a conservative change that may not affect the protein's function substantially. However, lysine residues can be sites of post-translational modification and thereby regulate protein function. For example, in tumor suppressor p53 sumoylation of a specific lysine residue activates its transcriptional response (Rodriguez et al. 1999). K-to-R mutation of this site blocks sumoylation of the protein while preserving the local charge in the protein (Sampson et al. 2001). Furthermore, another specific lysine residue in p53 has been found to be subject to methylation, which downregulates the protein's transcriptional activation activity (Shi et al. 2007). It will be interesting to see if the editing invoked K-to-R change in SRp25 also has a regulatory impact on SRp25 function.
The second gene that was detected in this study as a target for RNA editing is IGFBP7. Although editing in this gene had been postulated previously for two of the three sites (Eisenberg et al. 2005), we provide experimental validation that the observed A/G discrepancy is in fact due to RNA editing by analyzing matched cDNA and genomic DNA sequences from the same tissue sample.
IGFBP7 was initially identified as a gene differentially expressed in cancerous cells, and has been implicated in various forms of cancer, either as putative tumor suppressor (Sprenger et al. 2002; Wilson et al. 2002; Mutaguchi et al. 2003) with functions in apoptosis and senescence, or as a promoter of angiogenesis in human tumor endothelium (St Croix et al. 2000; van Beijnum et al. 2006), and it is overexpressed in circulating endothelial cells (CECs) of metastatic cancer patients (Smirnov et al. 2006).
The IGFBP7 protein comprises several functional domains in its N-terminal half, such as a leucine-rich sequence, a cysteine-rich domain (CRD), a heparin binding site, and a Kazal-type trypsin inhibitor domain (Collet and Candy 1998). The two editing sites A and B affect amino acid positions 78 (R-to-G) and 95 (K-to-R) of the full-length protein.
Interestingly, the core sequence K89SRKRRKGK97 (edited site in bold) has been proposed to function as a heparin binding site (Sato et al. 1999), and it was observed that cell-binding and cell-adhesion activities of IGFBP7 are indeed inhibited by heparin (Akaogi et al. 1996).
IGFBP7 is proteolytically cleaved after K97, which results in a two-chain form of the protein cross-linked by disulfide bridges. Proteolytic processing of IGFBP7 has been shown to modulate its growth-stimulatory activity (Ahmed et al. 2003). Futhermore, the heparin-binding activity of IGFBP7 is decreased upon proteolysis.
The main editing site (K95R) not only lies within the proposed heparin-binding site of IGFBP7, but is also part of the recognition sequence for proteolytic cleavage. It will be interesting to explore the potential functional implications of RNA editing on heparin binding and/or proteolytic processing and its downstream effects regarding apoptosis, regulation of cell growth and angiogenesis.
For both SRp25 and IGFBP7, the RNA fold-back structures that are predicted to mediate RNA editing, involve solely exonic RNA sequences. Interestingly, almost all known characterized recoding editing sites involve folds where the editing site complementary sequence is located within an intron. As more edited genes are identified, it will be interesting to see how often exon-only structures mediate editing compared to exon–intron fold-back structures, since it could have implications for the evolutionary mechanisms that lead to the emergence of novel editing sites and the increase or decrease of editing extents at individual sites over evolutionary time. Furthermore, RNAs that do not require the presence of intronic sequences for editing to occur could continue to undergo editing after the completion of nuclear pre-mRNA splicing.
Importantly, the results of our limited screen indicate that the strategy is successful in identifying novel recoding targets. The algorithms for deriving each individual score, as well as the weighted combined score value reflect the current knowledge of the A-to-I editing mechanism and the properties of known targets. In previous database-driven studies only A/G discrepancies that appear both in human sequences of a given gene as well as at the same position in another mammalian species were investigated (Clutterbuck et al. 2005; Levanon et al. 2005b). The latter is a valuable strategy for initial screens with little data on known targets. However, for a more comprehensive search the approach that is presented here is more suitable. In particular, current cDNA databases do not cover all genes and often do not have sufficient coverage across editing sites to reveal low-level editing events. Over time, improved and extended databases as well as additional insights into the RNA editing mechanism will allow refining the search algorithm. Biochemical approaches for performing target screens (Morse and Bass 1997; Ohlson et al. 2005) come with their separate set of biases that may favor the identification of certain types of editing targets but select against others.
At this point the presented screen represents the most unbiased search for edited sequences in the human transcriptome with a reasonable signal-to-noise ratio. In the present study several of the selection steps were performed in a nonautomated manner. A largely automated procedure will be needed to apply this approach to the complete transcriptome. Ultimately, it is expected that many more recoding RNA editing targets will be revealed, further shedding light on the impact of RNA editing on proteome diversity.
MATERIALS AND METHODS
Databases and data analysis
Annotations for human SNPs from the dbSNP database build 125 (Sherry et al. 2001) were downloaded using the UCSC genome table browser (Kuhn et al. 2007). For subsequent analysis of candidate genes the UCSC human genome browser (assembly May 2004) was used.
Cross-species conservation was analyzed on two levels. Initially, conservation was evaluated for all 554 candidate genes using the UCSC genome browser conservation track, which is based on the phastCons program designed to identify conserved elements in multiply aligned sequences (Siepel et al. 2005). PhastCons is based on a phylogenetic hidden Markov model (phylo-HMM), a type of statistical model that considers both the process by which nucleotide substitutions occur at each site in a genome and how this process changes from one site to the next (Siepel et al. 2005). PhastCons produces a continuous valued “conservation score” for each base of the reference genome. The conservation score at each base in the reference genome is defined as the posterior probability that the corresponding alignment column was generated by the conserved state (rather than the nonconserved state) of the phylo-HMM, given the model parameters and the multiple alignment. Therefore, the scores range between 0 and 1, corresponding to 0%–100% conservation.
All 554 candidate genes were grouped into five bins according to the PhastCons score covering the region of 15 nucleotides (nt) upstream of and 15 nt downstream from each candidate site for editing. The bins were: high (H), for conservation of higher than 90%; high-medium (HM) for conservation between 75% and 90%; medium (M) for conservation of 50%–75%; medium-low (ML) for conservation of 25%–50%; and low (L) for conservation <25%. Only candidates from the H and HM bins were used for further analysis.
The second level of cross-species conservation taken into consideration was the conservation of the potentially edited adenosine. Candidates where only human and mouse homologous carry an adenosine at the predicted editing position, but not the rat counterpart (and/or chicken if available for the gene), were eliminated from further analysis even if previously grouped into the H or HM bin. This two-step evaluation of cross-species conservation is based on the data from known editing sites where the sequence surrounding the editing site as well as the edited adenosine itself are conserved to a higher degree than the general conservation of exonic, coding sequences, since in addition to encoding amino acids, the sequences also participate in forming a functional RNA structure.
Two hundred ninety-three of the 554 candidate sites remained for further analysis, whereas 261 entries were filtered out at this step. Next, evidence for in silico editing was analyzed for each of the 293 sites using the BLASTN program (NCBI). To this end 30 nt upstream of and 30 nt downstream from the predicted sites were successively blasted against the nr (NCBI) and the human EST databases (NCBI) and the percentage of sequences that carry a G instead of an A at the predicted site was recorded. For 204 candidates in silico editing was equal to or higher than 1%, whereas for 89 entries no evidence of editing was detected in silico.
The possibility of a RNA fold-back structure was then investigated for each of the 204 remaining candidate genes. In known cases of RNA editing, the RNA fold-back structure usually involves the exonic sequence immediately surrounding the edited adenosine and an editing site complementary sequence (ECS), which is often located in the downstream intron in mammalian targets. For fold-back analysis we used the MFOLD program (Zuker 2003) in the batch mode, which allows for the folding of up to 800 nt of RNA sequence. Initially, 700 nt upstream of and 100 nt downstream from, or 100 nt upstream of and 700 nt downstream from, the predicted editing site were run and the resulting secondary structures were inspected for fold-back substructures that included the immediate region surrounding the predicted site. If no distinctive structure was found, additional sequences were folded using MFOLD by selecting ∼100 nt upstream of and downstream from the predicted site together with up to 600 nt of sequences from another region within the gene and < 2.5 kb upstream of or downstream from the predicted site. This selection is based on known edited genes, where the ECS was found to be located in intronic regions up to a few kilobases away from the exonic editing site. Only those sequences were selected that showed a high degree of conservation according to the PhastCon track of the UCSC human genome browser.
The substructure or substructures covering the sequence region around the predicted editing site that showed the highest double-stranded character for each candidate were then grouped into bins 1–5 based on a calculated structural score (STR).
The structural score STR was obtained from values for three different features determined for each evaluated candidate. First, the base-pairing (BP) score was calculated, which corresponds to the number of base pairs present in the structure multiplied by the fraction of nonbase-paired nucleotides [BP = bp(1-bp/nt)]. The value for this feature reflects the fraction of nucleotides that are base paired in the structure, and also accounts for the total lengths of the structure including base-paired as well as nonbase-paired nucleotides.
Second, the GC content of the base pairs was analyzed (the GC score) by determining the sum of base pair values using a value of 3 for a G/C base pair and a value of 2 for an A/T or a G/T base pair.
Third, a penalty value (IS score) was determined for the length of intervening sequence between the two base-paired regions, as our previous study of intramolecular folding and editing of Alu-element-containing sequences showed that the level of editing decreases with an increasing size of the intervening sequence. The individual IS score bins were: Intervening sequence of >100 nt: penalty reducing score by 10%; >500 nt: 18%; >750 nt: 23%; >1000 nt: 30%; >1250 nt: 38%; >1500 nt: 45%; >1750 nt: 51%; >2000 nt: 60%; and >2500 nt: 80%.
The overall structural score STR follows as:
Candidate structures with a STR score <100 were placed in bin 5; scores between 100 and 300 in bin 4; scores between 300 and 900 in bin 3; scores between 900 and 1800 in bin 2 and scores >1800 in bin 1.
Our scoring of fold-back structures is uniquely tailored to identify folds that are more likely functional in supporting RNA editing and does not simply select the most thermodynamically stable structures. This is, for example, reflected in that the penalty for intervening sequences between the base-pairing regions is based on the known and characterized editing targets
For each of the molecular features analyzed (identity of −1 and +1 nucleotide; conservation; structure, as described in the Results section) we then computed a comparative score. For each feature I with a value xi we calculated a log-odds score:
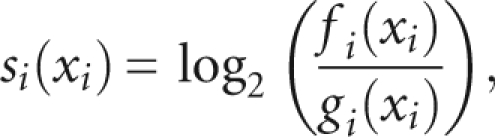 |
based on a relative entropy approach (Lim et al. 2003). fi(xi) corresponds to the frequency of the parameter value xi in the reference set of known edited exons, and gi(xi) being the frequency of xi among the sample set of all pre-mRNA sequences in our prefiltered database. Finally, a combined score for each candidate editing site is derived from the sum of the log-odds scores for each analyzed parameter:
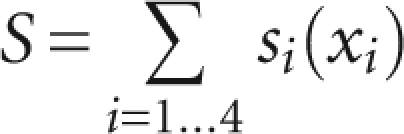 |
RNA editing analysis
For experimental validation, human brain total RNA and gDNA isolated from the same specimen (Biochain) were used and processed using standard protocols for reverse transcription and PCR (see Supplemental Table S1 for primer sequences used). For candidate genes that are tissue specifically expressed outside of the brain, tissue-specific cDNA and gDNA pairs were analyzed. Otherwise, brain cDNA was used for initial analysis even if the transcript for the gene in the database that carries the G was derived from another tissue because generally, brain tissue has been shown to express the highest levels of editing activity (Athanasiadis et al. 2004). Gene-specific fragments of cDNA as well as genomic regions were amplified by PCR and subjected to dideoxy sequencing as described previously (Athanasiadis et al. 2004). Initial analysis for editing at the predicted positions was done by inspecting the sequence traces of PCR products for double peaks with the ratio of the peak heights giving a first indication of potential editing levels. For SRp25 cDNA the PCR amplicon was subcloned and ligated into pBluescript vector (Stratagene). Individual recombinant clones were isolated and the purified plasmid DNAs were sequenced (Geneway).
Statistical analysis
To determine if the chance of finding a novel recoding editing site within the various scoring groups was significantly different from random chance we made use of Fisher's exact test.
SUPPLEMENTAL DATA
Supplemental material can be found at http://www.rnajournal.org.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.816908.
REFERENCES
- Ahmed, S., Yamamoto, K., Sato, Y., Ogawa, T., Herrmann, A., Higashi, S., Miyazaki, K. Proteolytic processing of IGFBP-related protein-1 (TAF/angiomodulin/mac25) modulates its biological activity. Biochem. Biophys. Res. Commun. 2003;310:612–618. doi: 10.1016/j.bbrc.2003.09.058. [DOI] [PubMed] [Google Scholar]
- Akaogi, K., Okabe, Y., Sato, J., Nagashima, Y., Yasumitsu, H., Sugahara, K., Miyazaki, K. Specific accumulation of tumor-derived adhesion factor in tumor blood vessels and in capillary tube-like structures of cultured vascular endothelial cells. Proc. Natl. Acad. Sci. 1996;93:8384–8389. doi: 10.1073/pnas.93.16.8384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athanasiadis, A., Rich, A., Maas, S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2004;2:e391. doi: 10.1371/journal.pbio.0020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass, B.L. RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 2002;71:817–846. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blow, M., Futreal, P.A., Wooster, R., Stratton, M.R. A survey of RNA editing in human brain. Genome Res. 2004;14:2379–2387. doi: 10.1101/gr.2951204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buetow, K.H., Edmonson, M.N., Cassidy, A.B. Reliable identification of large numbers of candidate SNPs from public EST data. Nat. Genet. 1999;21:323–325. doi: 10.1038/6851. [DOI] [PubMed] [Google Scholar]
- Clutterbuck, D.R., Leroy, A., O'Connell, M.A., Semple, C.A. A bioinformatic screen for novel A-I RNA editing sites reveals re-coding editing in BC10. Bioinformatics. 2005;21:2590–2595. doi: 10.1093/bioinformatics/bti411. 00: 000–000. [DOI] [PubMed] [Google Scholar]
- Collet, C., Candy, J. How many insulin-like growth factor binding proteins? Mol. Cell. Endocrinol. 1998;139:1–6. doi: 10.1016/s0303-7207(98)00078-1. [DOI] [PubMed] [Google Scholar]
- Eisenberg, E., Adamsky, K., Cohen, L., Amariglio, N., Hirshberg, A., Rechavi, G., Levanon, E.Y. Identification of RNA editing sites in the SNP database. Nucleic Acids Res. 2005;33:4612–4617. doi: 10.1093/nar/gki771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gommans, W.M., Dupuis, D.E., McCane, J.E., Tatalias, N.E., Maas, S. Diversifying exon code through A-to-I RNA editing. In: Smith H., editor. DNA RNA editing. Wiley & Sons, Inc; New York: 2008. pp. 3–30. [Google Scholar]
- Hoopengardner, B. Adenosine-to-inosine RNA editing: Perspectives and predictions. Mini Rev. Med. Chem. 2006;6:1213–1216. doi: 10.2174/138955706778742812. [DOI] [PubMed] [Google Scholar]
- Irizarry, K., Kustanovich, V., Li, C., Brown, N., Nelson, S., Wong, W., Lee, C.J. Genome-wide analysis of single-nucleotide polymorphisms in human expressed sequences. Nat. Genet. 2000;26:233–236. doi: 10.1038/79981. [DOI] [PubMed] [Google Scholar]
- Kim, D.D., Kim, T.T., Walsh, T., Kobayashi, Y., Matise, T.C., Buyske, S., Gabriel, A. Widespread RNA editing of embedded alu elements in the human transcriptome. Genome Res. 2004;14:1719–1725. doi: 10.1101/gr.2855504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn, R.M., Karolchik, D., Zweig, A.S., Trumbower, H., Thomas, D.J., Thakkapallayil, A., Sugnet, C.W., Stanke, M., Smith, K.E., Siepel, A., et al. The UCSC genome browser database: Update 2007. Nucleic Acids Res. 2007;35:D668–D673. doi: 10.1093/nar/gkl928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev-Maor, G., Sorek, R., Levanon, E.Y., Paz, N., Eisenberg, E., Ast, G. RNA-editing-mediated exon evolution. Genome Biol. 2007;8:R29. doi: 10.1186/gb-2007-8-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levanon, E.Y., Eisenberg, E., Yelin, R., Nemzer, S., Hallegger, M., Shemesh, R., Fligelman, Z.Y., Shoshan, A., Pollock, S.R., Sztybel, D., et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat. Biotechnol. 2004;22:1001–1005. doi: 10.1038/nbt996. [DOI] [PubMed] [Google Scholar]
- Levanon, E.Y., Hallegger, M., Kinar, Y., Shemesh, R., Djinovic-Carugo, K., Rechavi, G., Jantsch, M.F., Eisenberg, E. Evolutionarily conserved human targets of adenosine to inosine RNA editing. Nucleic Acids Res. 2005a;33:1162–1168. doi: 10.1093/nar/gki239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levanon, E.Y., Hallegger, M., Kinar, Y., Shemesh, R., Djinovic-Carugo, K., Rechavi, G., Jantsch, M.F., Eisenberg, E. Evolutionarily conserved human targets of adenosine to inosine RNA editing. Nucleic Acids Res. 2005b;33:1162–1168. doi: 10.1093/nar/gki239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, L.P., Lau, N.C., Weinstein, E.G., Abdelhakim, A., Yekta, S., Rhoades, M.W., Burge, C.B., Bartel, D.P. The microRNAs of Caenorhabditis elegans . Genes & Dev. 2003;17:991–1008. doi: 10.1101/gad.1074403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas, S., Kawahara, Y., Tamburro, K.M., Nishikura, K. A-to-I RNA editing and human disease. RNA Biol. 2006;3:1–9. doi: 10.4161/rna.3.1.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse, D.P. Identification of substrates for adenosine deaminases that act on RNA. Methods Mol. Biol. 2004;265:199–218. doi: 10.1385/1-59259-775-0:199. [DOI] [PubMed] [Google Scholar]
- Morse, D.P., Bass, B.L. Detection of inosine in messenger RNA by inosine-specific cleavage. Biochemistry. 1997;36:8429–8434. doi: 10.1021/bi9709607. [DOI] [PubMed] [Google Scholar]
- Morse, D.P., Aruscavage, P.J., Bass, B.L. RNA hairpins in noncoding regions of human brain and Caenorhabditis elegans mRNA are edited by adenosine deaminases that act on RNA. Proc. Natl. Acad. Sci. 2002;99:7906–7911. doi: 10.1073/pnas.112704299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutaguchi, K., Yasumoto, H., Mita, K., Matsubara, A., Shiina, H., Igawa, M., Dahiya, R., Usui, T. Restoration of insulin-like growth factor binding protein-related protein 1 has a tumor-suppressive activity through induction of apoptosis in human prostate cancer. Cancer Res. 2003;63:7717–7723. [PubMed] [Google Scholar]
- Nishikura, K. Editor meets silencer: Crosstalk between RNA editing and RNA interference. Nat. Rev. Mol. Cell Biol. 2006;7:919–931. doi: 10.1038/nrm2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlson, J., Enstero, M., Sjoberg, B.M., Ohman, M. A method to find tissue-specific novel sites of selective adenosine deamination. Nucleic Acids Res. 2005;33:e167. doi: 10.1093/nar/gni169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlson, J., Pedersen, J.S., Haussler, D., Ohman, M. Editing modifies the GABA(A) receptor subunit alpha3. RNA. 2007;13:698–703. doi: 10.1261/rna.349107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasanth, K.V., Prasanth, S.G., Xuan, Z., Hearn, S., Freier, S.M., Bennett, C.F., Zhang, M.Q., Spector, D.L. Regulating gene expression through RNA nuclear retention. Cell. 2005;123:249–263. doi: 10.1016/j.cell.2005.08.033. [DOI] [PubMed] [Google Scholar]
- Rodriguez, M.S., Desterro, J.M., Lain, S., Midgley, C.A., Lane, D.P., Hay, R.T. SUMO-1 modification activates the transcriptional response of p53. EMBO J. 1999;18:6455–6461. doi: 10.1093/emboj/18.22.6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson, D.A., Wang, M., Matunis, M.J. The small ubiquitin-like modifier-1 (SUMO-1) consensus sequence mediates Ubc9 binding and is essential for SUMO-1 modification. J. Biol. Chem. 2001;276:21664–21669. doi: 10.1074/jbc.M100006200. [DOI] [PubMed] [Google Scholar]
- Sasahara, K., Yamaoka, T., Moritani, M., Tanaka, M., Iwahana, H., Yoshimoto, K., Miyagawa, J., Kuroda, Y., Itakura, M. Molecular cloning and expression analysis of a putative nuclear protein, SR-25. Biochem. Biophys. Res. Commun. 2000;269:444–450. doi: 10.1006/bbrc.2000.2301. [DOI] [PubMed] [Google Scholar]
- Sato, J., Hasegawa, S., Akaogi, K., Yasumitsu, H., Yamada, S., Sugahara, K., Miyazaki, K. Identification of cell-binding site of angiomodulin (AGM/TAF/Mac25) that interacts with heparan sulfates on cell surface. J. Cell. Biochem. 1999;75:187–195. [PubMed] [Google Scholar]
- Sherry, S.T., Ward, M.H., Kholodov, M., Baker, J., Phan, L., Smigielski, E.M., Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, X., Kachirskaia, I., Yamaguchi, H., West, L.E., Wen, H., Wang, E.W., Dutta, S., Appella, E., Gozani, O. Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol. Cell. 2007;27:636–646. doi: 10.1016/j.molcel.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel, A., Bejerano, G., Pedersen, J.S., Hinrichs, A.S., Hou, M., Rosenbloom, K., Clawson, H., Spieth, J., Hillier, L.W., Richards, S., et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnov, D.A., Foulk, B.W., Doyle, G.V., Connelly, M.C., Terstappen, L.W., O'Hara, S.M. Global gene expression profiling of circulating endothelial cells in patients with metastatic carcinomas. Cancer Res. 2006;66:2918–2922. doi: 10.1158/0008-5472.CAN-05-4003. [DOI] [PubMed] [Google Scholar]
- Sprenger, C.C., Vail, M.E., Evans, K., Simurdak, J., Plymate, S.R. Over-expression of insulin-like growth factor binding protein-related protein-1(IGFBP-rP1/mac25) in the M12 prostate cancer cell line alters tumor growth by a delay in G1 and cyclin A associated apoptosis. Oncogene. 2002;21:140–147. doi: 10.1038/sj.onc.1205021. [DOI] [PubMed] [Google Scholar]
- St Croix, B., Rago, C., Velculescu, V., Traverso, G., Romans, K.E., Montgomery, E., Lal, A., Riggins, G.J., Lengauer, C., Vogelstein, B., et al. Genes expressed in human tumor endothelium. Science. 2000;289:1197–1202. doi: 10.1126/science.289.5482.1197. [DOI] [PubMed] [Google Scholar]
- Taylor, J.G., Choi, E.H., Foster, C.B., Chanock, S.J. Using genetic variation to study human disease. Trends Mol. Med. 2001;7:507–512. doi: 10.1016/s1471-4914(01)02183-9. [DOI] [PubMed] [Google Scholar]
- van Beijnum, J.R., Dings, R.P., van der Linden, E., Zwaans, B.M., Ramaekers, F.C., Mayo, K.H., Griffioen, A.W. Gene expression of tumor angiogenesis dissected: Specific targeting of colon cancer angiogenic vasculature. Blood. 2006;108:2339–2348. doi: 10.1182/blood-2006-02-004291. [DOI] [PubMed] [Google Scholar]
- Wilson, H.M., Birnbaum, R.S., Poot, M., Quinn, L.S., Swisshelm, K. Insulin-like growth factor binding protein-related protein 1 inhibits proliferation of MCF-7 breast cancer cells via a senescence-like mechanism. Cell Growth Differ. 2002;13:205–213. [PubMed] [Google Scholar]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker, M., Mathews, D.H., Turner, D.H. Algorithms and thermodynamics for RNA secondary structure prediction: A practical guide in rna biochemistry and biotechnology. Kluwer Academic Publishers; New York: 1999. [Google Scholar]