

Abstract
Background
Activation of the dopaminergic (DA) neurons of the ventral tegmental area (VTA) by ethanol has been implicated in its rewarding and reinforcing effects. At most central synapses, ethanol generally increases inhibitory synaptic transmission; however, no studies have explored the effect of acute ethanol on GABAergic transmission in the VTA.
Methods
Whole-cell patch clamp recordings of inhibitory postsynaptic currents (IPSCs) from VTA-DA neurons in midbrain slices from young rats.
Results
Acute exposure of VTA-DA neurons to ethanol (25 to 50 mM) robustly enhanced GABAergic spontaneous and miniature IPSC frequency while inducing a slight enhancement of spontaneous IPSC (sIPSC) amplitude. Ethanol (50 mM) enhanced paired-pulse depression of evoked IPSCs, further suggesting enhanced GABA release onto VTA-DA neurons. The frequency of sIPSCs was suppressed by the GABAB agonist, baclofen (1.25 μM) and enhanced by the antagonist, SCH50911 (20 μM); however, neither appeared to modulate or occlude the effects of ethanol on sIPSC frequency.
Conclusions
The present results indicate that ethanol increases postsynaptic GABAA receptor sensitivity, enhances action potential-independent GABA release onto VTA-DA neurons, and that this latter effect is independent of GABAB auto-receptor inhibition of GABA release.
Keywords: Reward, Mesolimbic, Dependence, Accumbal, Electrophysiology
DOPAMINERGIC (DA) NEURONS of the ventral tegmental area (VTA) constitute the central locus of the mesocorticolimbic dopamine system and send projections to the nucleus accumbens (NAc), prefrontal cortex, basolateral amygdala, and to a variety of other corticolimbic structures (Albanese and Minciacchi, 1983; Oades and Halliday, 1987). Substantial evidence suggests that these VTA-DA projections contribute to certain aspects of drug reinforcement and/or reward (Appel et al., 2004; Gatto et al., 1994; Koob et al., 1998; Robbins and Everitt, 1996; Schultz, 2002; Wise, 1996). Indeed, ethanol has been shown to increase the firing rate of DA neurons in the VTA both in vitro (Brodie et al., 1990) and in vivo (Gessa et al., 1985). Ethanol may be acting through direct effects on DA neuron excitability (Brodie and Appel, 1998; Brodie et al., 1999) that are at least partly attributable to an increase in the hyperpolarization-activated cation current (Ih) (Okamoto et al., 2006).
In addition to the principal DA neurons, the VTA also contains a major population of GABAergic neurons (Grace and Onn, 1989; Johnson and North, 1992b; Korotkova et al., 2003). The GABAergic neurons function importantly as local interneurons that regulate the firing of the DA neurons via tonic inhibition (Bonci and Malenka, 1999; Johnson and North, 1992a). The effect of acute ethanol on GABAergic transmission has been extensively explored in a variety of brain regions (Carta et al., 2004; Nie et al., 2004; Roberto et al., 2003; Wan et al., 1996; Weiner and Valenzuela, 2006); however, remarkably, virtually nothing is known about the direct effects of acute ethanol on GABAergic transmission in the VTA. One electrophysiological study measured prolonged enhancement of GABAergic transmission in mice 24 hours after a single in vivo exposure to ethanol (Melis et al., 2002).
Thus, while excitatory actions of ethanol in the VTA have been well-documented, the possibility that conventional stimulatory effects of ethanol on GABAergic transmission that also may modulate VTA-DA neuronal excitability has not been investigated. To determine whether ethanol modulates GABAergic transmission in this brain region, we studied GABAA-mediated inhibitory postsynaptic currents (IPSCs) recorded from VTA-DA neurons using conventional whole-cell patch clamp techniques in the presence of varying concentrations of ethanol.
Materials and Methods
Slice Preparation
All experiments were carried out in accordance with NIH guidelines. Slices used in this study were prepared from Sprague-Dawley rats of both sexes (postnatal day 21 to 30). Rats were anesthetized with halothane, decapitated, and the brain was rapidly removed and placed in ice-cold, oxygenated artificial cerebrospinal fluid (aCSF) containing (in mM): 120 NaCl, 25 NaHCO3, 3.3 KCl, 1.23 NaH2PO4, and 10 dextrose, 95%O2/5%CO2 (all chemicals obtained from Sigma, St Louis, MO). Cutting aCSF also contained 2.4 mM MgSO4 and 1.8 mM CaCl2. Horizontal midbrain slices (200 to 220 μm) were prepared using a vibratome (VT1000S; Leica, Nussloch, Germany). The slices were then maintained at 32°C in cutting aCSF buffer for a minimum of 60 minutes before electrophysiological recordings were performed.
Electrophysiological Recordings
Slices were transferred to a recording chamber and perfused with oxygenated aCSF (30 to 32°C) at a flow rate of ∼2 ml/min. Recording aCSF also contained 0.9 mM MgSO4 and 2 mM CaCl2. DA neurons were identified by the presence of a large hyperpolarization-induced Ih current (>200 pA) that was assayed immediately following break-in by application of a 1.5-s hyperpolarizing step from –60 to –110 mV (Johnson and North, 1992b). Only DA neurons identified in this manner from the VTA were used for this study. Recording electrodes were made from thin-walled borosilicate glass (TW 150F-4; WPI, Sarasota, FL; 1.5 to 2.5 MΩ). Recording electrodes for whole-cell recordings contained (in mM): 135 KCl, 12 NaCl, 0.5 EGTA, 10 HE-PES, 2 Mg-ATP, and 0.3 Tris-GTP, pH 7.3 with KOH (all chemicals obtained from Sigma). Access resistance was partially compensated and monitored throughout all experiments. Data were collected by an Axon Instruments Model 200B amplifier filtered at 1 kHz and digitized at 10 to 20 kHz with a Digidata interface using pClamp v9.2 (Axon Instruments, Foster City, CA).
Kynurenic acid (1 mM) was used in all whole-cell voltage clamp recordings to inhibit α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate and N-methyl-d-aspartate receptor-mediated currents. All GABA IPSCs were inward at a holding potential of –60 mV and completely blocked by application of picrotoxin (50 μM). Miniature IPCSs (mIPSCs) were recorded in the presence of 0.5 μM tetrodotoxin (TTX) to block action potentials. Spontaneous IPSCs (sIPSCs) were recorded in the absence of TTX. Following break-in and after a stable 5- to 10-minute baseline (control) recording, drugs were bath-applied through the aCSF and a continuous 5- to 15-minute recording was made to detect any changes in m/sIPSC frequency and amplitude. The number of neurons used per each treatment condition is represented as n with only one neuron used per slice. A 5-minute drug wash-on preceded the start of data collection in each treatment condition. A 10- to 15-minute washout period followed drug application. SCH50911 was obtained from Tocris Bioscience, Ellisville, MO. Baclofen, kynurenic acid, and picrotoxin were obtained from Sigma. TTX was obtained from Alomone Labs, Jerusalem, Israel.
A monopolar tungsten stimulating electrode was placed 50 to 100 μm rostral of the recording electrode for evoked IPSC recordings. Constant-current pulses (100-μs duration, 15 to 40 pA amplitude) were applied through a stimulus isolation unit driven by an analog stimulator (WPI). Paired-stimuli were delivered every 20 or 30 seconds with an interstimulus interval of 70 milliseconds.
Data Analysis
For m/sIPSC recordings, quantal events were detected (60 to 90 sweeps each condition, 5 seconds/sweep) and analyzed using Clamp-fit (pClamp v9.2 software; Axon Instruments). Access resistance for most experiments ranged from 6 to 20 MΩ and never exceeded 25 MΩ. To maintain the fidelity of the recordings, experiments where access resistance changed (>20% at anytime during the experiment) were not included in data analyses. To accurately determine the m/sIPSC amplitude, only those events that were >10 pA were accepted for analysis to maintain a high signal-to-noise ratio. Treatment and washout groups were normalized to the baseline (control) frequency or amplitude and represented as a percentage of the control. For evoked paired stimuli, the ratio between the second and the first IPSCs was calculated and averaged for a 10-minute baseline and a minimum 10-minute treatment condition. Averaged values for all datasets are expressed as mean ± SEM and were compared statistically using 2-tailed paired or unpaired student's t-test or ANOVA/Dunnett C or Tukey HSD posthoc where mentioned. Significant differences were considered as *p < 0.05 and **p < 0.01.
RESULTS
Baseline Parameters of sIPSCs on VTA Neurons
Data in this study were gathered using whole-cell voltage-clamp from VTA neurons that were identified as dopaminergic by the presence of a large Ih current (see Materials and Methods). The GABAA receptor blocker picrotoxin (50 μM) completely blocked the sIPSCs, indicating that they were GABA-mediated (data not shown, n = 3). The baseline frequency and amplitude of sIPSCs recorded in control conditions from all cells tested across every treatment group were 3.89 ± 0.30 Hz and 52.01 ± 1.31 pA, respectively (n = 52).
Ethanol Enhances GABAergic Transmission in the VTA
To measure the effect of varying concentrations of ethanol (15 to 50 mM; legal intoxication is ∼17 mM), GABAA-mediated sIPSCs were recorded across this range of ethanol concentrations in 2 separate experiments in separate groups of slices: 15 and then 25 mM (Fig. 1), and 25 and then 50 mM (Fig. 2) of ethanol. Thus, the inherent difficulties of maintaining very long-lasting whole-cell recordings for these concentration–response experiments were minimized. After application of ethanol (15 mM), no significant increases in sIPSC frequency (Fig. 1A,B) or amplitude (Fig. 1A,C) were observed. However, subsequent application of ethanol (25 mM) produced a significant increase in sIPSC frequency (Fig. 1A,B) but not amplitude (Fig. 1A,C).
Fig. 1.
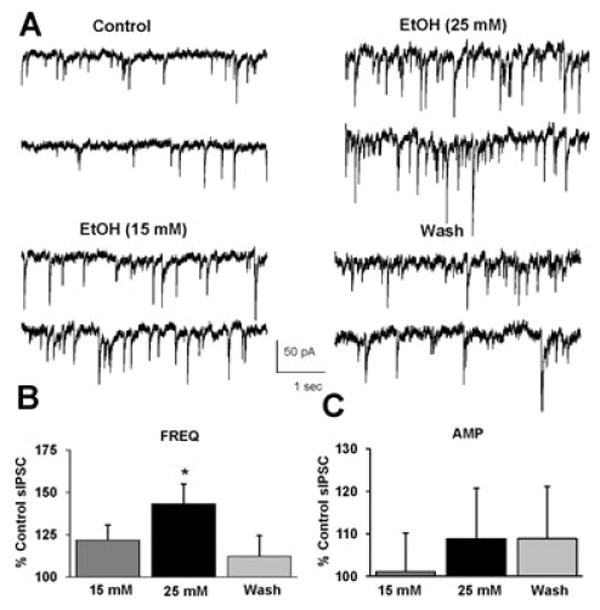
Ethanol (25 mM) potentiates sIPSC frequency. (A) sIPSCs recorded from a VTA-DA neuron under control conditions, in the presence of 15 and 25 mM ethanol, and after a washout. (B) A bar graph representing the percent change ± SEM above control sIPSC frequency for the conditions shown in (A). Event frequency under control conditions was 4.62 ± 0.75 Hz. (C) A bar graph representing the percent change ± SEM above control sIPSC amplitude for the conditions shown in (A). Event amplitude under control conditions was 52.82 ± 2.28 pA. (n = 7, *p < 0.05 by ANOVA/Dunnett C posthoc different from control).
Fig. 2.
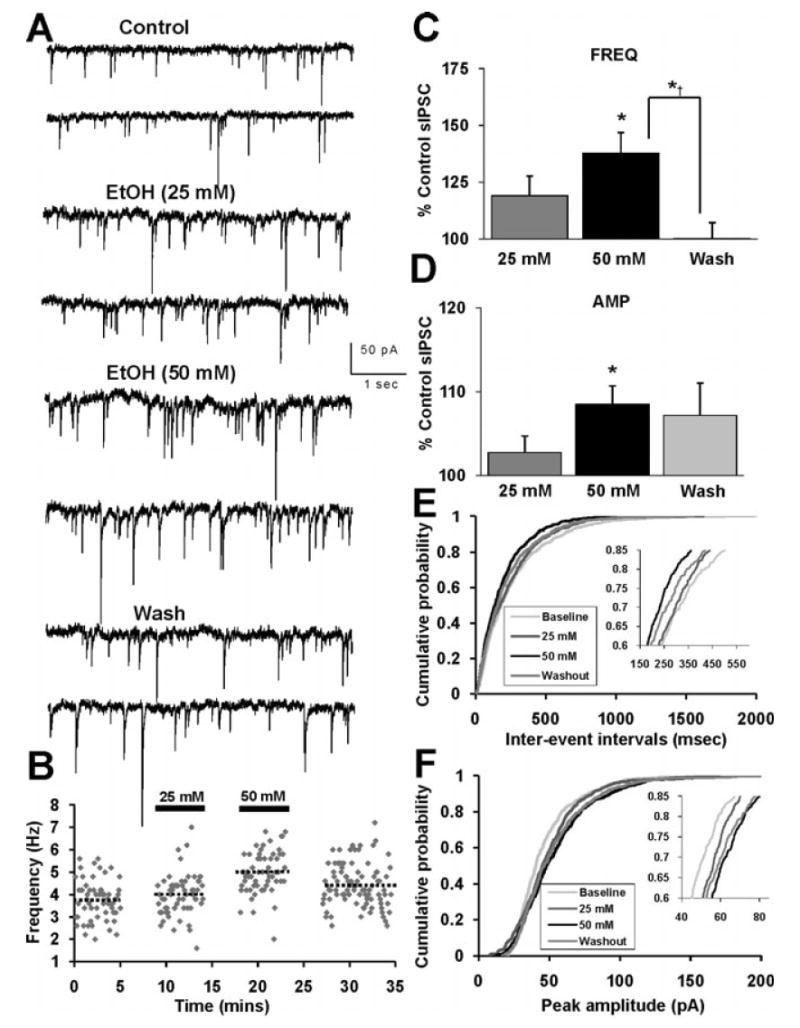
Ethanol (50 mM) potentiates sIPSC frequency and amplitude. (A) sIPSCs recorded from a VTA-DA neuron under control conditions, in the presence of 25 and 50 mM ethanol, and after a washout. (B) Time course for a sample representative neuron under conditions shown in (A). The dotted lines represent the average frequency under each condition for that neuron. (C) A bar graph representing the percent change ± SEM above control sIPSC frequency for the conditions shown in (A). Event frequency under control conditions was 5.48 ± 0.93 Hz. (D) A bar graph representing the percent change ± SEM above control sIPSC amplitude for the conditions shown in (A). Event amplitude under control conditions was 54.49 ± 5.56 pA. (E) Cumulative probability histogram of sIPSC inter-event intervals for epochs from same neuron shown in (B). (F) Cumulative probability histogram of sIPSC event amplitudes from the same neuron shown in (B), (E). (n = 8, *p < 0.05 by ANOVA/Dunnett C posthoc different from control, *†p < 0.05 by ANOVA/Dunnett C posthoc different from wash).
In the separate cohort of recordings, where 25 mM ethanol was initially applied, interesting differences in sIPSC event frequency and amplitude were observed (Fig. 2) compared to the effect presented in the previous 15 to 25 mM experiment. In this instance, application of ethanol (25 mM) did not result in a significant increase in either sIPSC frequency or amplitude (Fig. 2C,D). However, subsequent application of ethanol (50 mM) to the same neuron produced a significant increase in sIPSC frequency (Fig. 2C) and amplitude (Fig. 2D). The distributions of the sIPSC inter-event intervals and amplitude for control, ethanol (25, 50 mM) and wash conditions are shown in a sample representative neuron (Fig. 2E,F). There was a progressive leftward shift in the inter-event interval distribution, indicative of increased GABA release that at least partially reversed upon wash (Fig. 2E). Additionally, there was a progressive rightward shift in peak amplitude distribution, indicative of increased postsynaptic GABA sensitivity, which partially reversed upon wash (Fig. 2F). The cumulative changes in sIPSC frequency and amplitude recorded at each ethanol concentration were pooled from all experiments [15 mM (n = 7), 25 mM (n = 22), and 50 mM ethanol (n = 11)] (Fig. 3). Application of ethanol (25 and 50 mM) produced a significant increase above control in frequency (Fig. 3A) and amplitude (Fig. 3B). Ethanol at 15 mM did not produce a significant increase in either frequency or amplitude. No concentration above 50 mM was tested. It is conceivable that the observed ethanol-induced increase in sIPSC amplitude, although slight, may result in an apparent increase in sIPSC frequency due to the detections of small events under control conditions which were below detection threshold (10 pA) and thus not previously detected. Therefore, we calculated the percent enhancement induced by ethanol of the number of events for a defined recording duration binned across all amplitude ranges. As shown in Fig. 3C, from a neuron which showed a maximal increase in sIPSC frequency (∼86% above control) ethanol increases the number of events relatively equivalently across all amplitude bins.
Fig. 3.
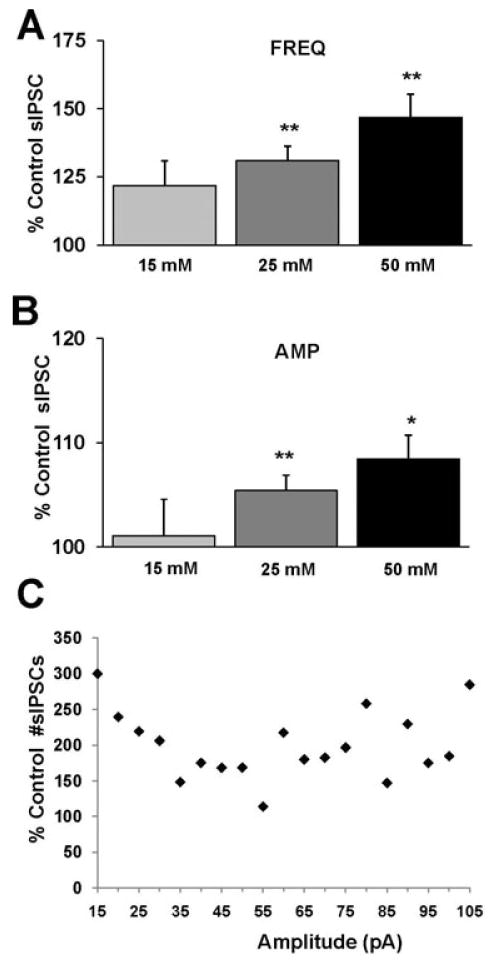
Cumulative results show ethanol-induced potentiation of sIPSC frequency and amplitude at varying concentrations. (A) A bar graph representing the percent change ± SEM above control sIPSC frequency in the presence of 15, 25, and 50 mM ethanol. Event frequency under control conditions was 4.92 ± 0.44 Hz. (B) A bar graph representing the percent change ± SEM above control sIPSC amplitude for the conditions shown in (A). Event amplitude under control conditions was 52.92 ± 2.06 pA. (n = 7 for 15 mM, n = 22 for 25 mM, n = 11 for 50 mM; *p < 0.05, **p < 0.01 by ANOVA/Dunnett C posthoc different from control) (C) A sample cell was analyzed that showed a maximal ethanol-induced (50 mM) enhancement in sIPSC frequency (∼86% above control) to determine the percent change in the number of events at their respective amplitude distribution. The events were binned into 5 pA groups, for example, the point at 30 pA represents all events >25 and <30 pA. All events at 105 pA represent those events greater than 100 pA in size. The total number of events was 1,148 and 2,135 in control and ethanol conditions, respectively, taken from a time course of 5 minutes for each condition (note: the 300% change in events <15 pA represents an increase from only 4 to 12 events).
To investigate further the ethanol-mediated enhancement in presynaptic release as demonstrated in the increase in sIPSC frequency, we measured the paired-pulse ratio of evoked IPSCs (as described in Materials and Methods) before and after application of ethanol (50 mM). In 5 of 6 VTA-DA neurons, application of 50 mM ethanol resulted in a significant enhancement of paired-pulse depression (PPD), indicative of enhanced GABA release (Fig. 4C). Additionally, we measured the decay time of the evoked IPSCs and found that ethanol prolonged the decay time constant, τ, indicative of an enhancement, although slight, in postsynaptic GABAA receptor sensitivity (Fig. 4D).
Fig. 4.
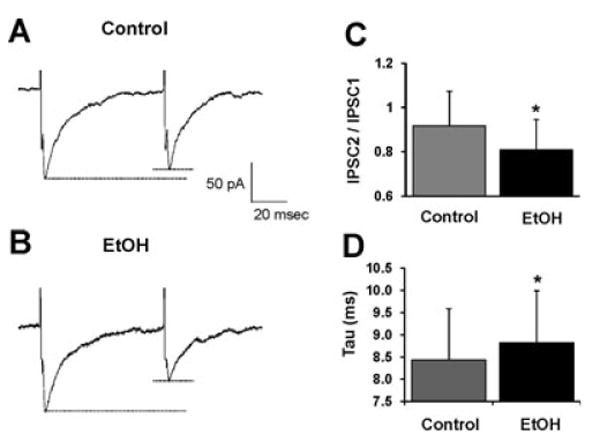
Ethanol enhances paired-pulse depression. (A), (B) Sample evoked IPSC recordings from a VTA-DA neuron prior to (A), and after exposure to ethanol (50 mM) (B). Each trace represents an averaged trace from 8 individual traces over a continuous 3.5-minute time period for each condition from a representative neuron. Both recordings exhibit paired-pulse depression (PPD); however after exposure to ethanol, the PPD is enhanced. (C) A bar graph representing the paired-pulse ratio (defined as IPS-C2/IPSC1) under control conditions and in the presence of ethanol (50 mM). (D) A bar graph representing the decay time as measured by the time constant, τ (milliseconds), under control conditions and in the presence of ethanol (50 mM). (n = 5 of 6, *p < 0.05 by a paired student's t-test different from control).
Ethanol Potentiation of sIPSCs Does Not Involve GABAB Receptors
Activation of GABAB receptors on midbrain GABAergic interneurons inhibits GABA release from these neurons (Bonci and Malenka, 1999; Rohrbacher et al., 1997), and inhibition of GABAB receptors on hippocampal interneurons can increase GABA release (Mott and Lewis, 1991). Therefore, to test the possibility that ethanol may act via presynaptic GABAB receptors to increase GABA release, we recorded sIPSCs from VTA-DA neurons in the presence of either the GABAB agonist, baclofen, or the antagonist, SCH50911. In separate experiments, sIPSCs were recorded from VTA-DA neurons under control conditions followed by bath application of either baclofen (1.25 μM) or SCH50911 (20 μM), followed again by co-application of baclofen or SCH50911 with ethanol (50 mM). Application of baclofen alone reversibly decreased sIPSC frequency (Fig. 5A,B) but not amplitude (Fig. 5A,C). Subsequent co-application of ethanol and baclofen resulted in approximately a 30% increase in frequency above the baclofen-induced decrease with no significant change in amplitude. Application of SCH50911 (20 μM) significantly increased sIPSC frequency (Fig. 6A,B) with no significant change in amplitude (Fig. 6A,C) with respect to control. Subsequent co-application of ethanol and SCH50911 resulted in a 23% increase in frequency above SCH50911 alone that was reversible upon washout, with no apparent change in amplitude. Therefore, the results displayed in Figs. 5 and 6 suggest that the ethanol-induced potentiation in sIPSC frequency is not affected by addition of a GABAB receptor agonist or antagonist.
Fig. 5.
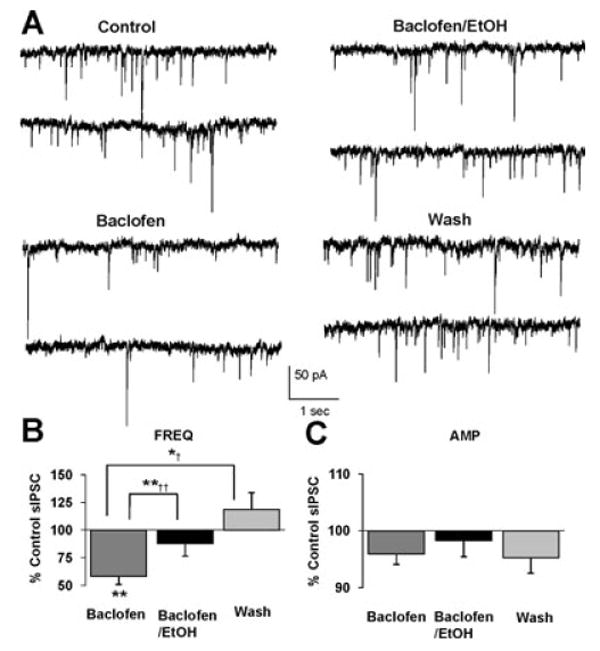
Baclofen does not inhibit ethanol-induced potentiation of sIPSC frequency. (A) sIPSCs recorded from a VTA-DA neuron under control conditions, in the presence of 1.25 μM baclofen, 50 mM ethanol with 1.25 μM baclofen, and after a washout. (B) A bar graph representing the percent change ± SEM above control sIPSC frequency for the conditions shown in (A). Event frequency under control conditions was 2.36 ± 0.23 Hz. (C) A bar graph representing the percent change ± SEM above control sIPSC amplitude for the conditions shown in (A). Event amplitude under control conditions was 49.48 ± 3.41 pA. (n = 6, **p < 0.01 by ANOVA/Dunnett C posthoc different from control, *†p < 0.05 by ANOVA/Dunnett C posthoc different from baclofen alone, **††p < 0.01 by a paired student's t-test different from baclofen alone).
Fig. 6.
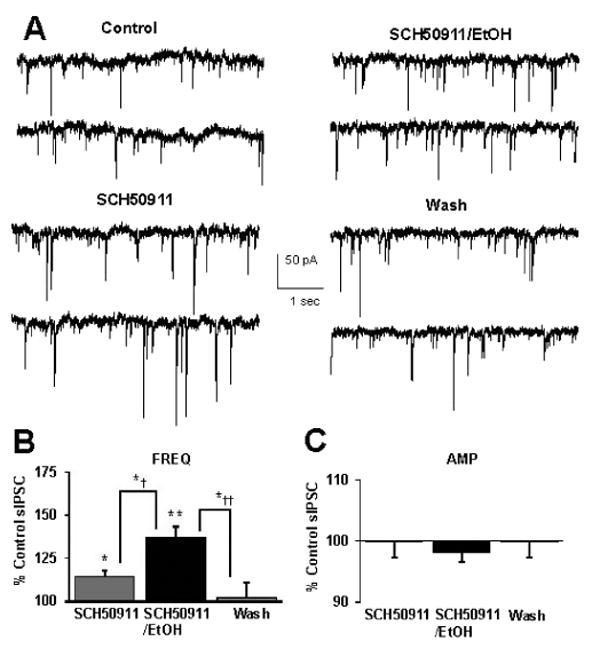
SCH50911 does not occlude ethanol-induced potentiation of sIPSC frequency. (A) sIPSCs recorded from a VTA-DA neuron under control conditions, in the presence of 20 μM SCH50911, 50 mM ethanol with 20 μM SCH50911, and after a washout. (B) A bar graph representing the percent change ± SEM above control sIPSC frequency for the conditions shown in (A). Event frequency under control conditions was 1.89 ± 0.40 Hz. (C) A bar graph representing the percent change ± SEM above control sIPSC amplitude for the conditions shown in (A). Event amplitude under control conditions was 51.58 ± 2.24 pA. (n = 6, *p < 0.05 by ANOVA/Tukey HSD posthoc different from control, **p < 0.01 by ANOVA/Tukey HSD posthoc different from control, *†p < 0.05 by ANOVA/Tukey HSD posthoc and by paired student's t-test different from SCH50911 alone, *††p < 0.05 by ANOVA/Tukey HSD posthoc different from wash).
Ethanol Potentiates GABA Release in the VTA
To further discern whether ethanol potentiates the frequency of spontaneous IPSCs by directly enhancing GABA release from presynaptic terminals or increasing the excitability of GABAergic interneurons, we recorded mIPSCs in the presence of TTX to block action potential-mediated release events. Application of ethanol (50 mM) produced a marked enhancement in mIPSC frequency (Fig. 7A,B), but not amplitude (Fig. 7A,C). A time course plot is shown in Fig. 7D to demonstrate the onset of ethanol action. These results support the notion that ethanol is acting presynaptically to enhance GABA release as evidenced by the sIPSC and eIPSC experiments.
Fig. 7.
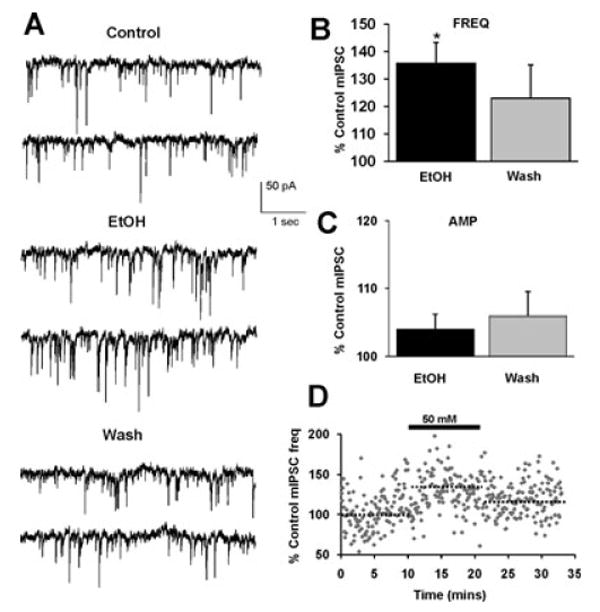
Ethanol (50 mM) potentiates mIPSC frequency. (A) mIPSCs recorded from a VTA-DA neuron under control conditions, in the presence of 50 mM ethanol, and after a washout. (B) A bar graph representing the percent change ± SEM above control mIPSC frequency for the conditions shown in (A). Event frequency under control conditions was 2.57 ± 0.45 Hz. (C) A bar graph representing the percent change ± SEM above control mIPSC amplitude for the conditions shown in (A). Event amplitude under control conditions was 43.13 ± 3.88 pA. (D) Cumulative time course for averaged cells. The dotted lines indicate the averaged value displayed in (B). (n = 11, *p < 0.05 by student's t-test different from control).
Discussion
Our results demonstrate that ethanol enhances GABAergic transmission onto VTA-DA neurons in a concentration-dependent manner in brain slices, and that this effect is independent of presynaptic somatic/axonal excitability and GABAB receptor modulation. To our knowledge, this is the first study to demonstrate a direct and acute enhancement of GABAergic transmission by ethanol in the VTA. Several labs have reported both pre- and postsynaptic enhancement of GABAergic transmission in a variety of other cortical and subcortical brain regions in response to acutely administered ethanol. In rat cerebellar slices, acute ethanol increases the frequency of GABAA-mediated sIPSCs recorded from granule cells (Carta et al., 2004). In the rat central amygdala, a brain region important in anxiety and stress responses, application of 44 mM ethanol facilitated GABAergic transmission via pre- and postsynaptic mechanisms (Roberto et al., 2003). Two recent reviews discuss the strong precedent for acute ethanol to enhance GABA release in the hippocampus, central amygdala, cerebellum, and NAc (Siggins et al., 2005; Weiner and Valenzuela, 2006). Thus, a strong precedent for ethanol modulation of GABAergic transmission exists in a variety of CNS structures.
Other labs have demonstrated a reduction in GABAergic neuron excitability in the presence of ethanol in the VTA (Stobbs et al., 2004; Xiao et al., 2007); however, neither lab explored GABA release specifically. It should be noted that a decrease in somatic excitability does not exclude a separate action of ethanol to enhance terminal excitability. Ethanol inhibits NMDA receptors in VTA-GABAergic neurons (Stobbs et al., 2004) and this inhibition may partially explain the ethanol-induced reduction in GABAergic neuron excitability observed by Xiao et al. Additionally, ethanol may increase GABA release by acting on terminals of GABAergic medium spiny neurons arising from the NAc and not on local VTA interneurons. Nonetheless, given the strong evidence of ethanol facilitation of GABA release in other CNS structures, we are confident that our results demonstrate specific and direct effects on GABA release.
It is notable that in the concentration–response experiments in Fig. 2, we typically did not observe a significant enhancement of GABAergic transmission after the initial exposure to 25 mM ethanol. However, that concentration of ethanol following a prior exposure to 15 mM ethanol did result in significant enhancement of sIPSC frequency but not amplitude. One possible explanation of this effect of successive applications of ethanol is that such intermediate concentrations of ethanol may elicit slowly developing alterations in GABAergic transmission and such a time course of slow onset may provide clues about potential mechanisms. On the other hand, an alternative explanation is that the lower ethanol concentrations may sensitize synapses to subsequent ethanol applications. Furthermore, it is important to note that we are using young rats (21 to 30 days old) in our experiments. In a study comparing the effect of ethanol on sIPSCs and mIPSCs between juvenile and adult rats, ethanol had a greater effect on sIPSCs in adults than juveniles (Li et al., 2006). However, there were no differences in mIPSCs between the adult and juvenile rats. Therefore, it is possible that the variability seen in our sIPSC recordings with these intermediate ethanol concentrations may reflect the slight variability in the ages of the rats used within each group of experiments.
The increase in sIPSC frequency in the presence of ethanol may indicate an increase in terminal GABA release probability or an increase in interneuron somatic/axonal excitability. To discern the former from the latter, we measured the paired-pulse ratio of evoked IPSCs and the frequency of mI-PSCs recorded in the presence of TTX. Under control conditions, we observed PPD; however, acutely applied ethanol further amplified the depression, suggesting an increased probability of GABA release from presynaptic terminals. Additionally, we saw a robust and reliable increase in mIPSC frequency in the presence of ethanol, also suggestive of enhanced GABA release independent of interneuron somatic/axonal excitability. We did not observe a change in mIPSC amplitude in the presence of ethanol. These findings are consistent with other reports in which ethanol increases mIPSC frequency while having no effect on postsynaptic GABAA receptor sensitivity (Kelm et al., 2007; Li et al., 2006; Ming et al., 2006; Zhu and Lovinger, 2006). Although we did observe variable changes in sIPSC amplitude in the presence of ethanol, the lack of change in postsynaptic sensitivity with mIPSCs may be a result of the elimination of larger action-potential induced events by TTX. Taken together, our electrophysiological recordings therefore strongly indicate that ethanol positively modulates GABA release onto VTA-DA neurons.
GABAB receptor activation has been implicated in some actions of ethanol (Ariwodola and Weiner, 2004) and therefore we assessed the potential involvement of GABAB receptors in ethanol modulation of GABA transmission. We observed a decrease in sIPSC frequency mediated by the GABAB receptor agonist, baclofen, alone which confirmed previous findings (Rohrbacher et al., 1997). Conversely, we noted an unexpected increase in frequency following application of the antagonist SCH50911 alone which suggests that tonic GABA release elicits tonic GABAB receptor-mediated auto-inhibition of GABAergic neurons. Application of the GABAB receptor antagonist presumably removes this tonic auto-inhibition, allowing for increased GABA release. Nevertheless, neither activation nor blockade of GABAB receptors appeared to occlude the ethanol enhancement in sIPSC frequency indicating that ethanol modulation of GABA release is independent of GABAB receptors. Interestingly, application of either the GABAB receptor agonist or antagonist completely blocked the enhancement in amplitude. The reason as to the occlusion of the postsynaptic effect is not clear; however, there is evidence of GABAB receptor modulation affecting postsynaptic GABAA receptor ethanol sensitivity (Wu et al., 2005). Nevertheless, the initial degree of sIPSC amplitude enhancement seen with ethanol (50 mM), while significant, is less than 10% over the control sIPSC amplitude value and this effect did not reverse upon ethanol washout. Thus, while we report these small changes in amplitude, we do not feel these constitute convincing evidence to indicate that ethanol modulation of sIPSC amplitude is nearly as significant as its effects on m/sIPSC frequency and thus its modulation of GABA release. In this regard, we can therefore unequivocally state that the robust presynaptic actions of ethanol are not modulated in any manner by activation or inhibition of GABAB receptors.
Multiple labs (including our own—data not shown) have demonstrated an enhancement in VTA-DA neuron firing rate and increased dopamine release into the NAc after exposure to ethanol, and this action is thought to underlie the addictive nature of this drug (Brodie et al., 1990, 1999; Gonzales and Weiss, 1998; Kohl et al., 1998; Okamoto et al., 2006). Our present observations of ethanol enhancement of GABAergic transmission therefore would appear to antagonize such stimulatory actions especially as GABAergic interneurons regulate DA neuron excitability (Johnson and North, 1992a). Furthermore, VTA-DA neurons undergo tonic modulation by GABAergic interneurons as prior studies have shown that GABAA receptor blockade increases extracellular dopamine levels in the NAc (Ikemoto et al., 1997). Thus, the potentiation of DA neuron excitability by ethanol may be self-limiting due to the simultaneous ethanol-induced enhancement in inhibitory drive onto those same neurons. Therefore, there potentially exists a biphasic action of ethanol first to increase VTA-DA excitability directly and secondly to enhance GABAergic inhibition of VTA-DA neurons. This inhibitory action of ethanol may in effect constitute an additional pharmacologic action of significant importance that results in a balance of inhibitory versus excitatory effects to regulate DA neuron firing rate and therefore may constitute an important target for further evaluation. A similar effect has recently been observed in cerebellar Purkinje neurons in which ethanol has dual actions on these neurons to increase GABA release pre-synaptically while simultaneously acting postsynaptically to increase cell firing rate (Ming et al., 2006).
While the increase in GABA release by ethanol is insufficient to prevent the excitatory effect(s) of ethanol on VTA-DA neurons, whether there exists a subset of DA neurons in the VTA that might display differential sensitivity to the excitatory and inhibitory effects of ethanol remains to be determined. This is quite possible considering a recent study that discerned 2 populations of VTA-DA neurons with differential sensitivity to opioid-mediated presynaptic inhibition (Ford et al., 2006). Nevertheless, our data clearly demonstrate that ethanol enhances GABA release in the VTA. The mechanism(s) underlying this action are currently under investigation, and preliminary results suggest that one potential route may involve ethanol enhancement of presynaptic Ca2+ mobilization via a 5-HT2C-dependent mechanism. As ethanol modulation of ventral tegmental DA output function is critically involved in reinforcement and reward, identification of such regulatory pathways is critical to construct an accurate picture of the neuropharmacological actions of ethanol.
Acknowledgments
This work was supported by F.M. Jones and H.L. Bruce Endowed Graduate Fellowship (JWT) and National Institutes of Health Grants RO1 AA 14874 (RAG) and RO1 AA 15167 (RAM).
References
- Albanese A, Minciacchi D. Organization of the ascending projections from the ventral tegmental area: a multiple fluorescent retrograde tracer study in the rat. J Comp Neurol. 1983;216:406–420. doi: 10.1002/cne.902160406. [DOI] [PubMed] [Google Scholar]
- Appel SB, McBride WJ, Diana M, Diamond I, Bonci A, Brodie MS. Ethanol effects on dopaminergic “reward” neurons in the ventral tegmental area and the mesolimbic pathway. Alcohol Clin Exp Res. 2004;28:1768–1778. [Google Scholar]
- Ariwodola OJ, Weiner JL. Ethanol potentiation of GABAergic synaptic transmission may be self-limiting: role of presynaptic GABA(B) receptors. J Neurosci. 2004;24:10679–10686. doi: 10.1523/JNEUROSCI.1768-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonci A, Malenka RC. Properties and plasticity of excitatory synapses on dopaminergic and GABAergic cells in the ventral tegmental area. J Neurosci. 1999;19:3723–3730. doi: 10.1523/JNEUROSCI.19-10-03723.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie MS, Appel SB. The effects of ethanol on dopaminergic neurons of the ventral tegmental area studied with intracellular recording in brain slices. Alcohol Clin Exp Res. 1998;22:236–244. [PubMed] [Google Scholar]
- Brodie MS, Pesold C, Appel SB. Ethanol directly excites dopaminergic ventral tegmental area reward neurons. Alcohol Clin Exp Res. 1999;23:1848–1852. [PubMed] [Google Scholar]
- Brodie MS, Shefner SA, Dunwiddie TV. Ethanol increases the firing rate of dopamine neurons of the rat ventral tegmental area in vitro. Brain Res. 1990;508:65–69. doi: 10.1016/0006-8993(90)91118-z. [DOI] [PubMed] [Google Scholar]
- Carta M, Mameli M, Valenzuela CF. Alcohol enhances GABAergic transmission to cerebellar granule cells via an increase in Golgi cell excitability. J Neurosci. 2004;24:3746–3751. doi: 10.1523/JNEUROSCI.0067-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP, Mark GP, Williams JT. Properties and opioid inhibition of mesolimbic dopamine neurons vary according to target location. J Neurosci. 2006;26:2788–2797. doi: 10.1523/JNEUROSCI.4331-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto GJ, McBride WJ, Murphy JM, Lumeng L, Li TK. Ethanol self-infusion into the ventral tegmental area by alcohol-preferring rats. Alcohol. 1994;11:557–564. doi: 10.1016/0741-8329(94)90083-3. [DOI] [PubMed] [Google Scholar]
- Gessa GL, Muntoni F, Collu M, Vargiu L, Mereu G. Low doses of ethanol activate dopaminergic neurons in the ventral tegmental area. Brain Res. 1985;348:201–203. doi: 10.1016/0006-8993(85)90381-6. [DOI] [PubMed] [Google Scholar]
- Gonzales RA, Weiss F. Suppression of ethanol-reinforced behavior by naltrexone is associated with attenuation of the ethanol-induced increase in dialysate dopamine levels in the nucleus accumbens. J Neurosci. 1998;18:10663–10671. doi: 10.1523/JNEUROSCI.18-24-10663.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA, Onn SP. Morphology and electrophysiological properties of immunocytochemically identified rat dopamine neurons recorded in vitro. J Neurosci. 1989;9:3463–3481. doi: 10.1523/JNEUROSCI.09-10-03463.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemoto S, Kohl RR, McBride WJ. GABA(A) receptor blockade in the anterior ventral tegmental area increases extracellular levels of dopamine in the nucleus accumbens of rats. J Neurochem. 1997;69:137–143. doi: 10.1046/j.1471-4159.1997.69010137.x. [DOI] [PubMed] [Google Scholar]
- Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci. 1992a;12:483–488. doi: 10.1523/JNEUROSCI.12-02-00483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SW, North RA. Two types of neurone in the rat ventral tegmental area and their synaptic inputs. J Physiol. 1992b;450:455–468. doi: 10.1113/jphysiol.1992.sp019136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelm MK, Criswell HE, Breese GR. Calcium release from presynaptic internal stores is required for ethanol to increase spontaneous gamma-aminobutyric acid release onto cerebellum Purkinje neurons. J Pharmacol Exp Ther. 2007;323:356–364. doi: 10.1124/jpet.107.126144. [DOI] [PubMed] [Google Scholar]
- Kohl RR, Katner JS, Chernet E, McBride WJ. Ethanol and negative feedback regulation of mesolimbic dopamine release in rats. Psychopharmacology (Berl) 1998;139:79–85. doi: 10.1007/s002130050692. [DOI] [PubMed] [Google Scholar]
- Koob GF, Roberts AJ, Schulteis G, Parsons LH, Heyser CJ, Hyytia P, Merlo-Pich E, Weiss F. Neurocircuitry targets in ethanol reward and dependence. Alcohol Clin Exp Res. 1998;22:3–9. [PubMed] [Google Scholar]
- Korotkova TM, Sergeeva OA, Eriksson KS, Haas HL, Brown RE. Excitation of ventral tegmental area dopaminergic and nondopaminergic neurons by orexins/hypocretins. J Neurosci. 2003;23:7–11. doi: 10.1523/JNEUROSCI.23-01-00007.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Wilson WA, Swartzwelder HS. Developmental differences in the sensitivity of spontaneous and miniature IPSCs to ethanol. Alcohol Clin Exp Res. 2006;30:119–126. doi: 10.1111/j.1530-0277.2006.00006.x. [DOI] [PubMed] [Google Scholar]
- Melis M, Camarini R, Ungless MA, Bonci A. Long-lasting potentiation of GABAergic synapses in dopamine neurons after a single in vivo ethanol exposure. J Neurosci. 2002;22:2074–2082. doi: 10.1523/JNEUROSCI.22-06-02074.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming Z, Criswell HE, Yu G, Breese GR. Competing presynaptic and postsynaptic effects of ethanol on cerebellar purkinje neurons. Alcohol Clin Exp Res. 2006;30:1400–1407. doi: 10.1111/j.1530-0277.2006.00167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott DD, Lewis DV. Facilitation of the induction of long-term potentiation by GABAB receptors. Science. 1991;252:1718–1720. doi: 10.1126/science.1675489. [DOI] [PubMed] [Google Scholar]
- Nie Z, Schweitzer P, Roberts AJ, Madamba SG, Moore SD, Siggins GR. Ethanol augments GABAergic transmission in the central amygdala via CRF1 receptors. Science. 2004;303:1512–1514. doi: 10.1126/science.1092550. [DOI] [PubMed] [Google Scholar]
- Oades RD, Halliday GM. Ventral tegmental (A10) system: neurobiology. 1. Anatomy and connectivity. Brain Res. 1987;434:117–165. doi: 10.1016/0165-0173(87)90011-7. [DOI] [PubMed] [Google Scholar]
- Okamoto T, Harnett MT, Morikawa H. Hyperpolarization-activated cation current (Ih) is an ethanol target in midbrain dopamine neurons of mice. J Neurophysiol. 2006;95:619–626. doi: 10.1152/jn.00682.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins TW, Everitt BJ. Neurobehavioural mechanisms of reward and motivation. Curr Opin Neurobiol. 1996;6:228–236. doi: 10.1016/s0959-4388(96)80077-8. [DOI] [PubMed] [Google Scholar]
- Roberto M, Madamba SG, Moore SD, Tallent MK, Siggins GR. Ethanol increases GABAergic transmission at both pre- and postsynaptic sites in rat central amygdala neurons. Proc Natl Acad Sci U S A. 2003;100:2053–2058. doi: 10.1073/pnas.0437926100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrbacher J, Jarolimek W, Lewen A, Misgeld U. GABAB receptor-mediated inhibition of spontaneous inhibitory synaptic currents in rat midbrain culture. J Physiol. 1997;500:739–749. doi: 10.1113/jphysiol.1997.sp022055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz W. Getting formal with dopamine and reward. Neuron. 2002;36:241–263. doi: 10.1016/s0896-6273(02)00967-4. [DOI] [PubMed] [Google Scholar]
- Siggins GR, Roberto M, Nie Z. The tipsy terminal: Presynaptic effects of ethanol. Pharmacol Ther. 2005;107:80–98. doi: 10.1016/j.pharmthera.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Stobbs SH, Ohran AJ, Lassen MB, Allison DW, Brown JE, Steffensen SC. Ethanol suppression of ventral tegmental area GABA neuron electrical transmission involves N-methyl-D-aspartate receptors. J Pharmacol Exp Ther. 2004;311:282–289. doi: 10.1124/jpet.104.071860. [DOI] [PubMed] [Google Scholar]
- Wan FJ, Berton F, Madamba SG, Francesconi W, Siggins GR. Low ethanol concentrations enhance GABAergic inhibitory postsynaptic potentials in hippocampal pyramidal neurons only after block of GABAB receptors. Proc Natl Acad Sci U S A. 1996;93:5049–5054. doi: 10.1073/pnas.93.10.5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner JL, Valenzuela CF. Ethanol modulation of GABAergic transmission: The view from the slice. Pharmacol Ther. 2006;111:533–554. doi: 10.1016/j.pharmthera.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Wise RA. Neurobiology of addiction. Curr Opin Neurobiol. 1996;6:243–251. doi: 10.1016/s0959-4388(96)80079-1. [DOI] [PubMed] [Google Scholar]
- Wu PH, Poelchen W, Proctor WR. Differential GABAB Receptor Modulation of Ethanol Effects on GABA(A) synaptic activity in hippocampal CA1 neurons. J Pharmacol Exp Ther. 2005;312:1082–1089. doi: 10.1124/jpet.104.075663. [DOI] [PubMed] [Google Scholar]
- Xiao C, Zhang J, Krnjevic K, Ye JH. Effects of ethanol on midbrain neurons: role of opioid receptors. Alcohol Clin Exp Res. 2007;31:1106–1113. doi: 10.1111/j.1530-0277.2007.00405.x. [DOI] [PubMed] [Google Scholar]
- Zhu PJ, Lovinger DM. Ethanol potentiates GABAergic synaptic transmission in a postsynaptic neuron/synaptic bouton preparation from basolateral amygdala. J Neurophysiol. 2006;96:433–441. doi: 10.1152/jn.01380.2005. [DOI] [PubMed] [Google Scholar]