

Abstract
Mice lacking 25-hydroxycholecalciferol [25(OH)D]-1α-hydroxylase (CYP27B1) are growth retarded, hypocalcemic, and have poor bone mineralization. We tested whether high dietary cholecalciferol (VD3) could exert effects in the absence of CYP27B1 in vivo. Weanling male wild-type (WT) and CYP27B1 knockout (KO) mice were fed either a 2% calcium (Ca), 20% lactose rescue diet or an AIN93G diet (0.5% Ca, 0.4% phosphorus) containing 1000 (1K, the rodent requirement, 25 μg), 10,000 (10K, 250 μg), or 20,000 (20K, 500 μg) IU VD3/kg diet until 12 wk when blood and tissues were taken. Serum 25(OH)D was >90 nmol/L in the 1K diet group and increased >4-fold in mice fed 10K and 20K diets. The 1K diet impaired growth and caused hypocalcemia in KO mice; the 10K and 20K diets were as effective as the high Ca rescue diet in preventing these outcomes. High VD3 restored expression of vitamin D-regulated genes in intestine (calbindin D9K) and kidney (CYP27B1, 24-hydroxylase, calbindin D9K) of KO mice. Micro-computed tomography of femora revealed complete recovery of cortical bone in KO mice fed either the rescue or 10K diets but only partial recovery of trabecular bone measures (e.g. 40% lower bone volume, 20% lower trabecular thickness, and 23% increase in trabecular separation). These data show that very high serum 25(OH)D can influence Ca and bone metabolism independent of its conversion to 1,25 dihydroxycholecalciferol. However, neither high dietary Ca nor high dietary VD3 is sufficient to fully recover the phenotype of CYP27B1 KO mice.
Introduction
The vitamin D endocrine system is indispensable for maintenance of systemic calcium homeostasis and skeletal growth. Cholecalciferol, or vitamin D3 (VD3)5, can be obtained from the diet and supplements or it can be generated in skin from 7-dehydrocholesterol upon exposure to UV radiation. In either case, VD3 must be further metabolized to bind to the nuclear vitamin D receptor (VDR) and exert its biological effects (1). Initial metabolism of VD3 in liver generates 25-hydroxychole-calciferol [25(OH)D3], a metabolite that displays low affinity for the VDR and has traditionally been considered biologically inactive. 25(OH)D3 is further metabolized by the renal 25(OH)D3-1α-hydroxylase (a mitochondrial cytochrome P450 enzyme also known as CYP27B1) to generate 1,25 dihydroxy-cholecalciferol [1,25(OH)2D3], the high affinity VDR ligand (1). The control of calcium (Ca) and bone metabolism by the VDR-1,25(OH)2D3 complex is mediated primarily via transcriptional regulation of genes in the intestine and kidney, leading to regulation of Ca uptake and transport, as well as in bone, leading to the regulation of bone formation and resorption (1).
The importance of VD3 metabolism and action for the regulation of calcium homeostasis has been confirmed in humans with type I and type II genetic rickets where loss-of-function mutations in either CYP27B1 or VDR, respectively, lead to severe hypocalcemia and osteomalacia (2,3). Recently, mice with targeted ablation of CYP27B1 have been developed as models for human vitamin D-dependent rickets type I. Mice with this disruption have impaired postnatal weight gain, progressive hypocalcemia, and skeletal disturbances secondary to the deficiency of 1,25(OH)2D3 (4,5). This phenotype can be completely prevented by exogenous administration of 1,25(OH)2D3, which induces VDR target genes in intestine (including calbindin D9K) to restore VD3-dependent Ca absorption (6). The rachitic phenotype of CYP27B1 knockout (KO) mice can also be improved, but not completely corrected, by feeding a high Ca/high lactose ‘‘rescue’’ diet from weaning (7,8). The inclusion of lactose promotes vitamin D-independent intestinal Ca absorption and it normalizes serum Ca without induction of VDR target genes such as calbindin D9K (6).
In addition to its central role in the vitamin D endocrine system, CYP27B1 is expressed at low levels in extra-renal tissues, including skin, prostate, breast, and colon (9). It has been hypothesized that extra-renal CYP27B1 generates 1,25(OH)2D3, which then acts locally to regulate Ca metabolism [e.g. suppression of parathyroid hormone (PTH) (10) and activation of Ca absorption (11)] and induce nonclassical vitamin D effects such as the regulation of cell proliferation and differentiation (12). This hypothesis predicts that reduced availability of 25(OH)D3 to the extra-renal CYP27B1 limits local vitamin D signaling and protection, whereas high serum 25(OH)D3 maximizes local 1,25(OH)2D3 production and beneficial effects. This model is supported by epidemiological studies that have linked indices of vitamin D status [i.e. vitamin D intake, latitude, UV exposure, and/or circulating 25(OH)D3] with the risk for various chronic diseases, including osteoporosis and cancer (13), as well as cell culture studies that demonstrate the conversion of 25(OH)D3 to 1,25(OH)2D3 in cell types such as activated immune cells and prostate epithelial cells (14,15). However, an alternative explanation for these observations that has yet to be rigorously tested is that 25(OH)D3 can mediate effects in the absence of conversion to 1,25(OH)2D3 by directly activating VDR. In this model, the high serum concentration of 25(OH)D3 [500–1000 times higher than 1,25(OH)2D3] overcomes its low affinity for the receptor [500 times lower than 1,25(OH)2D3]. In this study, we utilized the CYP27B1 KO mouse model to test the possibility that 25(OH)D3 can exert biological effects in the absence of CYP27B1 in vivo. As a straightforward proof of principle, we examined whether chronic feeding of diets high in VD3 would elevate serum 25(OH)D3 sufficiently to activate VDR in CYP27B1 KO mice. Our data show that high VD3 status can rescue the rachitic phenotype of CYP27B1 null mice and supports the hypothesis that high serum 25(OH)D3 can induce VDR-mediated molecular events in vivo.
Methods
Animals and diets
The CYP27B1 KO mice utilized in these studies were derived from animals originally obtained from Dr. René St. Arnaud (McGill University, Montreal, QC), who generated the CYP27B1 KO on the C57/BL6 background (4). In this model, exon 8, encoding the heme-binding region, was deleted. Because heme binding is essential for the hydroxylase activity of this enzyme, the altered gene produces a mutant protein lacking the ability to convert 25(OH)D3 to 1,25(OH)2D3. Male CYP27B1 KO and wild-type (WT) C57/BL6 mice were randomized to rescue diet [2% Ca, 1.25% phosphorus, and 20% lactose supplemented with 2200 IU (55 μg) VD3/kg] or semipurified AIN 93G diets [0.5% Ca, 0.4% phosphorus, (16)] containing 1000 IU (1K; 25 μg), 10,000 IU (10K; 250 μg), or 20,000 (20K; 500 μg) IU VD3/kg diet (n = 9–10 mice per dietary group for each genotype). The standard and modified AIN93G diets were prepared by Research Diets; the 2% Ca, 20% lactose rescue diet was prepared by Teklad (Diet TD96348) (Table 1). Mice consumed diets and water ad libitum. We recorded food intake and body weights weekly. At 12 wk, mice were anesthetized, at which time serum and tissues were collected for analysis and the mice were killed. The animal experiment was approved by the Notre Dame Animal Care and Use Committee.
TABLE 1.
Composition of the diets used in the experiment
| Rescue1 | AIN93G2 | |
|---|---|---|
| Ingredient | g/kg | |
| Casein, VFT | 192 | 200 |
| DL-Methionine | 3 | |
| Cysteine | 0 | 3 |
| Sucrose, cane | 264.78 | 100 |
| Lactose, monohydrate | 200 | |
| Dextrinized cornstarch | 132 | |
| Corn starch | 150 | 397.49 |
| Corn oil | 50 | |
| Soybean oil | 0 | 70 |
| Cellulose (fiber) | 50 | |
| Solka Floc (fiber) | 50 | |
| Choline bitrate | 2.50 | |
| t-Butylhydroquinone | 0.014 | |
| AIN93 vitamin mix3 | 10 | |
| AIN93 mineral mix4 | 35 | |
| Teklad 40060 vitamin mix5 | 10 | |
| AIN-76 mineral mix4 | 35 | |
| Calcium phosphate dibasic | 31.17 | |
| Calcium carbonate | 14.05 | |
| Composition | ||
| Protein, % | 17.74 | 19.30 |
| Carbohydrate, % | 59.82 | 64 |
| Fat, % | 5.05 | 16.70 |
| Ca, % | 2 | 0.50 |
| P, % | 1.25 | 0.40 |
| VD3, IU/kg | 2203 | 1000 |
| Energy density, kJ/kg | 1489 | 1576 |
Rescue diet = 2% Ca, 20% lactose diet from Teklad (TD 96348).
To make the 10,000 VD3/kg diets and 20,000 IU VD3/kg diets, the basal AIN93G diet was supplemented with 0.1 g/kg and 0.2 g/kg, respectively, of a 100,000 IU VD3/g stock solution.
AIN93G-MX and AIN93-VX (16).
AIN76 mineral mix (37).
Teklad 40060 vitamin mix (38).
Serum Ca and vitamin D metabolite analysis
Serum 1,25(OH)2D3 (n = 8–10 replicates per group) and 25(OH)D3 (n = 4–5) were analyzed via enzyme immunoassays using commercial kits (Immunodiagnostic Systems). The inter-assay CV for each assay is 10 and 8%, respectively, and the intra-assay CV for each assay is 15 and 10%, respectively. The stated cross-reactivity of 25(OH)D3 in the 1,25(OH)2D3 enzyme immunoassay is 0.0092% and this was confirmed by analysis of serum from CYP27B1 KO mice that was spiked to a concentration of 500 nmol/L with 25(OH)D3 (data not shown). Serum Ca (n = 4–5) was analyzed via a quantitative colorimetric assay using the QuantiChrom Ca assay kit (BioAssay Systems).
Real-time PCR
Total RNA was isolated from mucosal scrapings (collected from the first 2 cm of the duodenum) and minced kidney tissue using the TriReagent procedure (Molecular Research Center). The isolated RNA was reverse transcribed into cDNA as previously described (17). Real-time PCR was conducted on samples using the Bio-Rad My iQ RT-PCR system containing SYBR green (BioRad). Only samples whose gel pictures (0.5-μg sample on 1.5% agarose gel) confirmed that RNA degradation had not occurred were used for PCR analysis. Messenger RNA (mRNA) levels were normalized to the expression of glyceraldehyde 3-phosphate dehydrogenase within the sample and are expressed as relative to the WT mouse expression level. PCR conditions and primers for calbindin D9k, 25-hydroxy cholecalciferol, 24 hydroxylase (CYP24), and glyceraldehyde 3-phosphate dehydrogenase were previously reported by our group (18). Renal CYP27B1 mRNA levels were analyzed via RT-PCR using primer sets and conditions previously reported by Healy et al. (19). Six samples per group were analyzed for renal gene expression and 3–4 samples were analyzed per group for duodenal calbindin D9k mRNA levels.
Bone analyses
At the end of the experiment, the right and left femur from each mouse was stripped of all muscle. The right femur was fixed in 10% buffered formalin for 24 h and stored in 70% ethanol at 4°C until analysis. The left femur was examined using digital calipers for length and midshaft thickness. Afterwards, the femur was dried, ashed, and Ca content was examined by atomic absorption spectrometry as we have previously described (20).
Micro-computed tomography (μCT) was used to determine quantitative differences in bone structure using conditions and procedures described by Judex et al. (21). Because our assessment of bone Ca and ash showed that these parameters were not different in the WT groups and because we did not detect a difference between KO mice fed the 10K and 20K IU VD3/kg diets, only 4 representative groups were examined: WT fed the standard AIN93G diet (0.5% Ca, 1000 IU VD3/kg; reference group), CYP27B1 KO mice fed the standard AIN93G diet (negative control), and CYP27B1 KO mice fed either the rescue diet (partial recovery) or the AIN93G diet supplemented with 10K IU VD3/kg (test group). We scanned the midshaft and the proximal femur using a desktop μCT instrument (Scanco Medical uCT 40). Data sets were analyzed with Scanco μCT v.1.2 software. The distal point for the start of trabecular bone quantification, i.e. the point were distal condyles merge, was determined for each femur individually from reconstructed three-dimensional (3D) images. A manual contouring of 300 μm up the bone shaft from this point was performed to ensure that we included only trabecular bone in the analysis (21). Total and trabecular bone volume (BV,TV); connectivity density (Conn.D.; higher number = more connections between trabeculae); trabecular thickness (Tb.Th), number (Tb.N), and separation (Tb.Sp), and structure model index (a measure reflecting trabecular structure; 0 = plate-like to 3 = rod-like) were estimated using the direct model. Cortical bone analysis was conducted on 2 150-μm sections flanking the femur midpoint. We estimated midshaft BV and TV, bone surface, bone surface:BV ratio, and bone thickness using the direct model. The optimum threshold levels were determined from a random selection of bones from each group using an optimal threshold assessment program included in the Scanco uCT v.1.2 software.
Statistical analysis
Prior to the study, power calculations were completed to determine the proper sample size for the various analytical procedures. Based on the variability estimates from our previously published work (18,20), we determined that a sample size of 3 to 4 mice was sufficient to detect a 50% recovery of the KO phenotype for any of the analyses conducted. Data were analyzed by 1- or 2-way ANOVA. When the plots of predicted values vs. residuals demonstrated that the data were not normally distributed, a log transformation was conducted prior to statistical analysis. Samples identified as outliers by the statistical packages were excluded from the analysis. The analysis was conducted using the SYSTAT statistical analysis software (version 7.0, SAS Institute). Comparisons of multiple group means were conducted using Fisher’s protected least significant difference. For all analyses, differences between means were considered significant at P <0.05. Values in the text are reported as means ±SEM.
Results
High dietary VD3 does not fully normalize postnatal growth of CYP27B1 KO mice
Neither diet nor genotype affected food intake during the 12-wk study (data not shown). CYP27B1 KO mice fed the AIN 93G diet containing 1K VD3 were growth arrested compared with WT mice fed the same diet (Fig. 1). Final body weights of CYP27B1 KO mice fed this diet (20.7 ± 0.6 g) were 31% lower than that of WT controls (30.1 ± 1 g). CYP27B1 KO mice grew when fed the rescue diet, but final body weights (23.7 ± 0.4 g) remained lower than those of WT mice fed the rescue diet (30.0 ± 0.7 g). Consumption of the 10K or 20K diet did not adversely affect weight gain of WT mice and promoted postnatal growth of CYP27B1 KO mice. However, similar to the rescue diet, final body weights of CYP27B1 KO mice fed the high VD3 diets were significantly lower (25.5 ± 0.8 g) than those of WT mice fed the same diets (29.7 ± 0.6 g).
FIGURE 1.
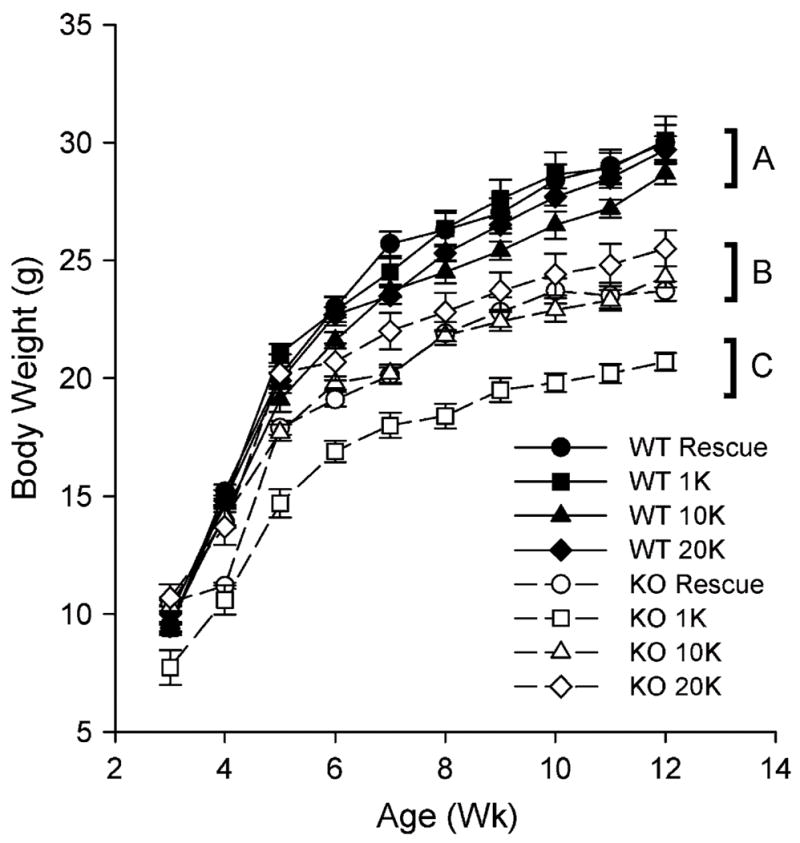
The effect of high calcium and high VD3 diets on growth of WT and KO mice. Values are means ± SEM, n = 9–10. Groups without a common letter differ, P < 0.05.
High dietary VD3 normalizes serum Ca
Consistent with previous reports (7), CYP27B1 KO mice fed the standard 0.5% Ca diet were severely hypocalcemic and the high Ca/high lactose rescue diet prevented this effect (Fig. 2A). Similarly, CYP27B1 KO mice fed 0.5% Ca diets containing 10K or 20K IU/kg VD3 were normocalcemic. Chronic feeding of high dietary VD3 did not influence serum calcium in WT mice.
FIGURE 2.
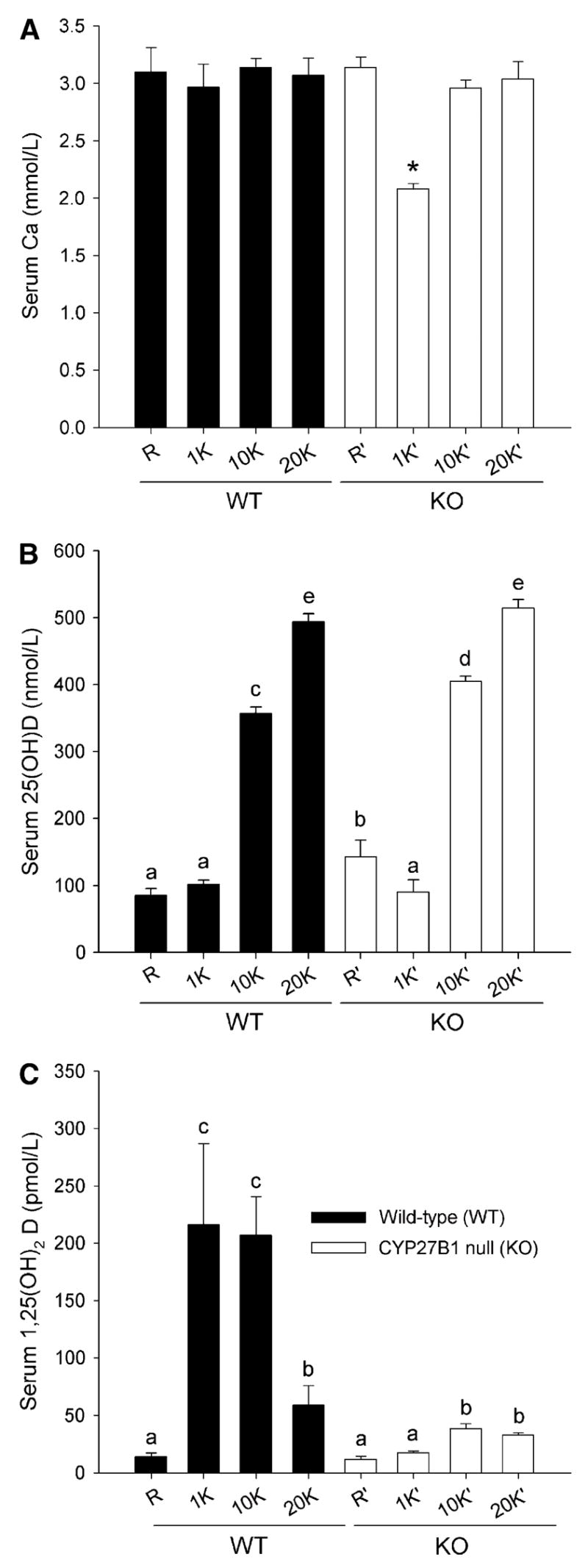
The effect of high Ca and high VD3 diets on serum Ca (A), (25(OH)D) (B), and 1,25(OH)2D (C) in WT and KO mice. Bars represent the mean ± SEM, 4 = 4–5 (A,B) or 8–10 (C). *Different from all other groups, P < 0.05 (A). Means without a common letter differ, P < 0.05 (B,C).
As expected, both WT and CYP27B1 KO mice fed the 10K or 20K diet exhibited significantly increased serum 25(OH)D3 (Fig. 2B). Circulating levels of 25(OH)D3 in mice fed the 20K diet were ~500 nmol/L, a 4-fold elevation compared with mice fed the standard AIN93G diet or the rescue diet.
In WT mice, chronic feeding of the 2% Ca rescue diet suppressed serum 1,25(OH)2D3 by 93% relative to the other diets, which all contained 0.5% Ca (Fig. 2C). WT mice fed the 20K diet also exhibited low serum 1,25(OH)2D3 (73% lower than mice fed the 1K diet) that was accompanied by a 70% reduction in renal CYP27B1 mRNA expression (data not shown). In CYP27B1 KO mice fed the 1K diet, 1,25(OH)2D3 was extremely low (5% of levels seen in WT mice fed the 1K diet) and consistent with the published assay cross-reactivity between 25(OH)D3 and 1,25(OH)2D3 (i.e. when 25(OH)D3 level is 100 nmol/L, serum 1,25(OH)2D3 level is predicted to be 9.2 pmol/L based upon the assay cross-reactivity). The absence of 1,25(OH)2D3 in the serum of these mice is consistent with the inability of these mice to produce the hormone.
In KO mice fed the high VD3 diets, serum 1,25(OH)2D3 levels were increased ~100% (e.g. to 35 pmol/L in KO mice fed the 10K diet, or just 16% of the level present in WT mice fed the same diet). The small increase in serum 1,25(OH)2D3 level detected in KO mice fed high dietary VD3, although significant, was equivalent to the calculated 1,25(OH)2D3 level expected from cross-reactivity of the assay for 25(OH)2D3 [400 nmol/L 25(OH)2D3 × 0.0092% = 37 pmol/L 1,25(OH)2D3 due to cross-reactivity].
Effect of diet and genotype on intestinal and renal gene expression
As another indicator of phenotypic rescue, we measured expression of CYP27B1 itself. Normally, hypocalcemia leads to elevated PTH levels and a strong stimulation of CYP27B1 gene expression. Using PCR primers designed to detect the 5′ end of the transcript produced from both the WT and mutant CYP27B1 allele, we found that CYP27B1 transcript levels were highly induced in CYP27B1 KO mice fed the 1K diet who were severely hypocalcemic (Table 2). This elevation in CYP27B1 expression was prevented in CYP27B1 KO mice fed either the rescue diet or the high dietary VD3 diets, reflecting the normalization of serum Ca levels in these groups.
TABLE 2.
The effect of high Ca and high VD3 diets on renal CYP27B1, calbindin D9k, and CYP24 mRNA levels in WT and KO mice
| Group | CYP27B11 | Calbindin D9k | CYP24 |
|---|---|---|---|
| WT 1K | 9.7 ± 3.5a | 86.3 ± 10.4ab | 8.2 ± 3.0c |
| KO 1K | 1769.1 ± 73.8c | 56.3 ± 8.5a | 0.1 ± 0.1a |
| KO 10K | 59.3 ± 18.5b | 143.5 ± 14.0c | 1.8 ± 0.7b |
| KO 20K | 7.2 ± 3.0a | 101.4 ± 13.4b | 4.9 ± 1.7c |
| KO R | 44.1 ± 32.7a | 59.1 ± 14.3a | 0.1 ± 0.1a |
Values are means ± SEM, n = 6. Means in a column without a common letter differ, P < 0.05.
To assess whether the recovery of growth and calcemia by high dietary VD3 in CYP27B1 KO mice was associated with activation of VDR, we measured expression of renal CYP24 mRNA as well as intestinal and renal calbindin D9K. Renal CYP24 mRNA levels were absent in CYP27B1 KO mice fed either the 1K diet or the rescue diet, consistent with the lack of hormone and no VDR activation in these mice (Table 2). Feeding the 10K or 20K diets caused a dose-dependent increase in renal CYP24 mRNA levels. There was a trend toward reduced renal calbindin D9K mRNA in CYP27B1 KO mice fed the normal VD3 diet (P = 0.062) and the rescue diet (P = 0.098) (Table 2). However, feeding the 10K or 20K diets significantly increased renal calbindin D9k mRNA 2- to 3-fold in CYP27B1 KO mice compared with KO mice fed the 1K diet. Duodenal calbindin D9K gene expression was largely unaffected by diet in WT mice and it tended to be reduced in CYP27B1 KO mice fed the 1K diet relative to WT mice (40%; P < 0.1; Fig. 3). When CYP27B1 KO mice were fed the high VD3 diets, duodenal calbindin D9K mRNA levels were significantly induced 4- to 6-fold.
FIGURE 3.

The effect of high Ca and high VD3 diets on duodenal calbindin D9k mRNA level in WT and KO mice. Bars represent the mean ±SEM, n = 3–4. Means without a common letter differ, P < 0.05.
Effect of high dietary VD3 on skeletal parameters in CYP27B1 KO mice
Only minor changes were detected in the percent bone ash and Ca content of the WT mice fed the rescue or the highest vitamin D-containing diet (data not shown). The critical hallmarks of rickets are: abnormal growth plate structure, excess osteoid surface and thickness, and markedly decreased mineralization (4). The dry weight, length, and mineral content of femur were all significantly reduced in CYP27B1 KO mice fed the 1K diet, reflecting the smaller, less mineralized bones characteristic of rickets. Both the high Ca rescue diet and high VD3 diets prevented this phenotype (Table 3).
TABLE 3.
The effect of high calcium and high VD3 diets on bone length, weight, and mineralization in WT and KO mice1
| Group | Dry weight | Ash | Ca | Femur length |
|---|---|---|---|---|
| mg2 | ——% dry bone weight —— | mm | ||
| WT 1K | 49.5 ± 1.9c | 56.1 ± 1.4b | 23.0 ± 0.5b | 16.5 ± 0.2b |
| KO 1K | 24.4 ± 1.5a | 44.6 ± 1.9a | 19.7 ± 0.4a | 11.9 ± 0.6a |
| KO 10K | 48.1 ± 2.0c | 55.6 ± 1.0b | 23.7 ± 0.5b | 15.8 ± 0.2b |
| KO 20K | 46.1 ± 3.6c | 55.8 ± 0.8b | 23.3 ± 0.3b | 15.5 ± 0.3b |
| KO R | 38.5 ± 1.3b | 57.6 ± 1.3b | 24.0 ± 0.4b | 15.8 ± 0.2b |
Values are means ± SEM, n = 6; means in a column without a common letter differ, P < 0.05.
Based on the changes observed in femur mineral content, a subset of groups was selected for a more comprehensive assessment of skeletal architecture using μCT scanning of cortical and trabecular sites (Table 4). This analysis is visually represented as 3D reconstructions of representative bones from each of the 4 groups examined (Fig. 4).
TABLE 4.
The effect of high Ca and high VD3 diets on cortical and trabecular bone quantity and structure in WT and KO mice assessed by μCT1
| Cortical bone
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Group | n | TV | BV | BV:TV | Cortical thickness | MDTV | MDBV | ||
| mm3 | mm3 | mm | mg/mm3 | mg/mm3 | |||||
| WT 1K | 3 | 0.38 ± 0.08b | 0.29 ± 0.03b | 0.96 ± 0.01b | 0.20 ± 0.01b | 1321.1 ± 16.2b | 1409.2 ± 7.3b | ||
| KO 1K | 4 | 0.17 ± 0.01a | 0.14 ± 0.01a | 0.84 ± 0.01a | 0.099 ± 0.01a | 1080.7 ± 25.3a | 1297.7 ± 13.6a | ||
| KO 10K | 3 | 0.26 ± 0.02b | 0.25 ± 0.02b | 0.95 ± 0.01b | 0.21 ± 0.01b | 1350.5 ± 12.9b | 1446.4 ± 8.9b | ||
| KO R | 3 | 0.25 ± 0.01b | 0.24 ±0.01b | 0.95 ± 0.01b | 0.21 ± 0.01b | 1327 ± 1.24b | 1421.3 ± 2.6b | ||
| Trabecular bone
| |||||||||
| Group | n | TV | BV | BV:TV | Structure model index | Conn.D. | Tb.N | Tb.Th | Tb.Sp |
|
| |||||||||
| mm3 | mm3 | 1/mm3 | 1/mm | mm | mm | ||||
| WT 1K | 4 | 0.80 ± 0.05c | 0.20 ± 0.03c | 0.25 ± 0.04b | 1.57 ± 0.19b | 346.2 ± 27.9c | 7.2 ± 0.3b | 0.051 ± 0.004c | 0.132 ± 0.005a |
| KO 1K | 4 | 0.38 ± 0.06a | 0.02 ± 0.01a | 0.05 ± 0.01a | 2.81 ± 0.08a | 71.2 ± 4.7a | 5.7 ± 0.1a | 0.027 ± 0.001a | 0.183 ± 0.005c |
| KO 10K | 3 | 0.76 ± 0.03c | 0.11 ± 0.04bc | 0.15 ± 0.05b | 2.17 ± 0.52b | 223.6 ± 75.8b | 6.2 ± 0.6ab | 0.041 ± 0.003b | 0.162 ± 0.015bc |
| KO R | 3 | 0.58 ± 0.03b | 0.09 ± 0.01ab | 0.16 ± 0.03b | 2.19 ± 0.14b | 237.3 ± 21.4bc | 6.3 ± 0.3ab | 0.043 ± 0.002bc | 0.155 ± 0.005ab |
Values are means ± SEM. Means in a column without a common letter differ, P < 0.05.
FIGURE 4.
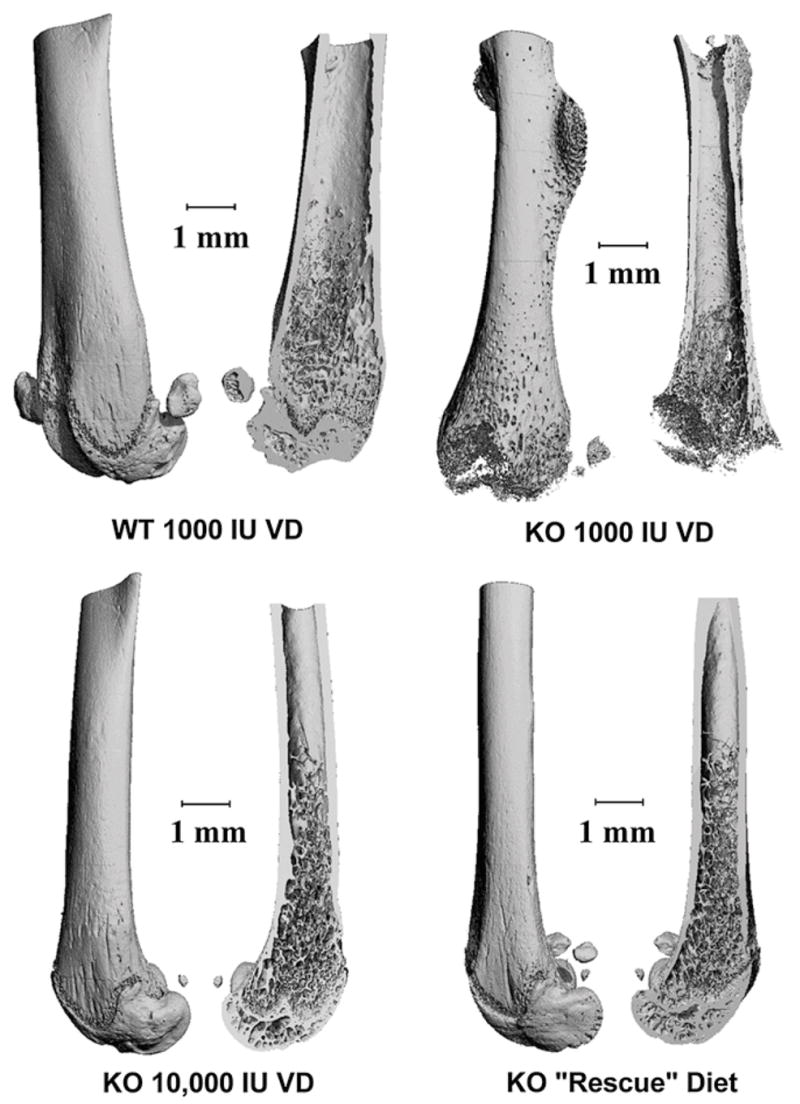
Representative μCT scans demonstrating the effect of high Ca and high VD3 diets on bone architecture in KO mice. Femora were harvest, fixed in formalin, and stored in ethanol. μCT scanning was conducted and 3D images were reconstructed for visualization. Exterior and interior views are presented for representative bones.
For cortical bone, all of the parameters measured were reduced in CYP27B1 KO mice fed the 1K diet. Feeding KO mice the 2% Ca rescue diet or the 10K diet restored midshaft femur cortical bone properties to normal.
Trabecular bone quality measures were significantly reduced in CYP27B1 KO mice fed the 1K diet. In particular, the 3D structure of the entire proximal femur was missing (reflecting undermineralization of the growth plate) and disruption of the trabecular structure was evident (Fig. 4). The rescue and high VD3 diets resulted in only a partial rescue of these phenotypes (presented visually in Fig. 4 and quantitatively in Table 4). The ratio of BV:TV (–40%), the Conn.D (–35%), Tb.N (–14%), and Tb.Th (–20%) were all significantly lower than WT levels in KO mice fed either the high VD3 or 2% Ca rescue diets. Similarly, the structural model index (+38%) and Tb.Sp (+23%) were both significantly higher than WT values in KO mice fed either the high VD3 or rescue diets.
Discussion
In these studies, we show that high dietary VD3 intake has biological effects in the absence of CYP27B1, the enzyme required for generation of the most potent VDR ligand, 1,25(OH)2D3. The question is whether this is due to direct activation of VDR by the elevated 25(OH)D3 levels resulting from high dietary VD3 intake. Serum 25(OH)D3 levels increased from ~90 nmol/L to >400 nmol/L in response to chronic feeding of the high VD3 diets. Thus, mice fed a standard AIN93G diet containing 1000 IU VD3/kg have serum 25(OH)D3 levels that are considered optimal for humans (22), whereas the high dietary VD3 levels we used raised serum 25(OH)D3 levels to ranges that have been reported to cause hypercalcemia in humans (23). However, in growing WT mice, high dietary VD3 did not negatively influence either serum Ca or growth. In CYP27B1 KO mice, the high level of 25(OH)D3 resulting from high dietary VD3 intake restored normal growth, prevented hypocalcemia, and limited osteomalacia in CYP27B1 KO mice and is consistent with what others have reported for ergocalciferol supplementation in humans with genetic pseudovitamin D-deficiency rickets (PDDR) (24). In these patients, daily oral doses of 20,000–100,000 IU ergocalciferol (0.52–1.3 IU/g body weight) are needed to heal rickets and 10,000–50,000 IU ergocalciferol (0.25–0.67 IU·g body weight−1·d−1) maintains normal Ca homeostasis. These doses are comparable to the VD3 levels that we found to be protective in the CYP27B1 KO mice (10K diet = 0.2 IU·g body weight−1·d−1). An argument against the direct activation of VDR by elevated 25(OH)D3 is that we observed a small increase in serum 1,25(OH)2D3 following consumption of the high VD3 diets in CYP27B1 KO mice [i.e. similar to findings previously reported in patients with PDDR given high daily doses of ergocalciferol (24)]. This level (35 pmol/L) is far below the normal serum 1,25(OH)2D3 levels observed in growing mice (150–220 pmol/L) and could reflect the ability of other enzymes to convert a small amount of prohormone to active hormone. However, we believe that the increase in serum 1,25(OH)2D3 we observed after high dietary VD3 feeding is due to cross-reactivity of the 1,25(OH)2D3 assay for 25(OH)D3. The increased serum 1,25(OH)2D3 is almost exactly the amount expected based on the observed increased serum 25(OH)D3 and the published cross-reactivity of the assay for 25(OH)D3. As a result, we do not think that there is any production of 1,25(OH)2D3 in CYP27B1 KO mice and that the role of enzymes other than CYP27B1 in the production of 1,25(OH)2D3 is negligible.
With this in mind, and despite the low affinity of 25(OH)D3 for the VDR, the direct activation of VDR by supraphysiologic serum levels of 25(OH)D3 mice fed high vitamin D diets is supported by several lines of evidence. First, WT mice fed the 20K diet had reduced serum 1,25(OH)2D3 (Fig. 2C) and reduced renal CYP27B1 mRNA (data not shown), suggesting there is suppression of the renal vitamin D endocrine system in normal mice when 25(OH)D3 is dramatically elevated (but serum Ca is not). Second, high serum 25(OH)D3 levels are also associated with suppression of the CYP27B1 gene in the kidney of CYP27B1 KO mice. The CYP27B1 gene promoter is normally upregulated by PTH through a cAMP-dependent mechanism (25) and inhibited by activation of VDR and binding of VDR to a negative vitamin D response element (26). Thus, our findings are consistent with enhanced 25(OH)D3-mediated gene repression. Finally, feeding the high VD3 diets strongly enhanced expression of the high vitamin D-responsive VDR target gene CYP24 in the kidney as well as calbindin-D9K levels in the intestine and kidney of CYP27B1 KO mice. This demonstrates that classical VDR target genes are activated when serum 25(OH)D3 is greatly elevated but serum 1,25(OH)2D3 is absent or well below normal levels.
Although we did not directly measure intestinal calcium absorption, our data are also consistent with an activation of this process in CYP27B1 mice fed the high VD3 diets. The loss of vitamin D-dependent active Ca absorption is responsible for the hypocalcemic, rachitic phenotype in VDR null mice (20), but low Ca absorption efficiency can be overcome in both VDR null and CYP27B1 null mice by feeding a highly bioavailable 2% Ca/high lactose rescue diet that is absorbed through a nonregulated diffusional pathway that does not require stimulation of intestinal gene expression through the VDR (7,27). However, here we found that calcium metabolism in the CYP27B1 KO mouse can be normalized by high VD3 intake in the context of a 0.5% Ca diet that is normally rachitogenic in these mice. Coupled with the enhanced expression of intestinal calbindin D9k that we observed [a marker for vitamin D-regulated Ca absorption (28,29)], our data indicate that VDR-mediated Ca absorption was activated by the high VD3 diets.
The mechanism by which 25(OH)D3 gains access to cellular VDR in mice fed high dietary VD3 remains an open question. 25(OH)D3 has a high affinity for the serum vitamin D-binding protein [DBP; >600-fold >1,25(OH)2D3] (30) that has been assumed to deliver vitamin D metabolites to tissues. However, uptake of DBP-bound vitamin D metabolites from the circulation into cells through a cubulin/megalin-dependent mechanism has been shown only for renal and mammary epithelial cells (31,32). Alternately, the high circulating concentrations of 25(OH)D3 achieved could theoretically have saturated serum DBP, increasing free 25(OH)D3 sufficiently to allow passive entry across cellular membranes. However, Rojanasathit and Haddad (33) estimated that only 1–2% of the serum binding sites for this sterol are occupied under physiological conditions, so the 4-fold increase in serum 25(OH)D3 that we observed with high vitamin D diets would not be enough to saturate serum DBP. Clearly, further studies are needed to determine the relative contribution of free, DBP-bound, lipid-bound, or albumin-bound 25(OH)D3 to the normalization of Ca metabolism in CYP27B1 KO mice fed high dietary VD3.
Dardenne et al. (7) previously showed that the rickets and osteomalacia in CYP27B1 KO mice was recovered by a high Ca/high lactose rescue diet but that femur size was still smaller in these mice. In our study, we extend these findings by showing that although overall mineral content was restored, bone microstructure is only partly recovered in CP27B1 KO mice fed either a high Ca/high lactose rescue diet or high VD3-containing diets. The mechanism underlying this incomplete rescue is unclear. At weaning, body weight was similar between WT and KO mice. However, although the high VD3-fed mice grew normally for the first 2 wk after weaning, they started to clearly diverge from WT mice after this point, which suggests several possibilities. First, the level of 25(OH)D3 reached by feeding the high VD3 diets may be inadequate for full recovery. This hypothesis seems unlikely given the fact that the 20K diet did not cause a further improvement of the phenotype even though serum 25(OH)D3 levels were 27–38% higher than in mice fed the 10K diet. Alternately, incomplete recovery with elevated 25(OH)D3 could reflect the fact that the prohormone is only a partial agonist for the VDR, leading to incomplete activation of vitamin D-responsive genes or selective activation of a subset of vitamin D-responsive genes. Finally, the partial recovery due to the prohormone may be revealing a unique role of 1,25(OH)2D3 that cannot be met by 25(OH)D3. It is tempting to speculate that this function may be a membrane-initiated action of vitamin D mediated through the membrane associated rapid response steroid-binding (MARRS) protein (34) or a unique function of the VDR at the membrane surface that requires an alternate ligand-binding pocket (35), as has been demonstrated for chondrocyte function in the growth plate (36). However, further studies using dynamic histomorphometry and cells isolated from CYP27B1 null cells will be necessary to determine whether the 25(OH)D3-mediated functions of bone cells are blunted relative to the effects of 1,25(OH)2D3.
In summary, semipurified AIN diets supplemented with high levels of VD3 supported postnatal growth, prevented hypocalcemia, and permitted nearly normal skeletal growth in CYP27B1 KO mice independent of elevated serum 1,25(OH)2D3. This dietary vitamin D-based recovery is associated with increased levels of the prohormone, 25(OH)D3, from those thought to be optimal in humans (>90 nmol/L) to supraphysiologic levels (>400 nmol/L). In CYP27B1 KO mice, the beneficial effects of VD3 supplementation were as good as or better than those obtained with the high Ca/high lactose rescue diet routinely used for maintenance of these mice. Thus, using a standard diet (0.5% Ca, 0.4% P) supplemented with VD3 appears to be an effective alternative approach for maintenance of CYP27B1 KO mice. However, although Ca metabolism appears to be normalized in these mice and others have shown that normalization of Ca metabolism is sufficient to restore reproductive vigor in the mice (5), we have not examined the impact of the diets in female mice nor have we formally examined reproductive performance in the mice fed the high vitamin D diets. Further studies should examine the benefits of high vitamin D intake under these conditions. Finally, although previous studies demonstrate that VD3 supplements would be an inexpensive treatment for human PDDR, clinicians have expressed concerns over the potential toxicity of the vitamin at therapeutic levels due to progressive accumulation of the vitamin in fat and muscle (24). Our data suggest that high VD3 treatment in CYP27B1 KO mice could be exploited to directly test the validity of those concerns.
Acknowledgments
We thank Dr. Mathieu Renouf, Dr. Judy Narvaez, Theresa Sikorska, Sarah Morden-McCombs, and David Taffany for assistance with animal handling, genotyping, and serum measurements, Ms. Pam Lachcik for assistance with the μCTanalysis, and Ms. Kate Barzan for assisting with the PCR analysis of samples. In addition, we also thank Dr. Rene St-Arnaud for providing the Welsh laboratory with breeding pairs of the CYP27B1 KO mice.
Footnotes
Supported by funds from NIH awards CA10113 (J.C.F.), DK054111 (J.F.), CA103018 (J.W.), and CA69700 (J.W.).
Author disclosures: M. J. Rowling, C. Gliniak, J. E. Welsh, no conflicts of interest; J. C. Fleet has received lecture fees from Wyeth Consumer Healthcare and Eli Lilly and Company.
Abbreviations used: 1K, 1,000 IU cholecalciferol/kg; 3D, three-dimensional; 10K, 10,000 IU cholecalciferol/kg; 20K, 20,000 IU cholecalciferol/kg; 25(OH)D, 25-hydroxycholecalciferol; 1,25 (OH)2 D3, 1,25 dihydroxycholecalciferol; BV, bone volume; Conn.D., trabecular bone connectivity density; CYP24, 25-hydroxy cholecalciferol, 24 hydroxylase; CYP27B1, 25-hydroxycholecalciferol-1α-hydroxylase; DBP, vitamin D-binding protein; KO, CYP27B1 knockout; MDTV, mean density of total volume; MDBV, mean density of bone volume; mRNA, messenger RNA; PDDR, pseudovitamin D-deficiency rickets; PTH, parathyroid hormone; Tb.N, trabecular number; Tb.Sp, trabecular separation; Tb.Th, trabecular thickness; TV, total volume; μCT, micro-computed tomography; VDR, vitamin D receptor; VD3, cholecalciferol; WT, wild-type.
Literature Cited
- 1.Fleet JC. Molecular regulation of calcium metabolism. In: Weaver CM, Heaney RP, editors. Calcium in human health. Totowa (NJ): Humana Press; 2006. pp. 163–90. [Google Scholar]
- 2.Fu GK, Portale AA, Miller WL. Complete structure of the human gene for the vitamin D 1alpha-hydroxylase, P450c1alpha. DNA Cell Biol. 1997;16:1499–507. doi: 10.1089/dna.1997.16.1499. [DOI] [PubMed] [Google Scholar]
- 3.Haussler MR, Whitfield GK, Haussler CA, Hsieh JC, Thompson PD, Selznick SH, Dominguez CE, Jurutka PW. The nuclear vitamin D receptor: biological and molecular regulatory properties revealed. J Bone Miner Res. 1998;13:325–49. doi: 10.1359/jbmr.1998.13.3.325. [DOI] [PubMed] [Google Scholar]
- 4.Dardenne O, Prud’homme J, Arabian A, Glorieux FH, St Arnaud R. Targeted inactivation of the 25-hydroxyvitamin D3-1α-hydroxylase gene (CYP27B1) creates an animal model of pseudovitamin D-deficiency rickets. Endocrinology. 2001;142:3135–41. doi: 10.1210/endo.142.7.8281. [DOI] [PubMed] [Google Scholar]
- 5.Panda DK, Miao D, Tremblay ML, Sirois J, Farookhi R, Hendy GN, Goltzman D. Targeted ablation of the 25-hydroxyvitamin D 1 α-hydroxylase enzyme: evidence for skeletal, reproductive, and immune dysfunction. Proc Natl Acad Sci USA. 2001;98:7498–503. doi: 10.1073/pnas.131029498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoenderop JG, Dardenne O, Van Abel M, van der Kemp AW, Van Os CH, Arnaud R, Bindels RJ. Modulation of renal Ca2+ transport protein genes by dietary Ca2+ and 1,25-dihydroxyvitamin D3 in 25-hydroxyvitamin D3-1 α -hydroxylase knockout mice. FASEB J. 2002;16:1398–406. doi: 10.1096/fj.02-0225com. [DOI] [PubMed] [Google Scholar]
- 7.Dardenne O, Prud’homme J, Hacking SA, Glorieux FH, St Arnaud R. Correction of the abnormal mineral ion homeostasis with a high-calcium, high-phosphorus, high-lactose diet rescues the PDDR phenotype of mice deficient for the 25-hydroxyvitamin D-1 α -hydroxylase (CYP27B1) Bone. 2003;32:332–40. doi: 10.1016/s8756-3282(03)00023-1. [DOI] [PubMed] [Google Scholar]
- 8.Dardenne O, Prudhomme J, Hacking SA, Glorieux FH, St Arnaud R. Rescue of the pseudo-vitamin D deficiency rickets phenotype of CYP27B1-deficient mice by treatment with 1,25-dihydroxyvitamin D3: biochemical, histomorphometric, and biomechanical analyses. J Bone Miner Res. 2003;18:637–43. doi: 10.1359/jbmr.2003.18.4.637. [DOI] [PubMed] [Google Scholar]
- 9.Zehnder D, Bland R, Williams MC, McNinch RW, Howie AJ, Stewart PM, Hewison M. Extrarenal expression of 25-hydroxyvitamin D3-1 α -hydroxylase. J Clin Endocrinol Metab. 2001;86:888–94. doi: 10.1210/jcem.86.2.7220. [DOI] [PubMed] [Google Scholar]
- 10.Thomas MK, Lloyd-Jones DM, Thadhani RI, Shaw AC, Deraska DJ, Kitch BT, Vamvakas EC, Dick IM, Prince RL, et al. Hypovitaminosis D in medical inpatients. N Engl J Med. 1998;338:777–83. doi: 10.1056/NEJM199803193381201. [DOI] [PubMed] [Google Scholar]
- 11.Heaney RP, Dowell MS, Hale CA, Bendich A. Calcium absorption varies within the reference range for serum 25-hydroxyvitamin D. J Am Coll Nutr. 2003;22:142–6. doi: 10.1080/07315724.2003.10719287. [DOI] [PubMed] [Google Scholar]
- 12.Verstuyf A, Verlinden L, Segaert S, Van Etten E, Mathieu C, Bouillon R. Nonclassical effects of 1 α,25-dihydroxyvitamin D3 and its analogs. Miner Electrolyte Metab. 1999;25:345–8. doi: 10.1159/000057472. [DOI] [PubMed] [Google Scholar]
- 13.Bischoff-Ferrari HA, Giovannucci E, Willett WC, Dietrich T, Dawson-Hughes B. Estimation of optimal serum concentrations of 25-hydroxyvitamin D for multiple health outcomes. Am J Clin Nutr. 2006;84:18–28. doi: 10.1093/ajcn/84.1.18. [DOI] [PubMed] [Google Scholar]
- 14.Gottfried E, Rehli M, Hahn J, Holler E, Andreesen R, Kreutz M. Monocyte-derived cells express CYP27A1 and convert vitamin D3 into its active metabolite. Biochem Biophys Res Commun. 2006;349:209–13. doi: 10.1016/j.bbrc.2006.08.034. [DOI] [PubMed] [Google Scholar]
- 15.Hsu JY, Feldman D, McNeal JE, Peehl DM. Reduced 1 α-hydroxylase activity in human prostate cancer cells correlates with decreased susceptibility to 25-hydroxyvitamin D3-induced growth inhibition. Cancer Res. 2001;61:2852–6. [PubMed] [Google Scholar]
- 16.Reeves PG, Nielsen FH, Fahey GC. AIN-93 purified diets for laboratory rodents: final report of the American Institute of Nutrition Ad Hoc writing committee on the reformulation of the AIN-76A rodent diet. J Nutr. 1993;123:1939–51. doi: 10.1093/jn/123.11.1939. [DOI] [PubMed] [Google Scholar]
- 17.Fleet JC, Wood RJ. Specific 1,25(OH)2 D3-mediated regulation of trans-cellular calcium transport inCaco-2 cells. Am J Physiol. 1999;276:G958–64. doi: 10.1152/ajpgi.1999.276.4.G958. [DOI] [PubMed] [Google Scholar]
- 18.Song Y, Peng X, Porta A, Takanaga H, Peng JB, Hediger MA, Fleet JC, Christakos S. Calcium transporter 1 and epithelial calcium channel messenger ribonucleic acid are differentially regulated by 1,25 dihydroxyvitamin D3 in the intestine and kidney of mice. Endocrinology. 2003;144:3885–94. doi: 10.1210/en.2003-0314. [DOI] [PubMed] [Google Scholar]
- 19.Healy KD, Zella JB, Prahl JM, DeLuca HF. Regulation of the murine renal vitamin D receptor by 1,25-dihydroxyvitamin D3 and calcium. Proc Natl Acad Sci USA. 2003;100:9733–7. doi: 10.1073/pnas.1633774100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song Y, Kato S, Fleet JC, Vitamin D. Receptor (VDR) knockout mice reveal VDR-independent regulation of intestinal calcium absorption and ECaC2 and calbindin D9k mRNA. J Nutr. 2003;133:374–80. doi: 10.1093/jn/133.2.374. [DOI] [PubMed] [Google Scholar]
- 21.Judex S, Garman R, Squire M, Donahue LR, Rubin C. Genetically based influences on the site-specific regulation of trabecular and cortical bone morphology. J Bone Miner Res. 2004;19:600–6. doi: 10.1359/JBMR.040101. [DOI] [PubMed] [Google Scholar]
- 22.Vieth R, Bischoff-Ferrari H, Boucher BJ, Dawson-Hughes B, Garland CF, Heaney RP, Holick MF, Hollis BW, Lamberg-Allardt C, et al. The urgent need to recommend an intake of vitamin D that is effective. Am J Clin Nutr. 2007;85:649–50. doi: 10.1093/ajcn/85.3.649. [DOI] [PubMed] [Google Scholar]
- 23.Hathcock JN, Shao A, Vieth R, Heaney R. Risk assessment for vitamin D. Am J Clin Nutr. 2007;85:6–18. doi: 10.1093/ajcn/85.1.6. [DOI] [PubMed] [Google Scholar]
- 24.Glorieux FH, St-Arnaud R. Vitamin D pseudodeficiency. In: Feldman D, Pike JW, Glorieux FH, editors. Vitamin D. 2. Amsterdam: Elsevier; 2005. pp. 1197–205. [Google Scholar]
- 25.Armbrecht HJ, Hodam TL, Boltz MA. Hormonal regulation of 25-hydroxyvitamin D3-1 α -hydroxylase and 24- hydroxylase gene transcription in opossum kidney cells. Arch Biochem Biophys. 2003;409:298–304. doi: 10.1016/s0003-9861(02)00636-7. [DOI] [PubMed] [Google Scholar]
- 26.Murayama A, Takeyama KI, Kitanaka S, Kodera Y, Kawaguchi Y, Hosoya T, Kato S. Positive and negative regulations of the renal 25-hydroxyvitamin D3 1 α -hydroxylase gene by parathyroid hormone, calcitonin, and 1a,25(OH)2D3 in intact animals. Endocrinology. 1999;140:2224–31. doi: 10.1210/endo.140.5.6691. [DOI] [PubMed] [Google Scholar]
- 27.Amling M, Priemel M, Holzmann T, Chapin K, Rueger JM, Baron R, Demay MB. Rescue of the skeletal phenotype of vitamin D receptor ablated mice in the setting of normal mineral ion homeostasis: formal histomorphometric and biomechanical analysis. Endocrinology. 1999;140:4982–7. doi: 10.1210/endo.140.11.7110. [DOI] [PubMed] [Google Scholar]
- 28.Bronner F, Pansu D, Stein WD. An analysis of intestinal calcium transport across the rat intestine. Am J Physiol. 1986;250:G561–9. doi: 10.1152/ajpgi.1986.250.5.G561. [DOI] [PubMed] [Google Scholar]
- 29.Pansu D, Bellaton C, Roche C, Bronner F. Duodenal and ileal calcium absorption in the rat and effects of vitamin D. Am J Physiol. 1983;244:695–700. doi: 10.1152/ajpgi.1983.244.6.G695. [DOI] [PubMed] [Google Scholar]
- 30.Bishop JE, Collins ED, Okamura WH, Norman AW. Profile of ligand specificity of the vitamin D binding protein for 1 α, 25-dihydroxy-vitamin D3 and its analogs. J Bone Miner Res. 1994;9:1277–88. doi: 10.1002/jbmr.5650090818. [DOI] [PubMed] [Google Scholar]
- 31.Nykjaer A, Fyfe JC, Kozyraki R, Leheste JR, Jacobsen C, Nielsen MS, Verroust PJ, Aminoff M, de la Chapelle CA, Moestrup SK, et al. Cubilin dysfunction causes abnormal metabolism of the steroid hormone 25(OH) vitamin D3. Proc Natl Acad Sci USA. 2001;98:13895–900. doi: 10.1073/pnas.241516998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rowling MJ, Kemmis CM, Taffany DA, Welsh J. Megalin-mediated endocytosis of vitamin D binding protein correlates with 25-hydroxy-cholecalciferol actions in human mammary cells. J Nutr. 2006;136:2754–9. doi: 10.1093/jn/136.11.2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rojanasathit S, Haddad JG. Ontogeny and effect of vitamin D deprivation on rat serum 25-hydroxyvitamin D binding protein. Endocrinology. 1977;100:642–7. doi: 10.1210/endo-100-3-642. [DOI] [PubMed] [Google Scholar]
- 34.Teillaud C, Nemere I, Boukhobza F, Mathiot C, Conan N, Oboeuf M, Hotton D, Macdougall M, Berdal A. Modulation of 1alpha,25-dihydroxyvitamin D3-membrane associated, rapid response steroid binding protein expression in mouse odontoblasts by 1 α,25-(OH)2D3. J Cell Biochem. 2005;94:139–52. doi: 10.1002/jcb.20275. [DOI] [PubMed] [Google Scholar]
- 35.Mizwicki MT, Keidel D, Bula CM, Bishop JE, Zanello LP, Wurtz JM, Moras D, Norman AW. Identification of an alternative ligand-binding pocket in the nuclear vitamin D receptor and its functional importance in 1 α,25(OH)2-vitamin D3 signaling. Proc Natl Acad Sci USA. 2004;101:12876–81. doi: 10.1073/pnas.0403606101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boyan BD, Sylvia VL, Dean DD, Del Toro F, Schwartz Z. Differential regulation of growth plate chondrocytes by 1 α,25-(OH)2 D3 and 24R,25-(OH)2 D3 involves cell-maturation-specific membrane-receptor-activated phospholipid metabolism. Crit Rev Oral Biol Med. 2002;13:143–54. doi: 10.1177/154411130201300205. [DOI] [PubMed] [Google Scholar]
- 37.AIN. Report of the American Institute of Nutrition ad hoc committee on standards for nutritional studies. J Nutr. 1977;107:1340–8. doi: 10.1093/jn/107.7.1340. [DOI] [PubMed] [Google Scholar]
- 38.Park KS, Kim SK, Kim MS, Cho EY, Lee JH, Lee KU, Pak YK, Lee HK. Fetal and early postnatal protein malnutrition cause long-term changes in rat liver and muscle mitochondria. J Nutr. 2003;133:3085–90. doi: 10.1093/jn/133.10.3085. [DOI] [PubMed] [Google Scholar]