

Abstract
T cell activation rapidly and transiently regulates the functional activity of integrin receptors. Stimulation of CD3/T cell receptor, CD2 or CD28, as well as activation with phorbol esters, can induce within minutes an increase in β1 integrin-mediated adhesion of T cells to fibronectin. In this study, we have produced and utilized a mutant of the Jurkat T cell line, designated A1, that lacks protein and mRNA expression of the β1 integrin subunit but retains normal levels of CD2, CD3, and CD28 on the cell surface. Activation-dependent adhesion of A1 cells to fibronectin could be restored upon transfection of a wild-type human β1 integrin cDNA. Adhesion induced by phorbol 12-myristate 13-acetate-, CD3-, CD2-, and CD28 stimulation did not occur if the carboxy-terminal five amino acids of the β1 tail were truncated or if either of two well-conserved NPXY motifs were deleted. Scanning alanine substitutions of the carboxy-terminal five amino acids demonstrated a critical role for the tyrosine residue at position 795. The carboxy-terminal truncation and the NPXY deletions also reduced adhesion induced by direct stimulation of the β1 integrin with the activating β1 integrin-specific mAb TS2/16, although the effects were not as dramatic as observed with the other integrin-activating signals. These results demonstrate a vital role for the amino-terminal NPXY motif and the carboxy-terminal end of the β1 integrin cytoplasmic domain in activation-dependent regulation of integrin-mediated adhesion in T cells. Furthermore, the A1 cell line represents a valuable new cellular reagent for the analysis of β1 integrin structure and function in human T cells.
INTRODUCTION
The functional activity of adhesion receptors expressed on T lymphocytes can be rapidly modulated by signals that T cells receive from the external environment. These adhesion regulatory signals can result in rapid changes in both adhesion receptor expression and function. One example of activation-induced changes in adhesion receptor expression is the rapid proteolytic cleavage of the L-selectin receptor upon activation of T cells and neutrophils (Kishimoto et al., 1989; Jung and Dailey, 1990). In contrast, adhesion mediated by integrin receptors expressed on T cells is regulated by rapid changes in the functional activity of integrins, rather than changes in levels of integrin expression on the cell surface. Thus, resting T lymphocytes express integrins that bind poorly to extracellular matrix (ECM) proteins or cell surface molecules. However, activation of T cells via the antigen-specific CD3/T cell receptor (CD3/TCR) complex, several different coreceptors, or inflammatory chemokines results in increased integrin-mediated adhesion within seconds to minutes (Dustin and Springer, 1989; van Kooyk et al., 1989; Shimizu et al., 1990; Diamond and Springer, 1994; Campbell et al., 1998). This rapid, but transient, change in integrin-mediated adhesiveness allows T cells to adhere to endothelial cells lining the blood vessel wall, interact with and respond to antigen-presenting cells and other cellular targets in tissue, and to navigate through, and respond to, the rich microenvironment in tissue sites. The significance of activation-dependent regulation of integrin function is further illustrated by recent reports of novel forms of leukocyte adhesion deficiency characterized by defective activation of β2 integrin-mediated adhesion (Kuijpers et al., 1997 and Harris, unpublished observations).
Integrin receptors consist of two noncovalently associated subunits, an α subunit and a β subunit. Integrin β subunit cytoplasmic domains are essential for activation-induced changes in cellular adhesion, as well as for transmitting signals back inside the cell upon interaction with ligand. Expression of β subunit cytoplasmic domains alone is often sufficient to transmit signals, and overexpression of integrin β subunit cytoplasmic domain sequences can inhibit endogenous integrin-mediated function (Akiyama et al., 1994; Chen et al., 1994; Finkelstein et al., 1997; Tahiliani et al., 1997). The use of integrin chimeras has been particularly fruitful, since the role of the integrin cytoplasmic domain can be analyzed in the context of a background of endogenous β1 integrin expression found in nucleated cells. A number of studies have utilized β1 chimeric proteins and nonhuman cells, such as CHO cells, to characterize effects of mutations in the β1 cytoplasmic domain on integrin function. These studies have demonstrated critical roles for specific regions of the β1 integrin cytoplasmic domain, particularly two well-conserved NPXY motifs, for localization of β1 integrins to focal contacts (Reszka et al., 1992; Vignoud et al., 1997). Antibodies that recognize activation-dependent epitopes on integrin β subunits have also been used to demonstrate a role for NPXY motifs in the β1 and β3 cytoplasmic domains in the regulation of integrin conformation (Bazzoni et al., 1995; Hughes et al., 1995; O’Toole et al., 1995; Luque et al., 1996; Puzon-McLaughlin et al., 1996). However, it remains unclear whether novel epitopes are precise predictors of the activation-induced changes in adhesion mediated by β1 integrins (Bazzoni and Hemler, 1998).
The expression of significant levels of endogenous β1 integrins on essentially all nucleated cells has complicated further attempts to study the role of the β1 cytoplasmic domain in β1 integrin function. Given the critical role that activation plays in regulating β1 integrin-mediated adhesion in T lymphocytes, we sought to develop an experimental system that would allow for rapid structure/function analysis of the β1 integrin subunit in T cells without the complications of endogenous β1 integrin subunit expression. Jurkat T cells express the α4β1 and α5β1 integrins and exhibit activation-dependent adhesion to the β1 integrin ECM ligand fibronectin (FN) (Mobley et al., 1994). We report in this study the isolation of a mutant of the Jurkat T cell line that does not express the β1 integrin subunit and does not adhere to FN. Transient expression of the integrin β1 subunit by DNA-mediated gene transfer yields transfectants that express the β1 integrin subunit and demonstrate activation-induced adhesion to FN. By transient expression of β1 integrin cytoplasmic mutants in this cell line, we demonstrate a critical role for the carboxy-terminal end of the β1 integrin cytoplasmic domain, as well as the NPXY motifs, in activation-dependent regulation of integrin-mediated adhesion in human T cells.
MATERIALS AND METHODS
Cells
The Jurkat E6–1 cell line was obtained from the American Type Tissue Collection (ATCC, Rockville, MD). Jurkat 6A, a subclone of E6–1, and the A1 mutant were maintained in RPMI 1640 (Life Technologies, Grand Island, NY) supplemented with 10% FCS (Atlanta Biological, Norcross, GA), l-glutamine, and penicillin/streptomycin (complete medium).
Generation and Isolation of A1 Cells
Mutant A1 was isolated as previously described (Mobley et al., 1994, 1996). Briefly, Jurkat 6A cells were irradiated with 300 rads of γ-rays, and mutants were selected by collecting cells not adherent to FN upon stimulation with the phorbol ester phorbol dibutyrate (Sigma, St. Louis, MO). After several rounds of selection, cells were analyzed for expression of CD2, CD3, CD28, α4 integrin, α5 integrin, and β1 integrin. The A1 mutant lacked surface expression of the β1 integrin subunit, as assessed by flow cytometric analysis. This mutant was cloned several times by limiting dilution to obtain a clonal population of β1 integrin-deficient cells.
Antibodies and Other Reagents
The anti-glycophorin mAb 10F7, the β1 integrin-specific mAb TS2/16, the α1 integrin-specific mAb TS2/7, the CD3-specific mAbs 38.1 and OKT3, and αL-specific mAb TS1/22 were from ATCC (Rockville, MD). The β1 integrin-specific mAb B-D15 was purchased from Biosource International (Camarillo, CA). The β1 integrin-specific mAb P4C10, the α2 integrin-specific mAb P1E6, the α3 integrin-specific mAb P1B5, and the α5 integrin-specific mAb P1D6 were obtained from Life Technologies/BRL (Grand Island, NY). The phycoerythrin (PE)-conjugated β1 integrin-specific mAb 4B4-RD1 was purchased from Coulter Immunology (Hialeah, FL). The α4 integrin-specific mAb NIH49d-1 was provided by Dr. S. Shaw (National Institutes of Health, Bethesda, MD). The β7-specific mAb Fib504 was provided by Dr. G. van Seventer (University of Chicago, Chicago, IL). The α6-specific mAb GoH-3 was provided by Dr. A. Sonnenberg (Central Laboratory of the Netherlands Red Cross Blood Transfusion Service, Amsterdam). The CD2-specific mAb 95–5-49 was provided by Dr. R. Gress (National Institutes of Health, Bethesda, MD). The CD2-specific mAb 9–1 was provided by Dr. S.Y. Yang (Memorial-Sloan Kettering Cancer Center, New York, NY). The CD28-specific mAb 9.3 was a gift from Dr. J. Ledbetter (Bristol-Myers Squibb, Seattle, WA). Anti-HA mAbs were purchased from BAbCo (Richmond, CA) and Boehringer-Mannheim (Indianapolis, IN). Goat anti-mouse immunoglobulin G (IgG)-FITC and goat anti-rat IgG-FITC were obtained from Southern Biotechnology (Birmingham, AL). Goat anti-mouse IgG was purchased from Cappel/Organon-Teknika (Durham, NC). Phorbol 12-myristate 13-acetate (PMA) was purchased from Sigma Chemical, dissolved in DMSO (100 μg/ml), and stored at −70°C. Human FN was generously provided by Dr. J. McCarthy (University of Minnesota, Minneapolis, MN).
DNA Constructs
The expression vector pHSX was derived from pMH-Neo (provided by Dr. B. Bierer, Dana-Farber Cancer Institute, Boston, MA) as follows. A HindIII site at position 1028 of the multiple cloning site was deleted by restriction enzyme digest and converted to blunt ends. The SalI (position 1042) to XhoI (position 1063) sequence was deleted by restriction enzyme digest and ligation of cohesive ends. The 3.6-kilobase (kb) EcoRI fragment from pECE.β1 encoding human β1 integrin (provided by Dr. E. Ruoslahti, Burnham Institute, La Jolla, CA) was cloned into the EcoRI site (at position 1070) of the multiple cloning site of the pHSX vector to create the pHSX-β1 vector.
An XhoI site at position 3590 (position 2520 of β1) was introduced by site-directed mutagenesis (Transformer Site-directed mutagenesis kit, CLONTECH, Palo Alto, CA). A HindIII site at position 3844 (position 2774 of the β1 sequence) was also deleted by site-directed mutagenesis as described above. Sequences were verified by the dideoxy method using Sequenase T7 DNA Polymerase (United States Biochemical, Cleveland, OH) according to the manufacturer’s instructions. The resulting vector, pHSX-β1X, contains a single HindIII site at position 3427, which lies immediately 5′ of the β1 cytoplasmic domain sequence, and an XhoI site at position 3590, just 3′ of the cytoplasmic domain sequence.
Constructs encoding the mutants β1(793), β1(ΔNPIY), and β1(ΔNPKY) were generated by gene synthesis, as described previously (Zell et al., 1996), using the following oligonucleotides: β1(793) primer 3: (5′-TCA-TAT-CAC-GGA-TTG-ACC-ACA-GTT-GTT-ACGGCA-CTC-TTA-TAA-ATA-GGA-TTT-TCA-CCC-GTG-TCC-CAT-TTG GCA-3′); and β1(793) primer 4: (5′-TCG-CTC-GAG-TCA-TTT-TCC-CTCATA-TCA-CGG-ATT-GAC-CAC-3′). β1(ΔNPIY) primer 3: TCA-TACTTC-G GA-TTG-ACC-ACA-GTT-GTT-ACG-GCA-CTC-TTT-TCA-CCCGTG-TCC-CAT-TTG-GCA (primers 1, 2 and 4 were as used for β1(793). β1(ΔNPKY) primer 3:(5′-TCG-ACC-ACA-GTT-GTT-ACG-GCA-CTCTTA-TAA-ATA-GGA-TTT-TCA-CCC-GTG-TCC-CAT-TTG-GCA-3′) and β1(ΔNPKY) primer 4: (5′-TCG-CTC-GAG-TCA-TTT-TCC-CTC-GAC-CAC-AGT-TGT-TAC-GGC-AC-3′). Oligonucleotides were synthesized on a Perkin Elmer-Cetus-Applied Biosystems (Foster City, CA) 392 Oligonucleotide Synthesizer. Primer 4 was used directly from the crude eluate, whereas primer 3 was purified on OPC columns (Perkin Elmer-Cetus-Applied Biosystems). PCR reactions were performed using the method of Gibbs et al. (1989) as described by Zell et al. (1996). Reaction products were fractionated by agarose gel electrophoresis. The appropriate bands were excised from the gel, purified using Wizard PCR preps (Promega, Madison, WI), digested with HindIII and XhoI, and cloned into the HindIII/XhoI site of the pHSX-β1X expression vector. All pHSX-β1 constructs were confirmed by automated DNA sequencing.
The alanine substitutions at positions 794, 795, 796, 797, and 798 and the phenylalanine substitution at position 795 of the β1 cytoplasmic domain were made by site-directed mutagenesis (QuickChange Site-Directed Mutagenesis kit, Stratagene, La Jolla, CA) according to the manufacturer’s instructions. Each mutation was confirmed by automated DNA sequencing of plasmid DNA. Primers for the point mutations K794A, Y795A, Y795F, G797A, and K798A were synthesized by Amitof Biotech (Boston, MA), and primers for the E796A mutation were synthesized by Life Technologies (Grand Island, NY). Primer sequences for the substitutions are listed below.
K794A primer A: 5′-CTG-TGG-TCA-ATC-CGG-CGT-ATC-AGG-GAA-AAT-GAG-TAC-TGC-CC-3′; K794A primer B: 5-GAC-ACC-AGT-TAG-GCC-GCA-TAC-TCC-CTT-TTA-CTC-ATG-ACG-GG-3′. Y795A primer A: 5′-CTG-TGG-TCA-ATC-CGA-AGG-CTG-AGG-GAA-AAT-GAG-TAC-TGC-CCG-TGC-3′; Y795A primer B: 5′-GCA-CGG-GCA-GTA-CTC-ATT-TTC-CCT-CAG-CCT-TCG-GAT-TGA-CCA-CAG-3′. Y795F primer A: 5′-GCA-CGG-GCA-GTA-CTC-ATT-TTC-CCT-CAA-ACT-TCG-GAT-TGA-CCA-CAG-3′; Y795F primer B: 5′-CTG-TGG-TCA-ATC-CGA-AGT-TTG-AGG-GAA-AAT-GAG-TAC-TGC-CCG TGC-3′. E796A primer A: 5′-CTG-TGG-TCA-ATC-CGA-AGT-ATGCGG-GAA-AAT-GAG-TAC-TGC-CC-3′; E796A primer B: 5′-GGGCAG-TAC-TCA-TTT-TCC-CGC-ATA-CTT-CGG-ATT-GAC-CAC-AG 3′. G797A primer A: 5′-CTG-TGG-TCA-ATC-CGA-AGT-ATG-AGGCAA-AAT-GAG-TAC-TGC-CC-3′; G797A primer B: 5′-GGG-CAG-TACTCA-TTT-TGC-CTC-ATA-CTT-CGG-ATT-GAC-CAC-AG-3′. K798A primer A: 5′-CTG-TGG-TCA-ATC-CGA-AGT-ATG-AGG-GAG-CAT-GAG-TAC-TGC-CCG-TGC-3′; K798A primer B: 5′-GCA-CGG-GCA-GTA-CTC-ATG-CTC-CCT-CAT-ACT-TCG-GAT-TGA-CCA-CAG-3′.
Transient Transfections
A1 cells were transfected by electroporation using a BTX Electro Square Porator T820 (BTX, San Diego, CA). Ten million cells in log-phase growth were harvested, washed twice with Opti-MEM (Life Technologies), and suspended in 0.2 ml of Opti-MEM containing 30 μg of the indicated pHSX-β1x construct and 20 μg of pHook2 (Invitrogen, San Diego, CA). Cells and DNA were incubated 10 min at room temperature, and then electroporated at 240 mV with a single 25-ms pulse. Cells were allowed to recover after transfection for 30 min at room temperature, and then seeded in complete medium and incubated 16–20 h at 37°C in 5% CO2.
Selection of Transfectants
A1 cells transiently expressing pHSX-β1 and hemagglutinin (HA)-tagged pHook2 were positively selected with detachable magnetic beads. Cells were harvested from culture 16–20 h posttransfection, and viable cells were recovered by Ficoll-Hypaque (Sigma Chemical) density gradient centrifugation, washed twice in PBS, and incubated with saturating amounts of mouse anti-HA mAb (clone 16B12) in PBS containing 0.1% BSA (PBS/0.1%BSA), for 30 min at 4°C. Cells were washed to remove unbound antibody and suspended in PBS/0.1% BSA (4 × 106 cells/ml). Magnetic beads conjugated with rat anti-mouse IgG1 via a DNA linker (rat anti-mouse IgG1 Cellection kit, Dynal, Lake Success, NY) were used to isolate the transfectants according to the manufacturer’s instructions. Briefly, rat anti-mouse beads were added to antibody-coated cells at 10 beads per target cell in the above cell suspension. Cells and beads were incubated for 1 h at 4°C on a rotator. The bead-rosetted cells were captured with a magnet, washed in 1 ml RPMI/1% FCS, and collected as per the manufacturer’s instructions. The antibody-coated cells were detached from the magnetic beads by DNase treatment, and the cells were collected and counted.
Flow Cytometry
Single-color flow cytometric analysis was performed as described previously (Mobley et al., 1996) by staining cells with saturating amounts of mouse or rat anti-human antibodies, followed by appropriate FITC-conjugated secondary antibody for detection. In some cases PE-conjugated anti-β1 antibody (4B4-RD1) was used. Stained cells were analyzed on a FACScan (Becton Dickinson, San Jose, CA).
Adhesion Experiments
Adhesion assays were performed as described by Mobley et al. (1995). Briefly, 96-well microtiter plates (Costar, Cambridge, MA) were incubated with the indicated concentrations of FN overnight at 4°C. Unbound binding sites were blocked with PBS/2.5% BSA. Cells were labeled with 2 μg/ml calcein-AM (Molecular Probes, Eugene OR) for 20 min at 37°C, washed, and added to wells containing the appropriate stimuli. PMA was used at 10 ng/ml; CD2 was stimulated with a 1:10 dilution of 95-5-49 hybridoma culture supernatant and a 1:2000 dilution of mAb 9–1 ascites fluid. Direct β1 stimulation was with a 1:10 dilution of TS2/16 hybridoma culture supernatant. For CD3 stimulation, wells contained 3 μg/ml mAb 38.1. For CD28 stimulation, cells were incubated for 30 min on ice with saturating amounts of 9.3, washed, and added to wells containing 1 μg/well goat anti-mouse IgG. The cells were allowed to settle in the plates for 60 min at 4°C, and then warmed rapidly for the indicated timepoints. Nonadherent cells were washed off, and adherent cells were quantitated using a fluorescence plate reader (Biotek). Percent adhesion was assessed as:
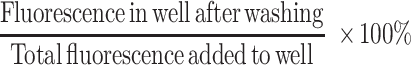 |
All data are averages of triplicate wells for each condition.
Northern Blotting Analysis
Poly-A RNA was isolated using the FastTrack 2.0 mRNA isolation system (Invitrogen). Poly-A RNA (2 μg) was separated on a formaldehyde gel and transferred to nylon membrane (Hybond-N, Amersham, Arlington Heights, IL). Probes used were a 1.3-kb BglII human β1 fragment from pECE.β1 (provided by Dr. E. Ruoslahti, Burnham Institute, La Jolla, CA) and the 1.0-kb BamHI cyclophilin fragment from pGEM4Z (provided by Dr. V. Dixit, University of Michigan, Ann Arbor, MI).
Biotinylation and Immunoprecipitation
Jurkat and A1 cells were biotin labeled, and immunoprecipitations were performed as previously described (Finkelstein et al., 1997). Precleared lysates were incubated with goat anti-mouse IgG-coupled Sepharose beads (Zymed, South San Francisco, CA) precoated with either the αL-specific mAb TS1/22 or the β1-specific mAb P4C10. Immunoprecipitates were washed, boiled 5 min, and then separated on a 5% SDS-polyacrylamide gel. Proteins were transferred to polyvinylidene difluoride membrane (Millipore, Bedford, MA). The membrane was blocked with PBS/4% BSA, and incubated with a 1:1000 dilution of horseradish peroxidase-conjugated streptavidin (Life Technologies), and protein was detected by enhanced chemiluminescence (Pierce, Rockford, IL).
RESULTS
Generation and Characterization of A1, a β1 Integrin-deficient T Cell Line
We have previously used Jurkat T cells to analyze activation-dependent regulation of β1 integrin-mediated adhesion (Mobley et al., 1994, 1996). One approach that we have employed with this cell line is the use of low-dose γ-irradiation and selection by panning on FN-coated dishes to isolate mutants with defects in β1 integrin expression and/or function. Three classes of mutants were isolated using this technique. The first class of mutants expressed normal levels of β1 integrin receptors, but had lost expression of specific integrin-regulatory receptors, such as CD3/TCR or CD2 (Mobley et al., 1994). The second class of mutants expressed normal levels of cell surface β1 integrins and integrin regulators, such as CD2, CD3, and CD28, but had defects in integrin activation (Mobley et al., 1996). Finally, we isolated a class of mutant that lacks expression of the β1 integrin subunit. This mutant is described in this report. The Jurkat mutant, A1, was isolated due to its inability to bind to FN after stimulation with the phorbol ester phorbol dibutyrate. Flow cytometric analysis shows that the A1 mutant does not express the integrin β1 subunit, as assessed with three different β1 integrin-specific mAbs (Figure 1, A and B). Consequently, A1 cells also lack expression of the α1, α2, α3, α5, and α6 subunits, which normally associate with the β1 subunit. Cell surface expression of the α4 subunit was also dramatically reduced in comparison to wild-type Jurkat cells but was still present at low levels. This is likely due to pairing of the α4 subunit with the β7 subunit, since Jurkat cells express the β7 subunit, as assessed by staining with the β7 integrin-specific mAb Fib504 (Figure 1B). The expression of the β7 subunit on wild-type Jurkat appears to be broader, containing a higher expressing population that is not seen in the A1 cells. Expression of the β2 integrin LFA-1 was not affected by the loss of β1 integrin cell surface expression in the A1 mutant (Figure 1A). Furthermore, receptors that regulate integrin activity, including CD3/TCR, CD2, and CD28, were expressed on A1 cells at levels comparable to wild-type Jurkat cells.
Figure 1.

Flow cytometric analysis of wild-type Jurkat cells and β1 integrin-deficient A1 Jurkat mutant. (A) Negative control (open profiles) staining with the anti-glycophorin mAb 10F7 is shown in each panel for ease of comparison. Solid profiles indicate staining with the following indicated mAbs: β1-specific mAbs B-D15 (left β1 panel) and P4C10 (right β1 panel), the α1-specific mAb TS2/7, the α2-specific mAb P1E6, the α3-specific mAb P1B5, the α4-specific mAb NIH49d-1, the α5-specific mAb P1D6, the α6-specific mAb GoH-3, the CD2-specific mAb 95–5-49, the CD3-specific mAb OKT3, the CD28-specific mAb 9.3, and the αL (LFA-1)-specific mAb TS1/22. (B) Negative control (open profiles) staining with secondary goat anti-mouse IgG alone is shown for comparison. Solid profiles represent staining with either a β7-specific mAb FIB504 or β1-specific mAb TS2/16 followed by appropriate secondary antibody.
The lack of cell surface expression of the β1 subunit in A1 cells was also assessed by immunoprecipitation from biotin-labeled cell lysates. Figure 2 shows that while the β1 subunit was efficiently immunoprecipitated from wild-type Jurkat cells, β1 could not be precipitated from A1 cell lysates. In contrast, the β2 integrin subunit could be immunoprecipitated from both wild-type Jurkat and A1 cell lysates (Figure 2). Northern blotting analysis was also performed to determine whether the loss of β1 integrin protein expression in A1 cells was due to loss of β1 integrin mRNA. Using a probe specific for human β1 integrin, we determined that A1 cells expressed less than 2% of the β1 mRNA detected in wild-type Jurkat cells, as assessed by densitometric analysis of Northern blots (our unpublished results). This minimal amount of β1 mRNA could only be detected after prolonged exposure of the autoradiograph. We conclude from these results that the A1 mutant does not express the integrin β1 subunit on the cell surface and expresses minimal amounts of β1 mRNA.
Figure 2.

The A1 mutant does not express β1 integrin protein. Jurkat cells and the A1 mutant were biotinylated and lysed in 1% Triton detergent, and lysates were subjected to immunoprecipitation (IP) with goat anti-mouse Sepharose beads conjugated with the β1-specific mAb P4C10 or the β2-specific mAb TS1/22. IPs were washed, run on 5% acrylamide gel, and transferred to nitrocellulose. Protein was detected by incubating the membrane with Streptavidin-HRP and visualized by chemiluminescence.
Functional Analysis of A1 Cells
We next compared the adhesion of the β1 integrin-deficient A1 cells and wild-type Jurkat cells to FN. As previously described, the basal level of wild-type Jurkat cell adhesion to FN can be enhanced within minutes by treatment with the phorbol ester PMA, mitogenic pairs of CD2-specific mAbs, or antibody cross-linking of the CD3/TCR or CD28. Direct stimulation of β1 integrins with the activating β1 integrin-specific mAb TS2/16 also resulted in increased adhesion of wild-type Jurkat cells (Figure 3). In contrast, A1 cells were unable to adhere to FN, even in the presence of these various integrin-activating signals. Although FN has been reported to be a ligand for the α4β7 integrin (Postigo et al., 1993), the low level of α4β7 expressed on A1 cells was not able to mediate the adhesion of A1 cells to FN. Therefore, the loss of β1 integrin expression on A1 cells is associated with an inability to adhere to the β1 integrin ligand FN.
Figure 3.

A1 cells do not adhere to the β1 integrin ligand FN. Jurkat and A1 cells were analyzed for adhesion to the indicated concentrations of FN for 20 min at 37°C without stimulation (□), or after stimulation with PMA (○), the activating β1-specific mAb TS2/16 (▵), the CD3-specific mAb 38.1 (▪), mitogenic pairs of CD2-specific mAbs (•), or the CD28-specific mAb 9.3 (▴). The data are represented as the mean percent adhesion of triplicate wells ± SD. Results shown are one of at least three representative experiments.
Transfection of Mutant A1 with Human β1 Integrin cDNA Restores Activation-dependent β1 Integrin-mediated Adhesion to FN
We transiently expressed cDNA encoding the human β1 integrin subunit in A1 cells by electroporation. Expression of the β1 subunit was generally observed in 10–30% of the electroporated cells (Figure 4D). Two-color flow cytometry was used to analyze the expression of the α4 and α5 integrin subunits, as well as CD3, CD2, and CD28, on the β1-negative and β1-positive subpopulations of A1 cells after electroporation (Figure 4). Expression of both the α4 and α5 subunits increased upon reexpression of the β1 subunit, but the level of expression of CD3, CD2, and CD28 was not affected.
Figure 4.

The β1 integrin subunit is transiently expressed in A1 cells without altered expression of CD2, CD3, or CD28. A1 cells were transiently transfected with pHSX-β1x DNA. Sixteen hours posttransfection, viable cells were isolated and stained for two-color flow cytometry using PE-conjugated β1-specific mAb 4B4 (x-axis) and mAbs against the indicated surface molecules followed by goat anti-mouse-FITC (y-axis). Quadrant boundaries were set from cells treated with nonspecific secondary antibody only. Panel D represents cells stained with anti-β1 mAb 4B4-PE alone. The β1+ population in this experiment was 30.2% of total, and is shown in the lower right quadrant of panel D. The expression of CD2, CD3, CD28, α4 integrin, or α5 integrin on β1− or β1+ cells is shown in panels A, B, C, E, and F, respectively.
We developed an enrichment strategy to determine the adhesive capabilities of A1 transfectants expressing the β1 subunit. A1 cells were cotransfected with the β1 expression vector and pHook2, a plasmid that encodes for a hapten-specific single chain antibody molecule that is expressed on the cell surface as an integral membrane protein (Chesnut et al., 1996). The antibody region is expressed extracellularly and also contains a hemagglutinin (HA) tag. Two-color flow cytometry with anti-HA and anti-β1 mAbs was used to demonstrate that A1 cells transfected with pHook2 only expressed the HA-tagged pHook2 molecule but remained β1-negative. Upon transfection with both pHook2 and β1, the subpopulation of A1 cells expressing pHook2 also expressed the β1 subunit (Figure 5). The subpopulation of A1 transfectants expressing pHook2 was then isolated with an anti-HA mAb and magnetic beads. As shown in Figure 5, this selection procedure enriched for a population of A1 cells expressing the transfected β1 cDNA. Expression of β1 in this selected population was typically broad and did not approach the high level of endogenous β1 integrin expressed on the wild-type parental Jurkat cells. Thus, this method allowed us to rapidly select and isolate β1+ transfectants without using anti-β1 mAbs that might alter integrin function.
Figure 5.

Coexpression of β1 and pHook2 in A1 cells. A1 cells were transiently transfected with pHook2 alone (left column), or with pHook2 and pHSX-β1x together (right column). Cells were collected 16 h after transfection and analyzed by two-color flow cytometry using an anti-HA mAb and GAR-FITC (y-axis) and PE-conjugated β1-specific mAb 4B4 (x-axis). Transfectants were analyzed before (top and middle panels) or after (bottom panels) magnetic bead selection. Coexpression of β1 and the HA tag is represented as percentage of total cells in the upper right quadrant (top panels).
To measure the ability of β1+ A1 cells to adhere to FN, we isolated A1 cells transiently transfected with pHook2 alone or cotransfected with β1 cDNA (Figure 6A). Similar to what was observed with untransfected A1 cells, A1 cells transfected with pHook2 alone did not adhere to FN, even after stimulation. Thus, expression of the pHook2 molecule did not alter the adhesion of A1 cells to FN. A1 cells expressing wild-type human β1 exhibited increased basal adhesion to FN when compared with control A1 cells expressing pHook2 only. Furthermore, stimulation of β1+ A1 cells with PMA, CD2 ligation, or cross-linking of CD3/TCR or CD28 resulted in a rapid, dose-dependent increase in adhesion to FN (Figure 6A). Like the adhesion response observed with wild-type Jurkat cells (Mobley et al., 1994), the adhesion of β1+ A1 transfectants to FN was rapid but transient: adhesion peaked within 10–20 min of stimulation and began to decrease within 40 min after stimulation (our unpublished results). The adhesion of β1+ A1 transfectants to FN was inhibited by the β1 integrin-specific mAb P5D2, or by a combination of α4- and α5-specific mAbs, indicating that the increased adhesion of these cells was mediated by endogenous α4 and α5 pairing with the β1 subunit encoded by the transfected cDNA (Figure 6B and our unpublished results). Together, these results show that A1 cells are capable of transiently expressing functional β1 integrins, and that β1+ transfectants exhibit activation-induced cell adhesion to a β1 integrin ligand.
Figure 6.

Adhesion to FN of A1 cells transfected with pHook2 alone or with β1. A1 cells were transiently transfected with pHook2 alone, or together with pHSX-β1x DNA. Cells were recovered after transfection, and β1-expressing transfectants were isolated as described in MATERIALS AND METHODS. (A) A1 cells expressing pHook2 alone or with β1 were analyzed for adhesion to the indicated concentrations of FN for 20 min at 37°C without stimulation (- - - - -), or after stimulation with PMA (▪), the activating β1-specific mAb TS2/16 (*), the CD3-specific mAb 38.1 (○), mitogenic pairs of CD2-specific mAbs (▴), or the CD28-specific mAb 9.3 (•). (B) A1 cells expressing pHook2 alone or with β1 were analyzed for adhesion as in panel A to 1 μg/well of FN in the absence or presence of a 1:200 dilution of ascites of the anti-β1 mAb P5D2. Cells were either unstimulated (left bar in each set of bars) or stimulated with PMA, mitogenic pairs of CD2-specific mAbs, the activating β1-specific mAb TS2/16, or the CD3-specific mAb 38.1 (left to right in each set of bars). The data are represented as the mean percent adhesion of triplicate wells ± SD. Results shown are one of at least three representative experiments.
Deletion of the Carboxy-terminal Five Amino Acids of the Cytoplasmic Domain Abolishes Adhesion of β1+ A1 Cells to FN
To study the structural requirements within the β1 integrin subunit cytoplasmic domain that are responsible for activation-dependent adhesion to FN, we generated a panel of cDNA clones encoding for β1 integrin subunits with specific cytoplasmic domain mutations (Figure 7). Each mutant β1 integrin subunit was then expressed transiently in A1 cells and analyzed for adhesion to FN (Figure 8 and 9). Although the level of β1 expression in transient transfectants varied from experiment to experiment, each β1 integrin cytoplasmic mutant tested was expressed on the cell surface at levels comparable to wild-type β1 in any given experiment (Figures 8A and 9). A deletion of the carboxy-terminal five amino acids (KYEGK) of the β1 cytoplasmic domain, designated β1(793), was unable to mediate adhesion to FN after stimulation (Figure 8B). Even at concentrations of immobilized FN that mediated maximal adhesion of Jurkat cells to FN, both basal and stimulated adhesion of A1 transfectants expressing the β1(793) was reduced when compared with the wild-type β1 subunit. Interestingly, adhesion induced by direct stimulation of the β1 activating mAb TS2/16 was also inhibited by deletion of the carboxy-terminal five amino acids. To establish whether the β1(793) transfectants exhibited delayed adhesion to FN, we tested the adhesion up to 40 min after stimulation. At all time points tested, the β1(793) transfectants were unable to adhere to FN (our unpublished results).
Figure 7.

Amino acid sequence of the wild-type β1 integrin cytoplasmic domain and the various cytoplasmic domain mutations analyzed in this study. The amino acid sequences of the wild-type β1 integrin cytoplasmic domain and each mutant construct are written in single letter code. Amino acid substitutions are boxed.
Figure 8.

Lack of adhesion to FN of A1 cells expressing the β1(793) mutant. A1 cells were transiently transfected with pHook2 alone, or together with pHSX-β1x or pHSX-β1(793) DNA. Cells were recovered after transfection, and β1-expressing transfectants were isolated as described in MATERIALS AND METHODS. (A) Cells were stained with PE-conjugated β1-specific mAb 4B4 as described in Figure 4 legend. (B) A1 cells expressing pHook2 alone or together with β1 or β1(793) were analyzed for adhesion to the indicated concentrations of FN for 20 min at 37°C without stimulation (- - - - -), or after stimulation with PMA (▪), the activating β1-specific mAb TS2/16 (▴), or the CD3-specific mAb 38.1 (▨). For each transfectant tested, adhesion after stimulation with CD2-specific mAbs or the CD28-specific mAb 9.3 was comparable to that seen for CD3 stimulation (our unpublished results). The data are represented as the mean percent adhesion of triplicate wells ± SD. Results shown are one of at least three representative experiments.
Figure 9.

Substitution of the tyrosine at position 795 with alanine, but not phenylalanine, reduces adhesion of A1 transfectants to FN. A1 cells were transfected with pHook2 alone, or together with the indicated β1 construct. Cells were recovered after transfection, and β1-expressing transfectants were isolated as described in MATERIALS AND METHODS. (A–E) A1 cells expressing pHook2 alone or together with the indicated β1 construct were analyzed for adhesion to FN (1 μg/well in panels A–D, 3 μg/well in panel E) for 20 min at 37°C without stimulation (░⃞), or after stimulation with PMA (▪), the activating β1-specific mAb TS2/16 (open bars), or the CD3-specific mAb 38.1 (hatched bars). For each transfectant tested, adhesion after stimulation with CD2-specific mAbs or the CD28-specific mAb 9.3 was comparable to that seen for CD3 stimulation (our unpublished results). Each panel represents a separate experiment. Transfectants were stained with PE-conjugated anti-β1 antibody 4B4 followed by FACS analysis to determine the β1 expression on transfectants for that particular experiment. Histograms representing β1 expression are shown. The data are represented as the mean percent adhesion of triplicate wells ± SD. Results shown are one of at least three representative experiments with each mutant β1 cytoplasmic domain construct.
Mutation of the Tyrosine in the NPKY Motif to Alanine Inhibits Activation-dependent β1 Integrin-mediated Adhesion to FN
Since deletion of the carboxy-terminal five amino acids in the β1 cytoplasmic domain prevented β1 integrin function in A1 cells, we performed alanine scanning mutagenesis in this region of the β1 cytoplasmic tail (Figure 7). Each of the five amino acids in the carboxy-terminal end of the β1 cytoplasmic domain was mutated to an alanine, and the effect of these mutations on activation-dependent adhesion of A1 transfectants to FN was assessed. As shown in Figure 9, mutation of amino acids 794, 796, 797, and 798 to alanine did not affect either basal or stimulated adhesion to FN after expression in A1 cells. In contrast, mutation of the tyrosine residue at position 795 to an alanine reduced the ability of the β1 subunit to mediate adhesion to FN in A1 cells (Figure 9). The Y795A mutation reduced both the basal level of adhesion in the absence of stimulation, as well as the increased adhesion induced by stimulation of the integrin regulators CD3, CD2, or CD28 (Figure 9 and our unpublished results). Interestingly, adhesion induced by the activating β1 integrin-specific mAb TS2/16 was minimally affected by the Y795A mutation. When the tyrosine at position 795 was changed to a phenylalanine, both basal and stimulated adhesion were comparable to that observed with transfectants expressing the wild-type β1 integrin subunit. Thus, these results suggest a critical role for the tyrosine residue in the NPKY motif in the carboxy-terminal end of the β1 cytoplasmic domain in activation-dependent adhesion of Jurkat T cells to FN.
Deletion of the Conserved NPXY Motifs in the β1 Integrin Cytoplasmic Domain Inhibits Activation-dependent Adhesion of Jurkat T Cells to FN
To determine the role of the conserved NPXY motifs within the β1 cytoplasmic tail in mediating β1 integrin regulation in T cells, we generated β1 cytoplasmic mutants containing a deletion of either the amino-terminal NPIY sequence or the carboxy-terminal NPKY sequence (Figure 7). In each case, surface expression of the deletion mutants was similar to that seen for full-length β1 (Figure 10A). However, a deletion of either the NPIY sequence or the NPKY sequence resulted in the expression of a β1 integrin subunit that was unable to mediate adhesion to FN (Figure 10B). Both basal and stimulated adhesion of A1 cells to FN were reduced in transfectants expressing either the β1ΔNPIY or β1ΔNPKY mutant when compared with the wild-type β1 subunit. However, while CD3- and CD28-mediated activation of adhesion was inhibited completely by these two deletions, direct stimulation of β1 integrins by the β1 integrin-specific mAb TS2/16 was only partially reduced when compared with wild-type β1 (Figure 10B).
Figure 10.

A1 cells expressing NPXY deletions exhibit reduced adhesion to FN. A1 cells were transfected with pHook2 alone, or together with the indicated β1 construct. Cells were recovered after transfection, and β1-expressing transfectants were isolated as described in MATERIALS AND METHODS. (A) Transfectants were stained with PE-conjugated 4B4 antibody followed by FACS analysis to determine the β1 expression on transfectants for that particular experiment. Histograms representing β1 expression are shown. (B) Transfectants were analyzed for adhesion to FN (3 μg/well) for 20 min at 37°C without stimulation (□), or after stimulation with the activating β1-specific mAb TS2/16 (░⃞), the CD3-specific mAb 38.1 (▪), or the CD28-specific mAb 9.3 (⊠). The data are represented as the mean percent adhesion of triplicate wells ± SD. Results shown are one of at least three representative experiments with each mutant β1 cytoplasmic domain construct.
DISCUSSION
Although β1 integrin-mediated adhesion of T cells has been shown to be dynamically regulated by activation signals that increase β1 integrin avidity, the structural basis for this regulation of integrin function remains incompletely defined. In this study, we sought to define the residues in the β1 cytoplasmic domain that are critical for activation-dependent regulation in T lymphocytes. The approach that we developed was made possible by the isolation of a β1 integrin-deficient variant of the Jurkat T cell line. This cell line, designated A1, does not express the β1 integrin subunit on the cell surface. Consequently, α subunits that associate with the β1 subunit, with the exception of low levels of the α4 subunit, are also not expressed on the surface of A1 cells. The A1 cell line has little detectable β1 mRNA, suggesting a defect in endogenous β1 integrin expression in A1 cells at the RNA level. The lack of β1 integrin expression on the cell surface results in loss of adhesion of A1 cells to FN when compared with wild-type β1+ Jurkat cells, even after stimulation with agonists that activate β1 integrin-mediated adhesion. Upon reexpression of the β1 integrin subunit in A1 cells, β1+ A1 cells adhered to immobilized FN in an activation- and β1-dependent manner, suggesting that the integrin-regulatory signaling cascades are still intact. Thus, the A1 cell line represents a valuable new cellular tool for the analysis of β1 integrin structure and function.
We used the β1 integrin-deficient A1 cell line to identify the regions of the β1 integrin cytoplasmic domain that are critical for activation-dependent regulation of β1 integrin-mediated adhesion. This question has been difficult to address, since all nucleated hematopoietic cells express the β1 integrin subunit. Although some prior studies have used integrin chimeric proteins expressed in nonhematopoietic cell lines (Vignoud et al., 1994; O’Toole et al., 1995), these cell lines do not exhibit the rapid changes in β1 integrin function that occur upon activation of T lymphocytes. Furthermore, these studies have, in general, involved the use of reporter antibodies that detect the expression of novel epitopes upon cell stimulation (Vignoud et al., 1994; O’Toole et al., 1995). Although these novel epitopes are clearly indicative of changes in β1 integrin conformation, the relationship between these conformational changes and adhesion has not been definitively established for many of these epitopes (Stewart et al., 1996; Yauch et al., 1997; Bazzoni and Hemler, 1998).
Since the A1 cell line does not express the β1 integrin subunit, we could directly determine whether specific mutations in the β1 integrin cytoplasmic domain affect activation-dependent adhesion of T cells to a β1 integrin ligand. We used a transient transfection approach to demonstrate that two highly conserved NPXY motifs in the human β1 integrin cytoplasmic domain are necessary for mediating both basal and stimulated adhesion of T cells to FN. Deletion of either the amino-terminal NPIY sequence or the carboxy-terminal NPKY sequence, or truncation of the carboxy-terminal five amino acids of the β1 integrin cytoplasmic domain, abolished adhesion to FN. These defects in adhesion were not due to effects of the β1 integrin cytoplasmic domain mutations on cell surface expression, since all of the mutant β1 integrin subunits were expressed on the cell surface at levels comparable to wild-type β1. Scanning alanine mutagenesis of the carboxy-terminal five amino acids revealed a critical role for the tyrosine residue at position 795 in the NPKY motif in β1 integrin function in T cells. While alanine substitutions at positions 794, 796, 797, and 798 did not affect β1 integrin-mediated adhesion, a change of the tyrosine at position 795 to alanine resulted in reduced basal and stimulated adhesion to FN compared with A1 cells expressing comparable levels of wild-type β1. However, a more conservative substitution of this tyrosine for phenylalanine did not affect β1 integrin-mediated adhesion in A1 cells, suggesting that phosphorylation of the tyrosine residue at position 795 is not required for adhesion of Jurkat T cells to FN. Since NPXY motifs have been shown to form tight turns in protein structure, and mutation of the tyrosine in the NPKY to alanine resulted in reduced β1 integrin-mediated adhesion in A1 cells, the NPKY motif may be a critical structural component of the β1 cytoplasmic domain.
The NPXY amino acid motifs are conserved among many integrin β chains. In the β1 cytoplasmic domain, the two NPXY motifs and a third region have been shown to be critical for localizing β1 integrins to focal contact sites (Reszka et al., 1992; LaFlamme et al., 1994; Vignoud et al., 1997). Studies of the NPXY motif in the β3 integrin subunit have also suggested a critical role for this motif in β3 integrin function (Ylänne et al., 1995; Filardo et al., 1995). Mutation of the amino-terminal NPXY motif in the β3 subunit cytoplasmic domain abolished the constitutive adhesion and spreading of αvβ3-expressing melanoma cells to vitronectin. In contrast to our results, a truncation of the carboxy-terminal 11 amino acids in the β3 subunit, a region that includes the carboxy-terminal NPXY motif, did not affect adhesion to vitronectin (Filardo et al., 1995). Deletion of the amino-terminal NPXY motif in the β1 cytoplasmic domain, when expressed in the context of a chimeric integrin expressing the β3 extracellular domain, also abolished the binding of PAC1, a mAb that recognizes the activated form of the αIIbβ3 integrin (O’Toole et al., 1995). Interestingly, substitutions of the asparagine or tyrosine residues in the carboxy-terminal NPXY motif also affected PAC-1 binding, although the level of inhibition was not as dramatic as that seen for mutations in the amino-terminal NPXY motif (O’Toole et al., 1995). Our results also suggest a role for the tyrosine residue in the carboxy-terminal NPXY motif in β1 integrin function, since the Y795A mutation in the β1 integrin cytoplasmic domain severely reduced adhesion of A1 cells to FN. A role for the carboxy-terminal end of the β1 integrin cytoplasmic domain in regulating β1 integrin-mediated cell adhesion is also suggested by the ability of a cell-permeable peptide representing the carboxy-terminal half of the β1 cytoplasmic tail to inhibit fibroblast adhesion (Liu et al., 1996).
Similar to β1-mediated adhesion, αLβ2-mediated adhesion of lymphocytes requires activation for interaction with intercellular adhesion molecules (ICAMs) (Dustin and Springer, 1989; van Kooyk et al., 1989). When expressed in COS cells, deletion of the carboxy-terminal five amino acids of the β2 cytoplasmic domain, including a lysine and phenylalanine in a NPKF motif, abolished constitutive adhesion to purified ICAM-1 (Hibbs et al., 1991b). Furthermore, this study showed that expression of a β2 mutant in which the carboxy-terminal 5 amino acids were replaced with Glu-Val-Cys in a β2 integrin-deficient B lymphoblastoid cell line resulted in a loss of adhesion of transfectants to immobilized ICAM-1 upon phorbol ester stimulation. Additional studies have implicated the phenylalanine residue in the carboxy-terminal NPKF motif in the β2 cytoplasmic domain as being critical in LFA-1–mediated adhesion to purified ICAM-1 in COS cells (Hibbs et al., 1991a). Although mutation of this phenylalanine to tyrosine had no effect on adhesion, a change to alanine or leucine abolished adhesion. Thus, these results with a related integrin β subunit also support a central role for the carboxy-terminal end of integrin β subunits in integrin function.
Our studies with the A1 cell line to some extent parallel recent analyses of β1 integrin structure and function using the GD25 cell line, which was derived from β1-null embryonic stem cells (Wennerberg et al., 1996). There are several important differences between these two β1-negative cell lines that should be noted. Unlike A1 cells, GD25 cells express other FN-binding integrins, notably αVβ3. This complicates the analysis of effects of β1 cytoplasmic domain mutations on β1-mediated interactions with FN using GD25 cells. In addition, the basal activity of β1 integrins on GD25 fibroblasts and A1 T cells differs, since β1 integrin-mediated adhesion of A1 cells is dependent to a large extent on activation signals that increase β1 integrin activity. Despite these differences, some common effects of β1 cytoplasmic domain mutations are observed. Most notably, the Y795F mutation, which did not affect A1 adhesion to FN, also did not affect β1 integrin-mediated attachment of GD25 cells to laminin-1 (Wennerberg et al., 1998) or to FN (Sakai et al., 1998). However, a dramatic effect of the Y795F mutation on directed migration of GD25 cells was observed, suggesting a critical role for this residue in movement rather than attachment. Mutation of the proline residues in either NPXY motifs, either singly or in combination, also impaired cell adhesion as well as expression of the β1 integrin epitope defined by mAb 9EG7 (Sakai et al., 1998). However, it is difficult to directly assess the effect of these mutations on cell adhesion relative to wild-type β1 in GD25 cells, since these mutations also affected cell surface expression of these mutant β1 subunits. Adhesion of GD25 cells to laminin-1 was profoundly affected by the mutation of two highly conserved threonines at positions 788 and 789 to alanine (Wennerberg et al., 1998). However, mutation of these two threonines did not abolish β1 integrin-dependent adhesion to FN, suggesting that the impact of mutating these threonine residues in GD25 cells is ligand-dependent. It remains to be determined whether these threonine substitutions would have a similar impact on activation-dependent adhesion to A1 Jurkat cells to FN or another β1 integrin ligand, such as VCAM-1.
Our studies also demonstrated a reduced ability of the β1 integrin-activating mAb TS2/16 to enhance the adhesion of cells expressing the β1(793), β1(ΔNPIY), or β1(ΔNPKY) mutant subunits. Although TS2/16 and other activating β1 integrin-specific antibodies can induce or stabilize the active form of β1 integrins in the absence of other modes of stimulation (Arroyo et al., 1992), our results suggest that optimal adhesion induced by direct activation of β1 integrins by TS2/16 does, in fact, require the integrity of the β1 integrin cytoplasmic tail. This is consistent with studies demonstrating that deletion of the carboxy-terminal 16 amino acids in the human β1 integrin cytoplasmic domain inhibited soluble FN binding by α5β1 expressed in CHO cells induced by the activating β1 integrin-specific mAb 8A2 (Puzon-McLaughlin et al., 1996). Our studies also demonstrate that the β1(793), β1(ΔNPIY), and β1(ΔNPKY) mutant subunits were unable to respond to all of the other stimuli that have been shown to activate β1 integrins in Jurkat cells, including phorbol ester stimulation and activation via CD3/TCR, CD2, and CD28. Furthermore, the β1(K794A), β1(Y795F), β1(E796A), β1(G797A), and β1(K798A) mutants were able to respond to all stimuli. This suggests that at the level of the β1 integrin cytoplasmic domain, all of these integrin-activating signals require common structural integrity of the β1 cytoplasmic domain.
In summary, we have produced and characterized a variant of the Jurkat T cell line that lacks expression of the β1 integrin subunit. This β1-deficient cell line was used to demonstrate a critical role for the carboxy-terminal end of the β1 cytoplasmic domain, and two conserved NPXY motifs, in the activation-dependent regulation of β1 integrin-mediated adhesion. Thus, this β1 integrin-deficient cell line provides a valuable new cellular reagent for the analysis of β1 integrin structure and function in the context of a cell that exhibits dynamic regulation of integrin adhesiveness without the complication of endogenous β1 integrin subunit expression.
ACKNOWLEDGMENTS
The authors thank Drs. G. van Seventer, S. Shaw, A. Sonnenberg, R. Gress, S.Y. Yang, J. Ledbetter, B. Bierer, E. Ruoslahti, V. Dixit, and J. McCarthy for providing valuable mAbs and other reagents; and Dr. S. Kellermann for critical reading of this manuscript. This work was supported by National Institutes of Health grant AI-38474. Y.S. is the Harry Kay Chair of Cancer Research at the University of Minnesota.
REFERENCES
- Akiyama SK, Yamada SS, Yamada KM, LaFlamme SE. Transmembrane signal transduction by integrin cytoplasmic domains expressed in single-subunit chimeras. J Biol Chem. 1994;269:15961–15964. [PubMed] [Google Scholar]
- Arroyo AG, Sánchez-Mateos P, Campanero MR, MartínPadura I, Dejana E, Sánchez-Madrid F. Regulation of the VLA integrin-ligand interactions through the β1 subunit. J Cell Biol. 1992;117:659–670. doi: 10.1083/jcb.117.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzoni G, Hemler ME. Are changes in integrin affinity and conformation overemphasized? Trends Biochem Sci. 1998;23:30–34. doi: 10.1016/s0968-0004(97)01141-9. [DOI] [PubMed] [Google Scholar]
- Bazzoni G, Shih DT, Buck CA, Hemler ME. Monoclonal antibody 9EG7 defines a novel β1integrin epitope induced by soluble ligand and manganese, but inhibited by calcium. J Biol Chem. 1995;270:25570–25577. doi: 10.1074/jbc.270.43.25570. [DOI] [PubMed] [Google Scholar]
- Campbell JJ, Hedrick J, Zlotnik A, Siani MA, Thompson DA, Butcher EC. Chemokines and the arrest of lymphocytes rolling under flow conditions. Science. 1998;279:381–384. doi: 10.1126/science.279.5349.381. [DOI] [PubMed] [Google Scholar]
- Chen Y-P, O’Toole TE, Shipley T, Forsyth J, LaFlamme SE, Yamada KM, Shattil SJ, Ginsberg MH. “Inside-out” signal transduction inhibited by isolated integrin cytoplasmic domains. J Biol Chem. 1994;269:18307–18310. [PubMed] [Google Scholar]
- Chesnut JD, Baytan AR, Russell M, Chang MP, Bernard A, Maxwell IH, Hoeffler JP. Selective isolation of transiently transfected cells from a mammalian cell population with vectors expressing a membrane anchored single-chain antibody. J Immunol Methods. 1996;193:17–27. doi: 10.1016/0022-1759(96)00032-4. [DOI] [PubMed] [Google Scholar]
- Diamond MS, Springer TA. The dynamic regulation of integrin adhesiveness. Curr Biol. 1994;4:506–517. doi: 10.1016/s0960-9822(00)00111-1. [DOI] [PubMed] [Google Scholar]
- Dustin ML, Springer TA. T-cell receptor cross-linking transiently stimulates adhesiveness through LFA-1. Nature. 1989;341:619–624. doi: 10.1038/341619a0. [DOI] [PubMed] [Google Scholar]
- Filardo EJ, Brooks PC, Deming SL, Damsky C, Cheresh DA. Requirement of the NPXY motif in the integrin β3 subunit cytoplasmic tail for melanoma cell migration in vitro and in vivo. J Cell Biol. 1995;130:441–450. doi: 10.1083/jcb.130.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelstein LD, Reynolds PJ, Hunt SW, III, Shimizu Y. Structural requirements for β1 integrin-mediated tyrosine phosphorylation in human T cells. J Immunol. 1997;159:5355–5363. [PubMed] [Google Scholar]
- Gibbs RA, Nguyen PN, McBride LJ, Koepf SM, Caskey CT. Identification of mutations leading to the Lesch-Nyhan syndrome by automated direct DNA sequencing of in vitro amplified cDNA. Proc Natl Acad Sci USA. 1989;86:1919–1923. doi: 10.1073/pnas.86.6.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbs ML, Jakes S, Stacker SA, Wallace RW, Springer TA. The cytoplasmic domain of the integrin lymphocyte function- associated antigen 1 β subunit: sites required for binding to intercellular adhesion molecule 1 and the phorbol ester-stimulated phosphorylation site. J Exp Med. 1991a;174:1227–1238. doi: 10.1084/jem.174.5.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbs ML, Xu H, Stacker SA, Springer TA. Regulation of adhesion to ICAM-1 by the cytoplasmic domain of LFA-1 integrin β subunit. Science. 1991b;251:1611–1613. doi: 10.1126/science.1672776. [DOI] [PubMed] [Google Scholar]
- Hughes PE, O’Toole TE, Ylänne J, Shattil SJ, Ginsberg MH. The conserved membrane-proximal region of an integrin cytoplasmic domain specifies ligand binding affinity. J Biol Chem. 1995;270:12411–12417. doi: 10.1074/jbc.270.21.12411. [DOI] [PubMed] [Google Scholar]
- Jung TM, Dailey MO. Rapid modulation of homing receptors (gp90MEL-14) induced by activators of protein kinase C: receptor shedding due to accelerated proteolytic cleavage at the cell surface. J Immunol. 1990;144:3130–3136. [PubMed] [Google Scholar]
- Kishimoto TK, Jutila MA, Berg EL, Butcher EC. Neutrophil Mac-1 and MEL-14 adhesion proteins inversely regulated by chemotactic factors. Science. 1989;245:1238–1241. doi: 10.1126/science.2551036. [DOI] [PubMed] [Google Scholar]
- Kuijpers TW, Van Lier RAW, Hamann D, De Boer M, Thung LY, Weening RS, VerHoeven AJ, Roos D. Leukocyte adhesion deficiency type 1 (LAD-1)/variant — a novel immunodeficiency syndrome characterized by dysfunctional β2integrins. J Clin Invest. 1997;100:1725–1733. doi: 10.1172/JCI119697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFlamme SE, Thomas LA, Yamada SS, Yamada KM. Single subunit chimeric integrins as mimics and inhibitors of endogenous integrin functions in receptor localization, cell spreading and migration, and matrix assembly. J Cell Biol. 1994;126:1287–1298. doi: 10.1083/jcb.126.5.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XY, Timmons S, Lin YZ, Hawiger J. Identification of a functionally important sequence in the cytoplasmic tail of integrin β3by using cell-permeable peptide analogs. Proc Natl Acad Sci USA. 1996;93:11819–11824. doi: 10.1073/pnas.93.21.11819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luque A, Gómez M, Puzon W, Takada Y, Sánchez-Madrid F, Cabañas C. Activated conformations of very late activation integrins detected by a group of antibodies (HUTS) specific for a novel regulatory region (355–425) of the common β1 chain. J Biol Chem. 1996;271:11067–11075. doi: 10.1074/jbc.271.19.11067. [DOI] [PubMed] [Google Scholar]
- Mobley JL, Ennis E, Shimizu Y. Differential activation-dependent regulation of integrin function in cultured human T-leukemic cell lines. Blood. 1994;83:1039–1050. [PubMed] [Google Scholar]
- Mobley JL, Ennis E, Shimizu Y. Isolation and characterization of cell lines with genetically distinct mutations downstream of protein kinase C that result in defective activation-dependent regulation of T cell integrin function. J Immunol. 1996;156:948–956. [PubMed] [Google Scholar]
- Mobley JL, Romzek NC, Shimizu Y. Integrin activation in lymphocyte adhesion. In: Weir DM, Herzenberg LA, Blackwell C, Herzenberg Le A, editors. Handbook of Experimental Immunology. Vol. 68. Cambridge, MA: Blackwell Scientific; 1995. pp. 1–68.11. [Google Scholar]
- O’Toole TE, Ylanne J, Culley BM. Regulation of integrin affinity states through an NPXY motif in the β subunit cytoplasmic domain. J Biol Chem. 1995;270:8553–8558. doi: 10.1074/jbc.270.15.8553. [DOI] [PubMed] [Google Scholar]
- Postigo AA, Sánchez-Mateos P, Lazarovits AI, SánchezMadrid F, De Landázuri MO. α4β7 integrin mediates B cell binding to fibronectin and vascular cell adhesion molecule-1: expression and function of α4 integrins on human B lymphocytes. J Immunol. 1993;151:2471–2483. [PubMed] [Google Scholar]
- Puzon-McLaughlin W, Yednock TA, Takada Y. Regulation of conformation and ligand binding function of integrin α5β1 by the β1 cytoplasmic domain. J Biol Chem. 1996;271:16580–16585. doi: 10.1074/jbc.271.28.16580. [DOI] [PubMed] [Google Scholar]
- Reszka AA, Hayashi Y, Horwitz AF. Identification of amino acid sequences in the integrin β1cytoplasmic domain implicated in cytoskeletal association. J Cell Biol. 1992;117:1321–1330. doi: 10.1083/jcb.117.6.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai T, Zhang QH, Fässler R, Mosher DF. Modulation of β1A integrin functions by tyrosine residues in the β1 cytoplasmic domain. J Cell Biol. 1998;141:527–538. doi: 10.1083/jcb.141.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu Y, van Seventer GA, Horgan KJ, Shaw S. Regulated expression and binding of three VLA (β1) integrin receptors on T cells. Nature. 1990;345:250–253. doi: 10.1038/345250a0. [DOI] [PubMed] [Google Scholar]
- Stewart MP, Cabañas C, Hogg N. T cell adhesion to intercellular adhesion molecule-1 (ICAM- 1) is controlled by cell spreading and the activation of integrin LFA-1. J Immunol. 1996;156:1810–1817. [PubMed] [Google Scholar]
- Tahiliani PD, Singh L, Auer KL, LaFlamme SE. The role of conserved amino acid motifs within the integrin β3cytoplasmic domain in triggering focal adhesion kinase phosphorylation. J Biol Chem. 1997;272:7892–7898. doi: 10.1074/jbc.272.12.7892. [DOI] [PubMed] [Google Scholar]
- van Kooyk Y, van de Wiel-van Kemenade P, Weder P, Kuijpers TW, Figdor CG. Enhancement of LFA-1-mediated cell adhesion by triggering through CD2 or CD3 on T lymphocytes. Nature. 1989;342:811–813. doi: 10.1038/342811a0. [DOI] [PubMed] [Google Scholar]
- Vignoud L, Albigés-Rizo C, Frachet P, Block MR. NPXY motifs control the recruitment of the α5β1 integrin in focal adhesions independently of the association of talin with the β1 chain. J Cell Sci. 1997;110:1421–1430. doi: 10.1242/jcs.110.12.1421. [DOI] [PubMed] [Google Scholar]
- Vignoud L, Usson Y, Balzac F, Tarone G, Block MR. Internalization of the α5β1integrin does not depend on “NPXY” signals. Biochem Biophys Res Commun. 1994;199:603–611. doi: 10.1006/bbrc.1994.1271. [DOI] [PubMed] [Google Scholar]
- Wennerberg K, Fässler R, Wärmegård B, Johansson S. Mutational analysis of the potential phosphorylation sites in the cytoplasmic domain of integrin β1A. Requirement for threonines 788–789 in receptor activation. J Cell Sci. 1998;111:1117–1126. doi: 10.1242/jcs.111.8.1117. [DOI] [PubMed] [Google Scholar]
- Wennerberg K, Lohikangas L, Gullberg D, Pfaff M, Johansson S, Fässler R. β1 integrin-dependent and -independent polymerization of fibronectin. J Cell Biol. 1996;132:227–238. doi: 10.1083/jcb.132.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yauch RL, Felsenfeld DP, Kraeft SK, Chen LB, Sheetz MP, Hemler ME. Mutational evidence for control of cell adhesion through integrin diffusion/clustering, independent of ligand binding. J Exp Med. 1997;186:1347–1355. doi: 10.1084/jem.186.8.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylänne J, Huuskonen J, O’Toole TE, Ginsberg MH, Virtanen I, Gahmberg CG. Mutation of the cytoplasmic domain of the integrin β3subunit. Differential effects on cell spreading, recruitment to adhesion plaques, endocytosis, and phagocytosis. J Biol Chem. 1995;270:9550–9557. doi: 10.1074/jbc.270.16.9550. [DOI] [PubMed] [Google Scholar]
- Zell T, Hunt SW, III, Finkelstein LD, Shimizu Y. CD28-mediated upregulation of β1 integrin-mediated adhesion involves phosphatidylinositol 3-kinase. J Immunol. 1996;156:883–886. [PubMed] [Google Scholar]