

Abstract
Small GTPases control key cellular events, including formation of cell–cell junctions and gene expression, and are regulated by activating and inhibiting factors. Here, we characterize the junctional protein paracingulin as a novel regulator of the activity of two small GTPases, Rac1 and RhoA, through the functional interaction with their respective activators, Tiam1 and GEF-H1. In confluent epithelial monolayers, paracingulin depletion leads to increased RhoA activity and increased expression of mRNA for the tight junction protein claudin-2. During tight junction assembly by the calcium-switch, Rac1 shows two transient peaks of activity, at earlier (10–20 min) and later (3–8 h) time points. Paracingulin depletion reduces such peaks of Rac1 activation in a Tiam1-dependent manner, resulting in a delay in junction formation. Paracingulin physically interacts with GEF-H1 and Tiam1 in vivo and in vitro, and it is required for their efficient recruitment to junctions, based on immunofluorescence and biochemical experiments. Our results provide the first description of a junctional protein that interacts with GEFs for both Rac1 and RhoA, and identify a novel molecular mechanism whereby Rac1 is activated during junction formation.
INTRODUCTION
In multicellular organisms, apicobasal polarity and cell–cell junctions are required for the functional integrity of all epithelial tissues. In vertebrate epithelia, tight junctions (TJs) are essential for the maintenance of polarity and the barrier function of epithelia (Anderson and Van Itallie, 1995; Shin et al., 2006), and adherens junctions (AJs) play key roles in morphogenesis and tissue sorting (Gumbiner, 2005). TJs comprise membrane proteins, which function as adhesion receptors and as pores/channels for the passage of molecules through the paracellular pathway, and cytoplasmic proteins, which comprise scaffolding and signaling proteins (Schneeberger and Lynch, 2004; Van Itallie and Anderson, 2006; Guillemot et al., 2008). AJs contain transmembrane adhesion receptors of the cadherin and Ig-like cell adhesion molecule families, and cytoplasmic proteins that regulate actin cytoskeleton dynamics and signaling pathways (Perez-Moreno et al., 2003).
Assembly and function of TJs and AJs are dependent on the activities of the Rho family GTPases Cdc42, RhoA, and Rac1 (Nusrat et al., 1995; Jou et al., 1998; Fukata and Kaibuchi, 2001; Braga, 2002), which act as molecular switches that control actin polymerization and dynamics through a variety of effector proteins, including kinases and scaffold proteins (Jaffe and Hall, 2005). In recent years, details have emerged about the roles of Rho family GTPases at epithelial junctions. Rac1 becomes activated at sites of cell–cell adhesion after cadherin-mediated interactions between adjacent cells (Braga et al., 1997; Noren et al., 2001), but it is rapidly down-regulated during expansion of the adhesive contact (Yamada and Nelson, 2007). RhoA activity concentrates at the outer edges of the expanding contact, and it is down-regulated when the monolayer reaches confluence (Coleman et al., 2004; Yamada and Nelson, 2007). Thus, a precise spatial and temporal fine-tuning of the activity of Rho family GTPases is critically important in the establishment and maintenance of junctions. However, little is known about the molecular mechanisms that control RhoA and Rac1 activities during the different phases of junction assembly, and in confluent cells.
Rho family GTPases are regulated by activating factors (guanine nucleotide exchange factors [GEFs]) and inhibiting factors (GTPase activation proteins and guanosine diphosphate-dissociation inhibitors), which in turn bind to adaptor proteins, which may act either to restrict their spatial localization, and/or influence their activity (Mertens et al., 2003; Jaffe and Hall, 2005; Rossman et al., 2005). So far, few junctional proteins have been shown to influence Rho family GTPase activity in epithelial cells (Guillemot et al., 2008). These proteins include Par3, angiomotin, p120catenin, and cingulin (Anastasiadis et al., 2000; Aijaz et al., 2005; Chen and Macara, 2005; Mertens et al., 2005; Sakurai et al., 2006; Wells et al., 2006; Wildenberg et al., 2006). For example, Par3 assembly into junctions is promoted by the Rac1 GEF Tiam1 (Mertens et al., 2005), and in turn Par3 acts to negatively regulate Rac1 activity, by interacting with Tiam1 (Chen and Macara, 2005). At epithelial junctions, the RhoA activator GEF-H1 is inhibited by binding to the TJ protein cingulin (Aijaz et al., 2005). Cingulin is a cytoplasmic TJ protein (Citi et al., 1988), which interacts with zonula occludens (ZO) proteins (ZO-1, ZO-2, and ZO-3) and the actomyosin cytoskeleton (Cordenonsi et al., 1999; D'Atri and Citi, 2001; Guillemot and Citi, 2006b; Guillemot et al., 2008). In cingulin-depleted cells, GEF-H1 sequestration at junctions is decreased (Aijaz et al., 2005), and this results in increased RhoA activity, increased expression of the TJ component claudin-2, and increased cell proliferation (Guillemot and Citi, 2006a).
The recently identified protein JACOP/paracingulin (also identified as cingulin-like1/CGNL1) has a domain organization similar to cingulin, with globular head and tail domains, and a central coiled-coil rod domain, which shows ∼40% sequence identity to cingulin (Ohnishi et al., 2004; Guillemot and Citi, 2006b; Guillemot et al., 2008). However, unlike cingulin, which is a TJ-specific protein, paracingulin has been localized at both TJs and AJs (Ohnishi et al., 2004). Because the function of paracingulin is unknown, we decided to explore it using a short hairpin RNA (shRNA)-mediated approach in Madin-Darby canine kidney (MDCK) epithelial cells. Our results show that paracingulin regulates both Rac1 and RhoA activities, through a mechanism involving interaction with and junctional recruitment of Tiam1 and GEF-H1.
MATERIALS AND METHODS
Cell Culture and Transfection
MDCK cell clones stably expressing shRNA were obtained by transfection with pTER constructs designed to target the following sequences: CGNL1, 5′-CGGAAAGTCAACCTGGTCT-3′; control, 5′TCTACGACCCTTCTTCCAT-3′; Tiam1, 5′-GCGAAGGAGCAGGTTTTCT-3′. Cell culture, clone selection, calcium-switch assays, measurement of transepithelial resistance (TER), and 5-bromo-2′-deoxyuridine (BrdU) incorporation assays were as described previously (Guillemot and Citi, 2006a; Paschoud and Citi, 2008). All experiments were carried out at least in triplicate, and values show means and SD. For immunoblots and immunofluorescence data, one representative example is shown.
Antibodies and Plasmids
Polyclonal antibodies to paracingulin were raised in rabbits against bacterially expressed glutathione transferase (GST) fusion proteins encoding either residues 582-940 or residues 493-633 of canine paracingulin, and were used at dilutions of 1:1000 and 1:10,000 for immunofluorescence and immunoblotting, respectively. These antibodies label a single polypeptide of ∼150 kDa in lysates of MDCK cells, and they do not cross-react with cingulin (Mr 140 kDa), as determined by immunoblotting analysis of cingulin recombinant proteins and immunoblotting and immunofluorescence analysis of cells overexpressing cingulin. Antibodies against α-catenin, β-catenin, actin, and vesicular stomatitis virus (VSV) were from Sigma-Aldrich (St. Louis, MO). Monoclonal anti-cadherin and anti-Rac1 antibodies were from BD Biosciences (San Jose, CA). Other antibodies were as described previously (Guillemot and Citi, 2006a; Paschoud and Citi, 2008). Monoclonal antibodies against GEF-H1 and a construct encoding VSV-tagged GEF-H1 were a kind gift from K. Matter (University College, London, United Kingdom). Polyclonal antibodies to Tiam1 were a kind gift from J. Collard (Netherlands Cancer Institute, Amsterdam, Holland) and were also obtained from Santa Cruz Biotechnology (Santa Cruz, CA). However, because these antibodies did not immunolocalize endogenous Tiam1 at junctions or label a polypeptide of correct size in MDCK cell lysates, we were obliged to use exogenous hemagglutinin (HA)-tagged Tiam1. Constructs encoding fluorescently- and myc-tagged canine paracingulin and cingulin, and myc-tagged yellow fluorescent protein (YFP) were generated in the mammalian expression vector pcDNA3.1myc-His (Paschoud and Citi, unpublished data) (Paschoud and Citi, 2008). Constructs for the production of His-tagged recombinant proteins in baculovirus-infected Sf29 insect cells were generated in pFastBacHT vectors (Citi et al., 2001). Construct for bacterial expression [in the BL21(DE3) strain] of GST fused to different regions of paracingulin, and the DH/PH domains of Tiam1 were generated by polymerase chain reaction (PCR) amplification and subcloning into the pGEX4T1 vector. Constructs for the expression of the GST-rhotekin/Rho-binding domain and GST-Pak1/p21 binding domain fusion proteins were a kind gift of K. Burridge (University of North Carolina, Chapel Hill, NC). Recombinant proteins were expressed and purified as described previously (Citi et al., 2001). The construct encoding HA-tagged dominant-negative RhoA (RhoAN19) was a kind gift from E. Olson (University of Texas Southwestern Medical Center, Dallas, TX). The construct encoding full-length, HA-tagged Tiam1 was a kind gift from J. Collard (Netherlands Cancer Institute). All new constructs were verified by sequencing.
Cell Fractionation
Cells were grown in 60-mm dishes, washed twice with ice-cold phosphate-buffered saline (PBS), and lysed in 0.25 ml of either radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 40 mM Tris-HCl, pH 7.5, 2 mM EDTA, 10% glycerol, 1% Triton X-100, 0.5 sodium deoxycholate, 0.2% SDS, 5 μg/ml antipain-leupeptin-pepstatin cocktail, and 1 mM phenylmethylsulfonyl fluoride [PMSF]), or cytoskeleton stabilizing buffer (CSK; 10 mM HEPES, pH 6.8, 250 mM sucrose, 150 mM KCl, 1 mM EGTA, 3 mM MgCl2, 0.5% Triton X-100, 1 mM PMSF, and protease inhibitor cocktail). The CSK lysate was centrifuged for 15 min at 13,000 × g, and the pellet was resuspended, washed with CSK buffer, centrifuged again, and taken as “low-speed” cytoskeleton fraction. The soluble fraction was centrifuged for 120 min at 100,000 × g, and the pellet was washed, and taken as the “high-speed” Triton X-100–insoluble cytoskeleton fraction. The supernatant from high-speed centrifugation was taken as “soluble” fraction. Equivalent protein loadings from each fraction were analyzed by SDS-polyacrylamide gel electrophoresis (PAGE) and immunoblotting.
Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
mRNA levels were analyzed by SYBR Green-based real-time PCR as described previously (Guillemot and Citi, 2006b), by using the same primers, except for CGNL1 forward, 5′-CTCAAGGACCTGGAATACGAGC-3′ and CGNL1 reverse, 5′-TCCGAGAGCAAATCCGAGTT-3′; and Tiam1 forward, 5′-GGATGCCCAGAACCCGA-3′ and Tiam1 reverse, 5′-GGATGGGCTTGATGAGGTAAGA-3′. Student's t tests were performed using the GraphPad Prism 4 software (GraphPad Software, San Diego, CA).
Immunochemical Techniques
Immunoblotting and immunofluorescence were carried out as described previously (Guillemot and Citi, 2006a; Paschoud and Citi, 2008). Cold methanol fixation was used for immunofluorescence, and cells were routinely counterstained with 4.6-diamidino-2-phenylindole to visualize nuclei. For immunoprecipitation, cells in 60-mm dishes were washed twice with ice-cold PBS and lysed in 1 ml of coimmunoprecipitation (CoIP) buffer, pH 7.8 (20 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, and 10 μg/ml antipain-leupeptin-pepstatin cocktail) for 15 min at 4°C. Lysates were centrifuged at 12,000 × g for 13 min at 4°C, and supernatants were incubated with polyclonal anti-CGNL1 or anti-cingulin antiserum overnight at 4°C. Dynabeads protein G (Invitrogen, Carlsbad, CA) were added for 1 h at 4°C. After washing with CoIP buffer, pH 4.8, proteins were eluted by boiling beads in SDS sample buffer, and immunoprecipitates were analyzed by SDS-PAGE and immunoblotting.
Imaging Techniques
Samples were imaged using an AxiovertS100TV microscope (Carl Zeiss, Jena, Germany), equipped with a Planapochromat 63× objective (1.4 numerical aperture) and excitation/emission filters to detect fluorescein isothiocyanate, tetramethylrhodamine B isothiocyanate, and 4,6-diamidino-2-phenylindole. Images were acquired with a C4742 digital camera (Hamamatsu, Bridgewater, NJ), operated with OpenLab imaging software and exposure times between 100 and 600 ms. Images were processed using Adobe Photoshop (Adobe Systems, Mountain View, CA) to adjust levels and crop images.
GST Pull-Down Assays
GST pull-down assays to isolate active RhoA (using GST-rhotekin/Rho-binding domain) and Rac1 (using GST-Pak1/p21 binding domain) from MDCK cell lysates were carried out using 20 μg of recombinant GST fusion protein and 0.5 to 1 ml of cell lysate per time point, as described previously (Guillemot and Citi, 2006b). For GST pull-down assays with proteins expressed in baculovirus-infected Sf29 insect cells, 5 μg of recombinant GST fusion protein was coupled for 1 h at room temperature to 20 μl of glutathione-Sepharose beads (GE Healthcare, Chalfont St. Giles, United Kingdom), beads were washed with PBS containing 2% bovine serum albumin and 1% NP-40, and incubated for 1 h at 4°C either with 5 μl of purified protein or 5–200 μl of insect cell lysate, followed by washing three times with ice-cold high-stringency wash buffer (0.5 M NaCl, 20 mM Tris-HCl, pH 7.5, 5 mM EDTA, 1% Triton X-100, 5% NP-40, and 0.1% SDS), and once with PBS. Proteins bound to beads were eluted with SDS sample buffer containing freshly added dithiothreitol (0.1 M), and analyzed by SDS-PAGE and immunoblotting. Loadings for immunoblotting were normalized for protein content with total RhoA/Rac1.
RESULTS
Paracingulin Depletion Up-Regulates the Expression of mRNAs Coding for the TJ Proteins Claudin-2 and ZO-3
We obtained stably transfected clonal lines of MDCK cells, in which paracingulin expression was down-regulated by expression of shRNA. We first investigated whether paracingulin depletion affects the expression of junctional proteins, at the protein and transcript levels. Immunoblot analysis showed that although paracingulin protein levels were dramatically reduced in paracingulin-depleted [CGNL1(-)] cells, levels of other junctional proteins were essentially unchanged, except for ZO-3, where they were consistently increased in CGNL1(-) cells (Figure 1A). Quantitative real-time PCR showed that in CGNL1(-) cells mRNA levels were approximately 5-fold decreased for paracingulin, ∼1.6-fold increased for claudin-2, and ∼1.8-fold increased for ZO-3 (Figure 1B). The phenotype was specifically due to paracingulin depletion, because the changes in ZO-3 (protein and mRNA) and claudin-2 (mRNA) expression were rescued by expression of the tetracycline repressor (TetR), which blocks the expression of shRNA (Figure 1, C and D), and restores paracingulin localization at junctions (Figure 1E). The normal levels of claudin-2 protein, despite the increase in the mRNA levels, may be due to regulatory mechanisms that operate at the level of translation efficiency or protein stability.
Figure 1.

Paracingulin depletion leads to specific increases in the protein levels of ZO-3, and mRNA levels of claudin-2 and ZO-3. (A) CGNL1 depletion leads to up-regulation of ZO-3 protein levels. Immunoblot analysis of paracingulin (CGNL1) and different TJ proteins (cingulin, CGN; ZO-1; ZO-3; claudin-2; GEF-H1; and Par3) in wild-type MDCK cells (WT), in a MDCK cell clone expressing a control shRNA (control), and in two independent clones (A and B) expressing shRNA against paracingulin [CGNL1(-)]. Loadings were normalized by immunoblotting with anti-β-tubulin antibodies. (B) CGNL1 depletion leads to up-regulation of mRNA levels for claudin-2 and ZO-3. The histogram shows relative mRNA levels, calculated as the ratio of the mRNA in experimental samples (determined by qRT-PCR) to that in WT cells (taken as 100%). Values represent the means ± SD of three independent RNA preparations. *p < 0.05, compared with mRNA levels of WT cells. (C–E) Up-regulation of ZO-3 protein levels, and claudin-2 and ZO-3 mRNA levels are specifically due to paracingulin depletion. Immunoblotting (C) and qRT-PCR (D) analysis of WT and CGNL1(-) cells expressing or not the TetR, which blocks the expression of shRNA. *p < 0.05, compared with mRNA levels of nontransfected WT cells. (E) Immunofluorescence analysis of WT, control and CGNL1(-) cells with anti-cingulin and anti-paracingulin antibodies, showing that paracingulin labeling is decreased in CGNL1(-) cells, and that expression of TetR rescues the junctional accumulation of paracingulin, without affecting cingulin. (F) CGNL1 depletion does not affect the junctional localization of TJ and AJ proteins at steady state. Immunofluorescence localization of selected TJ and AJ proteins (indicated above the images; also see text) in WT, control, and CGNL1(-) cells. Bar, 10 μm.
To characterize the effects of paracingulin depletion on the structural organization of cell–cell junctions, confluent cells were analyzed by immunofluorescence with antibodies against TJ and AJ markers. Whereas the labeling for paracingulin was significantly decreased in CGNL1(-) cells (Figure 1, E and F), no changes were observed in the distribution of the TJ-associated proteins cingulin, ZO-1, ZO-3, occludin, and claudin-2, and the AJ-associated proteins α-catenin, β-catenin, and E-cadherin (Figure 1F), indicating that paracingulin depletion does not alter the organization of TJs and AJs in confluent monolayers.
Paracingulin Regulates Claudin-2 mRNA Levels and Cell Proliferation in a RhoA-dependent Manner, and Interacts with GEF-H1
To test the hypothesis that the effects of paracingulin depletion on gene expression are correlated with its effects on RhoA activity, as it is the case for cingulin depletion (Guillemot and Citi, 2006a), we measured RhoA activity in lysates of confluent wild-type (WT) and CGNL1(-) cells. RhoA activity was increased in CGNL1(-) cells (Figure 2A), suggesting that one function of paracingulin is to down-regulate RhoA activity at steady state. Importantly, expression of a dominant-negative mutant of RhoA, which decreased RhoA activity (Supplemental Figure S1A) decreased claudin-2 protein levels in both WT and CGNL1(-) cells (Figure 2B) and reversed the increase in claudin-2 mRNA levels observed in CGNL1(-) cells (Figure 2C), whereas it did not affect ZO-3 protein and mRNA levels (Figure 2, B and C). The close correlation between cellular RhoA activity levels and claudin-2 expression indicates that the effects of paracingulin depletion on claudin-2 expression (but not on ZO-3 expression) are dependent on RhoA. Furthermore, a cell proliferation assay showed that paracingulin depletion induced a significant increase in G1/S phase transition in proliferating cells, which was also reversed by inhibition of RhoA activity (Figure 2D).
Figure 2.

Paracingulin controls claudin-2 expression and cell proliferation through RhoA. (A and B) Immunoblot analyses showing that paracingulin depletion results in increased RhoA activity in confluent monolayers, as detected by GST-RBD pull-down assay (A) and that expression of dominant-negative RhoA (RhoAN19) results in decreased claudin-2 (but not ZO-3) protein levels in WT and CGNL1(-) cells (B). Expression of HA-tagged dominant-negative RhoA was confirmed by immunoblotting, and results in decreased RhoA activity (Supplemental Figure S1A). (C and D) Histograms showing that expression of dominant-negative RhoA, but not empty vector, rescues the up-regulation of claudin-2 (but not ZO-3) mRNA levels (C) and the increase in cell proliferation, as determined by BrdU incorporation (D), in CGNL1(-) cells. The effect of expression of RhoAN19 in WT cells is not shown here, because it was the same as described previously (Guillemot and Citi, 2006a). *p < 0.05, compared with nontransfected WT cells.
To determine whether the increase in RhoA activity observed in CGNL1(-) cells may be due to an interaction between paracingulin and the RhoA activator GEF-H1, we immunoprecipitated paracingulin from lysates of WT cells expressing exogenous paracingulin and GEF-H1 (Supplemental Figure S1B). We were obliged to use exogenously expressed proteins because the yield of endogenous proteins was too low with available antibodies. GEF-H1 was specifically detected by immunoblot in immunoprecipitates of paracingulin (Figure 3A), demonstrating that GEF-H1 and paracingulin can form a complex in vivo. To test whether paracingulin and GEF-H1 interact directly, and to identify the regions in paracingulin that interact with GEF-H1, GST fusion proteins making up different regions of paracingulin (Figure 3B) were incubated with recombinant full-length His-tagged GEF-H1, and isolated by affinity purification on glutathione-Sepharose beads. Immunoblot analysis showed a strong signal for GEF-H1 when using fusion proteins containing either residues 250-420 (construct B) or residues 591-882 (construct D) of paracingulin, whereas a weak signal was obtained with the fusion protein containing residues 884-1302 (construct E) (Figure 3C). The GST fusion proteins B and D appeared to interact with GEF-H1 with a similar affinity (Figure 3D). Thus, paracingulin and GEF-H1 not only can form a complex in vivo but can also interact directly in vitro, through at least two distinct regions of paracingulin. Finally, to test whether the interaction between these two proteins is important in the physiological recruit-ment of GEF-H1 to junctions, the localization of endogenous paracingulin and GEF-H1 was examined in CGNL1(-) cells expressing the TetR, either in the absence of doxycycline (Dox), when paracingulin expression is rescued by the TetR, or in the presence of Dox, when TetR is repressed, and paracingulin expression is silenced. An obvious decrease in the intensity of junctional staining for both paracingulin and GEF-H1 was observed in the presence of Dox (Figure 3E), demonstrating that silencing paracingulin expression results in decreased junctional recruitment of GEF-H1 in vivo.
Figure 3.

Paracingulin interacts in vivo and in vitro with GEF-H1, and recruits GEF-H1 to junctions. (A) Paracingulin forms a complex with GEF-H1 in vivo. Immunoblot analysis (using either anti-VSV or anti-myc antibodies) of immunoprecipitates, prepared either with anti-paracingulin polyclonal antiserum, or with preimmune serum, from lysates of WT MDCK cells that were cotransfected with YFP- and myc-tagged full-length canine paracingulin, with (+) or without (-) VSV-tagged GEF-H1. Control immunoprecipitations were carried out from cells expressing YFP-myc. Expression of the exogenous proteins was verified by immunoblotting (Supplemental Figure S1B). Immunoprecipitation of paracingulin from nontransfected cells was attempted; however, the yield of paracingulin was below the detection threshold. (B–D) Paracingulin interacts directly with GEF-H1 through regions located in the head and rod domains. (B) Scheme of five different GST fusion constructs of human paracingulin, in which numbers indicate amino acid residue boundaries. The head domain is between residues 1 and 598, and the coiled-coil rod plus tail domains are between residues 599 and 1302 of human paracingulin. (C) Immunoblot, using anti-His antibody, of His-tagged full-length canine GEF-H1 produced in baculovirus-infected insect cells, after incubation with the GST-fusion protein constructs shown in B. (D) Immunoblot analysis of GEF-H1 (increasing amounts) after pull-down with constructs B, D, or GST alone. Bottom (Ponceau red staining), equivalent amounts of the GST fusion proteins were used in the pull-downs. (E) Paracingulin depletion reduces junctional localization of GEF-H1. Immunofluorescence localization of endogenous GEF-H1 in cells depleted of paracingulin expressing the TetR [CGNL1(-) + TetR], either in the absence or in the presence of Dox, which reduces paracingulin expression. Note the significant decrease in junctional labeling for GEF-H1 in cells in the presence of Dox, which also reduces paracingulin labeling. Bar, 10 μm. (F) Paracingulin depletion results in decreased association of GEF-H1 with the membrane-cytoskeleton fraction. Immunoblot analysis with anti-paracingulin, anti-GEF-H1 antibodies (to detect endogenous GEF-H1), and anti-actin antibodies (to normalize for protein concentration) of lysates of cells depleted of paracingulin expressing the TetR [CGNL1(-) + TetR], either in the absence or in the presence of Dox, which reduces paracingulin expression. Cells were either lysed with RIPA buffer, to visualize total protein, or they were fractionated into Triton-insoluble fractions, after centrifugation either at 13,000 × g (low, low-speed pellet) or at 100,000 × g (high, high-speed pellet), and Triton-soluble (soluble, supernatant after centrifugation at 100,000 × g) fraction. Note that upon depletion of paracingulin, there is a significant decrease in the amount of GEF-H1 detected in both low-speed and high-speed pellets, and an increase in the amount of soluble GEF-H1.
To establish the molecular mechanism through which paracingulin helps to recruit GEF-H1 to junctions, we examined the association of endogenous GEF-H1 with the membrane-cytoskeleton fraction in CGNL1(-) cells expressing TetR, in the absence or in the presence of Dox. Cells were lysed either with RIPA buffer, to determine total GEF-H1 levels, or with CSK buffer, followed by fractionation into low-speed and high-speed pellets, and soluble fractions (Figure 3F). Analysis of total cell lysates showed that GEF-H1 was expressed at similar levels regardless of the presence or absence of Dox, whereas paracingulin levels were substantially decreased in the presence of Dox. However, in cells depleted of paracingulin there was a significant decrease in the amount of GEF-H1 in both the low-speed and the high-speed pellet fractions, and an increase in the soluble fraction (Figure 3F), consistent with the observation that GEF-H1 localization at junctions is decreased in CGNL1(-) cells.
Together, these results demonstrate that paracingulin and GEF-H1 physically and functionally interact and that paracingulin helps to recruit and inactivate GEF-H1 at junctions, resulting in a down-regulation of RhoA activity in confluent monolayers.
Paracingulin Depletion Delays TJ Assembly during the Calcium Switch in a Tiam1-dependent and RhoA-independent Manner
To test whether paracingulin regulates TJ assembly, CGNL1(-), control and WT cells were analyzed by measuring the TER in the calcium-switch assay. In this assay, cells are incubated in low calcium for several hours, to induce junction disassembly, and simultaneous formation of junctions is initiated by calcium readdition (Gonzalez-Mariscal et al., 1985). WT cells, and cells expressing a control shRNA, showed the typical pattern in the development of TER, with a rapid increase to a peak of ∼180 ohm.cm2 8 h after the addition of calcium, followed by a decrease, and stabilization after 24 h (Figure 4A). The molecular mechanism leading to the peak in the TER value after the calcium-switch is unknown, despite that it was first described over 20 years ago (Gonzalez-Mariscal et al., 1985). Strikingly, in CGNL1(-) cells, the characteristic peak of TER at 8 h was abolished, since the TER value only reached ∼75 ohm.cm2 (Figure 4B). This phenotype was rescued by expression of the TetR (Figure 4B), showing that it was specifically due to paracingulin depletion. Three distinct CGNL1(-) clones showed this phenotype, and expression of the TetR did not affect TER values in WT cells (Supplemental Figure S2).
Figure 4.

Paracingulin depletion affects TJ barrier development and delays TJ assembly. (A and B) Establishment of the TJ barrier, as determined by measurement of the TER (expressed as ohms per square centimeter) during the calcium-switch assay. (A) In WT cells and cells expressing control shRNA (control), the TER shows a peak value at ∼180 ohm.cm2 8 h after the calcium-switch, followed by a decrease, and stabilization after 24 h. A very similar profile was obtained in WT cells expressing the TetR (Supplemental Figure S2B). (B) In CGNL1(-) cells, this peak is abolished, and the phenotype is rescued by expression of the TetR. The same phenotype was observed in three separate CGNL1(-) clones (Supplemental Figure S2A). (C) Immunofluorescence localization of occludin before the calcium-switch (time 0 h) and at 1, 8, and 24 h after the calcium-switch. Occludin is localized diffusely in cells in low calcium and rapidly accumulates at sites of cell–cell contact in WT cells and in cells expressing control shRNA, resulting in a continuous junctional labeling at 8 h. However, paracingulin depletion results in a delay in the junctional recruitment of occludin, as shown by the lack of continuous junctional labeling at 8 h. (D) immunofluorescence localization of E-cadherin before the calcium-switch (time 0 h) and at 1, 4, 8, and 24 h after the calcium-switch. Note that E-cadherin accumulation at junctions is inhibited in CGNL1(-) cells up to the 4-h time point, but it seems indistinguishable from WT at the 8-h time point. Bar, 10 μm.
Analysis of the immunofluorescent distribution of occludin showed a delay in its accumulation at junctions, since at 1 and 8 h after calcium-switch most WT and control cells showed continuous labeling for occludin at junctions, whereas most CGNL1(-) cells showed discontinuous labeling (Figure 4C). Expression of the TetR reverted this phenotype, demonstrating that it was specifically due to paracingulin depletion (Figure 4C). A delay in junctional accumulation was observed also for E-cadherin, up to the 4-h time point, although at the 8-h time point the localization of E-cadherin along junctions appeared very similar in WT, control and CGNL1(-) cells (Figure 4D). However, 24 h after beginning of the calcium-switch, the TER values and the localizations of occludin and E-cadherin in WT, control or CGNL1(-) cells were similar (Figure 4, A–D), indicating that paracingulin plays a role only in the initial phase of junction assembly, but not in the regulation of the barrier function in confluent monolayers, that have mature junctions.
TJ assembly and TER development are known to be dependent on the activities of the Rho family GTPases Rac1 and RhoA (Jou et al., 1998; Bruewer et al., 2004). To test whether the effect of paracingulin depletion on TJ assembly correlates with changes in either Rac1 or RhoA activities, we measured these activities both in confluent cells, and during the calcium-switch, by GST pull-down assays. In confluent monolayers, paracingulin depletion had no detectable effect on Rac1 activity (Figure 5A), whereas it increased RhoA activity (Figure 2A). During the calcium-switch, WT cells showed two distinct peaks of Rac1 activation, one peak occurring very early (10–20 min) and one peak at ∼3–8 h after addition of calcium (Figure 5, B and C; see also Supplemental Figure S3). In CGNL1(-) cells, these peaks of Rac1 activation were either abolished (early peak) or strongly reduced (late peak), as the fraction of active Rac1 remained more uniform throughout the calcium-switch assay (Figure 5, B and C, and Supplemental Figure S3). With regard to RhoA activity, it was high in both WT and CGNL1(-) cells up to the 3-h time point. At 8 h after the calcium-switch, RhoA activity was down-regulated in WT cells, whereas it remained high in CGNL1(-) cells (Figure 5B). In summary, during the calcium-switch paracingulin depletion resulted in down-regulation of Rac1 activity, and no change in RhoA activity.
Figure 5.

Paracingulin depletion down-regulates Rac1 activity during the calcium-switch. (A–C) Immunoblot analysis of Rac1 (A) and Rac1 and RhoA pull-down assays (B) showing active versus total Rac1 and RhoA in WT and CGNL1(-) cells at confluence (A) and during the calcium-switch (B and C). The graph in C shows a quantitative densitometric analysis of the Rac1 data shown in B.
To test whether up-regulation of Rac1 activity could rescue the TER phenotype of CGNL1(-) cells, we first expressed dominant-active Rac1 in CGNL1(-) cells. However, since in our hands overexpression of dominant-active Rac1 resulted in cell death, we overexpressed Tiam1, a GEF activator of Rac1, which has been shown to promote junction formation in keratinocytes, by activation of the Par polarity complex (Habets et al., 1994; Mertens et al., 2005). We also tested whether down-regulation of RhoA activity affected the phenotype of CGNL1(-) cells, by expressing a dominant-nega-tive mutant of RhoA in CGNL1(-) cells. Notably, CGNL1(-) cells expressing Tiam1 showed increased Rac1 activity (Figure 6A) and in the TER assay they displayed a profile similar to that of WT or control cells, with a peak at ∼150 ohm·cm2 at 8 h (Figure 6B), indicating that increased Rac1 activity driven by Tiam1 overexpression can rescue the TER phenotype of CGNL1(-) cells, and that the peak in TER is due to Rac1 activation. Conversely, expression of the dominant-negative form of RhoA did not rescue the TER phenotype of CGNL1(-) cells (Figure 6B). In agreement with these observations, occludin localization in CGNL1(-) cells expressing Tiam1 was similar to WT and control cells, whereas in cells expressing the dominant-negative RhoA mutant there was still an impairment of occludin accumulation into junctions (Figure 6C).
Figure 6.

Paracingulin affects TER development in a Tiam1-dependent and RhoA-independent manner. (A) Increased Rac1 activity in cells expressing HA-tagged full-length Tiam1, as determined by a GST pull-down assay. (B) Measurement of TER shows that CGNL1(-) cells expressing exogenous Tiam-1, but not expressing either empty vector or a dominant-negative RhoA, show a peak of TER of ∼150 ohm.cm2 8 h after the calcium-switch, rescuing the TER phenotype of CGNL1(-) cells. (C) Immunofluorescence localization of occludin, showing that expression of HA-tagged Tiam1, but not expression of either empty vector or HA-tagged dominant-negative RhoA, rescues the delay in the junctional accumulation of occludin in CGNL1(-) cells. HA-labeled cells show the localization of RhoA, which is cytoplasmic and diffuse, and Tiam1, which is junctional, in cells expressing the respective constructs. (D) Measurement of TER shows that Tiam1 depletion results in the disappearance of the TER peak at 8 h, and the appearance of a peak at ∼24 h after the calcium-switch. (E) Immunofluorescence localization of occludin shows that Tiam1 depletion results in a delay in the junctional accumulation of occludin. Bar, 10 μm.
To further confirm that Tiam1 is required to develop a peak of TER at 8 h after the calcium-switch, we prepared clones of MDCK cells where Tiam1 was depleted by expression of shRNA, resulting in a reduction of Tiam1 mRNA levels between 40 and 60% of WT (Supplemental Figure S4A). Rac1 activity was decreased in these clones (Supplemental Figure S4B), which showed no peak in TER at 8 h after the calcium-switch (Figure 6D and Supplemental Figure S4C). In addition, occludin assembly into junctions was delayed, since at 8 h after the calcium-switch there was discontinuous junctional labeling, similarly to what observed in CGNL1(-) cells (Figure 6E). Together, these data confirm that normal levels of Tiam1 are required to ensure efficient junction assembly, in agreement with previous data on Tiam1 KO cells (Mertens et al., 2005). However, unlike Tiam1 KO cells and CGNL1(-) cells, the Tiam1-depleted clones showed a peak in TER at ∼24 h after the calcium-switch (Figure 6D and Supplemental Figure S4C). We interpret this as due to the presence of residual Tiam1 protein, which requires longer time to accumulate at junctions in sufficient amounts to induce the peak.
Together, these results provide strong evidence that paracingulin depletion delays TJ assembly by interfering with Tiam1-dependent Rac1 signaling at junctions, independently of RhoA.
Paracingulin Interacts with Tiam1, and Recruits Tiam1 to Junctions
We next investigated the molecular mechanism through which paracingulin regulates Rac1 activity. Because overexpression of Tiam1 rescued the functional and structural effects of paracingulin depletion on TJ assembly (Figure 6, B and C), we hypothesized that paracingulin functions by helping to recruit Tiam1 to junctions, and thus promote Rac1 activation during the early phases of junction formation. To test this hypothesis, we first studied the in vivo interaction of paracingulin with Tiam1. Because endogenous Tiam1 could not be immunoprecipitated from MDCK lysates with any of the antibodies we tested, and the yield of endogenous paracingulin was very low, WT MDCK cells were cotransfected with paracingulin and Tiam1 (Supplemental Figure S1B), and paracingulin immunoprecipitates were analyzed by immunoblotting. A similar experiment was carried out with cells expressing cingulin plus either GEF-H1 or Tiam1 (Supplemental Figure S1B), to check whether cingulin, which does not affect TER development (Guillemot and Citi, 2006a), interacts or not with Tiam1. Tiam1 was specifically detected in paracingulin immunoprecipitates (Figure 7A), whereas only GEF-H1, but not Tiam1, was detected in cingulin immunoprecipitates (Figure 7B). Thus, paracingulin but not cingulin can form a complex in vivo with Tiam1. To test whether paracingulin and Tiam1 can interact directly in vitro, we first expressed recombinant full-length His-tagged Tiam1 in insect cells, and we incubated it with GST fusion proteins encoding different regions of paracingulin (Figure 3B). Tiam1 was detected by immunoblotting in GST pull-down experiments by using either construct B (residues 250-420) or construct D (residues 591-882) of paracingulin (Figure 7, C and D). The GST fusion protein D seemed to interact with Tiam1 with higher affinity than the protein B (Figure 7D). Next, we designed and expressed a GST fusion protein with the region comprising the Dbl homology (DH) and pleckstrin homology (PH) domains of Tiam1 (residues 1037-1404). We speculated that this region of Tiam1 may be involved in its interaction with paracingulin, based on our previous observation that interaction of cingulin with GEF-H1 occurs through the DH/PH domains of GEF-H1 (Aijaz et al., 2005). Indeed, the GST fusion protein making up the DH/PH domains of Tiam1 associated specifically with recombinant full-length paracingulin (Figure 7E), showing that the two proteins interact directly in vitro through at least this region of Tiam1.
Figure 7.

Paracingulin interacts in vivo and in vitro with Tiam1 and recruits Tiam1 to junctions. (A) Paracingulin forms a complex with Tiam1 in vivo. Immunoblot analysis (using either anti-HA or anti-myc antibodies) of immunoprecipitates, prepared either with anti-paracingulin polyclonal antiserum, or with preimmune serum, from lysates of WT MDCK cells that were cotransfected with YFP- and myc-tagged full-length canine paracingulin, with (+) or without (-) HA-tagged Tiam1. Control immunoprecipitations were carried out from cells expressing YFP-myc. Exogenous protein expression was verified by immunoblotting (Supplemental Figure S1B). Immunoprecipitation of endogenous proteins was not possible due to very low amounts of endogenous paracingulin that could be immunoprecipitated, and to the lack of anti-Tiam1 antibodies that could in our hands reliably detect Tiam1 in immunoprecipitates. (B) Cingulin forms a complex with GEF-H1 but not with Tiam1 in vivo. Immunoblot analysis (using either anti-HA, anti-VSV, or anti-myc antibodies) of immunoprecipitates prepared with anti-cingulin polyclonal antiserum, from lysates of MDCK cells expressing YFP- and myc-tagged cingulin, that were cotransfected either with VSV-tagged GEF-H1 or with HA-tagged Tiam1. Exogenous protein expression was verified by immunoblotting (Supplemental Figure S1C). (C and D) Paracingulin interacts directly with Tiam1, through regions located in its head and rod domains. (C) Immunoblot analysis, with anti-His antibodies, of full-length His-tagged Tiam1 expressed in baculovirus-infected insect cells, after incubation of the five different GST fusion constructs of human paracingulin (Figure 3B), or with GST alone. Note strong Tiam1 labeling in pull-downs with constructs B and D and weaker labeling with constructs C and E. (D) Immunoblot of increasing amounts of full-length Tiam1, after incubation with the GST-fusion proteins constructs B and D, and GST alone. Bottom, GST fusion protein amounts, visualized by Ponceau red staining. (E) Tiam1 interacts directly with paracingulin, through a region comprising its DH/PH domains. Immunoblot analysis, with anti-paracingulin antibodies, of full-length, His-tagged paracingulin expressed in baculovirus-infected insect cells, after incubation with GST-Tiam1DH/PH (residues 1037-1404), or with GST alone. Note that the GST fusion protein of Tiam1 interacts strongly with full-length paracingulin. (F) Paracingulin depletion reduces Tiam1 accumulation at junctions. The micrographs show the immunofluorescence localization of exogenously expressed, HA-tagged Tiam1 in CGNL1(-) cells, either without or with expression of the TetR, which rescues the expression of paracingulin. Junctional labeling for Tiam1 is dramatically reduced in cells depleted of paracingulin. Numbers in each micrograph indicate exposure time. Note that exposure time in CGNL1(-) cells was increased to 400 ms, to visualize weak Tiam1 staining. Bar, 10 μm. (G) Paracingulin depletion results in decreased association of Tiam1 with the membrane-cytoskeleton fraction. Immunoblot analysis with anti-paracingulin, anti-HA antibodies (to detect exogenous Tiam1), and anti-actin antibodies (to normalize for protein concentration) of lysates of cells depleted of paracingulin [CGNL1(-)], either in the absence or in the presence of TetR, which rescues paracingulin expression. Cells were either lysed with RIPA buffer, to visualize total protein, or were fractionated into Triton-insoluble fractions, after centrifugation either at 13,000 × g (low, low-speed pellet), or at 100,000 × g (high, high speed pellet), and Triton-soluble (soluble, supernatant after centrifugation at 100,000 × g) fraction. Note that upon expression of paracingulin there is a dramatic increase in the amount of Tiam1 detected in both low-speed and high-speed pellets, and a decrease in the amount of soluble Tiam1.
To establish whether the mechanism through which paracingulin activates Rac1 is by recruiting Tiam1 to junctions, and to demonstrate a physiological interaction between paracingulin and Tiam1, we asked whether Tiam1 localization at junctions depends on paracingulin. The localization of exogenous Tiam1 at junctions was examined by immunofluorescence in CGNL1(-) cells, either in the absence of the TetR, or in its presence, which rescues paracingulin expression (Figure 7F). In the presence of paracingulin, Tiam1 was clearly detectable and distributed linearly along all junctions, whereas in CGNL1(-) cells there was a dramatic reduction in the presence and intensity of the immunofluorescence signal (Figure 7F). Thus, efficient accumulation of Tiam1 at junctions requires paracingulin, demonstrating a functional interaction in vivo between these two proteins.
Finally, to confirm the molecular mechanism through which paracingulin helps to recruit Tiam1 to junctions, we examined the association of Tiam1 with the membrane–cytoskeleton fraction in CGNL1(-) cells expressing Tiam1, and expressing or not the TetR. Cells were lysed either with RIPA buffer, to determine total Tiam1 levels, or with CSK buffer, followed by fractionation into low-speed and high-speed pellets, and soluble fractions (Figure 7G). Analysis of total cell lysates showed that Tiam1 was expressed at similar levels in CGNL1(-) cells, and in CGNL1(-) cells where paracingulin expression was rescued by the TetR. However, depletion of paracingulin was associated with a significant decrease in the amount of Tiam1 in both the low-speed and the high-speed pellet fractions, and an increase in the soluble fraction (Figure 7G), consistent with the observation that Tiam1 localization at junctions is decreased in CGNL1(-) cells (Figure 7F). Together, these results demonstrate that paracingulin functions by favoring the junctional recruitment of Tiam1, leading to activation of Rac1 at junctions.
DISCUSSION
Here, we characterize paracingulin, a junctional protein with a previously unknown function, as a regulator of the activity of both Rac1 and RhoA. In confluent monolayers paracingulin depletion results in increased RhoA activity, whereas during junction assembly it results in decreased Rac1 activity. Paracingulin interacts in vivo and in vitro with GEF-H1 and Tiam1, and promotes their recruitment to junctions, providing a molecular mechanism through which paracingulin controls the activity of RhoA and Rac1.
Regulation of RhoA Signaling by Paracingulin
Paracingulin depletion leads to increased RhoA activity in confluent cells. Because paracingulin levels are higher in confluent than in proliferating cells (Pulimeno and Citi, unpublished observations), and paracingulin interacts with GEF-H1, we conclude that paracingulin contributes to down-regulating RhoA activity as cells become confluent, by recruiting GEF-H1 to junctions and inactivating it. No up-regulation of RhoA during junction assembly was observed in paracingulin-depleted cells, probably due to the fact that maximal activation of RhoA is already occurring under these conditions.
A consequence of paracingulin depletion is the small, but statistically significant increase in cell proliferation and claudin-2 mRNA levels. Because inhibition of RhoA reverses this phenotype, we conclude that it is due, at least in part, to up-regulation of RhoA activity in paracingulin-depleted cells. However Rac1, which is also regulated by paracingulin, is not involved in the regulation of claudin-2 expression and cell proliferation, because the changes in claudin-2 mRNA levels and BrdU incorporation are not rescued by overexpression of Tiam1 (data not shown).
The precise mechanism by which cingulin, paracingulin, and microtubules (Krendel et al., 2002; Aijaz et al., 2005) inhibit GEF-H1 remains unclear. Microtubules bind to the PH domain of GEF-H1, and they are believed to favor its folding into an inhibited state, where access to the substrate is blocked (Birkenfeld et al., 2008). Because cingulin also binds to the PH domain of GEF-H1, it is likely that it also blocks GEF-H1 function by favoring the inhibited state conformation.
Regulation of Rac1 Signaling by Paracingulin
TER development during the calcium-switch assay is the assay of choice to investigate the role of specific proteins in junction assembly. The signaling mechanism leading to the peak in TER observed during junction assembly by the calcium-switch was unknown until now. We show that the peak in TER is due to Rac1 activation, based on the temporal overlap between the second wave of Rac1 activation and the peak in TER, and on the observation that paracingulin depletion, which strongly reduces the waves of Rac1 activation, also strongly reduces the peak in TER. Because this phenotype is rescued by overexpression of Tiam1, and paracingulin interacts with Tiam1, we conclude that Rac1 activation and peak in TER during the calcium-switch depend on the recruitment of Tiam1 to junctions by paracingulin. Interestingly, although Tiam1 recruitment to junctions is impaired in CGNL1(-) cells, overexpression of Tiam1 rescued the TER phenotype of these cells. This apparently contradictory observation can be explained by the following possibilities: 1) in cells overexpressing Tiam1 sufficient amounts of Tiam1 can be recruited to junctions by residual paracingulin, and by other factors (see below); and 2) the overall increase in cellular Rac1 activity is sufficient to drive normal TJ assembly. Conversely, the observation that dominant-negative RhoA did not rescue the TER phenotype of CGNL1(-) cells argues against a role of GEF-H1–paracingulin interaction in mediating the loss of the peak in TER observed in CGNL1(-) cells. Future studies should dissect the role of different Tiam1 domains in rescuing the phenotype of CGNL1(-) cells.
The biological significance of the peak in TER, and how the two transient waves of increased Rac1 activity translate into increased TER, is unclear. The pattern of expression of claudin isoforms is critical in determining the paracellular barrier properties of epithelial tissues (reviewed in Van Itallie and Anderson, 2006). Thus, it is possible that during TJ assembly specific isoforms of claudins that confer high TER are transiently concentrated at junctions, or that the function of the barrier unit is dynamically affected by the local reorganization of the actin cytoskeleton, which interacts with the proteins, such as ZO proteins, that provide a scaffold for claudins.
Our results provide new insights into the mechanism through which Rac1 becomes activated at new sites of cadherin-based cell–cell contact. There is evidence that Rac1 activation is triggered by activation of PI-3 kinase, which results in the formation of phosphoinositides, which could recruit GEFs to the membrane, through interaction with their PH domains (Michiels et al., 1997; Stam et al., 1997; Sander et al., 1998; Pece et al., 1999; Malliri et al., 2004). However, other studies indicate that Rac1 activation occurs independently of phosphatidylinositol 3-kinase activation (Nakagawa et al., 2001; Betson et al., 2002; Ehrlich et al., 2002). Thus, the precise mechanism of Rac1 activation at cadherin-based junctions is controversial. In epithelial cells, Tiam1 is the major GEF activator of Rac1, and it promotes AJ and TJ formation via activation of the Par3 polarity complex (Malliri et al., 2004; Mertens et al., 2005). In turn, Par3 inhibits Tiam1 function and Rac1 activity (Chen and Macara, 2005). Thus, one possibility to explain our results is that paracingulin depletion decreases Rac1 activity indirectly, by increasing the levels of Par3. However, our immunoblot analysis showed that levels of Par3 were unaffected in CGNL1(-) cells, ruling out this possibility. Although Tiam1 has been reported to interact with several proteins, most of these proteins have been identified in fibroblasts/neurons, and not epithelial cell model systems (Mertens et al., 2003; Ten Klooster et al., 2006). Our results provide the first evidence that depletion of a Tiam1-interacting protein results in decreased accumulation of Tiam1 at epithelial junctions, and increased Tiam1 solubility. This, together with the observation that paracingulin depletion inhibits Rac1 activation during junction assembly, identifies paracingulin as a key molecule involved in Rac1 activation during junction assembly, by recruiting Tiam1 to junctions and promoting its association with the junctional acto-myosin cytoskeleton.
CONCLUSIONS
Paracingulin regulates the activity of two distinct small GTPases, by interacting with their respective GEFs. To our knowledge, this is the first time that a junctional protein with such dual function has been described. Although p120catenin both activates Rac1 and inhibits RhoA (Anastasiadis and Reynolds, 2001) and interacts with the Rac1 GEF Vav (Noren et al., 2000), it is not clear whether it controls Vav junctional recruitment. In addition, inhibition of RhoA by p120 is thought to occur through direct sequestration of RhoA, rather than interaction with a GEF (Anastasiadis et al., 2000).
The formation of stable E-cadherin-based adhesion sites critically requires Rac1 activity (Braga et al., 1997; Nakagawa et al., 2001), and the formation of stable E-cadherin adhesions is necessary (although not sufficient; Denisenko et al., 1994) for TJ assembly (Gumbiner et al., 1988). Paracingulin is localized both at AJ and TJ in some tissues (Ohnishi et al., 2004), although the precise localizations of both paracingulin and Tiam1 in cultured MDCK cells are not known. We show here that paracingulin depletion delays both AJ and TJ assembly, based on immunofluorescence and TER assays. We propose that paracingulin functions early during the formation of primordial cadherin-based AJ, by helping to recruit Tiam1 and thus activate Rac1 at AJ (Figure 8). Rac1 activation leads to efficient AJ assembly, which in turns allows TJ assembly to occur, through the reorganization of the actin cytoskeleton and the recruitment of membrane and cytoplasmic junctional proteins, including Par3 (Mertens et al., 2005). As junctions mature, Par3 accumulates at junctions and inactivates Tiam1 (Chen and Macara, 2005), in a negative feedback loop (Figure 8). Par3 translocates to cell–cell contract regions later than the formation of the primordial AJ (Suzuki et al., 2002), consistent with our model that Rac1 activation mediated by the paracingulin–Tiam1 interaction at AJ precedes the subsequent inactivation of Tiam1 by Par3 at TJ. Once junctions have matured in confluent cells, Tiam1 activity is negatively regulated by Par3, whereas GEF-H1 and RhoA activity are negatively regulated by cingulin and paracingulin at TJ (Figure 8). It is noteworthy that mature TJ are not as sensitive as AJ to changes in Rac1 activity (Jou et al., 1998; Bruewer et al., 2004). Thus, we propose that the delay of TJ assembly observed in CGNL1(-) cells is a consequence of delayed AJ formation due to decreased Rac1 activation at AJ.
Figure 8.
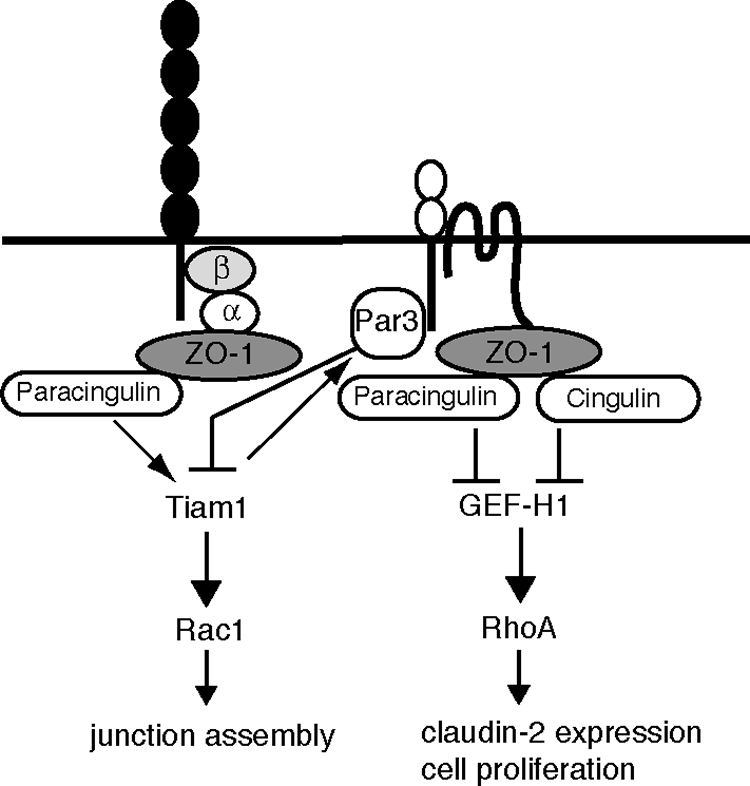
Schematic diagram illustrating a model for the roles of paracingulin at cadherin-based AJs and TJs. The horizontal line represents the plasma membrane, and selected proteins of AJ (cadherin, with associated cytoplasmic proteins α-catenin (α) and β-catenin (β), ZO-1) and TJ (JAM, claudin/occludin, Par3, ZO-1, and cingulin) are schematically represented. ZO-1 recruits cingulin (D'Atri et al., 2002) and paracingulin (Pulimeno and Citi, unpublished data) to junctions. Paracingulin activates Rac1 by recruiting Tiam1. Tiam1 promotes assembly of Par3 at junctions (Mertens et al., 2005). Par3, in turn, negatively regulates Tiam1 and Rac1 activity (Chen and Macara, 2005). Paracingulin and cingulin down-regulate RhoA activity through sequestration and inhibition of GEF-H1.
Coordination of RhoA and Rac1 activities is critical to ensure junction assembly and regulation of barrier function in developing and mature epithelia (Fukata and Kaibuchi, 2001). Diseases affecting the intestinal, renal, auditory epithelia, as well as invasion of epithelia by pathogens and toxins are associated with changes in the expression of claudins and Rho family GTPase activities (Mankertz and Schulzke, 2007; Birkenfeld et al., 2008). Thus, our results on the role of paracingulin in regulating Rho family GTPases and claudin-2 expression provide new insights and tools to understand the molecular mechanisms underlying the physiology and pathophysiology of epithelial tissues.
Supplementary Material
ACKNOWLEDGMENTS
We thank the colleagues cited in the text for gift of reagents. We thank the Swiss Cancer League, the Swiss National Foundation, the State of Geneva, and the Ministry of Italian University and Research for financial support.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E08-06-0558) on July 23, 2008.
REFERENCES
- Aijaz S., D'Atri F., Citi S., Balda M. S., Matter K. Binding of GEF-H1 to the tight junction-associated adaptor cingulin results in inhibition of Rho signaling and G1/S phase transition. Dev. Cell. 2005;8:777–786. doi: 10.1016/j.devcel.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Anastasiadis P. Z., Moon S. Y., Thoreson M. A., Mariner D. J., Crawford H. C., Zheng Y., Reynolds A. B. Inhibition of RhoA by p120 catenin. Nat. Cell Biol. 2000;2:637–644. doi: 10.1038/35023588. [DOI] [PubMed] [Google Scholar]
- Anastasiadis P. Z., Reynolds A. B. Regulation of Rho GTPases by p120-catenin. Curr. Opin. Cell Biol. 2001;13:604–610. doi: 10.1016/s0955-0674(00)00258-1. [DOI] [PubMed] [Google Scholar]
- Anderson J. M., Van Itallie C. M. Tight junctions and the molecular basis for regulation of paracellular permeability. Am. J. Physiol. 1995;269:G467–G475. doi: 10.1152/ajpgi.1995.269.4.G467. [DOI] [PubMed] [Google Scholar]
- Betson M., Lozano E., Zhang J., Braga V. M. Rac activation upon cell-cell contact formation is dependent on signaling from the epidermal growth factor receptor. J. Biol. Chem. 2002;277:36962–36969. doi: 10.1074/jbc.M207358200. [DOI] [PubMed] [Google Scholar]
- Birkenfeld J., Nalbant P., Yoon S. H., Bokoch G. M. Cellular functions of GEF-H1, a microtubule-regulated Rho-GEF: is altered GEF-H1 activity a crucial determinant of disease pathogenesis? Trends Cell Biol. 2008;18:210–219. doi: 10.1016/j.tcb.2008.02.006. [DOI] [PubMed] [Google Scholar]
- Braga V. M. Cell-cell adhesion and signalling. Curr. Opin. Cell Biol. 2002;14:546–556. doi: 10.1016/s0955-0674(02)00373-3. [DOI] [PubMed] [Google Scholar]
- Braga V. M., Machesky L. M., Hall A., Hotchin N. A. The small GTPases Rho and Rac are required for the establishment of cadherin-dependent cell-cell contacts. J. Cell Biol. 1997;137:1421–1431. doi: 10.1083/jcb.137.6.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruewer M., Hopkins A. M., Hobert M. E., Nusrat A., Madara J. L. RhoA, Rac1, and Cdc42 exert distinct effects on epithelial barrier via selective structural and biochemical modulation of junctional proteins and F-actin. Am. J. Physiol. Cell Physiol. 2004;287:C327–C335. doi: 10.1152/ajpcell.00087.2004. [DOI] [PubMed] [Google Scholar]
- Chen X., Macara I. G. Par-3 controls tight junction assembly through the Rac exchange factor Tiam1. Nat. Cell Biol. 2005;7:262–269. doi: 10.1038/ncb1226. [DOI] [PubMed] [Google Scholar]
- Citi S., D'Atri F., Cordenonsi M., Cardellini P. Tight junction protein expression in early Xenopus development and protein interaction studies. In: Fleming T. P., editor. Cell Cell Interactions. vol. 256. Oxford, United Kingdom: IRL Press; 2001. pp. 153–176. [Google Scholar]
- Citi S., Sabanay H., Jakes R., Geiger B., Kendrick-Jones J. Cingulin, a new peripheral component of tight junctions. Nature. 1988;333:272–276. doi: 10.1038/333272a0. [DOI] [PubMed] [Google Scholar]
- Coleman M. L., Marshall C. J., Olson M. F. RAS and RHO GTPases in G1-phase cell-cycle regulation. Nat. Rev. Mol. Cell Biol. 2004;5:355–366. doi: 10.1038/nrm1365. [DOI] [PubMed] [Google Scholar]
- Cordenonsi M., D'Atri F., Hammar E., Parry D. A., Kendrick-Jones J., Shore D., Citi S. Cingulin contains globular and coiled-coil domains and interacts with ZO-1, ZO-2, ZO-3, and myosin. J. Cell Biol. 1999;147:1569–1582. doi: 10.1083/jcb.147.7.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Atri F., Citi S. Cingulin interacts with F-actin in vitro. FEBS Lett. 2001;507:21–24. doi: 10.1016/s0014-5793(01)02936-2. [DOI] [PubMed] [Google Scholar]
- D'Atri F., Nadalutti F., Citi S. Evidence for a functional interaction between cingulin and ZO-1 in cultured cells. J. Biol. Chem. 2002;277:27757–27764. doi: 10.1074/jbc.M203717200. [DOI] [PubMed] [Google Scholar]
- Denisenko N., Burighel P., Citi S. Different effects of protein kinase inhibitors on the localization of junctional proteins at cell-cell contact sites. J. Cell Sci. 1994;107:969–981. doi: 10.1242/jcs.107.4.969. [DOI] [PubMed] [Google Scholar]
- Ehrlich J. S., Hansen M. D., Nelson W. J. Spatio-temporal regulation of Rac1 localization and lamellipodia dynamics during epithelial cell-cell adhesion. Dev. Cell. 2002;3:259–270. doi: 10.1016/s1534-5807(02)00216-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata M., Kaibuchi K. Rho-family GTPases in cadherin-mediated cell-cell adhesion. Nat. Rev. Mol. Cell Biol. 2001;2:887–897. doi: 10.1038/35103068. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Mariscal L., Chavez de Ramirez B., Cereijido M. Tight junction formation in cultured epithelial cells (MDCK) J. Membr. Biol. 1985;86:113–125. doi: 10.1007/BF01870778. [DOI] [PubMed] [Google Scholar]
- Guillemot L., Citi S. Cingulin regulates claudin-2 expression and cell proliferation through the small GTPase RhoA. Mol. Biol. Cell. 2006a;17:3569–3577. doi: 10.1091/mbc.E06-02-0122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemot L., Citi S. Cingulin, a cytoskeleton-associated protein of the tight junction. In: Gonzalez-Mariscal L., editor. Tight Junctions. New York USA: Landes Bioscience-Springer Science; 2006b. pp. 54–63. [Google Scholar]
- Guillemot L., Paschoud S., Pulimeno P., Foglia A., Citi S. The cytoplasmic plaque of tight junctions: a scaffolding and signalling center. Biochim. Biophys. Acta. 2008;1778:601–613. doi: 10.1016/j.bbamem.2007.09.032. [DOI] [PubMed] [Google Scholar]
- Gumbiner B., Stevenson B., Grimaldi A. The role of the cell adhesion molecule uvomorulin in the formation and maintenance of the epithelial junctional complex. J. Cell Biol. 1988;107:1575–1587. doi: 10.1083/jcb.107.4.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbiner B. M. Regulation of cadherin-mediated adhesion in morphogenesis. Nat. Rev. Mol. Cell Biol. 2005;6:622–634. doi: 10.1038/nrm1699. [DOI] [PubMed] [Google Scholar]
- Habets G. G., Scholtes E. H., Zuydgeest D., van der Kammen R. A., Stam J. C., Berns A., Collard J. G. Identification of an invasion-inducing gene, Tiam-1, that encodes a protein with homology to GDP-GTP exchangers for Rho-like proteins. Cell. 1994;77:537–549. doi: 10.1016/0092-8674(94)90216-x. [DOI] [PubMed] [Google Scholar]
- Jaffe A. B., Hall A. Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- Jou T. S., Schneeberger E. E., Nelson W. J. Structural and functional regulation of tight junctions by RhoA and Rac1 small GTPases. J. Cell Biol. 1998;142:101–115. doi: 10.1083/jcb.142.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krendel M., Zenke F. T., Bokoch G. M. Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat. Cell Biol. 2002;4:294–301. doi: 10.1038/ncb773. [DOI] [PubMed] [Google Scholar]
- Malliri A., van Es S., Huveneers S., Collard J. G. The Rac exchange factor Tiam1 is required for the establishment and maintenance of cadherin-based adhesions. J. Biol. Chem. 2004;279:30092–30098. doi: 10.1074/jbc.M401192200. [DOI] [PubMed] [Google Scholar]
- Mankertz J., Schulzke J. D. Altered permeability in inflammatory bowel disease: pathophysiology and clinical implications. Curr. Opin. Gastroenterol. 2007;23:379–383. doi: 10.1097/MOG.0b013e32816aa392. [DOI] [PubMed] [Google Scholar]
- Mertens A. E., Roovers R. C., Collard J. G. Regulation of Tiam1-Rac signalling. FEBS Lett. 2003;546:11–16. doi: 10.1016/s0014-5793(03)00435-6. [DOI] [PubMed] [Google Scholar]
- Mertens A. E., Rygiel T. P., Olivo C., van der Kammen R., Collard J. G. The Rac activator Tiam1 controls tight junction biogenesis in keratinocytes through binding to and activation of the Par polarity complex. J. Cell Biol. 2005;170:1029–1037. doi: 10.1083/jcb.200502129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michiels F., Stam J. C., Hordijk P. L., van der Kammen R. A., Ruuls-Van Stalle L., Feltkamp C. A., Collard J. G. Regulated membrane localization of Tiam1, mediated by the NH2-terminal pleckstrin homology domain, is required for Rac-dependent membrane ruffling and C-Jun NH2-terminal kinase activation. J. Cell Biol. 1997;137:387–398. doi: 10.1083/jcb.137.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa M., Fukata M., Yamaga M., Itoh N., Kaibuchi K. Recruitment and activation of Rac1 by the formation of E-cadherin-mediated cell-cell adhesion sites. J. Cell Sci. 2001;114:1829–1838. doi: 10.1242/jcs.114.10.1829. [DOI] [PubMed] [Google Scholar]
- Noren N. K., Liu B. P., Burridge K., Kreft B. p120 catenin regulates the actin cytoskeleton via Rho family GTPases. J. Cell Biol. 2000;150:567–580. doi: 10.1083/jcb.150.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noren N. K., Niessen C. M., Gumbiner B. M., Burridge K. Cadherin engagement regulates Rho family GTPases. J. Biol. Chem. 2001;276:33305–33308. doi: 10.1074/jbc.C100306200. [DOI] [PubMed] [Google Scholar]
- Nusrat A., Giry M., Turner J. R., Colgan S. P., Parkos C. A., Carmes D., Lemichez E., Boquet P., Madara J. L. Rho protein regulates tight junctions and perijunctional actin organization in polarized epithelia. Proc. Natl. Acad. Sci. USA. 1995;92:10629–10633. doi: 10.1073/pnas.92.23.10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi H., Nakahara T., Furuse K., Sasaki H., Tsukita S., Furuse M. JACOP, a novel plaque protein localizing at the apical junctional complex with sequence similarity to cingulin. J. Biol. Chem. 2004;279:46014–46022. doi: 10.1074/jbc.M402616200. [DOI] [PubMed] [Google Scholar]
- Paschoud S., Citi S. Inducible overexpression of cingulin in stably transfected MDCK cells does not affect tight junction organization and gene expression. Mol. Membr. Biol. 2008;25:1–13. doi: 10.1080/09687680701474009. [DOI] [PubMed] [Google Scholar]
- Pece S., Chiariello M., Murga C., Gutkind J. S. Activation of the protein kinase Akt/PKB by the formation of E-cadherin-mediated cell-cell junctions. Evidence for the association of phosphatidylinositol 3-kinase with the E-cadherin adhesion complex. J. Biol. Chem. 1999;274:19347–19351. doi: 10.1074/jbc.274.27.19347. [DOI] [PubMed] [Google Scholar]
- Perez-Moreno M., Jamora C., Fuchs E. Sticky business: orchestrating cellular signals at adherens junctions. Cell. 2003;112:535–548. doi: 10.1016/s0092-8674(03)00108-9. [DOI] [PubMed] [Google Scholar]
- Rossman K. L., Der C. J., Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 2005;6:167–180. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- Sakurai A., Fukuhara S., Yamagishi A., Sako K., Kamioka Y., Masuda M., Nakaoka Y., Mochizuki N. MAGI-1 is required for Rap1 activation upon cell-cell contact and for enhancement of vascular endothelial cadherin-mediated cell adhesion. Mol. Biol. Cell. 2006;17:966–976. doi: 10.1091/mbc.E05-07-0647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander E. E., van Delft S., ten Klooster J. P., Reid T., van der Kammen R. A., Michiels F., Collard J. G. Matrix-dependent Tiam1/Rac signaling in epithelial cells promotes either cell-cell adhesion or cell migration and is regulated by phosphatidylinositol 3-kinase. J. Cell Biol. 1998;143:1385–1398. doi: 10.1083/jcb.143.5.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneeberger E. E., Lynch R. D. The tight junction: a multifunctional complex. Am. J. Physiol. Cell Physiol. 2004;286:C1213–C1228. doi: 10.1152/ajpcell.00558.2003. [DOI] [PubMed] [Google Scholar]
- Shin K., Fogg V. C., Margolis B. Tight junctions and cell polarity. Annu. Rev. Cell Dev. Biol. 2006;22:207–235. doi: 10.1146/annurev.cellbio.22.010305.104219. [DOI] [PubMed] [Google Scholar]
- Stam J. C., Sander E. E., Michiels F., van Leeuwen F. N., Kain H. E., van der Kammen R. A., Collard J. G. Targeting of Tiam1 to the plasma membrane requires the cooperative function of the N-terminal pleckstrin homology domain and an adjacent protein interaction domain. J. Biol. Chem. 1997;272:28447–28454. doi: 10.1074/jbc.272.45.28447. [DOI] [PubMed] [Google Scholar]
- Suzuki A., Ishiyama C., Hashiba K., Shimizu M., Ebnet K., Ohno S. aPKC kinase activity is required for the asymmetric differentiation of the premature junctional complex during epithelial cell polarization. J. Cell Sci. 2002;115:3565–3573. doi: 10.1242/jcs.00032. [DOI] [PubMed] [Google Scholar]
- Ten Klooster J. P., Evers E. E., Janssen L., Machesky L. M., Michiels F., Hordijk P., Collard J. G. Interaction between Tiam1 and the Arp2/3 complex links activation of Rac to actin polymerization. Biochem. J. 2006;397:39–45. doi: 10.1042/BJ20051957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Itallie C. M., Anderson J. M. Claudins and epithelial paracellular transport. Annu. Rev. Physiol. 2006;68:403–429. doi: 10.1146/annurev.physiol.68.040104.131404. [DOI] [PubMed] [Google Scholar]
- Wells C. D., et al. A Rich1/Amot complex regulates the Cdc42 GTPase and apical-polarity proteins in epithelial cells. Cell. 2006;125:535–548. doi: 10.1016/j.cell.2006.02.045. [DOI] [PubMed] [Google Scholar]
- Wildenberg G. A., Dohn M. R., Carnahan R. H., Davis M. A., Lobdell N. A., Settleman J., Reynolds A. B. p120-catenin and p190RhoGAP regulate cell-cell adhesion by coordinating antagonism between Rac and Rho. Cell. 2006;127:1027–1039. doi: 10.1016/j.cell.2006.09.046. [DOI] [PubMed] [Google Scholar]
- Yamada S., Nelson W. J. Localized zones of Rho and Rac activities drive initiation and expansion of epithelial cell-cell adhesion. J. Cell Biol. 2007;178:517–527. doi: 10.1083/jcb.200701058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.