

Abstract
Helicobacter pylori is the strongest known risk factor for gastric adenocarcinoma, yet only a fraction of infected persons develop cancer. One H. pylori constituent that augments disease risk is the cytotoxin-associated gene (cag) pathogenicity island, which encodes a secretion system that translocates bacterial effector molecules into host cells. Matrix metalloproteinase (MMP)-7, a member of a family of enzymes with tumor-initiating properties, is overexpressed in premalignant and malignant gastric lesions, and H. pylori cag+ strains selectively increase MMP-7 protein levels in gastric epithelial cells in vitro and in vivo. We now report that H. pylori-mediated mmp-7 induction is transcriptionally regulated via aberrant activation of p120-catenin (p120), a component of adherens junctions. H. pylori increases mmp-7 mRNA levels in a cag- and p120-dependent manner and induces translocation of p120 to the nucleus in vitro and in a novel ex vivo gastric gland culture system. Nuclear translocation of p120 in response to H. pylori relieves Kaiso-mediated transcriptional repression of mmp-7, which is implicated in tumorigenesis. These results indicate that selective and coordinated induction of mmp-7 expression by H. pylori cag+ isolates may explain in part the augmentation in gastric cancer risk associated with these strains.
INTRODUCTION
Helicobacter pylori induces an inflammatory response in the stomach that persists for decades, and biological costs incurred by this pathogen include an increased risk for gastric adenocarcinoma and non-Hodgkins lymphoma of the stomach (Nomura et al., 1991; Parsonnet et al., 1991; Peterson, 1991; Hansson et al., 1993; Correa, 1996; Uemura et al., 2001; Peek and Blaser, 2002; Moss and Sood, 2003). However, only a fraction of colonized persons ever develop neoplasia, and enhanced cancer risk is related to strain-specific differences, aberrant host responses, and/or specific interactions between microbial and host determinants.
H. pylori strains that possess the cytotoxin-associated gene (cag) pathogenicity island increase the risk for cancer compared with strains that lack this genetic locus (Peek and Blaser, 2002). The cag island encodes proteins, such as CagE, that form a type IV secretion system that translocates components of bacterial peptidoglycan and CagA, the product of the terminal gene of the island, into host cells (Asahi et al., 2000; Backert et al., 2000; Odenbreit et al., 2000; Stein et al., 2000; Selbach et al., 2002; Viala et al., 2004). After translocation, peptidoglycan initiates innate immune signaling via activation of the intracellular pattern recognition receptor, Nod-1, and the transcriptional activator nuclear factor-κB (NF-κB) (Viala et al., 2004). Intracellular CagA undergoes Src-dependent tyrosine phosphorylation and activates a eukaryotic phosphatase, leading to dephosphorylation of host cell proteins and cellular morphological changes (Backert et al., 2000; Higashi et al., 2002; Selbach et al., 2002; Stein et al., 2002). Recently, CagA has been shown to activate β-catenin and induce NF-κB–mediated interleukin-8 release from gastric epithelial cells (Brandt et al., 2005; Franco et al., 2005). The presence of the cag island also influences the topography of colonization in the stomach, because H. pylori cag− strains predominate within the mucus gel layer, whereas cag+ strains are found immediately adjacent to epithelial cells (Camorlinga-Ponce et al., 2004).
Matrix metalloproteinase (MMP)-7 is a member of a family of zinc-dependent proteolytic enzymes with tumor-initiating properties and is expressed and secreted by epithelial cells (Coussens et al., 2002; Egeblad and Werb, 2002). We and others have previously demonstrated that H. pylori cag+ strains selectively up-regulate MMP-7 protein levels in gastric epithelial cells (Crawford et al., 2003; Wroblewski et al., 2003). Overexpression of MMP-7 occurs in premalignant and malignant gastric lesions (McDonnell et al., 1991; Honda et al., 1996; Saarialho-Kere et al., 1996; Adachi et al., 1998; Senota et al., 1998; Yamashita et al., 1998; Ajisaka et al., 2001; Hippo et al., 2002), and genetic polymorphisms linked to increased MMP-7 expression are associated with H. pylori infection status, gastric ulceration (a precursor for gastric cancer), and tumor-related survival among gastric cancer patients (Hellmig et al., 2006; Kubben et al., 2006). In mice, overexpression of MMP-7 leads to hyperproliferation and increased cancer susceptibility (Rudolph-Owen et al., 1998), and cell lines that overexpress MMP-7 develop enhanced tumorigenic potential (Witty et al., 1994). Conversely, mice with a genetic predisposition for intestinal adenocarcinoma that are then bred onto a background of MMP-7 deficiency develop fewer cancers than wild-type mice (Wilson et al., 1997). Together, these data suggest that MMP-7 may play an important role early in gastric carcinogenesis.
A host molecule that has been implicated in regulation of MMP-7 expression is p120-catenin (p120). p120 was originally identified as a substrate for Src- and receptor-tyrosine kinases (Reynolds et al., 1989, 1992; Reynolds and Carnahan, 2004) and is a member of the catenin (ctn) family, an Armadillo domain protein subfamily whose members interact with the cadherin cytoplasmic tail and modulate cadherin function (Reynolds et al., 1994; Shibamoto et al., 1995; Staddon et al., 1995; Reynolds and Carnahan, 2004). Aberrant redistribution of p120 has been observed in several epithelial malignancies, including gastric cancer (Jawhari et al., 1999; Karatzas et al., 1999; Karayiannakis et al., 1999; Thoreson and Reynolds, 2002; Mayerle et al., 2003). Typically found at low levels in the nuclei of normal cells, increased levels of p120 have been observed in nuclei of tumor cells (Mayerle et al., 2003; Wijnhoven et al., 2005; Sarrio et al., 2006), and recent evidence has revealed that nuclear p120 acts to relieve transcriptional repression mediated by Kaiso, a member of the broad complex, tramtrak, bric a brac/pox virus, and zinc finger family (BTB/POZ) (Daniel and Reynolds, 1999). The Kaiso/p120 complex can modulate noncanonical Wnt signaling (Kim et al., 2004), and, along with T cell factor (TCF)/β-catenin complexes, coordinately regulate canonical Wnt gene targets such as cyclin D1 and mmp-7 (Park et al., 2005; Spring et al., 2005), both of which are up-regulated by H. pylori (Hirata et al., 2001; Bebb et al., 2003; Crawford et al., 2003; Wroblewski et al., 2003; Chang et al., 2006). Because MMP-7 exerts cancer-initiating properties and is specifically induced by contact with H. pylori in vitro and in vivo, we sought to define the molecular pathways underpinning increased MMP-7 expression to define a potential tumor-promoting response to this pathogen.
MATERIALS AND METHODS
H. pylori Strains
The H. pylori strains 7.13 or SS1 were grown in Brucella broth with 5% fetal bovine serum (FBS) for 18 h, with shaking in an atmosphere of 5% CO2 at 37°C. Then, they were harvested by centrifugation and added to gastric epithelial cells at a multiplicity of infection (MOI) of 100. The 7.13 isogenic cagA− and cagE− null mutant strains were constructed by insertional mutagenesis using aphA (conferring kanamycin resistance) (Franco et al., 2005) and were selected on Brucella agar containing 25 μg/ml kanamycin. For luciferase assays, H. pylori strains were cultured on agar plates containing 10% horse serum in an atmosphere of 5% CO2 at 37°C for 48 h. Bacteria were harvested in PBS, pH 7.4, added to host cells at an MOI of 100, and routinely monitored using an inverted microscope (model TS 100; Intas, Göttingen, Germany).
Cell Culture and Reagents
MKN28 human gastric epithelial cells (kindly provided by Dr. Robert Coffey, Vanderbilt University) were grown in RPMI 1640 medium (Mediatech, Herndon, VA) supplemented with 10% heat-inactivated FBS, 2 mM l-glutamine, and 20 μg/ml gentamicin in an atmosphere of 5% CO2 at 37°C. Phoenix 293 cells (generously provided by Dr. Todd Graham, Vanderbilt University) were grown in DMEM supplemented with 10% heat-inactivated FBS, 2 mM l-glutamine, 50 U of penicillin/ml, and 50 μg of streptomycin/ml in an atmosphere of 5% CO2 at 37°C.
Primary Gastric Cell Extraction and Culture
All animal studies were approved by the Vanderbilt Institutional Animal Care and Usage Committee. Stomachs were removed from killed 8-wk-old male FVB/n mice (Harlan, Indianapolis, IN), ligated at the pylorus and esophagus, inverted, and injected with 1 ml of 0.5 mg/ml collagenase A as described previously (Wroblewski et al., 2003). Stomachs were then washed in Hanks' balanced salt solution (HBSS) three times at 37°C. Tissue was incubated in 10 ml of 1 mM dithiothreitol for 15 min at 37°C with shaking, washed in HBSS three times at 37°C, and incubated in 0.37 mg/ml collagenase for 30 min at 37°C. After the first collagenase digestion, samples were washed again in HBSS (3 times at 37°C) and incubated for a further 30 min in collagenase (0.37 mg/ml; 37°C). Tissue was triturated using a wide-mouthed pipette, and larger fragments of tissue were allowed to settle under gravity for 45 s. The supernatant containing isolated gastric cell colonies was removed and transferred to a clean 50-ml conical tube, shaken vigorously to release additional cell colonies, and left on ice to sediment for 45 min. The supernatant was then carefully removed and discarded, and isolated cell colonies were plated in chamber slides. Colonies of gastric epithelial cells were cultured in DMEM NUT Mix F-12 (Ham's) supplemented with 10% FBS and 1% antibiotic-antimycotic solution. Then, colonies were incubated in a humidified incubator at 37°C under an atmosphere of 5% CO2. Cell colonies were cultured for up to 72 h, and the medium was changed every 24 h.
Small Interfering RNA (siRNA) Constructs
pSUPER.retro.puro (Oligoengine, Seattle, WA) plasmid containing a human-specific targeting sequence directed toward p120 was kindly provided by Dr. Albert Reynolds (Vanderbilt University) (Davis et al., 2003; Mariner et al., 2004). pSUPER.retro.puro (Oligoengine) plasmid containing a scrambled nontargeting sequence was kindly provided by Dr. Howard Crawford (SUNY, Stony Brook). Nontargeting siRNA oligos (D-001210-01) or ON-TARGETplus SMARTpool siRNA oligonucleotides (oligos) directed toward Kaiso (ZBTB33; L-019982-00) were purchased from Dharmacon RNA Technologies (Lafayette, CO).
Viral Production and Retroviral Transduction
Phoenix 293 packaging cell lines at 50% confluence were transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Fresh medium was added 24 h after transfection, and tissue culture medium was collected and filtered through a 0.45-μm filter 72 h after transfection. For retroviral transduction, MKN28 cells at 50% confluence were incubated overnight with freshly harvested virus containing 4 μg/ml Polybrene (American Bioanalytical, Georgetown, ON, Canada). To generate stable cell lines, cells transduced with the pSUPER.retro.puro virus were selected with 1.5 μg/ml puromycin for 48 h. Clonal populations were selected using cloning rings and limiting dilution techniques.
Luciferase Assay
The plasmids 4xKBS-pGL3 and p120-pCMV were kindly provided by Dr. Juliet M. Daniel (McMaster University) (Kelly et al., 2004). To monitor Kaiso-dependent gene expression, MKN28 cells were cultured in 24-well plates and cotransfected with 100 ng of 4xKBS-pGL3, 4 ng of phRL Null, and 100 ng of p120-pCMV, or 100 ng of the empty vector pCMV for 12 h by using GeneJuice (Novagen, Madison, WI) as recommended by the manufacturer's instructions. Transfection efficiency ranged from 10 to 15%. Eighteen hours after transfection, cells were cocultured with H. pylori or medium alone. After 48 h, cells were harvested in 100 μl of reporter lysis buffer (Promega, Madison, WI), and luciferase activity was determined in a dual channel luminometer. Results were normalized for transfection efficiency by cotransfection with the Renilla luciferase plasmid (phRL Null).
Transient Transfection of siRNA
MKN28 cells (1.5 × 105) in 12-well plates were transiently transfected using DharmaFECT 2 transfection reagent (Dharmacon RNA Technologies) according to the manufacturer's instructions. Briefly, transfection reagent (1.0 μl/well) was mixed with siRNA oligos (2.5 μl of 20 μm solution/well) in 100 μl of Opti-MEM (Invitrogen). Cells were incubated with the transfection mixture for 24 h, fresh medium was added, and bacterial cocultures were performed 24 h later.
Real-Time Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
MKN28 gastric epithelial cells were grown to confluence and then cocultured with H. pylori or medium alone for 2, 4, 8, or 12 h. RNA was prepared from cocultures using TRIzol reagent (Invitrogen) following the manufacturer's instructions. Reverse transcriptase-PCR was performed using TaqMan reverse transcription reagents (Applied Biosystems, Foster City, CA), which was followed by real-time quantitative PCR using the TaqMan gene expression assay and a 7300 real-time PCR system (Applied Biosystems). mmp-7, p120, and Kaiso cDNA were quantified using the human-specific mmp-7 TaqMan gene expression primer set (Hs00159163_m1), CTNND1 TaqMan gene expression primer set (Hs00609741_m1), and ZBTB33 TaqMan gene expression primer set (Hs00406811_m1), respectively, and expression levels were normalized to levels of 18S rRNA.
Immunofluorescence
Gastric cells were cultured in glass chamber slides and subsequently cocultured with H. pylori or medium alone. Six hours after infection, cells were washed twice with Dulbecco's phosphate-buffered saline (DPBS) containing calcium chloride, and then they were fixed in 4.7% paraformaldehyde in DPBS for 15 min at room temperature. Cells were then subjected to antigen retrieval by immersion in deionized water (diH2O), followed by immersion in citrate buffer, pH 6.0, 8 mM citric acid, and 2 mM sodium citrate tribasic dihydrate) and heating in a 1200-W microwave for 15 min at 20% power. After cooling for 30 min at room temperature, cells were rinsed in diH2O, incubated with DPBS containing 3% bovine serum albumin (BSA) (Sigma-Aldrich, St. Louis, MO) and 0.1% Triton X-100 for 20 min, followed by incubation in 3% BSA for 1 h at room temperature. Slides were immunostained with mouse monoclonal anti-p120 antibody (pp120; BD Biosciences, San Jose, CA), rabbit anti-p120 antibody (F1αSH), mouse monoclonal anti-Kaiso antibody (6F/6F8; BD Biosciences), mouse monoclonal anti-Kaiso antibody (11D), rabbit polyclonal anti-Kaiso antibody, or rabbit anti-H. pylori antibody (Dako North America, Carpinteria, CA) at a concentration of 1:100 overnight at 4°C. Washed slides were incubated with goat anti-mouse AlexaFluor 488-conjugated antibody (Invitrogen), goat anti-mouse AlexaFluor 546-conjugated antibody (Invitrogen), or goat anti-rabbit AlexaFluor 488-conjugated antibody (Invitrogen) at a concentration of 1:100 for 2 h at room temperature. Washed slides were then incubated with TOTO-3 dimeric cyanine nucleic acid dye at a concentration of 1:100 for 20 min at room temperature (Invitrogen). Slides were mounted using ProLong Gold antifade reagent (Invitrogen). Imaging was performed on an LSM 510 confocal microscope (Carl Zeiss, Thornwood, NY) by using a 63×/1.40 Pan-APOCHROMAT oil objective at room temperature, and acquisition was performed with the manufacturer's proprietary software. All three-dimensional reconstructions and fluorescence profile analyses were performed using LSM Image Examiner 3.2 software (Carl Zeiss).
Chromatin Immunoprecipitation
Chromatin immunoprecipitation experiments were performed as described previously (Hearnes et al., 2005). Briefly, MKN28 cells were grown to confluence in 10 cm dishes, and then incubated with 1.6% formaldehyde (Sigma-Aldrich) in PBS for 10 min at room temperature. The cross-linking reaction was stopped by adding glycine to a final concentration of 0.14 M. Cells were washed twice with PBS and harvested in 250 μl of chromatin immunoprecipitation (ChIP) radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, and 5 mM EDTA) containing protease inhibitor cocktail (P2714; Sigma-Aldrich). Cells were subjected to sonication consisting of eight 10-s pulses at 50% amplitude and centrifuged at 14,000 × g at 4°C for 15 min. For each immunoprecipitation, supernatant containing 1 mg of total protein as quantified by the Bradford assay (Pierce Chemical, Rockford, IL) was precleared by the addition of 10 μg of mouse immunoglobulin G (IgG) conjugated to protein A-Sepharose beads (PAS; Zymogen) with rocking at 4°C for 1 h. PAS beads were removed by centrifugation. Two micrograms of antibody and 30 μl of PAS beads were added for each immunoprecipitation, with rocking at 4°C overnight. PAS beads were concentrated by centrifugation and washed twice with 1 ml of ChIP RIPA buffer, four times with 1 ml of immunoprecipitation (IP) wash buffer (100 mM Tris, pH 8.5, 500 mM LiCl, 1% NP-40, and 1% deoxycholate), followed by two washes with ChIP RIPA buffer. During each wash, samples were rotated for 5 min at 4°C. Two hundred μl of cross-link reversal buffer (125 mM Tris, pH 6.8, 10% β-mercaptoethanol, and 4% SDS) was added directly to PAS beads from each sample and boiled for 30 min. DNA was purified by phenol-chloroform extraction and precipitated with ethanol. DNA pellets were resuspended in 20 μl of PCR-grade water. Five microliters of resuspended DNA was subjected to PCR to amplify the mmp-7 promoter region (forward primer, TAGAGGCAGTGTTCCCCATT; reverse primer, CCAAATCCTGTGGTTCTCC) as described previously (Spring et al., 2005). PCR products were electrophoresed on a 1.5% agarose gel at 100 V for 30 min.
Western Analysis
Cells were lysed in RIPA buffer (50 mM Tris, pH 7.2, 150 mM NaCl, 1% Triton X-100, and 0.1% SDS) containing protease inhibitor cocktail, and protein concentrations were quantified by the Bradford assay. Proteins (30 μg) were separated by SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to polyvinylidene difluoride membranes (PVDF, Pall, Ann Arbor, MI). Protein levels were assessed by Western blotting by using mouse monoclonal anti-p120 antibody (1:1000, pp120), anti-E-cadherin antibody (1:1000, BD Bioscience), anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (1:2000; Millipore Bioscience Research Reagents, Temecula, CA) or goat polyclonal anti-actin antibody (1:2000, C-11; Santa Cruz Biotechnology, Santa Cruz, CA). Primary antibodies were detected using goat anti-mouse or donkey anti-goat (1:2500; Santa Cruz Biotechnology) horseradish peroxidase-conjugated secondary antibodies and visualized by Western Lightning Chemiluminescence Reagent Plus (PerkinElmer Life and Analytical Sciences, Boston, MA) according to the manufacturer's instructions on a Chemigenius system (Syngene, Frederick, MD). For analysis of exogenous p120 expression, transfected cells were harvested in lysis buffer (20 mM Tris-HCl, pH 7.5, 1 mM ETDA, 100 mM NaCl, 1% Triton X-100, 0.5% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 1× complete protease inhibitors [Roche Diagnostics, Indianapolis, IN], 1 mM Na3VO4, 1 mM sodium molybdate, 20 mM NaF, 10 mM sodium pyrophosphate, and 20 mM β-glycerophosphate), separated by SDS PAGE, and transferred to PVDF membranes. Protein levels were assessed by Western blotting by using anti-p120 antibody (pp120) and anti-GAPDH antibody (Abcam, Cambridge, MA).
Subcellular Fractionation
Fractionation was performed as described by Frey et al. (1997). Briefly, cells were lysed in low-detergent buffer (20 mM HEPES, 2 mM EDTA, 2 mM EGTA, 0.5 mg/ml digitonin) containing protease inhibitors, centrifuged for 40 min at 100,000 × g, and the soluble fraction was collected. The residual pellet was resuspended in high detergent buffer (10 mM Tris, pH 7.4, 2 mM EDTA, 2 mM EGTA, 1% Triton, 0.2% deoxycholate, and 0.1% SDS) containing protease inhibitors, centrifuged for 5 min at 20,000 × g, and the membranous fraction was collected.
Immunoprecipitation
Before collection, MKN28 cells were treated with sodium pervanadate to inhibit endogenous phosphatase activity as described previously (Mariner et al., 2004). Cells were then lysed in IP lysis buffer (50 mM Tris, pH 7.2, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, and 0.2% sodium deoxycholate) containing protease inhibitor cocktail, phosphatase inhibitor cocktails 1 and 2 (Sigma-Aldrich), and 150 mM vanadate/300 mM H2O2; harvested; and protein concentrations were quantified by the Bradford assay. One milligram of total protein was isolated from each sample, and total volumes were equilibrated with IP lysis buffer. Twenty microliters of mouse IgG (Sigma-Aldrich) was added, followed by 30 min of incubation at 4°C with rolling. Then, 20 μl of protein A/G PLUS-agarose immunoprecipitation reagent (Santa Cruz Biotechnology) was added, followed by 30 min of incubation at 4°C with rolling. Samples were centrifuged at 4°C for 10 min at 10,000 × g. The supernatants were then isolated and incubated overnight with 1 μg of the appropriate IP antibody (pp120; BD Biosciences; or c-myc, 9E10; Santa Cruz Biotechnology) at 4°C with rolling. Thirty-five microliters of protein A/G PLUS-agarose immunoprecipitation reagent was added to each sample, followed by 1 h of incubation at 4°C with rolling. Samples were centrifuged at 10,000 × g at 4°C for 2 min, and the supernatant was discarded. The remaining protein A/G reagent was washed six times with 500 μl of cold temperature PBS containing 150 mM vanadate/300 mM H2O2. After the last wash, 75 μl of Laemmli buffer was added to each sample and boiled for 5 min. The samples were centrifuged at room temperature at 10,000 × g for 5 min, and the supernatants were analyzed by Western blot analysis. Immunoblotting was performed using a mouse monoclonal anti-phospho-tyrosine antibody (1:200, PY99, Santa Cruz Biotechnology) and anti-phospho-p120 antibodies (1:1000; pY96, pY228, pY291; BD Biosciences).
Quantitative Culture of Adherent Bacteria
H. pylori:MKN28 cell cocultures were washed after 24 h with PBS, pH 7.6, × 2 to remove nonadherent bacteria, and total cell extracts were harvested as described previously (Crawford et al., 2003). Serial 10-fold dilutions of 1-ml aliquots of cell extracts were cultured on 5% sheep blood agar plates, and incubated for 3–5 d under microaerobic conditions before H. pylori colonies were counted. Results are expressed as colony forming units (cfu) per milliliter.
Statistical Analysis
Student's t test was used to evaluate the data, and significance was defined as p < 0.05. Fluorescence intensity profiles were generated by creating Lowess spline curves of the XY data output from LSM image examiner software (Carl Zeiss).
RESULTS
H. pylori-mediated mmp-7 Expression Is Transcriptionally Regulated and Is Dependent on a Functional cag Pathogenicity Island
We and others demonstrated previously that MMP-7 protein levels are increased in gastric cells after coculture with H. pylori (Crawford et al., 2003; Wroblewski et al., 2003) and that this is dependent on specific genes within the cag island (e.g., cagE but not cagA) (Crawford et al., 2003). cagE encodes a homologue of VirB4, a protein involved in toxin export in Bordetella pertussis. Based on homology, CagE is postulated to function as a NTPase, which is required for energy-dependent delivery of CagA and other cag island substrates from the bacterial cytoplasm directly into host cells (Backert and Selbach, 2008). Inactivation of cagE, therefore, results in a nonfunctional type IV cag secretion system. To determine whether increased MMP-7 protein expression was transcriptionally mediated, MKN28 human gastric epithelial cells were cocultured with the H. pylori wild-type cag+ strain 7.13, a 7.13 cagA− mutant, a 7.13 cagE− mutant, or medium alone. At defined time points, steady-state mmp-7 mRNA levels were assessed by real-time RT-PCR. Levels of mmp-7 mRNA began to increase 4 h after infection with the wild-type strain 7.13 and peaked by 8 h at levels ∼12-fold above control (Figure 1). Inactivation of cagA had no significant effect on H. pylori-mediated mmp-7 induction (Figure 1), confirming our previous results for MMP-7 protein expression (Crawford et al., 2003). However, inactivation of cagE significantly attenuated mmp-7 expression (Figure 1), indicating that H. pylori strain 7.13 induces cag-dependent transcriptional up-regulation of mmp-7 in human gastric epithelial cells.
Figure 1.
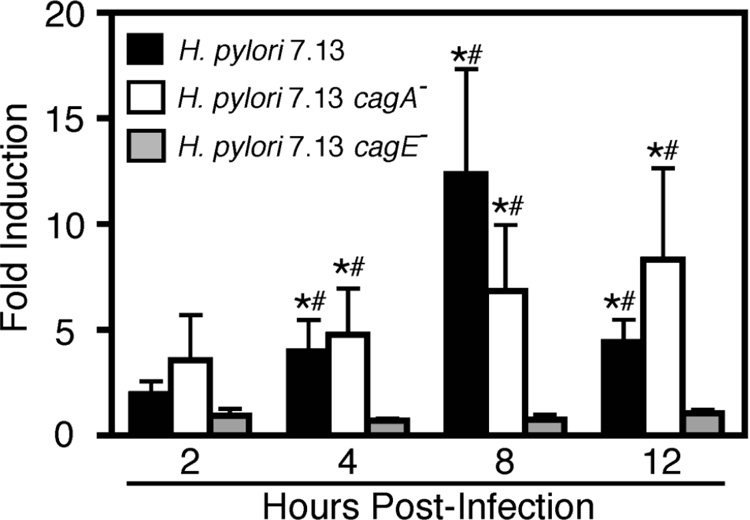
H. pylori infection of MKN28 cells results in an increase in mmp-7 steady-state mRNA levels in a cagE-dependent manner. MKN28 cells were cocultured with the H. pylori cag+ strain 7.13 (MOI = 100), the 7.13 cagA− or cagE− isogenic mutants, or medium alone. At defined time points, RNA was extracted, subjected to reverse transcription, and analyzed in triplicate by real-time PCR. Data represent -fold induction of mmp-7 mRNA in cells cocultured with H. pylori versus medium alone from experiments performed on at least four occasions. Error bars, SEM. *p < 0.05 versus medium alone. #p < 0.05 versus cagE−.
Kaiso Binds the mmp-7 Promoter Region in Gastric Epithelial Cells
Recent data have shown that Kaiso mediates transcriptional repression of mmp-7 in colonic epithelial cells (Spring et al., 2005); therefore, we used laser scanning immunofluorescent microscopy to determine whether gastric epithelial cells that produce MMP-7 in response to H. pylori (Figure 1) also express Kaiso. As shown in Figure 2A, Kaiso is expressed throughout the cell, including the nuclei, in MKN28 cells. These results were confirmed using additional monoclonal and polyclonal anti-Kaiso antibodies (Supplemental Figure 1).
Figure 2.

MKN28 cells express the transcriptional repressor, Kaiso, which binds the promoter region of mmp-7. (A) MKN28 cells were fixed and incubated with a monoclonal anti-Kaiso antibody (6F/6F8) or PBS alone, followed by incubation with an anti-mouse AlexaFluor-488 antibody and TOTO-3 nucleic acid dye. Cells were visualized by laser scanning immunofluorescent microscopy and are represented as projection images of a Z-stack. Kaiso, green; nuclei, red; 630× magnification. (B) MKN28 cells were subjected to chromatin immunoprecipitation analysis by using a monoclonal anti-Kaiso antibody (6F/6F8), mouse IgG (negative control), or input DNA alone (positive control) followed by PCR amplification by using human mmp-7 promoter-specific primers. Experiments were performed on at least two occasions. Products were visualized by agarose gel electrophoresis and a representative gel is shown.
The human mmp-7 promoter contains two conserved copies of the consensus Kaiso site, one of which overlaps an ETS transcription factor consensus site and is in proximity to activator protein-1 and TCF/Lef transcription factor binding sites (Spring et al., 2005). Kaiso has been shown previously to bind this region of the mmp-7 promoter in at least three human epithelial cell lines by ChIP assay (Spring et al., 2005). Although the immunofluorescence data in Figure 2A indicate that Kaiso is present in the requisite subcellular locale to function as a transcriptional repressor, it is not known whether Kaiso binds to the mmp-7 promoter in MKN28 cells. Therefore, primers specific for this region of the mmp-7 promoter were used for ChIP to confirm that Kaiso can bind and regulate mmp-7 in gastric epithelial cells. mmp-7 promoter fragments were specifically detected in input fractions and Kaiso immunocomplexes, but not in isotype control immunocomplexes (Figure 2B), indicating that Kaiso binds specifically to the promoter region of mmp-7 in MKN28 cells.
H. pylori Strain 7.13 Alters Subcellular Distribution of p120 and Induces Aberrant Localization of p120 to the Nucleus
Interactions between p120 and Kaiso can coordinately regulate genes implicated in carcinogenesis (Daniel and Reynolds, 1999; Daniel et al., 2002; Kelly et al., 2004; Kim et al., 2004; Park et al., 2005; Spring et al., 2005), and translocation of p120 to the nucleus relieves Kaiso-mediated transcriptional repression of mmp-7 (Kelly et al., 2004; Park et al., 2005; Spring et al., 2005). To determine whether H. pylori infection alters total p120 levels, MKN28 cells were cocultured with H. pylori strain 7.13 (MOI = 100) or medium alone and subjected to Western blot analysis (Figure 3A). Densitometric analysis of multiple immunoblot experiments indicated that total p120 levels were not significantly altered after infection compared with uninfected controls (data not shown). Similarly, mRNA levels of p120, as measured by real-time RT-PCR, remained unchanged by infection with H. pylori (Supplemental Figure 2A).
Figure 3.

H. pylori alters subcellular localization of p120 (A) MKN28 cells were cocultured with H. pylori strain 7.13 (MOI = 100) or medium alone. At defined time points, total protein was extracted and analyzed by Western blot by using a monoclonal anti-p120 antibody (pp120). Experiments were performed on at least three occasions. A representative blot is shown. Anti-actin blots served as normalization controls for MKN28 viability under different experimental conditions. (B) MKN28 cells were cocultured with H. pylori strain 7.13 (MOI = 100). At defined time points, total protein was extracted, subjected to subcellular fractionation, and analyzed by Western blot by using an anti-p120 antibody. Representative blots are shown. Anti-GAPDH and anti-E-cadherin antibodies served as normalization controls for purification of soluble and insoluble subcellular fractions, respectively. (C) Densitometric analysis of multiple Western blot repetitions performed on at least three occasions. Graph represents fold p120 expression in infected versus uninfected cells. Error bars, SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 versus time 0.
We next sought to determine whether H. pylori may induce alterations in subcellular localization of p120. MKN28 cells were cocultured with H. pylori strain 7.13, and, at defined time points, total cell lysates were fractionated into a soluble fraction containing cytosolic components and an insoluble fraction containing nuclear and membranous components. Immunoblot analysis of these fractions revealed that H. pylori significantly decreased soluble, and concomitantly increased insoluble levels of p120 over 24 h of infection (Figure 3B). Immunoblot analysis performed on cells cocultured with medium alone showed no alteration in soluble or insoluble p120 levels (Supplemental Figure 2B), indicating that increases in cell confluence over time did not contribute to changes in subcellular p120 levels. These results demonstrate that H. pylori alters the distribution, but not the total amount, of the cellular p120 pool.
Having identified alterations in the subcellular localization of p120, we next determined whether H. pylori strain 7.13 could induce nuclear translocation of p120 in MKN28 cells. Cells were cocultured with H. pylori strain 7.13 or medium alone for 6 h, and p120 localization was assessed by laser scanning immunofluorescent microscopy. Nuclear and perinuclear aggregations of p120 were observed in cells cocultured with H. pylori compared with medium alone (Figure 4A). As shown in Figure 4B, p120 specifically colocalized with Kaiso in the nuclei of infected but not uninfected cells, placing these molecules in position to potentially regulate genes implicated in carcinogenesis.
Figure 4.

H. pylori induces aberrant localization of p120 to the nucleus in MKN28 cells. MKN28 gastric epithelial cells were cocultured with H. pylori strain 7.13 or medium alone for 6 h. (A) Cells were fixed and incubated with a monoclonal anti-p120 antibody (pp120) and a polyclonal anti-H. pylori antibody, followed by incubation with an anti-mouse AlexaFluor-488 antibody, anti-rabbit AlexaFluor-546 antibody, and TOTO-3 nucleic acid dye. Cells were visualized by laser scanning immunofluorescent microscopy and are represented as projection images of a Z-stack. p120, green; nuclei, red; H. pylori, blue; 630× magnification. Localization of p120 in three dimensions is represented by orthogonal views of infected cells. (B) MKN28 cells were cocultured with H. pylori strain 7.13, or medium alone, respectively, for 6 h. Cells were fixed and incubated with a rabbit polyclonal anti-p120 antibody (F1αSH) and a monoclonal anti-Kaiso antibody (6F/6F8), followed by incubation with an anti-mouse AlexaFluor-546 antibody, anti-rabbit AlexaFluor-488 antibody, and TOTO-3 nucleic acid dye. Cells were visualized by laser scanning immunofluorescent microscopy and are represented as a single transverse image through the middle of the cell. p120, green; Kaiso, red; nuclei, blue. Uninfected, 630× magnification. Infected, 1260× magnification. Graph indicates fluorescence intensity as measured linearly through the nucleus, demonstrating colocalization of Kaiso and p120 within the nuclei of infected cells.
H. pylori Strain 7.13 Alters the Phosphorylation State of p120
Having demonstrated that infection with H. pylori strain 7.13 induces nuclear translocation of p120, we next sought to identify the mechanism underlying H. pylori-induced aberrant subcellular distribution of p120. Previous studies have suggested that phosphorylation of a number of tyrosine residues within the p120 regulatory domain may play a role in p120 signaling (Mariner et al., 2004; Shibata et al., 2004; Castano et al., 2007; Reynolds, 2007). Therefore, we investigated whether H. pylori infection similarly altered levels of p120 tyrosine phosphorylation. MKN28 cells were cocultured with H. pylori strain 7.13 and, at defined time points, cells were treated with phosphatase inhibitors, harvested, and subjected to immunoprecipitation by using an anti-p120 antibody. Of interest, immunoblot analysis of p120 immunocomplexes using a general anti-phosphotyrosine antibody revealed that H. pylori significantly decreased total tyrosine phosphorylation compared with uninfected controls (Figure 5), suggesting that dephosphorylation of p120 tyrosine residues may contribute to alterations in subcellular localization. To define which specific residues might be dephosphorylated, immunoblot analysis was performed using anti-phosphoantibodies targeting three specific p120 residues: Y228, Y291, and Y96. However, levels of phosphorylation at each of these residues were not significantly altered by infection (Figure 5A), indicating that other, as of yet uncharacterized, tyrosine phosphorylation sites represent targets for dephosphorylation by H. pylori.
Figure 5.

H. pylori infection of MKN28 cells results in reduced levels of p120 tyrosine phosphorylation. (A) MKN28 cells were cocultured with H. pylori strain 7.13 (MOI = 100) or medium alone. At defined time points, total protein was extracted, subjected to immunoprecipitation with an anti-p120 antibody (pp120), and analyzed by Western blot by using a total anti-phosphotyrosine antibody (pY99) or anti-phosphoantibodies specific to the p120 tyrosine residues 228, 96, or 291. Experiments were performed on at least three occasions. A representative blot is shown. Anti-p120 blots (pp120) served as normalization controls for MKN28 viability under different experimental conditions. As a negative control, 500 μg of total protein from uninfected and infected samples taken 6 h after infection was subjected to immunoprecipitation with an anti-c-myc antibody (9E10). (B) Densitometric analysis of multiple Western blot repetitions performed on at least three occasions. Graph represents percent total tyrosine phosphorylation as determined by Western blotting with pY99 antibody in infected versus uninfected cells. Error bars, SEM. *p < 0.05, **p < 0.001 versus uninfected cells treated with medium alone.
H. pylori Relieves Kaiso-mediated Transcriptional Repression in a cag Pathogenicity Island-dependent Manner
H. pylori induces an increase in transcription of mmp-7 in MKN28 gastric epithelial cells. Having also shown that Kaiso binds the promoter region of mmp-7 and that H. pylori induces aberrant localization of p120 to the nucleus, we next sought to determine whether inhibition of Kaiso-mediated transcriptional repression was required for mmp-7 transcriptional up-regulation. We used the pGL3–4xKBS plasmid (4xKBS), which carries four tandem copies of the consensus Kaiso binding site upstream of the luciferase reporter gene (Kelly et al., 2004). Previous studies suggest that the ability to inhibit Kaiso-mediated transcriptional repression of an artificial promoter such as 4xKBS requires p120 levels greater than those endogenously present within cells (Kelly et al., 2004). Therefore, we cotransfected MKN28 cells with 4xKBS and a p120 expression vector before H. pylori coculture (Figure 6A). Expression of exogenous p120 did not significantly relieve Kaiso-mediated transcriptional repression in uninfected cells (Supplemental Figure 3). However, coculture with H. pylori strain 7.13 resulted in a significant inhibition (∼2.5-fold) of Kaiso-mediated repression of luciferase expression compared with medium alone (Figure 6B). Inactivation of cagE, but not cagA, significantly attenuated the ability of H. pylori strain 7.13 to relieve Kaiso-mediated transcriptional repression (Figure 6B). These results demonstrate that H. pylori relieves Kaiso-mediated transcriptional repression in a cagE-dependent manner and that p120 is likely required for this inhibition.
Figure 6.
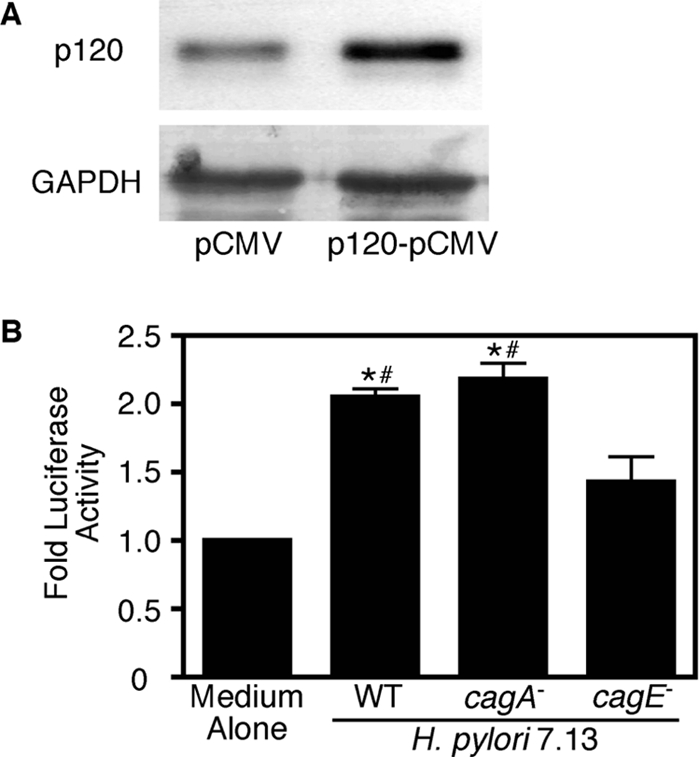
H. pylori infection of MKN28 cells mediates a release of Kaiso from its putative binding site on a luciferase reporter plasmid. MKN28 cells were cotransfected with 100 ng of 4xKBS-pGL3, 4 ng of phRL Null, and 100 ng of the empty vector pCMV or 100 ng of p120-pCMV for 12 h. (A) Eighteen hours after transfection, total protein was extracted and analyzed by Western blot by using a monoclonal anti-p120 antibody (pp120). A representative blot is shown. Anti-GAPDH blots served as normalization controls for MKN28 viability under different experimental conditions. (B) Eighteen hours after transfection with p120-pCMV, cells were cocultured with H. pylori strain 7.13 or its cagA− or cagE− isogenic mutants. After 48 h, cells were harvested and luciferase activity was determined in a dual-channel luminometer. Data are represented as -fold luciferase activity for experiments performed on at least three occasions. Error bars, SEM. *p < 0.01 versus medium alone. #p < 0.05 versus cagE−.
p120 Is Required for H. pylori-mediated Up-Regulation of mmp-7 Expression
To demonstrate coordinated regulation of H. pylori-induced mmp-7 expression by p120 and Kaiso, siRNA was used to suppress levels of endogenous p120. MKN28 cells were retrovirally transduced with pRetroSuper vector containing sequences encoding either scrambled siRNA or human-specific p120 siRNA. Clonal populations of transduced cells were isolated and expanded, and a >75% reduction in levels of p120 was achieved (Figure 7A). Because H. pylori has been shown previously to bind to components of epithelial junctional complexes, we established that p120 deficiency did not alter bacterial binding to MKN28 cells. As assessed by quantitative culture, H. pylori strain 7.13 bound to cells transduced with p120-specific siRNA as avidly as cells transduced with scrambled siRNA, thereby eliminating the possibility that differences in bacterial adherence mediate p120-dependent responses (Figure 7B).
Figure 7.
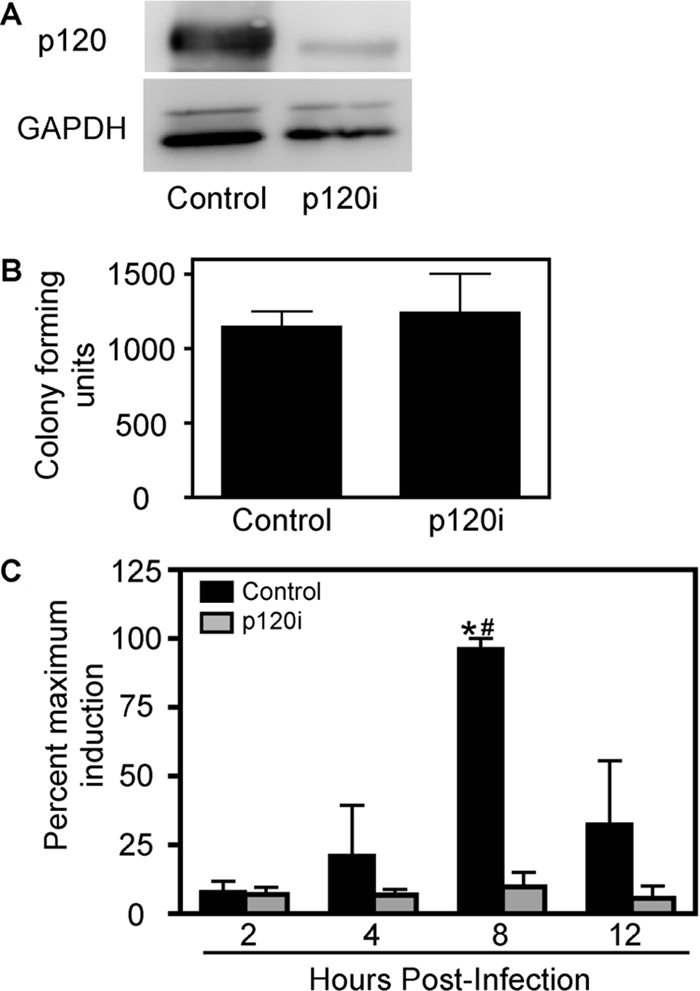
p120 is required for the H. pylori-mediated increase in steady-state mmp-7 mRNA levels. (A) MKN28 cells were retrovirally transduced with either scrambled (control) or human p120-specific siRNA (p120i), and clonal populations were selected. Total protein was extracted from control or p120i cells and analyzed by Western blot using a monoclonal anti-p120 antibody (pp120). GAPDH Western blots served as normalization controls for cell viability under different experimental conditions. (B) Control or p120i cells were incubated with H. pylori strain 7.13 (MOI = 100) for 24 h. Cells were washed to remove nonadherent bacteria, harvested, and plated in serial dilutions on blood agar plates. Data represent the number of colony forming units for experiments performed on at least three occasions. (C) Vector control or p120i cells were cocultured with H. pylori strain 7.13 (MOI = 100) or medium alone. At defined time points, total RNA was extracted, subjected to reverse transcription, and analyzed in triplicate by real-time PCR. Data are represented as the percentage of maximum induction of mmp-7 expression in infected versus uninfected cells for experiments performed on at least three occasions. Error bars, SEM. *p < 0.05 versus medium alone. #p < 0.05 versus p120i cells.
MKN28 cells stably transduced with control or p120-specific siRNA were then cocultured with H. pylori strain 7.13 or medium alone. At defined time points, steady-state mmp-7 mRNA levels were assessed by real-time RT-PCR. As expected, levels of mmp-7 mRNA were significantly increased by 8 h in control MKN28 cells infected with H. pylori, but this increase was attenuated in infected p120-deficient cells (Figure 7C), indicating that p120 is required for H. pylori to induce transcription of mmp-7 in this system. Results were confirmed in two other clonal cell populations transduced with p120-specific siRNA (data not shown).
Suppression of Kaiso in p120-deficient Cells Restores the Ability of H. pylori to Induce mmp-7
To more robustly demonstrate that p120 and Kaiso coordinately regulate H. pylori-induced mmp-7 expression, we transiently transfected control or p120 deficient MKN28 cells with scrambled or Kaiso-specific siRNA. Real-time RT-PCR analysis indicated that Kaiso expression was significantly reduced using Kaiso-specific, but not scrambled, siRNA (Figure 8A). H. pylori strain 7.13 was then cocultured with p120-deficient/Kaiso-deficient, p120-deficient/Kaiso-wild-type, or wild-type control MKN28 cells, and mmp-7 mRNA expression was quantified by real-time RT-PCR. Inhibition of Kaiso in p120-deficient cells restored the ability of H. pylori to induce expression of mmp-7 (Figure 8B), indicating a role for both of these transcriptional elements in H. pylori-mediated up-regulation of MMP-7.
Figure 8.
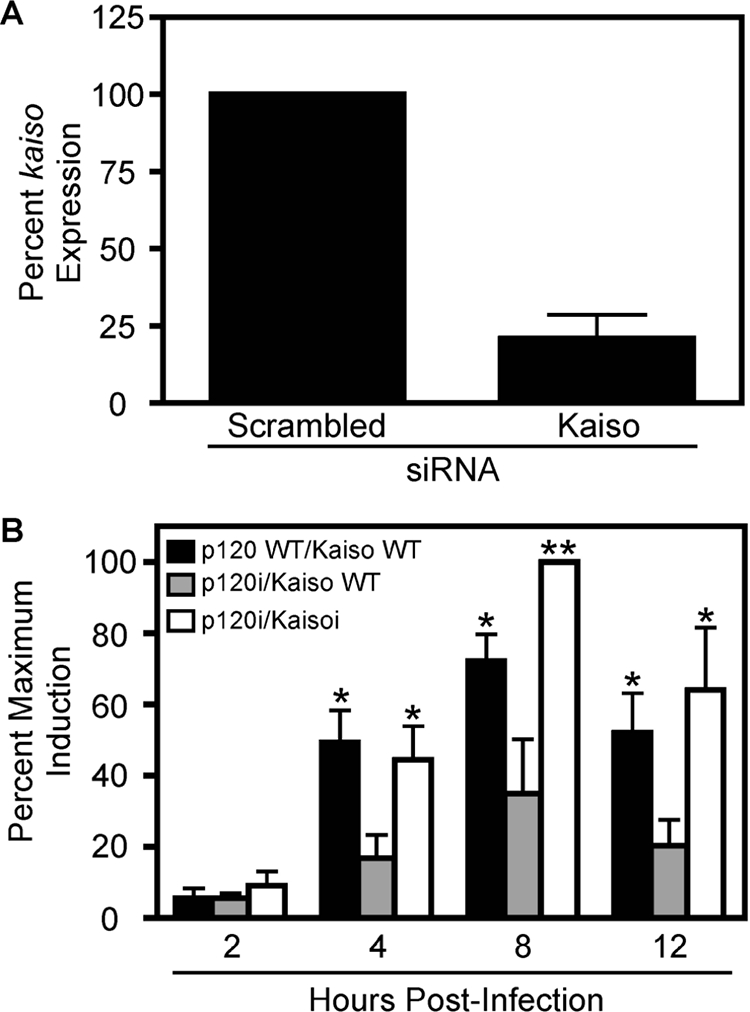
Suppression of Kaiso protein expression in p120i cells restores H. pylori-mediated increases in mmp-7 mRNA levels. Control or p120i cells transiently transfected with scrambled siRNA, and p120i cells transiently transfected with Kaiso-specific siRNA were cocultured with H. pylori strain 7.13 (MOI = 100) or medium alone. At defined time points, total RNA was extracted, subjected to reverse transcription, and analyzed in triplicate by real-time PCR. (A) Data are represented as an average percentage of kaiso expression in uninfected p120i cells at each time point for experiments performed on at least three occasions. (B) Data are represented as the percent maximum induction of mmp-7 expression in infected versus uninfected cells for experiments performed on at least three occasions. Error bars, SEM. *p < 0.05 and **p < 0.01 versus p120i cells.
H. pylori Induces Aberrant Localization of p120 to the Nucleus in Ex Vivo Gastric Cell Colonies
Our current data demonstrate that H. pylori induces increased transcription of mmp-7 through a p120-dependent mechanism involving inhibition of Kaiso-mediated transcriptional repression. We next extended these results using a model of H. pylori infection that more closely recapitulates cellular organization in the stomach. Gastric cell colonies were isolated from 8-wk-old male FVB/n mice, cocultured with the mouse-adapted H. pylori strain SS1 or medium alone for 6 h, and p120 localization was assessed by laser scanning immunofluorescent microscopy. Mouse-adapted H. pylori SS1 was used for these studies because gastric cell colonies were harvested from mice. Consistent with our results in MKN28 cells, nuclear aggregations of p120 were observed to specifically colocalize with Kaiso in cells cocultured with H. pylori, but not in cells incubated with medium alone (Figure 9, A and B). These results indicate that, similar to cultured gastric epithelial cells, H. pylori can alter p120 localization in a model system that approximates events within colonized gastric mucosa.
Figure 9.

H. pylori induces aberrant localization of p120 to the nucleus in ex vivo gastric cell colonies. (A) Primary murine gastric epithelial cell colonies were cocultured with H. pylori strain SS1 or medium alone for 6 h. Cells were fixed and incubated with a monoclonal anti-p120 antibody (pp120) and a polyclonal anti-H. pylori antibody, followed by incubation with an anti-mouse AlexaFluor-488 antibody, anti-rabbit AlexaFluor-546 antibody, and TOTO-3 nucleic acid dye. Cells were visualized by laser scanning immunofluorescent microscopy and are represented as projection images of a Z-stack. p120, green; H. pylori, blue; nuclei, red; 630× magnification. Localization of p120 in 3-dimensions is represented by orthogonal views of infected cells. (B) Primary murine gastric epithelial cell colonies were cocultured with H. pylori strain SS1 or medium alone for 6 h. Cells were fixed and incubated with a rabbit polyclonal anti-p120 antibody (F1αSH) and a monoclonal anti-Kaiso antibody (6F/6F8), followed by incubation with an anti-mouse AlexaFluor-546 antibody, anti-rabbit AlexaFluor-488 antibody, and TOTO-3 nucleic acid dye. Cells were visualized by laser scanning immunofluorescent microscopy and are represented as a single transverse image through the middle of the cell. p120, green; Kaiso, red; nuclei, blue. Uninfected, 630× magnification. Infected, 1260× magnification. Graph indicates fluorescence intensity as measured linearly through the nucleus demonstrating colocalization of Kaiso and p120 within the nuclei of infected cells.
DISCUSSION
MMP-7 is a host effector that mediates carcinogenesis and is induced by H. pylori within infected human gastric mucosa. Our current experiments identified the p120/Kaiso signaling pathway as a regulator of microbially induced expression of this carcinogenic factor by 1) demonstrating that H. pylori can alter the topography of p120 localization within gastric epithelial cells in vitro and in a physiologically relevant ex vivo primary gastric cell culture system, 2) establishing that H. pylori can inhibit Kaiso-mediated transcriptional repression of an artificial luciferase promoter, 3) capitalizing on a gastric cell model of p120 deficiency to demonstrate a requirement for p120 in the transcriptional upregulation of mmp-7, and 4) combining transient gene silencing techniques with a p120-deficient model system to delineate an interaction between p120 and Kaiso. Collectively, these studies indicate that H. pylori coopts p120 as a signaling molecule to relieve Kaiso-mediated repression of mmp-7.
p120 is a multidimensional protein that performs several distinct functions within host cells. p120 can regulate E-cadherin, a cell–cell adhesion molecule that functions as a component of the adherens junction of epithelial tissues, and turnover of E-cadherin is regulated by binding of p120 to the cadherin juxtamembrane domain (Reynolds et al., 1989; Reynolds et al., 1992, 1994; Shibamoto et al., 1995; Staddon et al., 1995; Anastasiadis et al., 2000; Thoreson et al., 2000; Anastasiadis and Reynolds, 2001; Ireton et al., 2002; Davis et al., 2003; Reynolds and Carnahan, 2004). Studies have demonstrated that loss of E-cadherin or overexpression of p120 results in mislocalization of p120 to the cytoplasm (Reynolds and Carnahan, 2004), where it induces a range of morphological changes that promote cell motility and metastasis. The effects of p120 seem to involve physical and functional interactions with Ras homolog (Rho) GTPases and their regulators, GTPase-activating proteins and guanine exchange factors (Noren et al., 2000; Anastasiadis and Reynolds, 2001; Grosheva et al., 2001). Based on our current data, a role for p120 in altering cell morphology and motility induced by H. pylori warrants further investigation, and such studies are ongoing in our laboratory.
Nuclear p120 relieves transcriptional repression exerted by Kaiso, which acts as a dual specificity repressor that recognizes both sequence-specific consensus sites (CTGCNA) and methylated CpG nucleotides (Prokhortchouk et al., 2001; Daniel et al., 2002; Park et al., 2005). Because H. pylori infection has been associated with gastric cancer and nuclear mislocalization of p120 in human gastric epithelium (Krueger et al., 2007), we investigated mislocalization of p120 in conjunction with altered expression of the oncogenic molecule MMP-7. Infection of p120-deficient gastric epithelial cells clearly demonstrated that p120 is required for H. pylori-mediated increases in mmp-7 transcription. Furthermore, silencing Kaiso expression in p120-deficient cells restored the ability of H. pylori to induce mmp-7 transcription, indicating that an interaction between p120 and Kaiso, whether direct or indirect, is also required.
To date, the signals that induce nuclear translocation of p120 remain undefined (Daniel, 2007), and delineation of these pathways is critical for understanding the relevance of p120/Kaiso-mediated transcriptional regulation in the context of carcinogenesis. For example, it will be important to determine which subcellular pool of p120 is responsible for relief of Kaiso-mediated transcriptional repression. Our subcellular fractionation data suggest that p120 from a free cytoplasmic pool translocates to the nucleus. However, work from Weydig et al. (2007) demonstrated that H. pylori mediates internalization of p120 found in adherens junctions at the plasma membrane, which may then be translocated to the nucleus. p120 is also phosphorylated at a number of sites by Src- and receptor tyrosine kinases (Reynolds, 2007), and multiple ligand–receptor pathways have been implicated in signaling to p120 through phosphorylation, including protein kinase C- and epidermal growth factor receptor-dependent pathways (Mariner et al., 2004; Xia et al., 2006), both of which are activated by H. pylori (Keates et al., 2001; Nozawa et al., 2004). However, our data demonstrate that H. pylori mediates a decrease in total p120 tyrosine phosphorylation, indicating a previously undescribed role for protein tyrosine phosphatases in H. pylori-mediated signaling to p120. It remains undefined whether signaling to p120 by H. pylori occurs directly or through another cellular intermediary.
Our current results have also provided insight into the augmentation in cancer risk exerted by cag+ strains and indicate that oncogenic epithelial responses such as MMP-7 expression may be regulated by different microbial effectors. Previous studies from our laboratory and others have demonstrated that intracellular events with carcinogenic potential, such as β-catenin nuclear translocation, are dependent upon the presence of CagA within host cells, which, in turn, is mediated by a functional cag secretion system (Franco et al., 2005; Suzuki et al., 2005; Murata-Kamiya et al., 2007). Our new data indicate that aberrant activation of p120 is dependent on a functional cag secretion system, but not CagA per se, suggesting that other substrates translocated by the cag island may mediate p120 signaling. One candidate is H. pylori peptidoglycan, which is translocated into host cells by the cag type IV secretion system and sensed by intracellular Nod1, which then activates NF-κB–dependent responses such as secretion of interleukin-8 and β-defensin 2 (Viala et al., 2004; Boughan et al., 2006). Another possibility based on a recent investigation (Kwok et al., 2007) is that binding of CagL to cell surface α5β1 integrins can alter local membrane dynamics and eventuate in the assembly of focal adhesions that trigger integrin signaling cascades (Backert and Selbach, 2008), which may aberrantly activate p120.
It is of interest that the H. pylori strain we used to infect murine gastric colonies (SS1) has been reported to contain a nonfunctional cag island. We specifically chose a mouse-adapted H. pylori strain for these studies because we were examining murine cells; hence, the choice of strain SS1. However, results from a recent study (Ferrero et al., 2008) have suggested that H. pylori can activate NF-κB signaling in mouse gastric epithelial cells via a cag pathogenicity island-independent pathway, suggesting that signaling pathways in murine cells may be activated by different microbial components than corresponding pathways in human cells. We also did not analyze the functional effects of p120/Kaiso interactions in murine gastric glands, only differences in localization. Differences in model systems may have contributed to these results. In contrast to MKN28 human gastric epithelial cell monolayers, our gastric gland culture model uses tissues isolated from mice that contain not only epithelial cells but also stromal and lamina propria cells. Thus, there are several potential reasons that may explain the ability of H. pylori strain SS1 to induce nuclear localization of p120 in murine gastric epithelial cells. Collectively, these data indicate that multiple H. pylori disease-related virulence constituents may be required for collaborative functional interactions between Kaiso, p120, and β-catenin in the nucleus to mediate expression of certain Wnt target genes such as mmp-7.
Gastric adenocarcinoma is strongly associated with the presence of H. pylori, and both microbial and host factors influence the risk for carcinogenesis. Interactions between H. pylori and epithelial cells play an important role in the development of gastric injury. p120 is a multifunctional host protein that orchestrates epithelial responses with carcinogenic potential. Our results indicate that p120 is aberrantly activated by H. pylori and regulates expression of the carcinogenic effector, mmp-7. Molecular delineation of pathways activated by host–microbe interactions will not only improve our understanding of H. pylori-induced carcinogenesis but also provide mechanistic insight into other malignancies that arise within the context of inflammatory states (e.g., ulcerative colitis and colon cancer).
Supplementary Material
ACKNOWLEDGMENTS
We thank the Vanderbilt Cell Imaging Shared Resource for use of the laser scanning immunofluorescent microscope and imaging software This study was supported by funding from National Institutes of Health grants DK-73902, DK-58587, and DK-77955 (to R.M.P.) and the Vanderbilt Digestive Diseases Research Center grant DK-058404.
Abbreviations used:
- cag
cytotoxin-associated gene
- MMP-7
matrix metalloproteinase-7
- p120
p120-catenin.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E08-03-0283) on July 23, 2008.
REFERENCES
- Adachi Y., et al. Matrix metalloproteinase matrilysin (MMP-7) participates in the progression of human gastric and esophageal cancers. Int. J. Oncol. 1998;13:1031–1035. doi: 10.3892/ijo.13.5.1031. [DOI] [PubMed] [Google Scholar]
- Ajisaka H., Fushida S., Yonemura Y., Miwa K. Expression of insulin-like growth factor-2, c-MET, matrix metalloproteinase-7 and MUC-1 in primary lesions and lymph node metastatic lesions of gastric cancer. Hepatogastroenterology. 2001;48:1788–1792. [PubMed] [Google Scholar]
- Anastasiadis P. Z., Moon S. Y., Thoreson M. A., Mariner D. J., Crawford H. C., Zheng Y., Reynolds A. B. Inhibition of RhoA by p120 catenin. Nat. Cell Biol. 2000;2:637–644. doi: 10.1038/35023588. [DOI] [PubMed] [Google Scholar]
- Anastasiadis P. Z., Reynolds A. B. Regulation of Rho GTPases by p120-catenin. Curr. Opin. Cell Biol. 2001;13:604–610. doi: 10.1016/s0955-0674(00)00258-1. [DOI] [PubMed] [Google Scholar]
- Asahi M., et al. Helicobacter pylori CagA protein can be tyrosine phosphorylated in gastric epithelial cells. J. Exp. Med. 2000;191:593–602. doi: 10.1084/jem.191.4.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backert S., Selbach M. Role of type IV secretion in Helicobacter pylori pathogenesis. Cell Microbiol. 2008;10:1573–1581. doi: 10.1111/j.1462-5822.2008.01156.x. [DOI] [PubMed] [Google Scholar]
- Backert S., Ziska E., Brinkmann V., Zimny-Arndt U., Fauconnier A., Jungblut P. R., Naumann M., Meyer T. F. Translocation of the Helicobacter pylori CagA protein in gastric epithelial cells by a type IV secretion apparatus. Cell Microbiol. 2000;2:155–164. doi: 10.1046/j.1462-5822.2000.00043.x. [DOI] [PubMed] [Google Scholar]
- Bebb J. R., Letley D. P., Thomas R. J., Aviles F., Collins H. M., Watson S. A., Hand N. M., Zaitoun A., Atherton J. C. Helicobacter pylori upregulates matrilysin (MMP-7) in epithelial cells in vivo and in vitro in a Cag dependent manner. Gut. 2003;52:1408–1413. doi: 10.1136/gut.52.10.1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boughan P. K., et al. Nucleotide-binding oligomerization domain-1 and epidermal growth factor receptor: critical regulators of beta-defensins during Helicobacter pylori infection. J. Biol. Chem. 2006;281:11637–11648. doi: 10.1074/jbc.M510275200. [DOI] [PubMed] [Google Scholar]
- Brandt S., Kwok T., Hartig R., Konig W., Backert S. NF-kappaB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc. Natl. Acad. Sci. USA. 2005;102:9300–9305. doi: 10.1073/pnas.0409873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camorlinga-Ponce M., Romo C., Gonzalez-Valencia G., Munoz O., Torres J. Topographical localisation of cagA positive and cagA negative Helicobacter pylori strains in the gastric mucosa; an in situ hybridisation study. J. Clin. Pathol. 2004;57:822–828. doi: 10.1136/jcp.2004.017087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castano J., Solanas G., Casagolda D., Raurell I., Villagrasa P., Bustelo X. R., Garcia de Herreros A., Dunach M. Specific phosphorylation of p120-catenin regulatory domain differently modulates its binding to RhoA. Mol. Cell. Biol. 2007;27:1745–1757. doi: 10.1128/MCB.01974-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y. J., Wu M. S., Lin J. T., Pestell R. G., Blaser M. J., Chen C. C. Mechanisms for Helicobacter pylori CagA-induced cyclin D1 expression that affect cell cycle. Cell Microbiol. 2006;8:1740–1752. doi: 10.1111/j.1462-5822.2006.00743.x. [DOI] [PubMed] [Google Scholar]
- Correa P. Helicobacter pylori and gastric cancer: state of the art. Cancer Epidemiol. Biomarkers Prev. 1996;5:477–481. [PubMed] [Google Scholar]
- Coussens L. M., Fingleton B., Matrisian L. M. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295:2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- Crawford H. C., Krishna U. S., Israel D. A., Matrisian L. M., Washington M. K., Peek R. M., Jr Helicobacter pylori strain-selective induction of matrix metalloproteinase-7 in vitro and within gastric mucosa. Gastroenterology. 2003;125:1125–1136. doi: 10.1016/s0016-5085(03)01206-x. [DOI] [PubMed] [Google Scholar]
- Daniel J. M. Dancing in and out of the nucleus: p120(ctn) and the transcription factor Kaiso. Biochim. Biophys. Acta. 2007;1773:59–68. doi: 10.1016/j.bbamcr.2006.08.052. [DOI] [PubMed] [Google Scholar]
- Daniel J. M., Reynolds A. B. The catenin p120(ctn) interacts with Kaiso, a novel BTB/POZ domain zinc finger transcription factor. Mol. Cell. Biol. 1999;19:3614–3623. doi: 10.1128/mcb.19.5.3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel J. M., Spring C. M., Crawford H. C., Reynolds A. B., Baig A. The p120(ctn)-binding partner Kaiso is a bi-modal DNA-binding protein that recognizes both a sequence-specific consensus and methylated CpG dinucleotides. Nucleic Acids Res. 2002;30:2911–2919. doi: 10.1093/nar/gkf398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. A., Ireton R. C., Reynolds A. B. A core function for p120-catenin in cadherin turnover. J. Cell Biol. 2003;163:525–534. doi: 10.1083/jcb.200307111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeblad M., Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- Ferrero R. L., Ave P., Ndiaye D., Bambou J. C., Huerre M. R., Philpott D. J., Memet S. NF-kappaB activation during acute Helicobacter pylori infection in mice. Infect. Immun. 2008;76:551–561. doi: 10.1128/IAI.01107-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco A. T., et al. Activation of beta-catenin by carcinogenic Helicobacter pylori. Proc. Natl. Acad. Sci. USA. 2005;102:10646–10651. doi: 10.1073/pnas.0504927102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey M. R., Saxon M. L., Zhao X., Rollins A., Evans S. S., Black J. D. Protein kinase C isozyme-mediated cell cycle arrest involves induction of p21(waf1/cip1) and p27(kip1) and hypophosphorylation of the retinoblastoma protein in intestinal epithelial cells. J. Biol. Chem. 1997;272:9424–9435. doi: 10.1074/jbc.272.14.9424. [DOI] [PubMed] [Google Scholar]
- Grosheva I., Shtutman M., Elbaum M., Bershadsky A. D. p120 catenin affects cell motility via modulation of activity of Rho-family GTPases: a link between cell-cell contact formation and regulation of cell locomotion. J. Cell Sci. 2001;114:695–707. doi: 10.1242/jcs.114.4.695. [DOI] [PubMed] [Google Scholar]
- Hansson L. E., Engstrand L., Nyren O., Evans D. J., Jr, Lindgren A., Bergstrom R., Andersson B., Athlin L., Bendtsen O., Tracz P. Helicobacter pylori infection: independent risk indicator of gastric adenocarcinoma. Gastroenterology. 1993;105:1098–1103. doi: 10.1016/0016-5085(93)90954-b. [DOI] [PubMed] [Google Scholar]
- Hearnes J. M., Mays D. J., Schavolt K. L., Tang L., Jiang X., Pietenpol J. A. Chromatin immunoprecipitation-based screen to identify functional genomic binding sites for sequence-specific transactivators. Mol. Cell. Biol. 2005;25:10148–10158. doi: 10.1128/MCB.25.22.10148-10158.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmig S., Ott S., Rosenstiel P., Robert Folsch U., Hampe J., Schreiber S. Genetic variants in matrix metalloproteinase genes are associated with development of gastric ulcer in H. pylori infection. Am. J. Gastroenterol. 2006;101:29–35. doi: 10.1111/j.1572-0241.2005.00348.x. [DOI] [PubMed] [Google Scholar]
- Higashi H., Tsutsumi R., Muto S., Sugiyama T., Azuma T., Asaka M., Hatakeyama M. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science. 2002;295:683–686. doi: 10.1126/science.1067147. [DOI] [PubMed] [Google Scholar]
- Hippo Y., Taniguchi H., Tsutsumi S., Machida N., Chong J. M., Fukayama M., Kodama T., Aburatani H. Global gene expression analysis of gastric cancer by oligonucleotide microarrays. Cancer Res. 2002;62:233–240. [PubMed] [Google Scholar]
- Hirata Y., Maeda S., Mitsuno Y., Akanuma M., Yamaji Y., Ogura K., Yoshida H., Shiratori Y., Omata M. Helicobacter pylori activates the cyclin D1 gene through mitogen-activated protein kinase pathway in gastric cancer cells. Infect. Immun. 2001;69:3965–3971. doi: 10.1128/IAI.69.6.3965-3971.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda M., Mori M., Ueo H., Sugimachi K., Akiyoshi T. Matrix metalloproteinase-7 expression in gastric carcinoma. Gut. 1996;39:444–448. doi: 10.1136/gut.39.3.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireton R. C., et al. A novel role for p120 catenin in E-cadherin function. J. Cell Biol. 2002;159:465–476. doi: 10.1083/jcb.200205115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jawhari A. U., Noda M., Pignatelli M., Farthing M. Up-regulated cytoplasmic expression, with reduced membranous distribution, of the src substrate p120(ctn) in gastric carcinoma. J. Pathol. 1999;189:180–185. doi: 10.1002/(SICI)1096-9896(199910)189:2<180::AID-PATH414>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Karatzas G., Karayiannakis A. J., Syrigos K. N., Chatzigianni E., Papanikolaou S., Riza F., Papanikolaou D. E-cadherin expression correlates with tumor differentiation in colorectal cancer. Hepatogastroenterology. 1999;46:232–235. [PubMed] [Google Scholar]
- Karayiannakis A. J., Syrigos K. N., Alexiou D., Kalahanis N., Rosenberg T., Bastounis E., Pignatelli M. Expression patterns of the novel catenin p120cas in gastrointestinal cancers. Anticancer Res. 1999;19:4401–4405. [PubMed] [Google Scholar]
- Keates S., Sougioultzis S., Keates A. C., Zhao D., Peek R. M., Jr, Shaw L. M., Kelly C. P. cag+ Helicobacter pylori induce transactivation of the epidermal growth factor receptor in AGS gastric epithelial cells. J. Biol. Chem. 2001;276:48127–48134. doi: 10.1074/jbc.M107630200. [DOI] [PubMed] [Google Scholar]
- Kelly K. F., Spring C. M., Otchere A. A., Daniel J. M. NLS-dependent nuclear localization of p120ctn is necessary to relieve Kaiso-mediated transcriptional repression. J. Cell Sci. 2004;117:2675–2686. doi: 10.1242/jcs.01101. [DOI] [PubMed] [Google Scholar]
- Kim S. W., Park J. I., Spring C. M., Sater A. K., Ji H., Otchere A. A., Daniel J. M., McCrea P. D. Non-canonical Wnt signals are modulated by the Kaiso transcriptional repressor and p120-catenin. Nat. Cell Biol. 2004;6:1212–1220. doi: 10.1038/ncb1191. [DOI] [PubMed] [Google Scholar]
- Krueger S., Hundertmark T., Kuester D., Kalinski T., Peitz U., Roessner A. Helicobacter pylori alters the distribution of ZO-1 and p120ctn in primary human gastric epithelial cells. Pathol. Res. Pract. 2007;203:433–444. doi: 10.1016/j.prp.2007.04.003. [DOI] [PubMed] [Google Scholar]
- Kubben F. J., Sier C. F., Meijer M. J., van den Berg M., van der Reijden J. J., Griffioen G., van de Velde C. J., Lamers C. B., Verspaget H. W. Clinical impact of MMP and TIMP gene polymorphisms in gastric cancer. Br. J Cancer. 2006;95:744–751. doi: 10.1038/sj.bjc.6603307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok T., et al. Helicobacter exploits integrin for type IV secretion and kinase activation. Nature. 2007;449:862–866. doi: 10.1038/nature06187. [DOI] [PubMed] [Google Scholar]
- Mariner D. J., Davis M. A., Reynolds A. B. EGFR signaling to p120-catenin through phosphorylation at Y228. J. Cell Sci. 2004;117:1339–1350. doi: 10.1242/jcs.01001. [DOI] [PubMed] [Google Scholar]
- Mayerle J., Friess H., Buchler M. W., Schnekenburger J., Weiss F. U., Zimmer K. P., Domschke W., Lerch M. M. Up-regulation, nuclear import, and tumor growth stimulation of the adhesion protein p120 in pancreatic cancer. Gastroenterology. 2003;124:949–960. doi: 10.1053/gast.2003.50142. [DOI] [PubMed] [Google Scholar]
- McDonnell S., Navre M., Coffey R. J., Jr, Matrisian L. M. Expression and localization of the matrix metalloproteinase pump-1 (MMP-7) in human gastric and colon carcinomas. Mol. Carcinog. 1991;4:527–533. doi: 10.1002/mc.2940040617. [DOI] [PubMed] [Google Scholar]
- Moss S. F., Sood S. Helicobacter pylori. Curr. Opin. Infect. Dis. 2003;16:445–451. doi: 10.1097/00001432-200310000-00011. [DOI] [PubMed] [Google Scholar]
- Murata-Kamiya N., et al. Helicobacter pylori CagA interacts with E-cadherin and deregulates the beta-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene. 2007;26:4617–4626. doi: 10.1038/sj.onc.1210251. [DOI] [PubMed] [Google Scholar]
- Nomura A., Stemmermann G. N., Chyou P. H., Kato I., Perez-Perez G. I., Blaser M. J. Helicobacter pylori infection and gastric carcinoma among Japanese Americans in Hawaii. N. Engl. J. Med. 1991;325:1132–1136. doi: 10.1056/NEJM199110173251604. [DOI] [PubMed] [Google Scholar]
- Noren N. K., Liu B. P., Burridge K., Kreft B. p120 catenin regulates the actin cytoskeleton via Rho family GTPases. J. Cell Biol. 2000;150:567–580. doi: 10.1083/jcb.150.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozawa Y., Nishihara K., Akizawa Y., Orimoto N., Nakano M., Uji T., Ajioka H., Kanda A., Matsuura N., Kiniwa M. Protein kinase C activation by Helicobacter pylori in human gastric epithelial cells limits interleukin-8 production through suppression of extracellular signal-regulated kinase. J. Pharmacol. Sci. 2004;94:233–239. doi: 10.1254/jphs.94.233. [DOI] [PubMed] [Google Scholar]
- Odenbreit S., Puls J., Sedlmaier B., Gerland E., Fischer W., Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science. 2000;287:1497–1500. doi: 10.1126/science.287.5457.1497. [DOI] [PubMed] [Google Scholar]
- Park J. I., Kim S. W., Lyons J. P., Ji H., Nguyen T. T., Cho K., Barton M. C., Deroo T., Vleminckx K., Moon R. T., McCrea P. D. Kaiso/p120-catenin and TCF/beta-catenin complexes coordinately regulate canonical Wnt gene targets. Dev. Cell. 2005;8:843–854. doi: 10.1016/j.devcel.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Parsonnet J., Friedman G. D., Vandersteen D. P., Chang Y., Vogelman J. H., Orentreich N., Sibley R. K. Helicobacter pylori infection and the risk of gastric carcinoma. N. Engl. J. Med. 1991;325:1127–1131. doi: 10.1056/NEJM199110173251603. [DOI] [PubMed] [Google Scholar]
- Peek R. M., Jr, Blaser M. J. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer. 2002;2:28–37. doi: 10.1038/nrc703. [DOI] [PubMed] [Google Scholar]
- Peterson W. L. Helicobacter pylori and peptic ulcer disease. N. Engl. J. Med. 1991;324:1043–1048. doi: 10.1056/NEJM199104113241507. [DOI] [PubMed] [Google Scholar]
- Prokhortchouk A., Hendrich B., Jorgensen H., Ruzov A., Wilm M., Georgiev G., Bird A., Prokhortchouk E. The p120 catenin partner Kaiso is a DNA methylation-dependent transcriptional repressor. Genes Dev. 2001;15:1613–1618. doi: 10.1101/gad.198501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds A. B. p120-catenin: Past and present. Biochim. Biophys. Acta. 2007;1773:2–7. doi: 10.1016/j.bbamcr.2006.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds A. B., Carnahan R. H. Regulation of cadherin stability and turnover by p120ctn: implications in disease and cancer. Semin. Cell Dev. Biol. 2004;15:657–663. doi: 10.1016/j.semcdb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Reynolds A. B., Daniel J., McCrea P. D., Wheelock M. J., Wu J., Zhang Z. Identification of a new catenin: the tyrosine kinase substrate p120cas associates with E-cadherin complexes. Mol. Cell. Biol. 1994;14:8333–8342. doi: 10.1128/mcb.14.12.8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds A. B., Herbert L., Cleveland J. L., Berg S. T., Gaut J. R. p120, a novel substrate of protein tyrosine kinase receptors and of p60v-src, is related to cadherin-binding factors beta-catenin, plakoglobin and armadillo. Oncogene. 1992;7:2439–2445. [PubMed] [Google Scholar]
- Reynolds A. B., Roesel D. J., Kanner S. B., Parsons J. T. Transformation-specific tyrosine phosphorylation of a novel cellular protein in chicken cells expressing oncogenic variants of the avian cellular src gene. Mol. Cell. Biol. 1989;9:629–638. doi: 10.1128/mcb.9.2.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph-Owen L. A., Chan R., Muller W. J., Matrisian L. M. The matrix metalloproteinase matrilysin influences early-stage mammary tumorigenesis. Cancer Res. 1998;58:5500–5506. [PubMed] [Google Scholar]
- Saarialho-Kere U. K., Vaalamo M., Puolakkainen P., Airola K., Parks W. C., Karjalainen-Lindsberg M. L. Enhanced expression of matrilysin, collagenase, and stromelysin-1 in gastrointestinal ulcers. Am. J. Pathol. 1996;148:519–526. [PMC free article] [PubMed] [Google Scholar]
- Sarrio D., Moreno-Bueno G., Sanchez-Estevez C., Banon-Rodriguez I., Hernandez-Cortes G., Hardisson D., Palacios J. Expression of cadherins and catenins correlates with distinct histologic types of ovarian carcinomas. Hum. Pathol. 2006;37:1042–1049. doi: 10.1016/j.humpath.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Selbach M., Moese S., Hauck C. R., Meyer T. F., Backert S. Src is the kinase of the Helicobacter pylori CagA protein in vitro and in vivo. J. Biol. Chem. 2002;277:6775–6778. doi: 10.1074/jbc.C100754200. [DOI] [PubMed] [Google Scholar]
- Senota A., Itoh F., Yamamoto H., Adachi Y., Hinoda Y., Imai K. Relation of matrilysin messenger RNA expression with invasive activity in human gastric cancer. Clin. Exp. Metastasis. 1998;16:313–321. doi: 10.1023/a:1006509312674. [DOI] [PubMed] [Google Scholar]
- Shibamoto S., et al. Association of p120, a tyrosine kinase substrate, with E-cadherin/catenin complexes. J. Cell Biol. 1995;128:949–957. doi: 10.1083/jcb.128.5.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata T., Kokubu A., Sekine S., Kanai Y., Hirohashi S. Cytoplasmic p120ctn regulates the invasive phenotypes of E-cadherin-deficient breast cancer. Am. J. Pathol. 2004;164:2269–2278. doi: 10.1016/S0002-9440(10)63783-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spring C. M., Kelly K. F., O'Kelly I., Graham M., Crawford H. C., Daniel J. M. The catenin p120ctn inhibits Kaiso-mediated transcriptional repression of the beta-catenin/TCF target gene matrilysin. Exp. Cell Res. 2005;305:253–265. doi: 10.1016/j.yexcr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Staddon J. M., Smales C., Schulze C., Esch F. S., Rubin L. L. p120, a p120-related protein (p100), and the cadherin/catenin complex. J. Cell Biol. 1995;130:369–381. doi: 10.1083/jcb.130.2.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein M., Bagnoli F., Halenbeck R., Rappuoli R., Fantl W. J., Covacci A. c-Src/Lyn kinases activate Helicobacter pylori CagA through tyrosine phosphorylation of the EPIYA motifs. Mol. Microbiol. 2002;43:971–980. doi: 10.1046/j.1365-2958.2002.02781.x. [DOI] [PubMed] [Google Scholar]
- Stein M., Rappuoli R., Covacci A. Tyrosine phosphorylation of the Helicobacter pylori CagA antigen after cag-driven host cell translocation. Proc. Natl. Acad. Sci. USA. 2000;97:1263–1268. doi: 10.1073/pnas.97.3.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M., Mimuro H., Suzuki T., Park M., Yamamoto T., Sasakawa C. Interaction of CagA with Crk plays an important role in Helicobacter pylori-induced loss of gastric epithelial cell adhesion. J. Exp. Med. 2005;202:1235–1247. doi: 10.1084/jem.20051027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreson M. A., Anastasiadis P. Z., Daniel J. M., Ireton R. C., Wheelock M. J., Johnson K. R., Hummingbird D. K., Reynolds A. B. Selective uncoupling of p120(ctn) from E-cadherin disrupts strong adhesion. J. Cell Biol. 2000;148:189–202. doi: 10.1083/jcb.148.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreson M. A., Reynolds A. B. Altered expression of the catenin p120 in human cancer: implications for tumor progression. Differentiation. 2002;70:583–589. doi: 10.1046/j.1432-0436.2002.700911.x. [DOI] [PubMed] [Google Scholar]
- Uemura N., Okamoto S., Yamamoto S., Matsumura N., Yamaguchi S., Yamakido M., Taniyama K., Sasaki N., Schlemper R. J. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 2001;345:784–789. doi: 10.1056/NEJMoa001999. [DOI] [PubMed] [Google Scholar]
- Viala J., et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat. Immunol. 2004;5:1166–1174. doi: 10.1038/ni1131. [DOI] [PubMed] [Google Scholar]
- Weydig C., Starzinski-Powitz A., Carra G., Lower J., Wessler S. CagA-independent disruption of adherence junction complexes involves E-cadherin shedding and implies multiple steps in Helicobacter pylori pathogenicity. Exp. Cell Res. 2007;313:3459–3471. doi: 10.1016/j.yexcr.2007.07.015. [DOI] [PubMed] [Google Scholar]
- Wijnhoven B. P., Pignatelli M., Dinjens W. N., Tilanus H. W. Reduced p120ctn expression correlates with poor survival in patients with adenocarcinoma of the gastroesophageal junction. J. Surg. Oncol. 2005;92:116–123. doi: 10.1002/jso.20344. [DOI] [PubMed] [Google Scholar]
- Wilson C. L., Heppner K. J., Labosky P. A., Hogan B. L., Matrisian L. M. Intestinal tumorigenesis is suppressed in mice lacking the metalloproteinase matrilysin. Proc. Natl. Acad. Sci. USA. 1997;94:1402–1407. doi: 10.1073/pnas.94.4.1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witty J. P., McDonnell S., Newell K. J., Cannon P., Navre M., Tressler R. J., Matrisian L. M. Modulation of matrilysin levels in colon carcinoma cell lines affects tumorigenicity in vivo. Cancer Res. 1994;54:4805–4812. [PubMed] [Google Scholar]
- Wroblewski L. E., Noble P. J., Pagliocca A., Pritchard D. M., Hart C. A., Campbell F., Dodson A. R., Dockray G. J., Varro A. Stimulation of MMP-7 (matrilysin) by Helicobacter pylori in human gastric epithelial cells: role in epithelial cell migration. J. Cell Sci. 2003;116:3017–3026. doi: 10.1242/jcs.00518. [DOI] [PubMed] [Google Scholar]
- Xia X., Carnahan R. H., Vaughan M. H., Wildenberg G. A., Reynolds A. B. p120 serine and threonine phosphorylation is controlled by multiple ligand-receptor pathways but not cadherin ligation. Exp. Cell Res. 2006;312:3336–3348. doi: 10.1016/j.yexcr.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Yamashita K., Azumano I., Mai M., Okada Y. Expression and tissue localization of matrix metalloproteinase 7 (matrilysin) in human gastric carcinomas. Implications for vessel invasion and metastasis. Int. J. Cancer. 1998;79:187–194. doi: 10.1002/(sici)1097-0215(19980417)79:2<187::aid-ijc15>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.