

Abstract
In the fission yeast, Schizosaccharomyces pombe, blocks to DNA replication elongation trigger the intra-S phase checkpoint that leads to the activation of the Cds1 kinase. Cds1 is required to both prevent premature entry into mitosis and to stabilize paused replication forks. Interestingly, although Cds1 is essential to maintain the viability of mutants defective in DNA replication elongation, mutants defective in DNA replication initiation require the Chk1 kinase. This suggests that defects in DNA replication initiation can lead to activation of the DNA damage checkpoint independent of the intra-S phase checkpoint. This might result from reduced origin firing that leads to an increase in replication fork stalling or replication fork collapse that activates the G2 DNA damage checkpoint. We refer to the Chk1-dependent, Cds1-independent phenotype as the rid phenotype (for replication initiation defective). Chk1 is active in rid mutants, and rid mutant viability is dependent on the DNA damage checkpoint, and surprisingly Mrc1, a protein required for activation of Cds1. Mutations in Mrc1 that prevent activation of Cds1 have no effect on its ability to support rid mutant viability, suggesting that Mrc1 has a checkpoint-independent role in maintaining the viability of mutants defective in DNA replication initiation.
INTRODUCTION
Before cell division, all eukaryotic cells replicate their DNA through a highly regulated process that involves the binding of several proteins to origin DNA during the G1 phase of the cell cycle (Bell and Dutta, 2002; Diffley and Labib, 2002; Diffley, 2004). The first step in the initiation of DNA replication requires the binding of a six-member hetero-protein complex called Orc to replication origins, marking these sites for subsequent assembly of prereplicative complexes (pre-RCs). The assembly of pre-RCs is believed to occur in early G1, and it requires the loading of Mcm proteins to origin DNA. Two additional proteins Cdc18 and Cdt1 recruit Mcms to DNA and conversion of the pre-RC to an active initiation complex requires activation of both Cdk1 and Hsk1 kinases. Phosphorylation of key components of the pre-RC is believed to result in a reorganization of the origin-associated complex that allows the binding of additional replication proteins, including DNA polymerases and single-strand DNA binding protein. The Mcm complex, with its intrinsic ATPase activity, is believed to participate in the unwinding of DNA during DNA synthesis (Ishimi, 1997; Lee and Hurwitz, 2001).
Analysis of cell cycle mutants in both Saccharomyces cerevisiae and Schizosaccharomyces pombe has led to the identification of a number of proteins that are involved in both the initiation and elongation of DNA replication. In addition to the previously mentioned Orc, Cdc18, Cdt1, and Mcm proteins, they include the Gins complex (Alberghina et al., 1983; Takayama et al., 2003; Gambus et al., 2006; Labib and Gambus, 2007), Sna41/Cdc45 (Miyake and Yamashita, 1998; Uchiyama et al., 2001a,b), Sld2, and Sld3 (Kamimura et al., 1998, 2001; Nakajima and Masukata, 2002; Kanemaki and Labib, 2006; Tanaka et al., 2007; Zegerman and Diffley, 2007). In addition, our studies on DNA polymerase epsilon have suggested that along with its presumed function in DNA replication elongation, the C-terminal half of this protein is required for assembly of the DNA replication initiation complex (Feng and D'Urso, 2001; Feng et al., 2003). Other proteins, including DNA polymerase δ (Pol δ), proliferating cell nuclear antigen, and the putative Pol δ-subunits, Cdc1, Cdc27, and Cdm1 that are believed to be important for the later stages of DNA replication elongation (MacNeill et al., 1996; Zuo et al., 1997; Reynolds et al., 1998, 2000; Bermudez et al., 2002).
It is well known that cells respond to DNA damage or stalled replication forks via cell cycle checkpoints that delay cell cycle progression to allow DNA repairs to be completed (Nyberg et al., 2002). In fission yeast, two distinct checkpoints operate during different stages of the cell cycle to arrest cell cycle progression in response to DNA damage, or blocks to DNA replication. For example, in S phase, inhibition of replication fork progression by treatment with hydroxyurea (HU) leads to activation of the protein kinase Cds1, which delays G2 and stabilizes stalled replication forks (Lindsay et al., 1998; Alcasabas, 2001; Tanaka and Russell, 2001; Zhao et al., 2003; Tanaka and Russell, 2004; Zhao and Russell, 2004). Activation of Cds1 occurs downstream of the Rad–Hus signaling pathway that recognizes stalled forks and leads to activation of the ataxia-telangiectasia-mutated and ATM- and Rad3-related (ATM/ATR) homologue Rad3.
In the G2 phase of the cell cycle, DNA damage resulting from environmental agents such as UV or ionizing radiation can lead to activation of a similar checkpoint; however, in this case, Chk1, rather than Cds1, is activated (Walworth et al., 1993; Furuya et al., 2004). At least one of the functions of Chk1 is to delay the onset of mitosis and similar to Cds1, the mechanism of action is believed to involve direct phosphorylation and inhibition of the mitotic activator, Cdc25 (Furnari et al., 1997, 1999; Blasina et al., 1999). Although it has been shown that phosphorylation of Cdc25 creates a binding site for the 14-3-3 protein Rad24, causing it to be excluded from the nucleus, this form of regulation does not seem to be essential for enforcement of the DNA damage checkpoint in response to ionizing radiation (Lopez-Girona et al., 1999, 2001).
Previously, we reported that cells deleted for the N- terminal half of DNA polymerase ε (Pol ε) are viable (Feng and D'Urso, 2001). Surprisingly, these cells require Chk1, but not Cds1, to maintain cell viability. Consistent with these results, cells lacking the N terminus of Pol ε display wild-type sensitivity to HU (unpublished observation), whereas cells defective in Pol δ are sensitive to loss of Cds1 and are hypersensitive to HU (Liu et al., 1999). We considered the possibility that the Chk1 dependency of the Pol ε mutant might reflect its unique role in DNA replication initiation and therefore set out to test whether other DNA replication mutants are sensitive to the loss of either Chk1 or Cds1 when grown at semipermissive temperatures. Our findings suggest that the viability of mutants defective in DNA replication initiation is dependent on the checkpoint kinase Chk1, whereas mutants defective in DNA replication elongation require both Cds1 and Chk1. Therefore, cells seem to respond differently to inhibition of initiation versus elongation. Moreover, our analysis of the checkpoint response to DNA replication initiation defects has revealed a novel function for Mrc1 in DNA replication that is independent of its checkpoint function in activating Cds1.
MATERIALS AND METHODS
Fission Yeast Strains and Methods
All fission yeast strains used for this study were derived from S. pombe strains 972 and 975, and they are listed in Supplemental Table S1. All media, growth conditions, and genetic manipulations were used as described previously (Moreno et al., 1991).
Camptothecin Treatment
Camptothecin (CPT) lactone was a gift from Dr. Nancy Walworth (University of Medicine and Dentistry of New Jersey) who obtained it from the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program (Division of Cancer Treatment, National Cancer Institute). Cells were grown to mid-log phase and treated with 40 μM CPT for 3 h as described previously (Capasso et al., 2002).
Cell Lysate Preparation and Immunoblotting Analysis
For lysate preparation, cells were collected by centrifugation and lysed in phosphate-buffered saline (PBS) containing complete protease inhibitor (Roche Diagnostics, Indianapolis, IN) by using acid-washed glass beads (425–600 μm; Sigma-Aldrich, St. Louis, MO) and a Savant FastPrep (Bio 101, Vista, CA) Bead Beater. Cells were centrifuged at 3000 rpm for 5 min in an Eppendorf tabletop microcentrifuge (Eppendorf North America, New York, NY), and the supernatant was transferred to a new tube and centrifuged for an additional 15 min. Total protein concentrations in supernatants were determined by Bradford protein assay. Aliquots were separated by SDS-polyacrylamide gel electrophoresis, transferred to nitrocellulose membrane (BA83; Whatman Schleicher and Schuell, Keene, NH), and probed with 12CA5 antibody (gift from Dr. Nancy Walworth) to detect the hemagglutinin (HA) epitope at the C terminus of Chk1. Blocking of the membranes and all antibody incubations and washes were performed in 1% nonfat dried milk and 0.05% Tween 20 in PBS. 12CA5 antibody was diluted 1:500, and a horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology) was used at 1:5000. For detection of proteins, we used the Western Lightening chemiluminescence detection system (PerkinElmer Life and Analytical Sciences, Boston, MA).
Synthetic Lethal Analysis (Viability Assay)
Double mutant strains were constructed by standard genetic procedures and grown to mid-log phase. For viability tests, cell cultures were adjusted to an initial density of 107 cells/ml. Five fivefold serial dilutions were then plated on YE-agar plates, starting with an initial plating of 5 μl or ∼5 × 104 cells. Cells were then incubated at permissive temperature (25°C), semipermissive temperatures (27–34°C), and restrictive temperature (36°C) for 3–5 d. Single mutant and wild-type (wt) cells were used as controls to determine the degree of synthetic lethality between any two single mutants.
Construct of rid Mutants Containing mrc1 Phosphorylation Site Mutations
A series of plasmids expressing different mrc1 mutations that are specifically disrupted for Rad3-dependent phosphorylation sites were provided by Drs. Y. J. Xu and T. J. Kelly (Sloan-Kettering Institute). These mrc1 mutations were constructed in the plasmid of pRep under the control of nmt1 promotor (Xu et al., 2006). A double mutant of cdc30Δmrc1 was transformed with each plasmid and grown in minimal media plus adenine to mid-log phase at 25°C. For HU sensitivity test, cells were plated on YEA agar containing hydroxyurea at various concentrations, and viability assays were conducted as described above. All plates were incubated at the permissive temperature of 25°C and the semipermissive temperature of 29°C for 3–5 d.
Flow cytometry analysis
To determine the length of S phase in rid mutants, rid mutant cells were grown to mid-log phase at 25° C, and then shifted to the critical temperature for 2 h, followed by the 12 mM hydroxyurea treatment for 4 h to arrest cells in early S phase. Upon release, cells were collected every 15 mins for 90 mins, and DNA content was analyzed by FACS (Fluorescence-activated cell sorting). For DNA content measurements, cells were stained with Sytox Green and analyzed by FACS as described previously (Moreno et al., 1991).
RESULTS
Viability of Mutants Defective in DNA Replication Initiation Require Chk1, but Not the Cds1 Kinase
Previously, we have shown that deletion of the N-terminal half of DNA polymerase ε, a protein implicated in DNA replication initiation, leads to a cell cycle delay that is dependent on the Chk1 kinase (Feng and D'Urso, 2001). In the absence of Chk1, these cells entered mitosis without completing DNA replication, resulting in a loss of viability. Interestingly, in the absence of Cds1 cell viability was maintained. We have now extended this analysis to a number of conditional mutants defective in either DNA replication initiation (referred to as rid mutants) or DNA replication elongation (referred to as red mutants).
Temperature-sensitive mutants of key replication proteins were crossed to either the cds1 or chk1 deletion strain, and double mutants were isolated. Serial dilutions of each double mutant and control strains were plated on yeast extract agar plates and incubated at several temperatures, ranging from the permissive temperature of 25°C to the restrictive temperature of 36°C, to determine the “critical” semipermissive temperature. The critical temperature was defined as the temperature that induces a slight elongation phenotype, indicating a cell cycle delay, but it does not result in any significant loss in cell viability. Depending on the mutant strain analyzed, this critical temperature ranged from 27 to 34°C. Our results show that the viability of mutants defective in DNA replication initiation, including cdc30 (encoding Orc1), cdc20 (encoding the catalytic subunit of DNA polymerase epsilon), orp2-2 (encoding Orc2), cdc18 (encoding Cdc18), and cdc21 (encoding Mcm4), when grown at their respective critical temperatures are dependent on the presence of the Chk1 kinase (Figure 1A). In contrast, mutants defective in DNA replication elongation, including cdc6 (encoding DNA polymerase δ), cdc24 (encoding a novel replication elongation factor), and cdc27 (encoding the third subunit of DNA polymerase δ) are sensitive to the loss of Cds1 and Chk1 (Figure 1B). Moreover, we have conducted a large-scale random mutagenesis screen for mutants that display the rid phenotype and have identified only two mutants, both of which are defective in proteins required for DNA replication initiation demonstrating that our screen is highly selective for initiation mutants (to be published elsewhere). Our data suggest that blocks to DNA replication initiation lead to activation of the DNA damage checkpoint and that this response is unique to mutants defective in DNA replication initiation. This implies that a reduction in origin firing might result in DNA damage that cannot be detected in S phase but that it can trigger the Chk1-dependent DNA damage response in G2. This might be due to either DNA damage at the site of initiation or to DNA damage that might result from an increase in replication fork stalling or replication fork collapse. Consistent with the rid mutants having an initiation-specific defect, all of the rid mutants showed wild-type sensitivity to HU, a known inhibitor of DNA replication elongation (data not shown).
Figure 1.

Analysis of the rid mutant phenotype. Fivefold serial dilutions of initiation or elongation defective mutants (as indicated) in the absence or presence of Δcds1 and Δchk1 were plated on YEA plates and incubated for 3–5 d at the indicated critical temperature. Wild-type, Δcds1, and Δchk1 cells were included as controls. After shift to the critical temperature in the absence of Chk1, the viability of rid mutants was dramatically decreased compared with rid mutants lacking Cds1 or control strains. In contrast, the viability of elongation mutants is dependent on Cds1.
We also observed that in addition to Cds1, many of the elongation mutants also required the Chk1 kinase. This dependency on Chk1 most likely reflects the accumulation of DNA damage during S phase as a result of defective DNA replication elongation and would therefore be expected to lead to activation of Chk1 in G2. Although both initiation and elongation mutants are sensitive to the loss of Chk1, it is important to emphasize that only the initiation mutants are viable in the absence of Cds1.
Additional Checkpoint and DNA Repair Genes Are Required for rid Mutant Viability
In addition to the checkpoint kinases Chk1 and Cds1, we also tested whether other checkpoint or repair genes are required to maintain rid mutant viability. Mutants deleted for several nonessential checkpoint or repair genes were crossed to the rid mutants cdc20 and cdc30 and double mutants were isolated and tested for cell viability by using the serial dilution assay (Figure 2). Our analysis included mutants deleted for genes required for sensing DNA damage or replication fork arrest (rad26, rad1, rad3, rad9, rad17, mrc1, and crb2) or genes believed to be involved in DNA repair (mus81, rad24, and hus2). We found that rid mutant viability is dependent on expression of all of these genes.
Figure 2.

Additional checkpoint genes are required for rid mutant viability. (A) The rid mutants cdc20 and cdc30 were crossed to mutants deleted for genes involved in either checkpoint or DNA repair including Δrad1, Δrad3, Δrad9, Δrad26, Δrad17, Δmrc1, Δcrb2, Δmus81, Δhus2, and Δrad24. Viability was assessed at the critical temperature for cdc20-M10 or cdc30-2H4 by using the serial dilution assay described in the text. In the absence of checkpoint/DNA repair genes, the viability of cdc20-M10 was decreased at the critical temperature compared with either the single mutant, cdc20-M10 or wild-type cells. (B) Similar analysis for cdc30-2H4. (C) Mrc1 is required for viability of rid mutants. The mutants cdc21-M68 and orp2-2 were crossed to the Δmrc1 mutant, and the double mutants were isolated and tested for viability at the critical temperature.
Collectively, these results suggest that activation of Chk1 in rid mutants is due to DNA damage that results from defective DNA replication initiation. Either the damage is not of sufficient levels to trigger the Cds1 pathway in S phase, or Chk1 has a specific role in response to the type of damage that occurs specifically within initiation zones. Interestingly, we find that mrc1 is required for rid mutant viability even though cds1 is dispensable (Figure 2C). This suggests that Mrc1 has additional functions in DNA replication that are independent of its normal role in the activation of Cds1 during replication fork arrest.
Chk1 Kinase Is Active in rid Mutants Grown under Semipermissive Temperatures
Our observation that Chk1 is required to maintain rid mutant viability suggests that defects in DNA replication initiation lead to activation of the Chk1 kinase. Most of the rid mutants we have characterized grow normally at the permissive temperature of 25°C and display no requirement for expression of either chk1 or cds1. However, when the temperature is raised to the critical temperature, cells begin to elongate indicating that cell cycle progression is delayed. Cells, on average, are 10–20% longer than wild-type cells grown at the same temperature. Under these conditions, all of the rid mutants tested required chk1 to maintain cell viability. To test whether the dependency of rid mutant viability on chk1 gene expression reflects a dependency on Chk1 kinase activity, we examined the Chk1 kinase by Western immunoblotting of cell extracts isolated from rid mutants following shift to the critical temperature. Chk1 is phosphorylated in response to DNA damage (Capasso et al., 2002), and phosphorylation is dependent on Rad3, a member of the ATM/ATR family of protein kinases, that is believed to directly phosphorylate and activate Chk1. All strains incorporated an HA-tagged copy of Chk1 replacing the endogenous Chk1 so that the Chk1 protein could be easily visualized by Western blot analysis by using HA antibodies. Extracts were prepared from the rid mutants cdc30-2H4, cdc20-M10, and cdc18-K46 at the permissive temperature of 25°C and 3 h after shift to their respective critical temperatures. The phosphorylated form of Chk1 runs with a slower mobility upon gel electrophoresis, and it is diagnostic of Chk1 activation (Palermo and Walworth, 2005). Western blot analysis using antibodies to HA clearly indicated that Chk1 is phosphorylated in rid mutants (Figure 3A, lanes 3–8). The level of Chk1 phosphorylation was similar to the levels detected after treatment with CPT, a DNA topoisomerase I inhibitor that causes DNA damage (Figure 3A, lane 2). These results suggest that Chk1 phosphorylation and presumably its activation are required for the observed cell cycle delay and maintenance of rid mutant viability.
Figure 3.
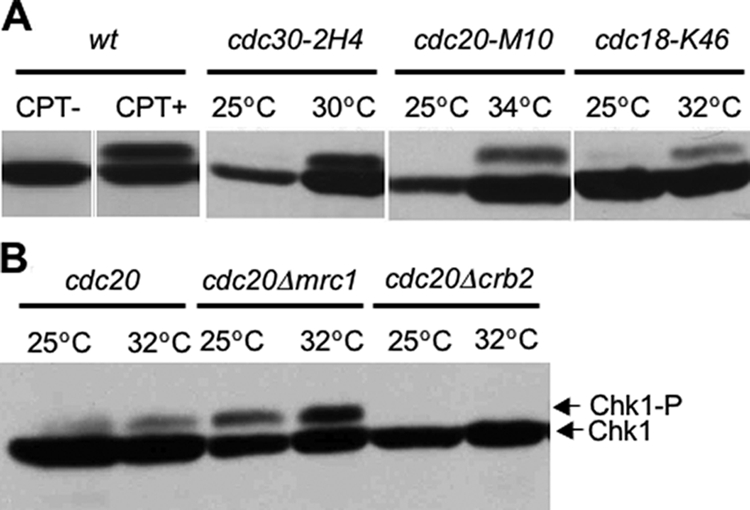
Chk1 kinase is activated in rid mutants when grown at the critical temperature, and Chk1 activation is not dependent on expression of mrc1. (A) Phosphorylation of HA-tagged Chk1-protein was detected by Western immunoblotting using anti-HA antibodies. On activation, Chk1 shows a characteristic mobility shift as indicated by the arrow. The rid mutants cdc20, cdc30, and cdc18 containing a HA-tagged version of Chk1 were shifted to their respective critical temperatures for 3 h and assayed for Chk1 activation. Chk1 is activated in all three rid mutant strains. (B) Mrc1 is not required for Chk1 activation in rid mutants. When shifted to the critical temperature of 32°C, Chk1 phosphorylation is observed in the rid mutant cdc20Δmrc1 (lane 4). In contrast, no Chk1 phosphorylation is observed in the strain cdc20Δcrb2 (lane 6).
Mrc1 Is Not Required for Chk1 Activation in rid Mutants
Our observation that mrc1 is essential for rid mutant viability at the critical temperature suggested that Mrc1 might have a role in the activation of Chk1, analogous to its role in the activation of Cds1 during DNA replication fork arrest. To test this possibility, the rid mutant cdc20-M10 containing the HA-tagged version of Chk1 was crossed to Δmrc1 and double mutants were isolated. After shift to the critical temperature, in the absence of mrc1, no decrease in Chk1 activity is observed, indicating that Mrc1 is not required for activation of Chk1 in rid mutants (Figure 3B, lane 4). We also tested whether Crb2, the adaptor protein implicated in the activation of Chk1 in response to DNA damage in G2, is required for activation of Chk1 in rid mutants. We find that deletion of Crb2 from rid strains abolishes activation of Chk1 when cells are shifted to the semipermissive temperature, indicating Chk1 activation in rid mutants is dependent on the expression of the crb2 gene (Figure 3B, lane 6). Interestingly, in the absence of Mrc1, phosphorylation of Chk1 is observed in rid mutants at the permissive temperature of 25°C, suggesting that loss of Mrc1 may lead to increased DNA damage in rid mutants even at the permissive temperature (Figure 3B, lane 3). These results suggest that Mrc1 has an additional function in rid mutants that is not dependent on its ability to activate Cds1 or Chk1.
Phosphorylation of Mrc1 Is Not Required to Maintain Rid Mutant Viability
It has previously been demonstrated that activation of the intra-S phase checkpoint involves a two-stage mechanism. First, Mrc1 recruits Cds1 to stalled replication forks through an interaction between the forkhead-associated (FHA) domain of Cds1 and the Rad3-phosphorylated sites of Mrc1, thus rendering Cds1 primed for activation by Rad3. Cds1 then dimerizes through interaction between the FHA domain and the phospho-specific site (T11) of Cds1 and is activated by autophosphorylation (Xu et al., 2006).
We obtained a series of plasmids expressing mrc1 mutants that have been specifically disrupted for phosphorylation sites targeted by the Rad3 kinase. These constructs convert a series of serine and threonine residues to alanine, blocking phosphorylation of these sites. These sites include the N-terminal group, containing four SQ/TQ residues (N4); the middle group, containing seven SQ/TQ residues (Mid7); and the C-terminal group, containing three SQ/TQ residues (C3) (Figure 4A). It has been reported that mutations in the SQ cluster and TQ repeats within the Mid7 group are essential for the checkpoint activity of Mrc1 and thus the activation of Cds1 kinase in response to the hydroxyurea treatment (Xu et al., 2006). We confirmed these results by showing that a Δmrc1 strain containing plasmids expressing mrc1 mutant proteins with defective phosphorylation sites at the SQ cluster or TQ repeats are sensitive to HU (Figure 4B).
Figure 4.

Phosphorylation sites in Mrc1 are not required to maintain rid mutant viability. (A) Locations of Rad3 and Tel1 consensus phosphorylation sites in Mrc1. TQ sites are labeled in red and SQ sites are labeled in blue. In mutant Δmrc1-AQ, all 14 S/T residues were changed to alanine. The mutant Δmrc1-N4, Δmrc1-Mid7, and Δmrc1-C3 are mutated for the serine or threonine residues located in these three specific regions only. The phosphorylation sites T645 and T653 are referred to as the TQ repeats, and they have been mutated to alanine in the mutant Δmrc-TQ. The phosphorylation sites from S572 to S614 are referred to as the SQ cluster (Xu et al., 2006), and these serines have been replaced with alanine in mutant Δmrc1-SQ. (B) The SQ cluster and TQ repeats within the Mid7 group are required for the checkpoint function of Mrc1 during DNA replication blocks. Deletions of the N-terminal and C-terminal SQ/TQ sites have a minor effect on HU sensitivity (Δmrc1-N4 and Δmrc1-C3), demonstrating that these sites are not essential for the intra-S phase checkpoint, as was reported previously (Xu et al., 2006). In contrast, in mutant Δmrc1-AQ, or in mutants within the Mid7 group (Δmrc1-Mid7, Δmrc-SQ, Δmrc1-TQ) hypersensitivity to HU is observed. (C) After shift to the critical temperature of 29°C, none of the mrc1 mutations are synthetically lethal with the cdc30 mutant and behave identically to the cdc30Δmrc1 strain containing wild-type mrc1. However, as expected, in the presence of HU, the viability of cdc30Δmrc1 strains containing the AQ, SQ, TQ, or Mid7 mutations are significantly reduced.
To test whether any of the Rad3-dependent phosphorylation sites identified on Mrc1 are required for maintaining rid mutant viability, a double mutant cdc30Δmrc1 was transformed with the plasmids containing the different mrc1 mutants, and double mutants containing each mrc1 mutation were isolated. The viability of these plasmid-containing strains was analyzed at 29°C, the critical temperature for cdc30. It showed that none of the phosphorylation sites in Mrc1 are required for rid mutant viability when shifted to the critical temperature, suggesting that the checkpoint function of Mrc1 in activating the Cds1 kinase is not essential for maintaining the viability of rid mutant strains (Figure 4C). This is consistent with our data that Mrc1 is not required for Chk1 activation in rid mutants.
Swi1 and Swi3 Are Essential to Maintain rid Mutant Viability
In fission yeast the swi1 gene has been implicated in sensing natural replication fork pause sites (Sommariva et al., 2005). Together with swi3, it has been hypothesized to be part of a replication fork protection complex (RFPC) (Schmidt et al., 1989; Noguchi et al., 2003; Sommariva et al., 2005). To test whether swi1 and swi3 are also required for rid mutant viability, we examined the phenotype of rid mutants lacking swi1 or swi3. Our experiments demonstrate that swi1 and swi3 are essential to maintain rid mutant (cdc20-M10, cdc30-2H4, and orp2-2) viability when shifted to the critical temperatures (Figure 5A), suggesting that the stability of replication complexes may be compromised in rid mutants. A previous study showed that Tof1, a Swi1-homologue in S. cerevisiae and Mrc1 are both required for normal fork progression (Tourrière et al., 2005). Therefore, we hypothesize that in fission yeast Swi1, Swi3, and Mrc1 function in a similar pathway to alleviate replication stress resulting from defects in DNA replication initiation. Therefore, we expect that like Mrc1, neither Swi1 nor Swi3 would be required for activation of Chk1. To confirm this, cdc20 containing the HA-tagged version of Chk1 was crossed to the swi1 and swi3 deletion strains, and double mutants were isolated. After shift to the critical temperature of 31°C, activation of Chk1 is still observed in both double mutants (Figure 5B, lanes 4 and 6), indicating that Swi1 and Swi3 are not required for activation of Chk1. However, similar to the cdc20Δmrc1 strain, loss of swi1 or swi3 in cdc20 mutants leads to Chk1 phosphorylation even at the permissive temperature of 25°C (Figure 5B, lanes 3 and 5). These data suggest that similar to Mrc1, Swi1, and Swi3 have a checkpoint-independent role in maintaining the viability of rid mutants.
Figure 5.

Swi1 and Swi3 are essential to maintain rid mutant viability. (A) Mutants defective in DNA replication initiation require swi1 and swi3 to maintain cell viability. Fivefold serial dilutions of the double mutant strains cdc30Δswi1, cdc20Δswi1, and orp2Δswi, cdc30Δswi3, cdc20Δswi3, orp2Δswi3, the rid single mutant strains, and wild-type were plated on YEA plates and incubated at their respective critical temperature. In the absence of Swi1 or Swi3, rid mutant viability was dramatically decreased compared with the single mutants or wild-type cells. (B) Neither Swi1 nor Swi3 is required for Chk1 activation in rid mutants. Phosphorylation of tagged Chk1-protein was detected by Western immunoblotting using anti-HA antibodies. On activation, Chk1 shows a characteristic mobility shift as indicated by the arrow. The double mutants of cdc20-M10Δswi1 and cdc20-M10Δswi3 were shifted to the critical temperature of 31°C for 3 h and analyzed for Chk1 activation. Chk1 activation was not abolished in rid mutants deleted for swi1 or swi3 at 31°C, indicating they are not required for Chk1 activation in rid mutants (lanes 3–6). (C) cdc20Δmrc1Δchk1 cells have a lower critical temperature than cdc20Δchk1 or cdc20Δmrc1. The strain cdc20Δmrc1 was crossed to the Δchk1 strain and triple mutants were isolated and assayed for cell viability at different temperatures. The critical temperature for cdc20Δmrc1Δchk1 was lower than the critical temperature for either of the two double mutants.
To better understand the potential role of Mrc1 (and potentially Swi1 and Swi3) in maintaining the viability of rid mutants we constructed rid mutants that were deleted for both mrc1 and chk1. We reasoned that if deletion of both of genes from a rid mutant (triple mutant) lowered the critical temperature as compared with either of the two double mutant strains (ridΔchk1 or ridΔmrc1), it would suggest that defects in DNA replication initiation lead to increased fork stalling or fork collapse that would be enhanced by loss of Mrc1. In contrast, if the primary defect in rid mutants is DNA damage at initiation sites, then it might be expected that loss of mrc1 would have no effect on the viability of the ridΔchk1 mutant. We observed that deletion of both mrc1 and chk1 from the rid strain cdc20 resulted in a decrease in the critical temperature, demonstrating that loss of mrc1 enhances the phenotype of the cdc20Δchk1 strain (Figure 5C). These results are consistent with the notion that maintenance of replication fork stability is critically important for rid mutant viability and that it is dependent on the expression of mrc1.
Inhibition of DNA Replication Initiation Does Not Increase the Length of S Phase
Recently, in both human and Drosophila S2 cells it has been shown that decreasing the levels of Mcm proteins has little effect on S phase progression or cell cycle kinetics, consistent with the observation that the amount of Mcm protein loaded on chromatin far exceeds the number of Mcm molecules required to replicate the genome (Edwards et al., 2002; Crevel et al., 2007; Ge et al., 2007). It has recently been shown in human cells that dormant origins that are normally replicated passively in early S phase can be activated when DNA replication is inhibited, thus ensuring that even under conditions of DNA replicative stress, the genome can still be efficiently replicated (Ge et al., 2007). It has also been demonstrated that assembly and therefore activation of these origins are dependent on the availability of excess Mcm protein (Ge et al., 2007). To test whether defects in DNA replication initiation in rid mutants increases the duration of S phase, we examined the rate of DNA synthesis in three rid mutants. Each mutant was shifted to its respective critical temperature, and hydroxyurea was then added to block cells in early S phase. After release from the hydroxyurea block progression through S phase was monitored by DNA content by using flow cytometry (Figure 6). In all three mutants examined, cdc20, cdc21, and cdc30, replication rates through S phase were identical to those observed for wild-type cells, suggesting that the rid mutations have no significant impact on the length of S phase. Although S phase remained unchanged, the cell generation time for these mutants increased by at least 30 min, consistent with activation of the DNA damage checkpoint in G2. These experiments suggest that defects in DNA replication initiation might lead to activation of dormant replication origins similar to what has been observed in human cells upon inhibition of DNA replication elongation. However, a small decrease in the number of active origins may not be easily detected by measuring the length of S phase by flow cytometry. An alternative and perhaps more likely explanation, is that there are less replication forks in rid mutants (as would be expected in mutants defective in DNA replication initiation), and this would result in increased stress to the replication machinery and the observed dependency on the presence of the replication fork protection complex.
Figure 6.

The length of S phase is unchanged in rid mutants. The rid mutants cdc20 (A), cdc21 (B), and cdc30 (C) were shifted to the critical temperature for 2 h and then treated with 12 mM hydroxyurea for 4 h to arrest cells in early S phase. On release, cells were collected every 15 min for 90 min, and DNA content analyzed by fluorescence-activated cell sorting analysis. The positions of 1C and 2C DNA control peaks are indicated. Progression through S phase is not affected by the shift to the critical temperature when compared with wild-type cells grown under identical conditions.
DISCUSSION
To ensure the integrity of the eukaryotic genome, two distinct checkpoint pathways have evolved in fission yeast to recognize stalled replication forks or DNA damage. The intra-S phase checkpoint pathway operates during S phase, and it responds to stalled replication forks. This pathway can be induced by treatment with HU, resulting in activation of Cds1 kinase via a Rad3-dependent pathway (Xu et al., 2006). The adaptor protein Mrc1, itself a target of the Rad3 kinase, is critical for maximal activation of Cds1 and therefore plays a pivotal role in the intra-S phase checkpoint response (Zhao et al., 2003). In both Xenopus and yeast, Rad3 targets Mrc1 for phosphorylation at multiple sites, but only two of these sites seem to be critical for checkpoint signaling (Osborn and Elledge, 2003; Zhao et al., 2003; Xu et al., 2006). In S. cerevisiae, phosphorylation of Mrc1 does not seem to be important for its function in stabilizing DNA replication fork progression or replication restart (Osborn and Elledge, 2003).
In G2, DNA damage from exposure to DNA-damaging agents leads to activation of the Chk1 kinase (Walworth and Bernards, 1996; O'Connell et al., 2000). Similar to Cds1, Chk1 phosphorylates and inhibits the mitotic activator Cdc25, thus delaying entry into mitosis. Similar to the role of Mrc1 in the activation of Cds1, full activation of Chk1 in G2 requires the adaptor protein Crb2 (Saka et al., 1997).
Our analysis of mutants of DNA polymerase ε in S. pombe led us to investigate how these different checkpoints respond to different types of DNA replication blocks. Based on an extensive series of experiments from several different laboratories, it is well known that blocks to DNA replication elongation cause activation of Cds1 kinase in S phase (Boddy and Russell, 1999). Therefore, we were surprised to find that mutations that disrupt the catalytic activity of DNA polymerase epsilon (an enzyme believed to be required for replication elongation) are viable in the absence of Cds1. Although insensitive to the loss of Cds1, these mutants were found to be inviable in the absence of Chk1. These observations led us to test whether other conditional mutants defective in DNA replication are sensitive to the loss of either Cds1 or Chk1 when grown at semipermissive temperatures. Our results show that all the initiation mutants tested thus far, including cdc20, orp1, orp2, mcm4, orp5, cdc18, and sna41 show a characteristic sensitivity to the loss of Chk1, but not Cds1. Also, a genetic screen for rid mutants identified only two mutants both of which are defective in DNA replication initiation (to be published elsewhere). Conversely, mutant strains defective in DNA replication elongation, including cdc6, cdc22, cdc24, and cdc27, are nonviable in the absence of Cds1. It should be noted that two of the rid mutants identified, mcm4 and sna41, are not only essential for initiation, but they have also been implicated in DNA replication elongation (Labib et al., 2000; Tercero et al., 2000). However, in these experiments replication elongation rates were monitored after complete destruction of the protein. We propose that when grown at the critical temperature, the rid mutants described here do not have a significant elongation defect but do have defects in initiation of DNA replication. Therefore, we suggest that the rid phenotype is a good indicator of an initiation defect, but we cannot rule out the possibility that the protein corresponding to the rid mutant may have additional roles in DNA replication elongation that would be revealed upon further evaluation.
We also examined the activation of Chk1 in rid mutants, and as expected, all rid mutants showed elevated levels of phosphorylated Chk1 after shift to the critical temperature. Analysis of the checkpoint and repair genes required for the viability of rid mutants revealed that several checkpoint and repair proteins are essential. This implies that DNA damage occurs in these mutants and is recognized by the normal DNA damage-sensing machinery. Interestingly, even though Cds1 is not essential for rid mutant viability, the Cds1 adaptor protein, Mrc1, is required. We confirmed that Chk1 is phosphorylated in the absence of Mrc1, as well as in the absence of Swi1 and Swi3; therefore, these proteins are not required for activation of Chk1. The most likely scenario is that Mrc1, as a component of the RFPC, is required for viability of the rid mutants at the critical temperatures.
It is interesting that defective DNA replication initiation fails to activate the Cds1 kinase even though Chk1 is activated. Although we have not yet identified the source of DNA damage that activates Chk1 under these conditions, we can speculate as to the nature of this damage. One possibility is that failed initiation events lead to damage at the site of the replication origin, and this is recognized by the DNA damage checkpoint. This damage would occur by interfering with the activity of the initiation proteins required for assembly of the helicase early in S phase. In contrast, when replication forks are blocked during elongation, as in the case of the DNA polymerase ε mutants, then the intra-S phase checkpoint would be activated in response to a different type of checkpoint signal emanating from a stalled fork structure. In this case, perhaps extensive single-stranded DNA generated by the paused fork provides the necessary signal to activate the Cds1-dependent pathway.
Finally, our results demonstrate that defects in DNA replication initiation can result in DNA damage that bypasses the intra-S phase checkpoint. If this holds true for human cells, it might have significant implications for the development of tumors. It has been shown that replication stress is one of the first markers for precancerous lesions and that loss of checkpoints might result in suppression of cell death pathways that contribute to tumor formation (Venkitaraman, 2005). If mutations that cause a decrease in replication initiation can bypass the S phase checkpoint, then mutations in the G2 checkpoint alone might be sufficient to trigger genomic instability and tumorigenesis.
Supplementary Material
ACKNOWLEDGMENTS
We thank A. Carr (University of Sussex, Sussex, United Kingdom), J. Dalgaard (Marie Curie Research Institute, Surrey, United Kingdom), S. Forsburg (University of Southern California, Los Angeles, CA), S. Kearsey (Oxford University, Oxford, United Kingdom), J. Leatherwood (Stony Brook University, Stony Brook, NY), P. J. McHugh (University of Oxford, Oxford, United Kingdom), Y. Murakami (Kyoto University, Kyoto, Japan), P. Nurse (Rockefeller University, New York, NY), P. Russell (Scripps Research Institute, La Jolla, CA), and S. Yamashita (Toho University School of Medicine, Tokyo, Japan) for providing strains. Special thanks to N. Walworth for providing strains and for protocols related to assaying Chk1 phosphorylation and to Y. Xu and T. Kelly for kindly providing the Mrc1 phosphorylation site mutant genes. We also thank W. Burhans, J. Diffley, and S. Huard for helpful discussions throughout the course of this work. G. D., L. Y, and A.M.D. are supported by National Institutes of Health grant R01 CA-099034. A. L. was a recipient of an American Heart Graduate Student Fellowship.
Abbreviations used:
- CPT
camptothecin
- HU
hydroxyurea
- Pol
polymerase
- red
replication elongation defective
- rid
replication initiation defective.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E08-01-0020) on July 30, 2008.
REFERENCES
- Alberghina L., Martegani E., Mariani L., Bortolan G. A bimolecular mechanism for the cell size control of the cell cycle. Biosystems. 1983;16:297–305. doi: 10.1016/0303-2647(83)90012-6. [DOI] [PubMed] [Google Scholar]
- Alcasabas A. A. Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat. Cell Biol. 2001;3:958–965. doi: 10.1038/ncb1101-958. [DOI] [PubMed] [Google Scholar]
- Bell S. P., Dutta A. DNA replication in eukaryotic cells. Annu. Rev. Biochem. 2002;71:333–374. doi: 10.1146/annurev.biochem.71.110601.135425. [DOI] [PubMed] [Google Scholar]
- Bermudez V. P., MacNeill S. A., Tappin I., Hurwitz J. The influence of the Cdc27 subunit on the properties of the Schizosaccharomyces pombe DNA polymerase delta. J. Biol. Chem. 2002;277:36853–36862. doi: 10.1074/jbc.M202897200. [DOI] [PubMed] [Google Scholar]
- Blasina A., de Weyer I. V., Laus M. C., Luyten W. H., Parker A. E., McGowan C. H. A human homologue of the checkpoint kinase Cds1 directly inhibits Cdc25 phosphatase. Curr. Biol. 1999;9:1–10. doi: 10.1016/s0960-9822(99)80041-4. [DOI] [PubMed] [Google Scholar]
- Boddy M. N., Russell P. DNA replication checkpoint control. Front. Biosci. 1999;4:D841–D848. doi: 10.2741/boddy. [DOI] [PubMed] [Google Scholar]
- Capasso H., Palermo C., Wan S., Rao H., John U. P., O'Connell M. J., Walworth N. C. Phosphorylation activates Chk1 and is required for checkpoint-mediated cell cycle arrest. J. Cell Sci. 2002;115:4555–4564. doi: 10.1242/jcs.00133. [DOI] [PubMed] [Google Scholar]
- Crevel G., Hashimoto R., Vass S., Sherkow J., Yamaguchi M., Heck M. M., Cotterill S. Differential requirements for MCM proteins in DNA replication in Drosophila S2 cells. PLoS ONE. 2007;2:e833. doi: 10.1371/journal.pone.0000833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diffley J. F. Regulation of early events in chromosome replication. Curr. Biol. 2004;14:R778–R786. doi: 10.1016/j.cub.2004.09.019. [DOI] [PubMed] [Google Scholar]
- Diffley J. F., Labib K. The chromosome replication cycle. J. Cell Sci. 2002;115:869–872. doi: 10.1242/jcs.115.5.869. [DOI] [PubMed] [Google Scholar]
- Edwards M. C., Tutter A. V., Cvetic C., Gilbert C. H., Prokhorova T. A., Walter J. C. MCM2-7 complexes bind chromatin in a distributed pattern surrounding the origin recognition complex in Xenopus egg extracts. J. Biol. Chem. 2002;277:33049–33057. doi: 10.1074/jbc.M204438200. [DOI] [PubMed] [Google Scholar]
- Feng W., D'Urso G. Schizosaccharomyces pombe cells lacking the amino-terminal catalytic domains of DNA polymerase epsilon are viable but require the DNA damage checkpoint control. Mol. Cell. Biol. 2001;21:4495–4504. doi: 10.1128/MCB.21.14.4495-4504.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng W., Rodriguez-Menocal L., Tolun G., D'Urso G. Schizosaccharomyces pombe Dpb2 binds to origin DNA early in S phase and is required for chromosomal DNA replication. Mol. Biol. Cell. 2003;14:3427–3436. doi: 10.1091/mbc.E03-02-0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furnari B., Blasina A., Boddy M. N., McGowan C. H., Russell P. Cdc25 inhibited in vivo and in vitro by checkpoint kinases Cds1 and Chk1. Mol. Biol. Cell. 1999;10:833–845. doi: 10.1091/mbc.10.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furnari B., Rhind N., Russell P. Cdc25 mitotic inducer targeted by chk1 DNA damage checkpoint kinase. Science. 1997;277:1495–1497. doi: 10.1126/science.277.5331.1495. [DOI] [PubMed] [Google Scholar]
- Furuya K., Poitelea M., Guo L., Caspari T., Carr A. M. Chk1 activation requires Rad9 S/TQ-site phosphorylation to promote association with C-terminal BRCT domains of Rad4TOPBP1. Genes Dev. 2004;18:1154–1164. doi: 10.1101/gad.291104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambus A., Jones R. C., Sanchez-Diaz A., Kanemaki M., van Deursen F., Edmondson R. D., Labib K. GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat. Cell Biol. 2006;8:358–366. doi: 10.1038/ncb1382. [DOI] [PubMed] [Google Scholar]
- Ge X. Q., Jackson D. A., Blow J. J. Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev. 2007;21:3331–3341. doi: 10.1101/gad.457807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimi Y. A DNA helicase activity is associated with an MCM4, -6, -7 protein complex. J. Biol. Chem. 1997;272:24508–24513. doi: 10.1074/jbc.272.39.24508. [DOI] [PubMed] [Google Scholar]
- Kamimura Y., Masumoto H., Sugino A., Araki H. Sld2, which interacts with Dpb11 in Saccharomyces cerevisiae, is required for chromosomal DNA replication. Mol. Cell. Biol. 1998;18:6102–6109. doi: 10.1128/mcb.18.10.6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamimura Y., Tak Y. S., Sugino A., Araki H. Sld3, which interacts with Cdc45 (Sld4), functions for chromosomal DNA replication in Saccharomyces cerevisiae. EMBO J. 2001;20:2097–2107. doi: 10.1093/emboj/20.8.2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanemaki M., Labib K. Distinct roles for Sld3 and GINS during establishment and progression of eukaryotic DNA replication forks. EMBO J. 2006;25:1753–1763. doi: 10.1038/sj.emboj.7601063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labib K., Gambus A. A key role for the GINS complex at DNA replication forks. Trends Cell Biol. 2007;17:271–278. doi: 10.1016/j.tcb.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Labib K., Tercero J. A., Diffley J. F. Uninterrupted MCM2–7 function required for DNA replication fork progression. Science. 2000;288:1643–1647. doi: 10.1126/science.288.5471.1643. [DOI] [PubMed] [Google Scholar]
- Lee J. K., Hurwitz J. Processive DNA helicase activity of the minichromosome maintenance proteins 4, 6, and 7 complex requires forked DNA structures. Proc. Natl. Acad. Sci. USA. 2001;98:54–59. doi: 10.1073/pnas.98.1.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay H. D., Griffiths D. J., Edwards R. J., Christensen P. U., Murray J. M., Osman F., Walworth N., Carr A. M. S-phase-specific activation of Cds1 kinase defines a subpathway of the checkpoint response in Schizosaccharomyces pombe. Genes Dev. 1998;12:382–395. doi: 10.1101/gad.12.3.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu V. F., Bhaumik D., Wang T. S. Mutator phenotype induced by aberrant replication. Mol. Cell. Biol. 1999;19:1126–1135. doi: 10.1128/mcb.19.2.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Girona A., Furnari B., Mondesert O., Russell P. Nuclear localization of Cdc25 is regulated by DNA damage and a 14-3-3 protein. Nature. 1999;397:172–175. doi: 10.1038/16488. [DOI] [PubMed] [Google Scholar]
- Lopez-Girona A., Kanoh J., Russell P. Nuclear exclusion of Cdc25 is not required for the DNA damage checkpoint in fission yeast. Curr. Biol. 2001;11:50–54. doi: 10.1016/s0960-9822(00)00026-9. [DOI] [PubMed] [Google Scholar]
- MacNeill S. A., Moreno S., Reynolds N., Nurse P., Fantes P. A. The fission yeast Cdc1 protein, a homologue of the small subunit of DNA polymerase delta, binds to Pol3 and Cdc27. EMBO J. 1996;15:4613–4628. [PMC free article] [PubMed] [Google Scholar]
- Miyake S., Yamashita S. Identification of sna41 gene, which is the suppressor of nda4 mutation and is involved in DNA replication in Schizosaccharomyces pombe. Genes Cells. 1998;3:157–166. doi: 10.1046/j.1365-2443.1998.00177.x. [DOI] [PubMed] [Google Scholar]
- Moreno S., Klar A., Nurse P. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 1991;194:795–823. doi: 10.1016/0076-6879(91)94059-l. [DOI] [PubMed] [Google Scholar]
- Nakajima R., Masukata H. SpSld3 is required for loading and maintenance of SpCdc45 on chromatin in DNA replication in fission yeast. Mol. Biol. Cell. 2002;13:1462–1472. doi: 10.1091/mbc.02-01-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi E., Noguchi C., Du L. L., Russell P. Swi1 prevents replication fork collapse and controls checkpoint kinase Cds1. Mol. Cell. Biol. 2003;23:7861–7874. doi: 10.1128/MCB.23.21.7861-7874.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyberg K. A., Michelson R. J., Putnam C. W., Weinert T. A. Toward maintaining the genome: DNA damage and replication checkpoints. Annu. Rev. Genet. 2002;36:617–656. doi: 10.1146/annurev.genet.36.060402.113540. [DOI] [PubMed] [Google Scholar]
- O'Connell M. J., Walworth N. C., Carr A. M. The G2-phase DNA-damage checkpoint. Trends Cell Biol. 2000;10:296–303. doi: 10.1016/s0962-8924(00)01773-6. [DOI] [PubMed] [Google Scholar]
- Osborn A. J., Elledge S. J. Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev. 2003;17:1755–1767. doi: 10.1101/gad.1098303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palermo C., Walworth N. C. Assaying cell cycle checkpoints: activity of the protein kinase Chk1. Methods Mol. Biol. 2005;296:345–354. doi: 10.1385/1-59259-857-9:345. [DOI] [PubMed] [Google Scholar]
- Reynolds N., Warbrick E., Fantes P. A., MacNeill S. A. Essential interaction between the fission yeast DNA polymerase delta subunit Cdc27 and Pcn1 (PCNA) mediated through a C-terminal p21(Cip1)-like PCNA binding motif. EMBO J. 2000;19:1108–1118. doi: 10.1093/emboj/19.5.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds N., Watt A., Fantes P. A., MacNeill S. A. Cdm1, the smallest subunit of DNA polymerase d in the fission yeast Schizosaccharomyces pombe, is non-essential for growth and division. Curr. Genet. 1998;34:250–258. doi: 10.1007/s002940050394. [DOI] [PubMed] [Google Scholar]
- Saka Y., Esashi F., Matsusaka T., Mochida S., Yanagida M. Damage and replication checkpoint control in fission yeast is ensured by interactions of Crb2, a protein with BRCT motif, with Cut5 and Chk1. Genes Dev. 1997;11:3387–3400. doi: 10.1101/gad.11.24.3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H., Kapitza F. P., Stephen E. R., Gutz H. Some of the swi genes of Schizosaccharomyces pombe also have a function in the repair of radiation damage. Curr. Genet. 1989;16:89–94. doi: 10.1007/BF00393400. [DOI] [PubMed] [Google Scholar]
- Sommariva E., Pellny T. K., Karahan N., Kumar S., Huberman J. A., Dalgaard J. Z. Schizosaccharomyces pombe Swi1, Swi3, and Hsk1 are components of a novel S-phase response pathway to alkylation damage. Mol. Cell. Biol. 2005;25:2770–2784. doi: 10.1128/MCB.25.7.2770-2784.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama Y., Kamimura Y., Okawa M., Muramatsu S., Sugino A., Araki H. GINS, a novel multiprotein complex required for chromosomal DNA replication in budding yeast. Genes Dev. 2003;17:1153–1165. doi: 10.1101/gad.1065903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K., Russell P. Mrc1 channels the DNA replication arrest signal to checkpoint kinase Cds1. Nat. Cell Biol. 2001;3:966–972. doi: 10.1038/ncb1101-966. [DOI] [PubMed] [Google Scholar]
- Tanaka K., Russell P. Cds1 phosphorylation by Rad3-Rad26 kinase is mediated by forkhead-associated domain interaction with Mrc1. J. Biol. Chem. 2004;279:32079–32086. doi: 10.1074/jbc.M404834200. [DOI] [PubMed] [Google Scholar]
- Tanaka S., Tak Y. S., Araki H. The role of CDK in the initiation step of DNA replication in eukaryotes. Cell Div. 2007;2:16. doi: 10.1186/1747-1028-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tercero J. A., Labib K., Diffley J. F. DNA synthesis at individual replication forks requires the essential initiation factor Cdc45p. EMBO J. 2000;19:2082–2093. doi: 10.1093/emboj/19.9.2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiyama M., Arai K., Masai H. Sna41goa1, a novel mutation causing G1/S arrest in fission yeast, is defective in a CDC45 homolog and interacts genetically with polalpha. Mol. Genet. Genomics. 2001a;265:1039–1049. doi: 10.1007/s004380100499. [DOI] [PubMed] [Google Scholar]
- Uchiyama M., Griffiths D., Arai K., Masai H. Essential role of Sna41/Cdc45 in loading of DNA polymerase α onto minichromosome maintenance proteins in fission yeast. J. Biol. Chem. 2001b;276:26189–26196. doi: 10.1074/jbc.M100007200. [DOI] [PubMed] [Google Scholar]
- Venkitaraman A. R. Medicine: aborting the birth of cancer. Nature. 2005;434:829–830. doi: 10.1038/434829a. [DOI] [PubMed] [Google Scholar]
- Walworth N., Bernards R. rad-dependent response of the chk1-encoded protein kinase at the DNA damage checkpoint. Science. 1996;271:353–356. doi: 10.1126/science.271.5247.353. [DOI] [PubMed] [Google Scholar]
- Walworth N., Davey S., Beach D. Fission yeast chk1 protein kinase links the rad checkpoint pathway to cdc2. Nature. 1993;363:368–371. doi: 10.1038/363368a0. [DOI] [PubMed] [Google Scholar]
- Xu Y. J., Davenport M., Kelly T. J. Two-stage mechanism for activation of the DNA replication checkpoint kinase Cds1 in fission yeast. Genes Dev. 2006;20:990–1003. doi: 10.1101/gad.1406706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zegerman P., Diffley J. F. Phosphorylation of Sld2 and Sld3 by cyclin-dependent kinases promotes DNA replication in budding yeast. Nature. 2007;445:281–285. doi: 10.1038/nature05432. [DOI] [PubMed] [Google Scholar]
- Zhao H., Russell P. DNA binding domain in the replication checkpoint protein Mrc1 of Schizosaccharomyces pombe. J. Biol. Chem. 2004;279:53023–53027. doi: 10.1074/jbc.M410449200. [DOI] [PubMed] [Google Scholar]
- Zhao H., Tanaka K., Nogochi E., Nogochi C., Russell P. Replication checkpoint protein Mrc1 is regulated by Rad3 and Tel1 in fission yeast. Mol. Cell. Biol. 2003;23:8395–8403. doi: 10.1128/MCB.23.22.8395-8403.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo S., Gibbs E., Kelman Z., Wang T. S., O'Donnell M., MacNeill S. A., Hurwitz J. DNA polymerase delta isolated from Schizosaccharomyces pombe contains five subunits. Proc. Natl. Acad. Sci. USA. 1997;94:11244–11249. doi: 10.1073/pnas.94.21.11244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.