

SUMMARY
Development of the prospective midbrain and cerebellum are coordinated by FGF ligands produced by the isthmic organizer. Previous studies have suggested that the midbrain and cerebellum require different levels of FGF signaling for their development. However, little is known about the extent to which specific regions within these two parts of the brain differ in their requirement for FGF signaling during embryogenesis. In this study, we have explored the effects of inhibiting FGF signaling within the embryonic midbrain (mesencephalon) and cerebellum (rhombomere 1) by misexpressing Sprouty2 (Spry2) specifically in the mouse mesencephalon and rhombomere 1 from an early stage. We show that such Spry2 misexpression moderately reduces FGF signaling, and that this reduction causes the death of cells in the anterior mesencephalon, the region furthest from the source of FGF ligands. Interestingly, the remaining cells in the posterior mesencephalon develop into anterior midbrain, indicating that a low level of FGF signaling is sufficient to promote only anterior midbrain development. Spry2 misexpression also affects development of the vermis, the medial part of the cerebellum that spans the midline. We found that whereas misexpression of Spry2 alone caused loss of the anterior vermis, reducing FGF signaling further, by decreasing Fgf8 gene dosage, resulted in loss of the entire vermis. We provide evidence that cell death is not responsible for this tissue loss. Instead, our data suggest that the vermis fails to develop because reducing FGF signaling perturbs the balance between vermis and roof plate development in rhombomere 1. We suggest a molecular explanation for this phenomenon by providing evidence that FGF signaling functions to inhibit the BMP signaling that promotes roof plate development.
Keywords: FGF, midbrain, cerebellum, Sprouty, apoptosis, vermis, roof plate
INTRODUCTION
Remarkable progress has been made in understanding the processes that regulate the development of the anatomically and functionally distinct subdivisions of the vertebrate brain. The midbrain and cerebellum have been particularly well-studied, and much is known about how they form from adjacent regions of the neural tube (Raible and Brand, 2004; Zervas et al., 2005). The midbrain develops from the mesencephalon (mes) (see Fig. 1D). The dorsal mes gives rise to the tectum, which consists of the superior colliculus (SC) anteriorly and inferior colliculus (IC) posteriorly, where visual and auditory stimuli are processed, respectively. The cerebellum develops from dorsal rhombomere 1 (r1), the anterior-most segment of the hindbrain (see Fig. 1D). In the mouse, the cerebellum consists of a medial vermis, flanked laterally by a pair of hemispheres (see Fig. 1E). The cerebellum, where sensory information and motor activity are coordinated, undergoes dramatic changes after birth that transform it into a highly foliated structure (Sillitoe and Joyner, 2007). The molecular pathways that subdivide the tectum and cerebellum into subregions are poorly understood.
Figure 1. Spry2-GOF, a conditional Spry2 gain-of-function allele, and its expression following recombination by En1Cre.
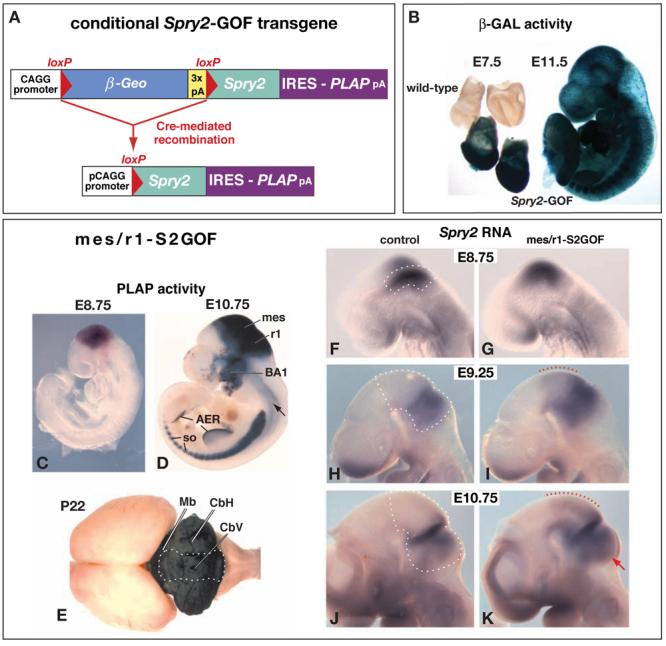
(A) Schematic diagram illustrating the Spry2-GOF transgene. Before Cre-mediated recombination, the CAGG promoter drives expression of the β-Geo gene, followed by a triple polyadenylation sequence (3x pA). After recombination of the loxP sites and the consequent deletion of β-Geo, mouse Spry2 and human placental alkaline phosphatase (PLAP) cDNAs are expressed as a bicistronic mRNA containing an internal ribosome entry site (IRES) that directs translation of PLAP. (B) Assays in whole mount for lacZ expression (β-Galactosidase [β-GAL] activity) in embryos hemizygous for Spry2-GOF. (C-K) Analysis of expression of the Spry2-GOF transgene after it has undergone recombination in En1Cre/+;Spry2-GOF (mes/r1-S2GOF) mutants. (C-E) Assays for PLAP activity in embryos and postnatal brain at the stages indicated. Blue staining identifies cells in which recombination of the transgene occurred. The arrow in panel D points to two ventrolateral stripes in the spinal cord. (F-K) RNA in situ hybridization assays in whole mount at the stages indicated, using a mouse Spry2 probe. The dashed white lines outline mes/r1 on the left side of the brain in the control embryos, where the probe detects endogenous Spry2 expression. In mes/r1-S2GOF embryos, the Spry2 RNA detected represents the sum of expression from the endogenous Spry2 gene and the recombined transgene. Regions in which there is ectopic Spry2 expression as a consequence of recombination of the transgene in mes and in r1 are delineated by red dotted lines in panels I and K and a red arrow in panel K, respectively. Abbreviations: AER, apical ectodermal ridge of the limb bud, BA1, first branchial arch, CbV, cerebellar vermis; CbH, cerebellar hemispheres; Mb, midbrain; mes; mesencephalon; r1, rhombomere 1; so, somites.
At early stages, the development of midbrain and cerebellum is coordinated because the signals that pattern them are produced by a single signaling center, the isthmic organizer (IsO), situated at the mes/r1 boundary (Echevarria et al., 2003; Nakamura et al., 2005; Partanen, 2007). FGF8, a member of the Fibroblast Growth Factor (FGF) family of secreted signaling molecules (Itoh and Ornitz, 2004), is a key component of the IsO signal (Crossley et al., 1996). In the mouse, Fgf8 is expressed in r1 from early neural plate (embryonic day [E] 8.25) through mid-gestation (E12.5) stages. By E10.0, Fgf8 expression is localized to a circular domain in r1, just posterior to the mes/r1 boundary, that is interrupted by Fgf8-negative regions at the dorsal and ventral midlines (roof and floor plates, respectively) (Crossley and Martin, 1995). When Fgf8 is inactivated in the early neural plate, all mes and r1 cells die between E8.5 and E10. However, when Fgf8 expression is only moderately reduced, the anterior midbrain appears normal, but posterior midbrain, isthmus, and vermis are lost (Chi et al., 2003). The reason for such tissue loss in Fgf8 hypomorphs is not known.
FGF17 is also required for midbrain and cerebellum development. Fgf17 expression is dependent on FGF8 (Chi et al., 2003) and is found in a broad domain that encompasses both prospective cerebellum and posterior midbrain (Xu et al., 2000). In Fgf17 null mice, part of the IC and anterior vermis are absent, but the remainder of the midbrain and cerebellum appear normal. The extent of vermis tissue loss is increased in these mutants by removing one copy of Fgf8, suggesting that the two FGF family members cooperate to control cerebellum development (Xu et al., 2000).
The level of FGF signaling can affect cell fate during mes/r1 development. Ectopic expression of the Fgf8b splice variant, which encodes an FGF8 isoform with high affinity for FGF receptors (Olsen et al., 2006), transforms mouse mes cells to a cerebellar fate (Liu et al., 1999). In contrast, ectopic expression of the Fgf8a splice variant, which encodes an FGF8 isoform with much lower affinity for FGF receptors (Olsen et al., 2006; Zhang et al., 2006), expands the mes and transforms posterior forebrain (diencephalon) progenitors to a midbrain fate (Lee et al., 1997; Liu et al., 1999). Importantly, when Fgf8b is ectopically expressed at a low rather than a high level in chicken embryos, the results are similar to those obtained with Fgf8a or Fgf17, indicating that specification of midbrain and cerebellum require different levels of FGF signaling (Sato et al., 2001; Liu et al., 2003).
Together, these studies highlight the importance of controlling FGF signaling during mes/r1 development. Such control can be achieved at the intracellular level by Sprouty proteins (Hacohen et al., 1998), whose expression is induced by signaling through FGF receptors (FGFRs) and other receptor tyrosine kinases (RTKs) (Mason et al., 2006). There are four Sprouty genes in mouse and human. Spry1 and Spry2 are strongly expressed in mes/r1 from early neural plate stages, in cells close to the IsO (Minowada et al., 1999). Studies in zebrafish (Furthauer et al., 2001) and chicken (Suzuki-Hirano et al., 2005) embryos have shown that increasing Sprouty gene expression above normal levels can antagonize FGF activity and interfere with mes/r1 development. Here we have employed a mouse line carrying a conditional Spry2 gain-of-function transgene in conjunction with an Fgf8 null allele to produce the equivalent of an FGF loss-of-function allelic series specifically in mes/r1. By studying the phenotypic effects of these genetic manipulations we have uncovered potential mechanism(s) by which FGF signaling differentially regulates the development of specific regions within the midbrain and cerebellum.
MATERIALS AND METHODS
Genotyping
All mutant alleles used in this study were maintained on a mixed genetic background. Genotypes of embryos and postnatal mice were determined by PCR assays using embryonic yolk sac or tail DNA as a template. The Spry2-GOF allele (Calmont et al., 2006), which was produced basically as described by Lu et al. (2006), was detected using primers that amplify lacZ (Lu et al., (2006). Spry2-GOF homozygotes were identified using a quantitative PCR assay for PLAP sequences (primers and conditions available upon request). The En1Cre allele was detected using primers that amplify cre (Sun et al., 2002). Genotyping for Fgf8 and Fgf17 alleles was performed as previously described by Sun et al. (Sun et al., 1999; 2002) and Xu et al. (Xu et al., 2000), respectively.
Histological analysis and assays for cell death and gene expression
Noon of the day when a vaginal plug was detected was considered E0.5. Embryos were staged more precisely by counting somites posterior to the forelimb bud and scoring the first one counted as somite 13. Histological analysis of late gestation and postnatal brains, and immunolocalization in brain tissue were performed as previously described by Chi et al. (2003).
Assays for cell death were performed in whole mount by staining with LysoTracker (Molecular Probes L-7528), as previously described by Grieshammer et al. (2005). We confirmed that LysoTracker staining gave results on En1Cre/+;Fgf8flox/− (referred to here as mes/r1-Fgf8-KO) embryos similar to those we previously obtained using Nile Blue Sulfate staining and TUNEL assays to detect cell death (Chi et al., 2003).
For gene expression analysis, embryos were collected in cold PBS, fixed in 4% paraformaldehyde, and stored in 70% ethanol at −20°C. Some embryos were embedded in paraffin and sectioned at 7μm. Standard protocols were employed for RNA in situ hybridization on sections and in whole mount, and for assaying β-GAL activity in whole mount.
RESULTS
Cre-mediated recombination of a conditional Spry2 gain-of-function transgene in midbrain and cerebellum progenitors
In order to reduce FGF signaling in mes/r1, we employed a mouse line that carries a conditional Sprouty2-gain-of-function allele, Spry2-GOF (Calmont et al., 2006). In these mice, cells expressing the transgene produce β-Geo, which encodes a fusion protein with both neomycin-resistance and β-Galactosidase (β-GAL) activities (Friedrich and Soriano, 1991)(Fig. 1A). Assays for β-GAL demonstrated that Spry2-GOF is expressed in most cells of the embryo from E7.5 to at least E11.5 (Fig. 1B, and data not shown). Cells in which the transgene has undergone Cre-mediated recombination express a bicistronic mRNA containing both Spry2 and PLAP coding sequences (Fig. 1A). PLAP protein, which can be detected by a histochemical stain, functions as a convenient reporter for expression of the recombined transgene.
To obtain mice in which the transgene was recombined in mes/r1, we crossed Spry2-GOF animals with mice carrying En1Cre, an En1 allele with cre inserted in the first exon (Kimmel et al., 2000). We detected PLAP activity in their En1Cre/+;Spry2-GOF offspring only in those domains in which En1Cre has previously been shown to function (Li et al., 2002; Chi et al., 2003). Thus, in the developing brain PLAP activity was not detected prior to E8.25 (not shown), but strong PLAP activity was observed throughout mes/r1 from E8.75 (Fig. 1C). Subsequently, the CAGG promoter remained active in the descendants of mes/r1 cells, and PLAP activity was therefore detected throughout the prospective midbrain and cerebellum at E10.75 (Fig. 1D), and in midbrain and cerebellum until at least postnatal day (P) 22 (Fig. 1E). In addition, by E10.75, PLAP activity was detected in cells in the first branchial arch (Fig. 1D). These cells are presumably mes/r1-derived neural crest cells, which normally cease expressing En1 after they begin migrating towards the first branchial arch, but continue to express the recombined Spry2-GOF transgene under the control of the CAGG promoter. PLAP activity was also detected in other domains in which En1 is normally expressed (Kimmel et al., 2000), including limb bud ventral ectoderm and apical ectodermal ridge, somites, and two ventrolateral stripes that run the length of much of the posterior hindbrain and spinal cord (Fig. 1D). No PLAP activity was detected in control littermates (wild-type, or mutants carrying only En1Cre/+ or only Spry2-GOF) (not shown). En1Cre/+;Spry2-GOF mutants will hereafter be referred to as mes/r1-S2GOF embryos, to indicate that the recombined transgene was expressed in mes/r1.
To directly assess Spry2 expression from the recombined Spry2-GOF transgene, we performed a whole mount RNA in situ hybridization analysis on mes/r1-S2GOF mutants and control littermates. Consistent with data from our previous analysis (Minowada et al., 1999), in control embryos we detected endogenous Spry2 RNA at E8.75 in a domain encompassing most of mes/r1 (Fig. 1F), which subsequently became progressively more restricted, such that by E9.25 and at E10.75 it encompassed the posterior mes and r1 but not the anterior mes (Fig. 1H,J). In mes/r1-S2GOF mutants at these stages (Fig. 1C,D), the level of Spry2 RNA appeared to be only slightly increased within the normal domains of Spry2 expression in these mutants (Fig. 1G,I,K). However, because transgene expression persisted in regions where endogenous Spry2 expression is no longer detected, we observed ectopic Spry2 expression in anterior mes and posterior r1 at later stages (E9.25 and E10.75; Fig. 1I,K). In situ hybridization analysis on sections of mes/r1-S2GOF mutants at 42 som demonstrated that Spry2 RNA was distributed uniformly within the ectopic expression domain (not shown). Together our data suggest that in mes/r1-S2GOF embryos, the level of Spry2 RNA is slightly elevated within its normal expression domain and is ectopically expressed throughout the remainder of the developing midbrain and cerebellum from at least E8.75.
Similar effects on midbrain and cerebellum are obtained by expressing a single copy of recombined Spry2-GOF in mes/r1 or by reducing Fgf17 and Fgf8 gene dosage
Histological analysis of mes/r1-S2GOF mutants shortly before birth revealed the effects of expressing the recombined transgene on midbrain and cerebellum development. In midsagittal sections of all E17.5 mes/r1-S2GOF mutants examined (n=4), the dorsal midbrain appeared normal at its anterior end, some tissue loss was observed at the posterior end of the SC, and the IC was entirely absent (Fig. 2A,B, and data not shown). Posterior to the midbrain, the dorsal isthmus (Fig. 2A,B) and tissue derived from it (velum medullare, Fig. 2C,D) appeared to be absent. The medial cerebellum (vermis) also appeared to have lost tissue, but only at its anterior end (Fig. 2A,B, and data not shown). Consistent with the latter conclusion, a population of cells that is normally detected in the anterior vermis by staining for Calretinin (Fig. 2C), was not observed (Fig. 2C,D, and data not shown). In contrast, basal plate derivatives in the midbrain (oculomotor nucleus [nIII], substantia nigra and ventral tegmental area [SN-VTA]), and the isthmus (trochlear nucleus [nIV]), were present, as was the r1-derived locus coeruleus, and all appeared normal (not shown).
Figure 2. Expression of a single copy of the recombined Spry2-GOF allele in mes/r1 causes absence of the inferior colliculus and anterior vermis.
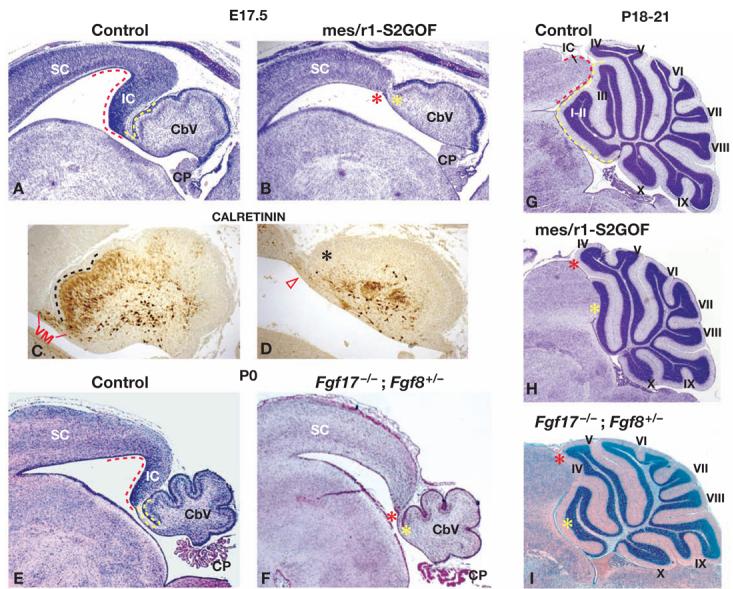
(A,B,E-I) Midsagittal sections of brains of the genotypes indicated, collected at the stages denoted. All sections were stained with Cressyl violet, except that in panel I, which was stained with hematoxylin and eosin. Anterior is to the left, posterior to the right. In control embryos (panels A and E), the posterior midbrain and anterior hindbrain regions that are absent in the mutant embryos are demarcated by red and yellow dashed lines, respectively; in the mutant embryos (panels B and F), the regions where the missing midbrain and anterior hindbrain tissue would normally be found are indicated by red and yellow asterisks, respectively. (C and D) Higher power view of the vermis shown in panels A and B, respectively, stained with an antibody against Calretinin. In the control vermis (panel C), the region that is missing in the mutant vermis is demarcated by a black dashed line; in the mutant vermis (panel D), the region where cells expressing Calretinin would normally be found is indicated by a black asterisk and the region where the velum medullare would normally be found is indicated by an open red arrowhead. (G-I) Comparison of vermis development in control, mes/r1-S2GOF, and Fgf17−/−; Fgf8+/− animals at approximately 3 weeks of age. In the control embryo (panel G), the regions of the posterior midbrain and anterior vermis that are missing in the mutant embryos are demarcated by red and yellow dashed lines, respectively; in the mutant embryos (panels H and I), the region where the missing midbrain and anterior lobule tissue would normally be found is indicated by red and yellow asterisks, respectively. Abbreviations: CP, choroid plexus; IC, inferior colliculus; SC, superior colliculus; VM, velum medullare. The lobules of the vermis are denoted by Roman numerals, as described by (Inouye and Oda, 1980).
Since mes/r1-S2GOF mutants were viable, we were able to exclude the possibility that the apparent tissue loss we observed at E17.5 was due to a developmental delay. We found that both the IC and anterior vermis were indeed absent in postnatal mes/r1-S2GOF animals (Fig. 2G,H). With respect to the vermis, in wild-type mice there are strain-specific variations in foliation pattern, such that some strains have three distinct lobules (I, II, and III) anterior to the lobule of culmen (IV-V), and other strains have only two (indicated as I-II and III in Fig. 2G) (Inouye and Oda, 1980). In all postnatal mes/r1-S2GOF mutants examined (n=5), it was clear that lobules I-III were either reduced or absent, whereas the remaining lobules appeared essentially normal (Fig. 2H, and data not shown). In contrast to these vermis defects, the cerebellar hemispheres appeared grossly normal (not shown). Thus, expressing a single copy of the recombined Spry2-GOF transgene in mes/r1 from an early stage of development resulted in absence of the posterior dorsal midbrain (IC), isthmus, and anterior vermis. There were no obvious abnormalities in these animals in other tissues that developed from cells in which the Spry2-GOF transgene was recombined by En1Cre.
The phenotype of mes/r1-S2GOF mutants described above was remarkably similar to, although slightly more severe than, that observed in Fgf17 null homozygotes carrying one copy of an Fgf8 null allele (Fgf17−/−; Fgf8+/− animals) (Xu et al., 2000). At birth, the IC, isthmus, and anterior tissue in the developing cerebellum appeared to be absent in such compound FGF mutant animals (Fig. 2E,F), and at 3-4 weeks after birth, only one major lobule was present anterior to lobules IV-V (Fig. 2I). These results suggest that expressing a single copy of the recombined Spry2-GOF allele results in a reduction in FGF signaling during midbrain and cerebellar development to approximately the same degree as in Fgf17−/−; Fgf8+/− mutants.
Reducing Fgf8 gene dosage in mes/r1-S2GOF mutants or expressing two copies of the recombined Spry2-GOF allele in mes/r1 results in loss of the cerebellar vermis
If expression of the recombined Spry2-GOF allele in mes/r1 does indeed function to reduce the level of FGF signaling, one might expect that reducing it even further, for example by reducing Fgf8 gene dosage, would result in a more severe phenotype. We therefore crossed mes/r1-S2GOF mutants with Fgf8+/− mice, in which brain development is normal, and examined their En1Cre/+;Spry2-GOF;Fgf8+/− progeny (mes/r1-S2GOF;F8 mutants) at E17.5. When viewed in whole mount, it was evident that the entire vermis was absent in these mutants (Fig. 3A,B). A histological analysis showed that only the anterior region of the dorsal midbrain, including the anterior-most mes-derived structure, the PPT (Lagares et al., 1994), appeared normal; tissue was missing at the posterior end of the SC, and the IC, the dorsal isthmus and the vermis were all absent (Fig. 3C-E). The mes/r1-S2GOF;F8 mutants could be classified in two groups: group I (n=3/5) had less tissue loss at the posterior end of the SC, and basal structures including nIII, nIV, and the SN-VTA were present although somewhat reduced; group II (n=2/5) had more tissue loss at the posterior end of the SC and the basal structures were all absent (Fig. 3C-K). Ventrolateral r1-derived tissue was present in both groups, as evidenced by the presence of the Locus coeruleus, but it was clearly reduced in group II (Fig. 3I-K).
Figure 3. Decreasing Fgf8 gene dosage in mes/r1-S2GOF embryos results in absence of the vermis.
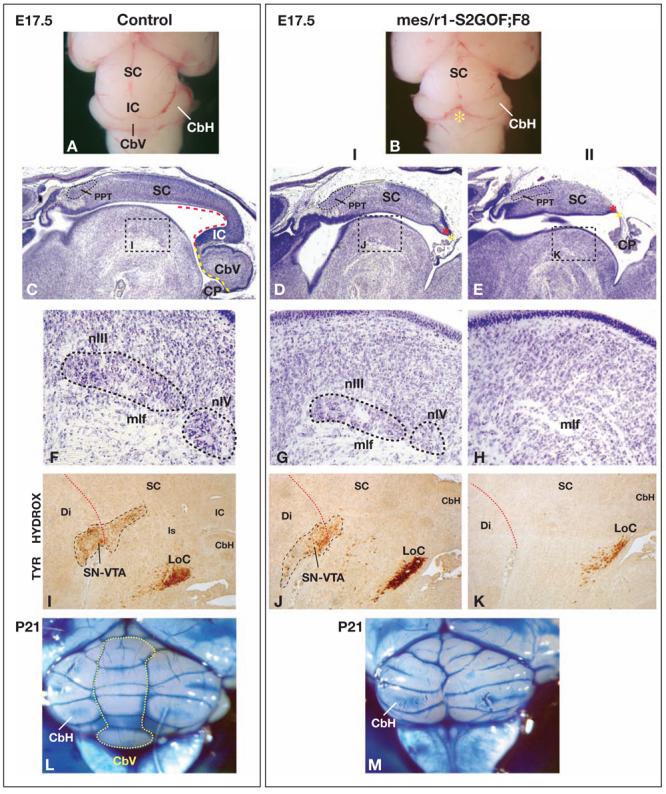
(A,B) Dorsal views of brains collected from E17.5 embryos of the genotypes indicated. In the mutant brain (panel B), the region where the vermis would normally be found is indicated by a yellow asterisk. (C-E) Low magnification views of midsagittal sections of E17.5 control and mes/r1-S2GOF;F8 mutants, stained with Cressyl violet. Panels D and E show examples of the group I (milder) and group II (more severe) brain phenotypes observed in the mutants. The dashed red and yellow lines and red and yellow asterisks are explained in the legend to Fig. 2A,B,E,F. The areas demarcated by dashed boxes in panels C, D, E are shown at higher magnification in panels F, G, H, respectively. (F-H) High magnification views of basal structures. When present, nIII and nIV are circled with dashed lines. (I-K) High magnification views of more lateral sagittal sections of the same brains, assayed by immunohistochemistry with an antibody against Tyrosine Hydroxylase, which specifically stains nuclei in the ventrolateral posterior diencephalon, midbrain, and anterior hindbrain. The dotted red line indicates the boundary between diencephalon and midbrain. (L,M) Dorsal views of intact brains collected from P21 mice of the genotypes indicated, stained with ink. The dotted line in panel L outlines the vermis, which is absent in the mutant brain. Abbreviations are the same as in the legends to Figs. 1 and 2, with the following additions: Di, diencephalon; LoC, locus ceruleus; mlf, medial longitudinal fascicle; nIII, oculomotor nucleus; PPT, posterior pretectal nucleus; nIV, trochlear nucleus; SN, substantia nigra; VTA, ventral tegmental area.
We next examined animals carrying En1Cre/+ and two copies of Spry2-GOF (mes/r1-S2GOF;S2GOF; n=3). These animals appeared similar to mes/r1-S2GOF;F8 mutants in group I (not shown). Thus, expressing a second copy of the recombined Spry2-GOF allele appeared to inhibit FGF signaling to approximately the same extent as removing one copy of the Fgf8 gene in mes/r1-S2GOF animals. The phenotype of the group I mes/r1-S2GOF;F8 and mes/r1-S2GOF;S2GOF mutants is similar to that observed in embryos homozygous for Fgf8neo (Chi et al., 2003), a hypomorphic allele of Fgf8, that has been estimated to express Fgf8 RNA at approximately 40% of the level in wild-type embryos (Meyers et al., 1998). Unlike Fgf8neo/neo animals, which have numerous developmental abnormalities caused by reduced FGF8 signaling in all tissues and die at birth, several mes/r1-S2GOF;F8 and mes/r1-S2GOF;S2GOF mutants survived to adulthood. Thus, we were able to determine the postnatal consequences of the midbrain and cerebellum defects that were detected just before birth. In P21 mes/r1-S2GOF;F8 animals, a complete loss of vermis was readily observed in intact brains, and the cerebellar hemispheres appeared slightly abnormal (Fig. 3L,M). Consistent with the known function of the vermis in controlling posture and locomotion, the mes/r1-S2GOF;F8 mutants exhibited a widened gait and pronounced ataxia that increased in severity with age (not shown).
Cell death occurs in the anterior mesencephalon in Spry2-GOF mutants and Fgf8 hypomorphs
We have previously shown that eliminating Fgf8 function in mes/r1 causes extensive cell death between the 11–28 somite stages, resulting in complete absence of the Mb, isthmus, and cerebellum (Chi et al., 2003). However, the effects of only moderately reducing the level of FGF gene expression on cell survival in mes/r1 were not examined. We therefore sought to determine whether abnormal cell death could account for the tissue loss in embryos that were homozygous for Fgf8neo or that expressed the recombined Spry2-GOF transgene, by staining with LysotrackerT (see Materials and Methods) at 4 - 6 hour intervals between E8.75 and E10.75 (12 to 38 som).
We detected a relatively small domain of abnormal cell death in Fgf8neo/neo and mes/r1-S2GOF mutants, but only at the 15-16 som and 18-20 som stages, respectively. A similar domain of abnormal cell death was observed at 18-20 som in mes/r1-S2GOF;F8 mutants, in which the level of FGF signaling is lower than in mes/r1-S2GOF mutants. Surprisingly, given that the anterior midbrain appears to develop normally in all these mutants (Fig. 2A,B, 3C-E and Chi et al., 2003), the domain of abnormal cell death was localized at the anterior end of the dorsal mes (Fig. 4A-E, and data not shown). To confirm the location of this abnormal cell death, we performed an in situ hybridization assay on the embryos labeled with Lysotracker using a probe for Pax6. This gene is expressed throughout the prospective forebrain, with the posterior limit of its expression domain defining the boundary between the developing diencephalons (di) and mes. The domain of abnormal cell death, as detected by Lysotracker staining, was indeed localized immediately posterior to the di/mes boundary (Fig. 4C,C', and data not shown). Importantly, in these assays we detected no abnormal cell death in the prospective posterior midbrain or cerebellum of the mes/r1-S2GOF and mes/r1-S2GOF;F8 mutants (Fig. 4A-C, and data not shown), suggesting that the loss of IC, isthmus, and vermis in these mutants is not due to cell death between E8.75 and E11.5.
Figure 4. Reducing FGF signaling from the isthmic organizer results in cell death in the anterior mesencephalon.

(A-E) Lateral views of embryos of the genotypes indicated, collected at the somite stages denoted and labeled with Lysotracker Red (LysoT), which marks regions in which dead cells are abundant. The approximate position of the isthmic constriction (Is) is indicated. (C') The embryo shown in panel C was fixed after labeling with LysoT and assayed by RNA in situ hybridization for Pax6 expression, which is detected throughout the developing forebrain, with a posterior limit at the border between the prospective forebrain (diencephalon; Di) and midbrain (mesencephalon, Mes) (yellow dotted line). Note that in control embryos (panels A and D), LysoT labeling is detected where neural crest (NC) is migrating into the first branchial arch (BA1), but there is relatively little labeling in the neuroepithelium. In contrast, in the mutant embryos shown in panels B and C, there is extensive LysoT labeling in the mes (white arrows).
The roof plate is expanded in anterior rhombomere 1 in mes/r1-S2GOF;F8 mutants
An alternative explanation for the loss of vermis in mes/r1-S2GOF;F8 mutants was suggested by an analysis of transverse sections through anterior r1 at E11.5 (see legend to Fig. 5A,B). In control embryos, the roof plate (dorsal midline) was characteristically thin, i.e. only 2-3 cell layers deep, across a medial-lateral domain 5-8 cell diameters in width (Fig. 5A,A'). In contrast, in mes/r1-S2GOF;F8 mutants at a comparable anterior-posterior (A-P) level, the roof plate was similarly thin but much wider than normal (Fig. 5B,B'). This region in the mutants appeared similar in width to the roof plate in more posterior sections of control and mutant embryos (see legend to Fig. 5C,D), where it comprised a single cell layer that was ∼30 cell diameters wide (Fig. 5C-D').
Figure 5. The roof plate is expanded and Grem1 expression is reduced in mes/r1-S2GOF;F8 embryos.
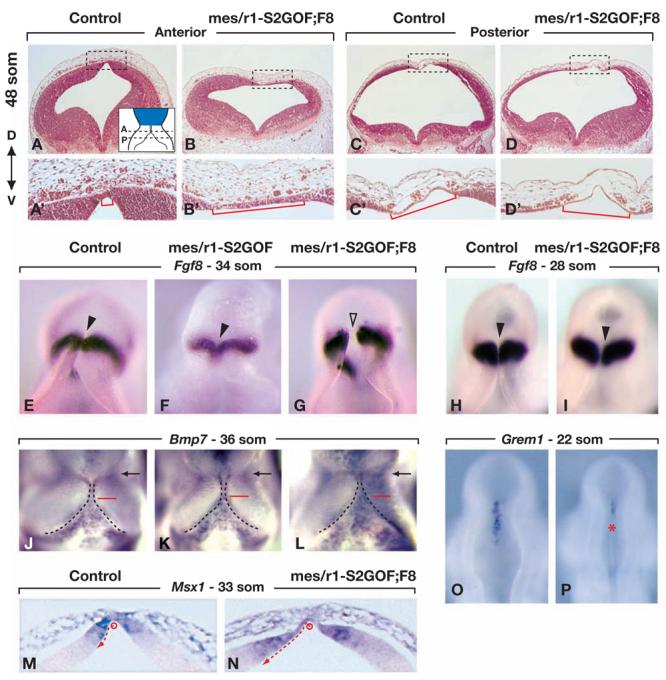
(A-D') Transverse sections through r1 of a control and a mes/r1-S2GOF;F8 embryo at 48 som (E11.75), stained with hematoxylin and eosin. Every third or fourth section in the series was assayed by RNA in situ hybridization with a probe for Otx2, to locate the posterior limit of the mesencephalon (not shown). The approximate levels at which the anterior (A) and posterior (P) sections were cut, i.e, within 40-100 and 90-150 μm of the posterior limit of the Otx2 expression domain (indicated in blue), respectively, are illustrated by dashed lines in the inset in panel A. The sections in all panels are shown at the same magnification. The regions demarcated by dashed boxes in panels A-D are shown at four-fold higher magnification in panels A'-D', respectively. The medial-lateral width of the dorsal midline domain in which the neuroepithelium is only two or three cell layers thick is demarcated by red brackets (panels A'-D'). (E-L, O, P) RNA in situ hybridization assays in whole mount using the probes indicated. Dorsal views of embryos of the genotypes indicated, collected at the stages denoted. Anterior is at the top, posterior at the bottom. (E-I) The roof plate that bisects the Fgf8 expression domain is indicated by arrowheads. In control and mes/r1-S2GOF embryos, the Fgf8-negative roof plate is difficult to discern; it is considerably expanded medial-laterally in the mes/r1-S2GOF;F8 embryo (open arrowhead in panel G). (J-L) The isthmic constriction at the mes/r1 boundary is indicated by a black arrow. In all three panels, a red line is drawn at the same distance posterior to the isthmic constriction. The dashed lines demarcate the lateral edges of the roof plate in r1. Note that Bmp7 expression marks the roof plate, and that the roof plate widens closer to the isthmic constriction in the mes/r1-S2GOF;F8 embryo than in the control and mes/r1-S2GOF embryos. (M,N) Transverse sections through anterior r1 at approximately the same A-P level as in panels A and B, hybridized with a probe for Msx1. The medial-lateral extent of the Msx1 expression domain from the center of the roof plate (open circle) is indicated by a dashed red arrow. (O,P) Grem1 expression is readily detected at the dorsal midline of r1 in the control embryo, but is barely detected in r1 of the mes/r1-S2GOF;F8 embryo (red asterisk).
To corroborate the conclusion from these histological observations that the roof plate was expanded in anterior r1 of mes/r1-S2GOF;F8 mutants, we examined gene expression patterns at earlier stages. At E10.5, the near circular Fgf8 expression domain in anterior r1 is normally interrupted in the small group of roof plate cells at the dorsal midline (Fig. 5E and data not shown). Although the Fgf8 expression pattern appeared almost normal in mes/r1-S2GOF;F8 embryos up to E9.5 (28 som) (Fig. 5H, I), we found that the Fgf8-negative domain in dorsal r1 was much wider than normal by E10.5 (34 som) (Fig. 5G, compare to 5E), supporting the conclusion that the roof plate is expanded in r1.
Unlike Fgf8 expression, which is excluded from the roof plate in r1, Bmp7 expression is a positive marker for the roofplate. Analysis of Bmp7 expression at E10.5 indicated that the roof plate in r1 indeed normally consisted of a narrow domain in anterior r1 that progressively widened towards posterior r1 (Alder et al., 1999) (Fig. 5J), and the same was the case in mes/r1-S2GOF embryos at this stage (Fig. 5K). In contrast, in mes/r1-S2GOF;F8 embryos, a wider Bmp7-positive roof plate domain was observed in anterior r1 (Fig. 5L, compare to 5J and K).
Together these data provide strong evidence that the roof plate had expanded laterally in anterior r1 in mes/r1-S2GOF;F8 embryos. Fate mapping studies in the mouse embryo have localized vermis progenitors to a small domain flanking the dorsal midline in the anterior-most region of r1 at E12.5, and there is evidence that at earlier stages these cells are likewise localized close to the dorsal midline (Sgaier et al., 2005). Thus, it appears that the region containing the vermis progenitors, which is normally positive for Fgf8 and negative for Bmp7 expression, has been replaced by Fgf8-negative, Bmp7-positive roof plate cells in mes/r1-S2GOF;F8 embryos. These results indicate that reducing FGF signaling to the level attained in mes/r1-S2GOF;F8 embryos causes an expansion of the roof plate between the 28 and 34 som stages, possibly at the expense of vermis progenitors.
BMP target gene expression is increased and BMP antagonist gene expression is decreased in mes/r1-S2GOF;F8 mutants
Previous studies have indicated that roof plate cells express several BMP family members in addition to Bmp7, and that BMP signaling is necessary and sufficient for roof plate development in the chick neural tube (Lee and Jessell, 1999; Chizhikov and Millen, 2004b; Chizhikov and Millen, 2004a; Liu et al., 2004). The transcription factor gene Msx1 is a downstream target of BMP signaling in the dorsal neural tube, and misexpression of Msx1 in the chick spinal cord results in expansion of the roof plate (Liu et al., 2004). Therefore, to determine whether excess BMP signaling might be responsible for the expansion of the roof plate that we observed in mes/r1-S2GOF;F8 embryos at E10.5, we assayed for Msx1 expression as a readout for BMP signaling in dorsal r1. The domain of Msx1 expression was indeed significantly expanded medial-laterally in anterior r1 in mes/r1-S2GOF;F8 embryos as compared with control embryos at 33 som (Fig. 5M,N). These data support the hypothesis that a decrease in FGF signaling in r1 results in an increase in BMP signaling and expansion of the roof plate.
In considering how FGF signaling in anterior r1 could function to inhibit BMP signaling, we investigated the possibility that it might induce or maintain the expression of genes that encode secreted BMP antagonists. One such gene is Gremlin1 (Grem1) (Hsu et al., 1998), which has been reported to be expressed in the roof plate of anterior r1 (Pearce et al., 1999; Louvi et al., 2003). We found that in control embryos at E9.25 (22 som), Grem1 RNA was readily detected in the roof plate and adjacent neuroepithelium (Fig. 5O, and data not shown), whereas in mes/r1-S2GOF;F8 embryos Grem1 expression was barely detected in anterior r1 (Fig. 5P). In contrast, Grem1 expression was only moderately reduced in mes/r1-S2GOF embryos (not shown). These results suggest that a normal function of FGF signaling from the IsO is to inhibit BMP signaling in dorsal r1 via a positive effect on Grem1 expression.
DISCUSSION
In this study, we produced the equivalent of an FGF loss-of-function allelic series specifically in mes/r1 and examined the effects of progressively reducing FGF signaling on midbrain and cerebellum development. By recombining one copy of a conditional Spry2 gain-of-function allele throughout mes/r1 at ∼E8.5, we obtained mes/r1-S2GOF mutants with a phenotype similar to that of Fgf17−/−;Fgf8+/− mutants (Xu et al., 2000). When we reduced Fgf8 gene dosage in mes/r1-S2GOF mutants, or produced animals with two copies of the recombined Spry2-GOF allele, we obtained a more severe phenotype, which resembled that of embryos homozygous for an Fgf8 hypomorphic allele (Fgf8neo) (Chi et al., 2003). Together, these data strongly support the hypothesis that expression of the recombined Spry2-GOF allele results in an inhibition of signaling via FGF receptors rather than other RTKs during midbrain and cerebellum development. A dorsal mes/r1 phenotype similar to that in mes/r1-S2GOF;F8 embryos has also been observed following conditional inactivation of Fgfr1 in mes/r1 (Trokovic et al., 2003), suggesting that the FGF signaling that is affected in the Spry2 gain-of-function mutants is primarily relayed by FGFR1.
The level of FGF signaling attained in mes/r1-S2GOF mutants resulted in loss of the posterior midbrain (IC) and anterior vermis, whereas the lower level attained in mes/r1-S2GOF;F8 and mes/r1-S2GOF;S2GOF mutants additionally caused a loss of more tissue at the posterior end of the midbrain as well as the entire vermis. The results of our analysis provide genetic evidence consistent with the proposal that the processes that shape the midbrain and cerebellum require distinct levels of FGF signaling (Liu et al., 1999; Sato et al., 2001; Liu et al., 2003), and further reveal that anatomically and functionally distinct regions within the midbrain and cerebellum require different levels of FGF signaling for their development. In addition, we present evidence that FGF signaling influences the balance between vermis and roof plate development in anterior r1 via an effect on BMP signaling.
The effects of Spry2 gain-of-function in mes/r1 differ in mouse and chicken embryos
The phenotypes we observed in our mutant mice differ from those reported in chicken embryos, in which ectopic expresssion of Spry2 caused cells in r1 to express Otx2, a marker for the mes, and to develop into midbrain tissue (Suzuki-Hirano et al., 2005). We did observe that Otx2 expression was abnormal in mes/r1-S2GOF;F8 embryos, in that the boundary between Otx2-positive and Otx2-negative cells was not as sharp as in control embryos and there were a few Otx2-positive cells scattered in r1 (not shown). However, we found no evidence that r1 cells took on a midbrain fate in any of the Spry2 gain-of-function mouse embryos we examined. Instead, misexpressing Spry2 in mes/r1 resulted in a failure of vermis development. Although there is evidence that the forced expression of Otx2 in r1 from E8.75 can transform anterior r1 into midbrain tissue in the mouse embryo (Broccoli et al., 1999), there is as yet no direct evidence that mouse r1 can give rise to midbrain tissue when FGF signaling is reduced. The disparity between our findings and those of Suzuki-Hirano et al. (2005) might be due to differences in the methods employed to obtain ectopic Sprouty gene expression, i.e., sustained expression of an inherited transgene in the mouse vs. transient expression of a transgene introduced by electroporation in the chicken embryo. Alternatively, it is possible that the different results reflect differences in the mechanisms that control the level of FGF signaling in mouse vs. chicken brain development.
Loss of the posterior tectum caused by reducing FGF signaling can be explained by death of anterior cells and mis-specification of posterior cells as anterior tectum
A specific loss of the IC, with normal development of the SC, is a feature common to mutants in which FGF signaling is moderately reduced in mes/r1, including Fgf17−/−;Fgf8+/− embryos (Xu et al., 2000), Fgf8neo/neo embryos (Chi et al., 2003), and embryos in which Fgfr1 has been inactivated in mes/r1 (Trokovic et al., 2003), but the mechanism by which this loss occurs is not known. Death of IC progenitors is one possible explanation, since we previously showed that FGF8 is a survival factor for cells in the mes, and that inactivation of Fgf8 in mes/r1 by the 10 somite stage, apoptosis rapidly ensues throughout the mes (Chi et al., 2003). However, cell death was not detected at E9.5 in the mesencephalon when Fgfr1 was inactivated (Trokovic et al., 2003).
Here we show that a moderate reduction in FGF signaling does cause cell death prior to E9.25, but that the dying cells are detected only in the anterior mes. To explain this localization, we propose that there is a minimum level of FGF signaling below which cells in the mes die. In normal embryos, there is sufficient FGF signaling to sustain survival, even of cells that are furthest from the source of FGFs in the IsO (Fig. 6A). However, when FGF signaling is moderately reduced, as for example in mes/r1-S2GOF mutants, only cells close to the IsO attain the level of FGF signaling required for survival. At early stages, when the mes is relatively small, all the cells are sufficiently close to the IsO. But as the mes increases in size and the anterior-most cells in such mutants are displaced progressively further from the IsO, they reach a point at which they are too far from the IsO to attain the level of FGF signaling required for survival, and therefore they die.
Figure 6. A model to explain the phenotypes obtained when FGF signaling is progressively reduced in mes/r1.
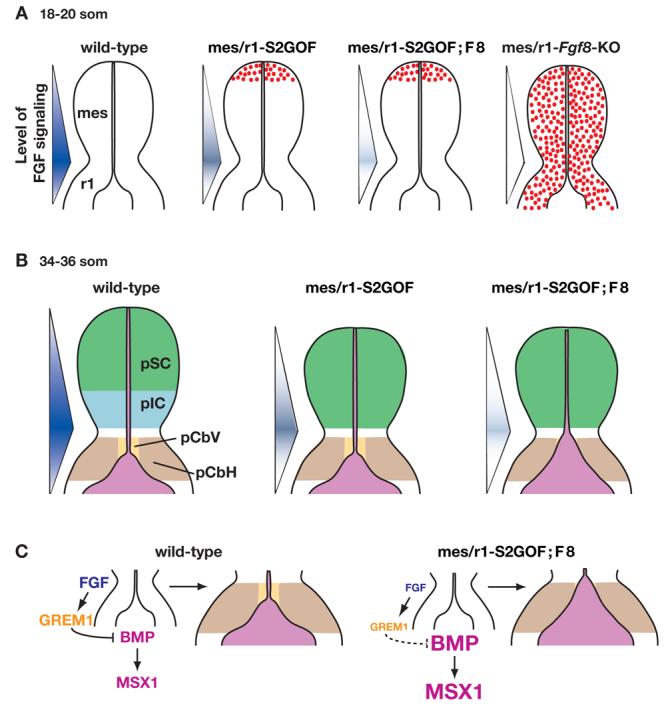
Schematic diagrams illustrate dorsal views of mes/r1 in embryos of the genotypes indicated at the somite stages denoted. The intensity of the blue color in the triangle to the left of a diagram illustrates the level of FGF signaling. (A) The regions in which cells are dying or have already died are indicated by red stippling. (B) The regions that contain the progenitors (p) of the superior colliculus (SC), inferior colliculus (IC), cerebellar vermis (CbV) and cerebellar hemispheres (CbH) are indicated. The phenotypes observed in mutants that attain progressively lower levels of FGF signaling are illustrated. In mes/r1-S2GOF embryos, the anterior mes has been lost, and the surviving posterior cells are specified to an anterior (SC) fate. Consequently the IC does not develop. In mes/r1-S2GOF;F8 embryos, the effects on the mes are similar, and in addition, the vermis fails to develop because roof plate (RP) expansion in r1 results in a loss of CbV progenitors. (C) In the wild-type embryo, FGF signaling induces/maintains the expression of Grem1, which functions to inhibit BMP signaling and expression of the BMP effector MSX1. This pathway maintains the normal balance of vermis and roof plate development. In the mes/r1-S2GOF;F8 embryo, the reduction in the level of FGF signaling results in a severe reduction in Grem1 expression. In the absence of this antagonist, the level of BMP signaling and therefore MSX1 expression increases and an expanded roof plate develops where the vermis progenitors normal reside.
The observation that cell death is restricted to the anterior mes suggests the following explanation for the loss of the IC in mutants with reduced FGF signaling in the mes: A-P cell fates have not yet been determined in the mes at the stage when the cells are dying, and the remaining posterior cells are subsequently specified as SC due to the low level of FGF signaling. Consistent with this hypothesis, it has been shown that fate changes can occur in explants of E9.5 mouse midbrains (Liu et al., 1999). Presumably, if cells in the anterior mes had died at a stage after the fate of all mes cells had been determined, a normal SC would not have formed. This model further suggests that specification of IC progenitors requires a higher level of FGF signaling than specification of SC progenitors. This idea is supported by data showing that increasing FGF signaling, by inserting beads loaded with FGF protein in the anterior mes of chicken embryos, induces anterior cells to take on a posterior fate (Martinez et al., 1999; Shamim et al., 1999).
Loss of the vermis caused by reducing FGF signaling can be explained by an increase in BMP signaling and expansion of the roofplate
A loss of the entire vermis is also observed when FGF signaling is reduced to a level lower than in Fgf17−/−;Fgf8+/− embryos and in mes/r1-S2GOF mutants (this study; Xu et al., 2000). Our data indicate that abnormal cell death is not responsible for the loss of the vermis in such mutant embryos. Instead, the vermis may be absent because a certain minimum level of FGF signaling is required for specification and/or expansion of vermis progenitors (Liu et al., 2003), and that level is not attained in such mutants. Another possibility is based on our observation that the roof plate in r1 is abnormally expanded in mes/r1-S2GOF;F8 embryos. Although this abnormality might be secondary to a failure of vermis development, we favor the hypothesis that the converse is true, and that the observed expansion of the roof plate is the primary cause of loss of the vermis (Fig. 6B).
The model we advocate is based on studies showing that in the spinal cord, the roof plate forms in response to BMP signaling from the adjacent epidermal ectoderm (Lee and Jessell, 1999; Chizhikov and Millen, 2004b; Chizhikov and Millen, 2005) and that BMP signaling can be inhibited by FGF signaling in numerous developmental settings, including the forebrain (Storm et al., 2003) and midbrain (Alexandre et al., 2006). We propose that in r1, roof plate formation is likewise controlled by BMP signaling from the ectoderm, which in turn is inhibited by FGF signaling from the IsO. According to this hypothesis, when FGF signaling is sufficiently reduced, as in mes/r1-S2GOF;F8 embryos, BMP signaling increases and stimulates roof plate development (Fig. 6C). In support of this idea, we found that in mes/r1-S2GOF;F8 embryos, BMP signaling is increased, as evidenced by an expansion of the Msx1 expression domain. Moreover, we observed that a dramatic reduction in expression of the BMP antagonist Grem1 precedes abnormal expansion of the roof plate by approximately 12 hours, suggesting a molecular mechanism by which FGF signaling could exert a negative effect on BMP signaling. However, GREM1 alone is unlikely to be the sole factor responsible for mediating the proposed inhibitory effect of FGF signaling on BMP signaling and roof plate expansion in r1, because we found that roof plate development appears normal in Grem1 null embryos (not shown).
An important question is how might the expansion of the roof plate observed in mes/r1-S2GOF;F8 mutants compromise vermis development? One possibility is based on the fact that the roof plate itself expresses BMPs (Alder et al., 1999; Lee and Jessell, 1999; Alexandre et al., 2006), and an increase in the size of the cell population producing these potent signaling molecules might in turn cause the nearby vermis progenitors to slow their proliferation or differentiate prematurely, leading ultimately to absence of the vermis (Alder et al., 1999; Krizhanovsky and Ben-Arie, 2006; Machold et al., 2007). Alternatively, since misexpression of the BMP effector MSX1 results in the conversion of spinal cord neuroepithelum into roof plate (Liu et al., 2004), it is possible that the increase in Msx1 expression in mes/r1-S2GOF;F8 mutants functions to stimulate roof plate development at the expense of the vermis by converting vermis progenitors to a roof plate fate. Further studies will be needed to distinguish between these possibilities.
Concluding remarks
Based on our analysis of the phenotypes and gene expression patterns in a set of mutants with reduced FGF signaling in mes/r1, we provide evidence that different levels of FGF signaling are required for the development of specific regions within the embryonic midbrain and cerebellum. In particular, our data indicate that posterior midbrain (IC) development requires a higher level of FGF signaling than is necessary for anterior midbrain (SC) or medial cerebellum (vermis) development. It is tempting to speculate that such differences underlie the anatomical variations between species. For example, future studies may reveal that the chicken tectum consists mainly of superior colliculus because the level of FGF signaling in the chicken mesencephalon is not high enough to support IC development.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Drs. George Minowada, who produced the Spry2-GOF mouse lines and initiated the studies described here, and Benjamin Yu for helpful suggestions. We are grateful to P. Ghatpande and Monica Rodenas for excellent technical assistance. We also thank Drs. Roy Sillitoe and Richard Wingate, and our laboratory colleagues for helpful comments on the manuscript. This work was supported by grants from the Wellcome Trust (080470) to M.A.B. and (072111) to M.A.B, and I.M, the Medical Research Council and a Leverhulme Trust Fellowship to I.M., the EU research program (LSHG-CT-2004-512003 and MEIF-CT-2006-025154), the Spanish Science Program (MEC BFU2005-09085, RD06/0011/0012), the ELA Foundation Research and TV3 LA (MARATO-062232) to D.E. and S.M., and the National Institutes of Health (R01 HD050767) to A.L.J and (R01 CA78711) G.R.M.
REFERENCES
- Alder J, Lee KJ, Jessell TM, Hatten ME. Generation of cerebellar granule neurons in vivo by transplantation of BMP-treated neural progenitor cells. Nat Neurosci. 1999;2:535–540. doi: 10.1038/9189. [DOI] [PubMed] [Google Scholar]
- Alexandre P, Bachy I, Marcou M, Wassef M. Positive and negative regulations by FGF8 contribute to midbrain roof plate developmental plasticity. Development. 2006;133:2905–2913. doi: 10.1242/dev.02460. [DOI] [PubMed] [Google Scholar]
- Broccoli V, Boncinelli E, Wurst W. The caudal limit of Otx2 expression positions the isthmic organizer. Nature. 1999;401:164–168. doi: 10.1038/43670. [DOI] [PubMed] [Google Scholar]
- Calmont A, Wandzioch E, Tremblay KD, Minowada G, Kaestner KH, Martin GR, Zaret KS. An FGF Response Pathway that Mediates Hepatic Gene Induction in Embryonic Endoderm Cells. Dev Cell. 2006;11:339–348. doi: 10.1016/j.devcel.2006.06.015. [DOI] [PubMed] [Google Scholar]
- Chi CL, Martinez S, Wurst W, Martin GR. The isthmic organizer signal FGF8 is required for cell survival in the prospective midbrain and cerebellum. Development. 2003;130:2633–2644. doi: 10.1242/dev.00487. [DOI] [PubMed] [Google Scholar]
- Chizhikov VV, Millen KJ. Control of roof plate formation by Lmx1a in the developing spinal cord. Development. 2004a;131:2693–2705. doi: 10.1242/dev.01139. [DOI] [PubMed] [Google Scholar]
- Chizhikov VV, Millen KJ. Mechanisms of roof plate formation in the vertebrate CNS. Nat Rev Neurosci. 2004b;5:808–812. doi: 10.1038/nrn1520. [DOI] [PubMed] [Google Scholar]
- Chizhikov VV, Millen KJ. Roof plate-dependent patterning of the vertebrate dorsal central nervous system. Dev Biol. 2005;277:287–295. doi: 10.1016/j.ydbio.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Crossley PH, Martin GR. The mouse Fgf8 gene encodes a family of polypeptides and is expressed in regions that direct outgrowth and patterning in the developing embryo. Development. 1995;121:439–451. doi: 10.1242/dev.121.2.439. [DOI] [PubMed] [Google Scholar]
- Crossley PH, Martinez S, Martin GR. Midbrain development induced by FGF8 in the chick embryo. Nature. 1996;380:66–68. doi: 10.1038/380066a0. [DOI] [PubMed] [Google Scholar]
- Echevarria D, Vieira C, Gimeno L, Martinez S. Neuroepithelial secondary organizers and cell fate specification in the developing brain. Brain Res Brain Res Rev. 2003;43:179–191. doi: 10.1016/j.brainresrev.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Friedrich G, Soriano P. Promoter traps in embryonic stem cells: a genetic screen to identify and mutate developmental genes in mice. Genes Dev. 1991;5:1513–1523. doi: 10.1101/gad.5.9.1513. [DOI] [PubMed] [Google Scholar]
- Furthauer M, Reifers F, Brand M, Thisse B, Thisse C. sprouty4 acts in vivo as a feedback-induced antagonist of FGF signaling in zebrafish. Development. 2001;128:2175–2186. doi: 10.1242/dev.128.12.2175. [DOI] [PubMed] [Google Scholar]
- Grieshammer U, Cebrian C, Ilagan R, Meyers E, Herzlinger D, Martin GR. FGF8 is required for cell survival at distinct stages of nephrogenesis and for regulation of gene expression in nascent nephrons. Development. 2005;132:3847–3857. doi: 10.1242/dev.01944. [DOI] [PubMed] [Google Scholar]
- Hacohen N, Kramer S, Sutherland D, Hiromi Y, Krasnow MA. sprouty encodes a novel antagonist of FGF signaling that patterns apical branching of the Drosophila airways. Cell. 1998;92:253–263. doi: 10.1016/s0092-8674(00)80919-8. [DOI] [PubMed] [Google Scholar]
- Hsu DR, Economides AN, Wang X, Eimon PM, Harland RM. The Xenopus dorsalizing factor Gremlin identifies a novel family of secreted proteins that antagonize BMP activities. Mol Cell. 1998;1:673–683. doi: 10.1016/s1097-2765(00)80067-2. [DOI] [PubMed] [Google Scholar]
- Inouye M, Oda SI. Strain-specific variations in the folial pattern of the mouse cerebellum. J Comp Neurol. 1980;190:357–362. doi: 10.1002/cne.901900209. [DOI] [PubMed] [Google Scholar]
- Itoh N, Ornitz DM. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004;20:563–569. doi: 10.1016/j.tig.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Kimmel RA, Turnbull DH, Blanquet V, Wurst W, Loomis CA, Joyner AL. Two lineage boundaries coordinate vertebrate apical ectodermal ridge formation. Genes Dev. 2000;14:1377–1389. [PMC free article] [PubMed] [Google Scholar]
- Krizhanovsky V, Ben-Arie N. A novel role for the choroid plexus in BMP-mediated inhibition of differentiation of cerebellar neural progenitors. Mech Dev. 2006;123:67–75. doi: 10.1016/j.mod.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Lagares C, Caballero-Bleda M, Fernandez B, Puelles L. Reciprocal connections between the rabbit suprageniculate pretectal nucleus and the superior colliculus: tracer study with horseradish peroxidase and fluorogold. Vis Neurosci. 1994;11:347–353. doi: 10.1017/s0952523800001681. [DOI] [PubMed] [Google Scholar]
- Lee KJ, Jessell TM. The specification of dorsal cell fates in the vertebrate central nervous system. Annu Rev Neurosci. 1999;22:261–294. doi: 10.1146/annurev.neuro.22.1.261. [DOI] [PubMed] [Google Scholar]
- Lee SM, Danielian PS, Fritzsch B, McMahon AP. Evidence that FGF8 signalling from the midbrain-hindbrain junction regulates growth and polarity in the developing midbrain. Development. 1997;124:959–969. doi: 10.1242/dev.124.5.959. [DOI] [PubMed] [Google Scholar]
- Li JY, Lao Z, Joyner AL. Changing requirements for Gbx2 in development of the cerebellum and maintenance of the mid/hindbrain organizer. Neuron. 2002;36:31–43. doi: 10.1016/s0896-6273(02)00935-2. [DOI] [PubMed] [Google Scholar]
- Liu A, Li JY, Bromleigh C, Lao Z, Niswander LA, Joyner AL. FGF17b and FGF18 have different midbrain regulatory properties from FGF8b or activated FGF receptors. Development. 2003;130:6175–6185. doi: 10.1242/dev.00845. [DOI] [PubMed] [Google Scholar]
- Liu A, Losos K, Joyner AL. FGF8 can activate Gbx2 and transform regions of the rostral mouse brain into a hindbrain fate. Development. 1999;126:4827–4838. doi: 10.1242/dev.126.21.4827. [DOI] [PubMed] [Google Scholar]
- Liu Y, Helms AW, Johnson JE. Distinct activities of Msx1 and Msx3 in dorsal neural tube development. Development. 2004;131:1017–1028. doi: 10.1242/dev.00994. [DOI] [PubMed] [Google Scholar]
- Louvi A, Alexandre P, Metin C, Wurst W, Wassef M. The isthmic neuroepithelium is essential for cerebellar midline fusion. Development. 2003;130:5319–5330. doi: 10.1242/dev.00736. [DOI] [PubMed] [Google Scholar]
- Lu P, Minowada G, Martin GR. Increasing Fgf4 expression in the mouse limb bud causes polysyndactyly and rescues the skeletal defects that result from loss of Fgf8 function. Development. 2006;133:33–42. doi: 10.1242/dev.02172. [DOI] [PubMed] [Google Scholar]
- Machold RP, Kittell DJ, Fishell GJ. Antagonism between Notch and bone morphogenetic protein receptor signaling regulates neurogenesis in the cerebellar rhombic lip. Neural Develop. 2007;2:5. doi: 10.1186/1749-8104-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez S, Crossley PH, Cobos I, Rubenstein JL, Martin GR. FGF8 induces formation of an ectopic isthmic organizer and isthmocerebellar development via a repressive effect on Otx2 expression. Development. 1999;126:1189–1200. doi: 10.1242/dev.126.6.1189. [DOI] [PubMed] [Google Scholar]
- Mason JM, Morrison DJ, Basson MA, Licht JD. Sprouty proteins: multifaceted negative-feedback regulators of receptor tyrosine kinase signaling. Trends Cell Biol. 2006;16:45–54. doi: 10.1016/j.tcb.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Meyers EN, Lewandoski M, Martin GR. An Fgf8 mutant allelic series generated by Cre- and Flp-mediated recombination. Nat Genet. 1998;18:136–141. doi: 10.1038/ng0298-136. [DOI] [PubMed] [Google Scholar]
- Minowada G, Jarvis LA, Chi CL, Neubuser A, Sun X, Hacohen N, Krasnow MA, Martin GR. Vertebrate Sprouty genes are induced by FGF signaling and can cause chondrodysplasia when overexpressed. Development. 1999;126:4465–4475. doi: 10.1242/dev.126.20.4465. [DOI] [PubMed] [Google Scholar]
- Nakamura H, Katahira T, Matsunaga E, Sato T. Isthmus organizer for midbrain and hindbrain development. Brain Res Brain Res Rev. 2005;49:120–126. doi: 10.1016/j.brainresrev.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Olsen SK, Li JY, Bromleigh C, Eliseenkova AV, Ibrahimi OA, Lao Z, Zhang F, Linhardt RJ, Joyner AL, Mohammadi M. Structural basis by which alternative splicing modulates the organizer activity of FGF8 in the brain. Genes Dev. 2006;20:185–198. doi: 10.1101/gad.1365406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partanen J. FGF signalling pathways in development of the midbrain and anterior hindbrain. J Neurochem. 2007;101:1185–1193. doi: 10.1111/j.1471-4159.2007.04463.x. [DOI] [PubMed] [Google Scholar]
- Pearce JJ, Penny G, Rossant J. A mouse cerberus/Dan-related gene family. Dev Biol. 1999;209:98–110. doi: 10.1006/dbio.1999.9240. [DOI] [PubMed] [Google Scholar]
- Raible F, Brand M. Divide et Impera--the midbrain-hindbrain boundary and its organizer. Trends Neurosci. 2004;27:727–734. doi: 10.1016/j.tins.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Sato T, Araki I, Nakamura H. Inductive signal and tissue responsiveness defining the tectum and the cerebellum. Development. 2001;128:2461–2469. doi: 10.1242/dev.128.13.2461. [DOI] [PubMed] [Google Scholar]
- Sgaier SK, Millet S, Villanueva MP, Berenshteyn F, Song C, Joyner AL. Morphogenetic and cellular movements that shape the mouse cerebellum; insights from genetic fate mapping. Neuron. 2005;45:27–40. doi: 10.1016/j.neuron.2004.12.021. [DOI] [PubMed] [Google Scholar]
- Shamim H, Mahmood R, Logan C, Doherty P, Lumsden A, Mason I. Sequential roles for Fgf4, En1 and Fgf8 in specification and regionalisation of the midbrain. Development. 1999;126:945–959. doi: 10.1242/dev.126.5.945. [DOI] [PubMed] [Google Scholar]
- Sillitoe RV, Joyner AL. Morphology, Molecular Codes, and Circuitry Produce the Three-Dimensional Complexity of the Cerebellum. Annu Rev Cell Dev Biol. 2007;23:549–577. doi: 10.1146/annurev.cellbio.23.090506.123237. [DOI] [PubMed] [Google Scholar]
- Storm EE, Rubenstein JL, Martin GR. Dosage of Fgf8 determines whether cell survival is positively or negatively regulated in the developing forebrain. Proc Natl Acad Sci U S A. 2003;100:1757–1762. doi: 10.1073/pnas.0337736100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Mariani FV, Martin GR. Functions of FGF signalling from the apical ectodermal ridge in limb development. Nature. 2002;418:501–508. doi: 10.1038/nature00902. [DOI] [PubMed] [Google Scholar]
- Sun X, Meyers EN, Lewandoski M, Martin GR. Targeted disruption of Fgf8 causes failure of cell migration in the gastrulating mouse embryo. Genes Dev. 1999;13:1834–1846. doi: 10.1101/gad.13.14.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki-Hirano A, Sato T, Nakamura H. Regulation of isthmic Fgf8 signal by sprouty2. Development. 2005;132:257–265. doi: 10.1242/dev.01581. [DOI] [PubMed] [Google Scholar]
- Trokovic R, Trokovic N, Hernesniemi S, Pirvola U, Vogt Weisenhorn DM, Rossant J, McMahon AP, Wurst W, Partanen J. FGFR1 is independently required in both developing mid- and hindbrain for sustained response to isthmic signals. Embo J. 2003;22:1811–1823. doi: 10.1093/emboj/cdg169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Liu Z, Ornitz DM. Temporal and spatial gradients of Fgf8 and Fgf17 regulate proliferation and differentiation of midline cerebellar structures. Development. 2000;127:1833–1843. doi: 10.1242/dev.127.9.1833. [DOI] [PubMed] [Google Scholar]
- Zervas M, Blaess S, Joyner AL. Classical embryological studies and modern genetic analysis of midbrain and cerebellum development. Curr Top Dev Biol. 2005;69:101–138. doi: 10.1016/S0070-2153(05)69005-9. [DOI] [PubMed] [Google Scholar]
- Zhang X, Ibrahimi OA, Olsen SK, Umemori H, Mohammadi M, Ornitz DM. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J Biol Chem. 2006;281:15694–15700. doi: 10.1074/jbc.M601252200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.