

Summary
MlotiK1 is a prokaryotic homolog of cyclic nucleotide-dependent ion channels which contains an intracellular C-terminal cyclic nucleotide binding domain (CNB domain). X-ray structures have been solved of the CNB domain in the absence of ligand and bound to cAMP. Both the full-length channel and CNB domain fragment are easily expressed and purified, making MlotiK1 a useful model system for dissecting activation by ligand binding. We have used X-ray crystallography to determine three new MlotiK1 CNB domain structures: a second apo configuration, a cGMP-bound structure, and a second cAMP-bound structure. In combination, the five MlotiK1 CNB domain structures provide a unique opportunity for analyzing, within a single protein, the structural differences between the apo and bound states and the structural variability within each state. With this analysis as a guide, we have probed the nucleotide selectivity and importance of specific residue side chains in ligand binding and channel activation. These data help to identify ligand-protein interactions that are important for ligand-dependence in MlotiK1 and more globally in the class of nucleotide-dependent proteins.
Keywords: Mesorhizobium loti, potassium channel, ligand-dependent gating, cyclic nucleotide, conformational variability
Introduction
Cyclic nucleotides play vital roles in signal transduction, altering the activity of proteins as diverse as protein kinase A and G, the transcription factor CAP, the guanine nucleotide-exchange factor Epac and cyclic nucleotide-regulated ion channels 1. Nucleotide-dependence is conferred by a similar mechanism in each case: the ligand binds to a conserved cyclic nucleotide binding (CNB) domain, which itself undergoes a conformational change that is ultimately propagated to and affects the activity of a catalytic domain 2. Biochemical experiments, particularly with the CAP and protein kinase CNB domains 3; 4; 5, together with the crystal structures of CNB domains bound to nucleotides have led to the identification of specific residues that are functionally critical for ligand binding and activation 6; 7; 8; 9. Yet, determining which interactions and conformational changes are unique to a specific protein, and which are more universally important for the whole set of CNB dependent proteins, remains an important challenge.
The prokaryotic MlotiK1 ion channel is the most recently characterized cyclic nucleotide-dependent protein 10; 11. The cyclic nucleotide-dependent ion channels all share a common architecture: the core transmembrane channel domain with the topology and four-fold symmetry of voltage-gated channels, attached to an intracellular C-terminal CNB domain 12. Eukaryotic cyclic nucleotide-channels are generally studied using electrophysiological approaches, and are either directly activated by cyclic nucleotide (cyclic nucleotide-gated or CNG channels) 13, or have basal activity that is further modulated by cyclic nucleotide binding (hyperpolarization activated cyclic nucleotide-gated or HCN channels) 14. The functional properties of the prokaryotic MlotiK1 are typically studied using a radioactive uptake assay 15; MlotiK1 displays basal activity which is further stimulated by sub-micromolar concentrations of cAMP and cGMP 10; 11.
MlotiK1 has proven particularly useful as a tool for understanding cyclic-nucleotide-dependence, largely because of its potential for structural work 16; 17. The CNB domain is easily purified as an independent soluble construct which binds nucleotide selectively with a relatively high affinity; cAMP actually co-purifies with the soluble CNB domain, but binding is weakened by the introduction of a point mutation, R348A 10. Recently Cukkemane, et al. measured a binding affinity of 100 nM for cAMP for refolded wild type CNB domain 18. We have been unable to confirm this value using equilibrium experiments of native CNB domain, since in our hands the off-rate of nucleotide is immeasurably slow. Importantly, the MlotiK1 CNB domain was the first CNB domain to be crystallized in both a cAMP-bound and an apo form (the apo form was stabilized by the introduction of the mutation R348A) 10. The two structures display the characteristic architecture of other CNB domains, including those of the prokaryotic transcription factor CAP, two regulatory subunits of protein kinase A, Epac and the HCN2 ion channel 6; 7; 8; 9. Both the cAMP-bound and apo MlotiK1 CNB domain structures are organized as dimers in the asymmetric unit and show a ligand binding pocket that is formed by a wide β-roll; the two structures diverge mainly in the position of the helices of the domain. Binding of ligand induces a rearrangement of the four helices on the surface of the β-roll which is thought to be propagated to the channel regions and affect the state of the gate. The comparison between the cAMP-bound and apo structures provides a valuable framework for understanding the physical basis of ligand-dependence in cyclic nucleotide-dependent proteins, however, as there is only a single structure in each conformation, the question remains as to the extent to which the two structures reflect “universal” bound and apo conformations.
In this study, we have used X-ray crystallography to determine three new MlotiK1 CNB domain structures. These include both a cGMP-bound structure, as well as a second apo configuration which is slightly different than that seen with R348A. Comparisons of these crystal structures with each other and with the two original structures were used to guide mutagenesis studies of ligand binding and channel activation. Taken together, these data help to identify ligand-protein interactions that are important for ligand-dependence in MlotiK1 and more globally in the class of nucleotide-dependent proteins.
Results
Nucleotide selectivity of binding and activation of Wild-Type MlotiK1
To fully understand ligand-protein interactions in MlotiK1, we first needed to establish the selectivity of ligand binding and channel activation. Although the high nucleotide affinity of the soluble CNB domain rendered direct equilibrium binding experiments impractical, we were able to examine ligand binding using a crude competition assay. With this approach, purified CNB domain was bound, through a long incubation, to cAMP-immobilized matrix. The protein was then selectively eluted by incubation with different test ligands, and the quantity of eluted protein was assessed by Coomassie staining of SDS-PAGE gels (Figure 1A). We tested a panel of ligands at 3 mM, since our initial trials showed that this concentration of cAMP was sufficient to release a substantial fraction of the bound domain. Among the purine nucleotides, cAMP yielded the most protein, followed by cGMP and cIMP. Both pyrimidine nucleotides, cCMP and cUMP, reproducibly yielded small quantities of CNB domain, while the non-cyclic nucleotides, 5’ AMP and 3’ AMP, failed to elute protein. Although the competition binding assay is not quantitative, it does provide a qualitative measure of the relative binding affinities of the different ligands, since the higher the affinity, the more protein should be eluted. Based on the relative yields observed in Figure 1A, the MlotiK1 CNB domain displays a binding selectivity of cAMP >cGMP >cIMP ≫cCMP ≈cUMP ⋙5’AMP, 3’AMP.
Figure 1. MlotiK1 nucleotide selectivity of binding and activation.
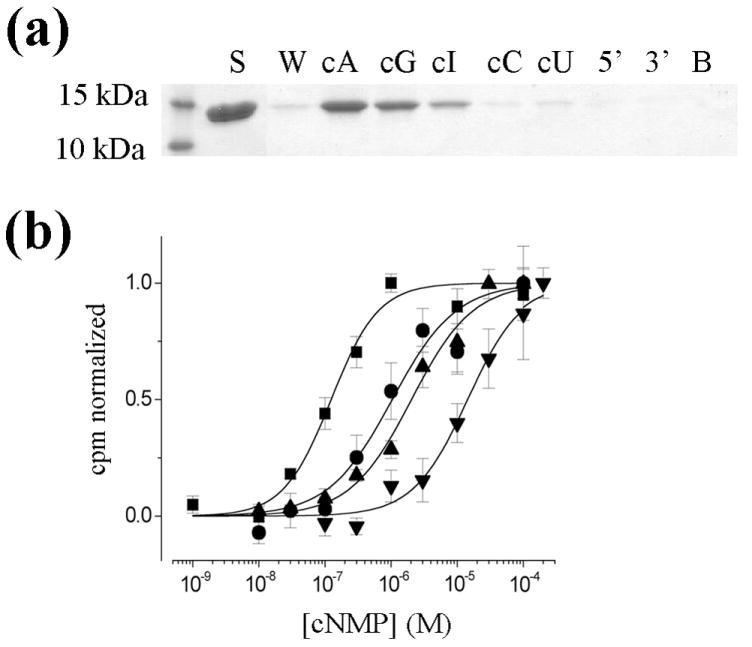
a). Immobilized agarose elution assay. Purified MlotiK1 CNB domain was bound to cAMP-immobilized agarose and 3 mM of the indicated nucleotide was added to the slurry to elute the protein from the agarose. Equal volumes of the elutions were separated by SDS-PAGE and visualized by Coomassie staining. Nucleotides tested were adenosine 3’:5’ cyclic monophosphate (cA), guanosine 3’:5’ cyclic monophosphate (cG), inosine 3’:5’ cyclic monophospate (cI), cytidine 3’:5’ cyclic monophosphate (cC), uridine 3’:5’ cyclic monophosphate (cU), 5’ adenosine monophosphate (5’) and 3’ adenosine monophosphate (3’). S refers to the starting material while W is the wash and B is a buffer-only control. b) Concentration dependence of nucleotide-mediated uptake in wild-type MlotiK1. The various concentrations of cNMPs (1nM-300μM) were added to vesicles reconstituted with MlotiK1. Uptake at 90 min was normalized to basal uptake in the absence of nucleotide (0.0) and the maximal uptake measured for each nucleotide (1.0). Symbols and errors show mean ± SEM for 3-11 independent determinations. Solid lines show Hill fits to the data. Parameters from the fits – cAMP (■): K½ = 110 ± 16 nM and a Hill coefficient (n) of 1.3 ± 0.23; cGMP (●): K½ = 920 ± 109 nM, n = 1.1 ± 0.13; cIMP (▲): K½ = 2.1 ± 0.20 μM, n = 0.95 ± 0.10; cCMP (▼): K½ = 15 ± 3 μM, n = 1.05 ± 0.11.
Since the binding experiment might identify antagonists as well as agonists, we used a functional assay to determine whether similar ligand specificity was observed in channel activation. MlotiK1 activity is studied using a concentrative 86Rb+ flux assay, in which channel-containing proteoliposomes are loaded with high internal [K+], and the resulting diffusion potential is used to drive uptake of 86Rb+ into the vesicles 15. We focused in on the three purine nucleotides and cCMP (as a representative pyrimidine) and examined MlotiK1 activity over a broad range of ligand concentrations (Figure 1B). Both cAMP and cGMP are known to activate MlotiK1; our data show K1/2 values for cAMP and cGMP of 0.11 (±0.02) and 0.92 (±0.11) μM, respectively, closely matching the published work (0.06 and 0.59 μM, respectively, for cAMP and cGMP). Here the third purine ligand cIMP activates MlotiK1 with slightly lower potency (K1/2 of 2.1 μM) than cAMP or cGMP. In addition, the pyrimidine cCMP also activates MlotiK1 with a K1/2 of 15 μM. The activation curves were nicely fitted by a simple 1:1 isotherm and thus showed no obvious signs of cooperativity.
The selectivity of channel activation mirrors that observed in the binding assay: cAMP > cGMP > cIMP > cCMP, and thus indicates that cGMP, cIMP and cCMP are all potential tools for structural studies of MlotiK1 activation.
Wild type MlotiK1 CNB domain in complex with cGMP
Obtaining crystallographic quantities of MlotiK1 CNB domain in complex with a second ligand presented a technical hurdle, as the cAMP that co-purifies with the domain is not easily dialyzed away. We applied the matrix-based approach described above to exchange cAMP, generating CNB domain bound to either cGMP, cIMP or cCMP. Although the yield was low, the resulting protein was free of residual cAMP as judged with a modified cell-based cAMP-specific detection system (see Methods). We attempted crystallization of the cGMP, cIMP and cCMP-bound CNB domains, and while all three yielded crystals, we obtained well-diffracting crystals of only the cGMP-exchanged protein.
We solved a 2.35 Å resolution structure of the wild type CNB domain bound to cGMP using molecular replacement with the wild type cAMP-bound structure as the search molecule (Table 1). Overall, the new structure is nearly identical to the cAMP-bound structure and, as in the original structures, there are two molecules in the asymmetric unit. No major changes were observed in either the positions of the protein backbone or side chains (RMS deviation between all α-carbons is 0.2 Å). Like our previous cAMP-bound structure of the MlotiK1 CNB domain, the ligand binding pocket of the new structures has the β-roll typical of CNB domains, and the αC-helix “lid” is closed over the binding pocket (Figure 2A). A ligand is bound within the pocket and the density of the purine moiety shows two clear lobes, as expected for the purine ring N2 and O6 groups of cGMP (Figure 2B). The relative rotation of the glycosidic bond (bond between ribose and base) places cGMP in the syn conformation, whereas cAMP is bound in the anti conformation in the original wild type structure 10 (Figure 2 C and 3A).
Table 1.
Summary of crystallographic data and refinement statistics.
| cGMP-bound wild type | R307E | R307W | |
|---|---|---|---|
| Space Group | P21 | P21 | P21212 |
| Resolution (Å) | 50.0-2.35 | 50.0-2.20* | 41.09-2.90 |
| Rwork (%) | 20.9 | 20.2* | 26.0 |
| Rfree (%) | 25.6 | 22.0* | 30.6 |
| Rmsd bond length (Å) | 0.009 | 0.01 | 0.006 |
| Rmsd bond angle (°) | 1.3 | 1.2 | 0.95 |
| Completeness (%) | 90.0 (94.0) | 89.9(96.0) * | 99.3 (97.7) |
| Rsym (%) | 8.0 (7.9) | 7.3 (28.4) | 6.6 (29.6) |
| I/Sig I | 10.0 (7.0) | 8.0 (2.3) | 24.0 (5.3) |
| Redundancy | 3.3 (3.2) | 4.1(2.9) | 4.8 (4.6) |
During refinement data included reflections to 2.0 Å. Due to outer shell incompleteness, refinement statistics and completeness are reported to 2.2 Å only.
Figure 2. Structure of cGMP bound MlotiK1 CNB domain.

a) The fold of the cGMP-bound wild type CNB domain mirrors that which has been observed for other CNB domains. The ligand sits in a β-roll which is topped by the C-helix. b) Structure and electron density map (simulated annealing omit map contoured at 1.0 sigma) of cGMP bound to the CNB domain. c) Overlay of the ligands from both cAMP and cGMP-bound structures. cAMP is in the anti conformation while cGMP is in the syn.
Figure 3. Ligand interactions.

a) Comparison of binding pocket interactions from the cGMP-bound (left) and cAMP-bound (right) structures. b) Dose response relations for S308V (left) and S308A (right) activation by cAMP and cGMP. Solid lines are fits to the Hill equation. For 308V: cAMP (■): K½ = 0.1 ± 0.03 μM, Hill coefficient (n) of 1.1 ± 0.4; cGMP (●): K½ = 1.6 ± 0.2 μM, n = 1.3 ± 0.2. For S308A: cAMP (■): K½ = 0.3 ± 0.1 μM, Hill coefficient (n) of 0.9 ± 0.2; cGMP (●): K½ = 4.1 ± 0.6 μM, n = 1.1 ± 0.1.
The conformational difference between nucleotides in MlotiK1 mirrors that previously observed in the HCN2 crystal structures, which also had cAMP and cGMP in the anti and syn conformations, respectively 9. In the HCN2 domain structures the two ligand conformations are related by a simple rotation around the glycosidic bond; as a result of this flipping, the bases of the two ligands occupy distinct areas of the binding pocket. In MlotiK1, cAMP and cGMP are also related by an equivalent rotation around the glycosidic bond, yet the bases of the two ligands occupy nearly overlapping volumes. Figure 2C shows the relative structures and positions of cAMP and cGMP molecules when their two protein structures are superimposed. In addition to the rotation of the glycosidic bond, the conformation of the ribose group appears to change and, as a consequence, the base occupies nearly the same physical space in the two structures.
Many of the contacts between the protein and the nucleotide are identical in the cAMP and GMP-bound structures. At the cyclic phosphate and ribose moieties for example, the side chain of Glu298 forms a hydrogen bond with the 2’ hydroxyl group of the ribose, while those of Arg307 and Ser308 interact directly with an exocylic oxygen from the cyclic phosphate (Figure 3A). Because the cAMP and cGMP bases occupy the same physical volume, the side chain of Arg348 still appears to stabilize the closure of the αC-helix “lid” through Van der Waal’s contacts with the nucleotide base of cGMP. The interaction surface between the bases and Arg348 is almost identical: the R348 side chain buries ~92 Å2 of the cAMP surface area compared to ~95 Å2 of cGMP.
Although the purine base of cAMP makes only Van der Waals contacts with the protein, cGMP is capable of Van der Waals interactions as well as specific hydrogen bonds with the binding pocket. In particular, the N1 nitrogen of the cGMP purine ring hydrogen bonds with the backbone carbonyl group of Arg348 while the N2 amine forms a hydrogen bond with the hydroxyl group of Ser308 (Figure 3A). A similar interaction was reported in the HCN2 CNB domain-cGMP crystal structure, where Thr592 makes a specific hydrogen bond with the N2 of cGMP. We investigated the functional impact of this interaction in MlotiK1 using the 86Rb+ uptake assay to study two mutant channels, S308A and S308V (Figure 3B). As expected, cAMP-activation was not dramatically altered by either mutation (K1/2 for wild-type, S308A and S308V of 0.11± 0.02, 0.3 ± 0.1 and 0.1 ± 0.03 μM, respectively). Although cGMP sensitivity was somewhat more affected (K1/2 of 0.92 ± 0.11, 1.6 ± 0.2, and 4.1 ± 0.6 μM for wild-type, S308V and S308A, respectively), the mutations had a relatively small effect overall on binding energy (< 1 kcal/ mol).
MlotiK1 CNB domain mutants
The portion of the binding pocket formed by the “phosphate binding cassette” contains highly conserved residues that interact directly with the cyclic phosphate moiety. In particular, one arginine (R307 in MlotiK1) is conserved in each of the CNB domain crystal structures to date and its side chain forms an ionic bond with the cyclic phosphate in each case; mutations of this side chain are known to decrease ligand binding in other CNB domain proteins. We evaluated the biochemical impact of three mutations that should interrupt the ionic interaction R307 has with nucleotide: R307A and R307E, which neutralize or substitute an acidic side chain, respectively, and R307W, which, in addition to being neutral, has increased side chain bulk and might have a steric effect on ligand binding.
All three R307 mutations substantially decreased ligand binding affinity. R307A was easily studied using standard binding studies; a direct binding experiment measured a Kd of 45.1 ± 5.8 μM for the fluorescent cAMP analogue 8-NBD-cAMP (Figure 4A), while a competition binding experiment showed a lower affinity for cAMP (205.1 ± 10.4 μM) (Figure 4B). By comparison, these affinities are over 1000-fold lower than recent analogous measurements on refolded wild-type domain (0.022 and 0.068 μM, respectively, for 8-NBD-cAMP and cAMP). Ligand binding by the other two mutants, R307E and R307W, was even more severely reduced. Both domains displayed negligible binding to 8-NBD cAMP over the dynamic range of the assay (Kd estimated to be > 1.5 mM); the weak 8-NBD-cAMP binding precluded competition assays with unlabelled ligands.
Figure 4. Binding and activation of R307A.

a) Fluorescence enhancement upon R307A CNB domain binding to 8-NBD cAMP. Purified protein was titrated (0 to 1mM) against 2 μM fluorescent ligand and a binding affinity of 45.1 ± 5.8 μM was determined. b) Competition by unlabelled cAMP. 50 μM of pure R307A was incubated with 150 μM 8-NBD cAMP and subsequently competed with unlabelled cAMP (0 to 10 mM). A binding affinity of 205.1 ± 10.4 μM was determined. c) Concentration dependence of nucleotide-mediated uptake as measured with the 86Rb+ flux assay. Uptake at 90 min was normalized to basal uptake in the absence of nucleotide (0.0) and the maximal uptake (1.0). K1/2 Activation of 30 ± 4.9 μM and a Hill coefficient (n) of 1.7 ± 0.4 was determined.
We turned to examine the R307A mutation in the context of the full-length MlotiK1 protein. The R307A mutant channel yielded protein, and its activity remains modulated by cAMP. The R307A mutation has a large effect on the K1/2 of activation, shifting it by 300-fold relative to wild type (K1/2 of 30 and 0.11 μM, for R307A and wild type, respectively) (Figure 4C). Although the qualitative effect of R307A is similar in the flux assay of full-length channel and cAMP binding to the isolated R307A mutant domain (right-shifting both dose-response curves with respect to the wild-type protein, as though destabilizing a bound state(s) relative to unliganded conformation(s)), there is a curious quantitative difference: the mutation had a larger effect on ligand binding than on channel activation (R307A changes the ligand binding Kd by at least 3000-fold, but the K1/2 of activation by only 300-fold). This latter observation has important consequences for the energetics of ligand-dependence in MlotiK1, as detailed in the discussion below. We were unable to obtain sufficient quantities of R307E to test its activity in the flux assay, and the R307W mutant channel displayed only nominal activity under control conditions that was not increased by additional nucleotide.
To better understand the ligand binding properties of the Arg307 mutants, we determined the crystal structures of R307E and R307W domains. R307E crystallized only in the presence of a high cAMP concentration (20 mM). We have solved a 2.2Å resolution structure by molecular replacement using the wild type structure (Table 1). Like in the original wild type structure, the R307E mutant crystals contain 2 molecules in the asymmetric unit and have bound cAMP in the anti conformation. The similarities between the wild type and R307E mutant structures are striking (RMS deviation between all α-carbons is 0.3 Å), with just very small distortions of the binding pocket (less than 1Å shift in the relative position of Cα of PBC residues). This observation demonstrates that mutations of the highly conserved Arg307 in the binding pocket are well tolerated at the structural level, and that changes in ligand binding properties are not the result of severe structural distortions of the bound state. With the exception of the interactions formed with the side chain of residue 307, all molecular interactions between the ligand and the CNB domain are conserved between the two structures. The density of the R307E glutamate side chain is well defined. The side chain is placed close to the phosphate of the cyclic nucleotide (Figure 5A), but the carboxylic group is not within hydrogen bonding distance (>3.5 Å).
Figure 5. Binding pockets of Arg 307 mutant structures.
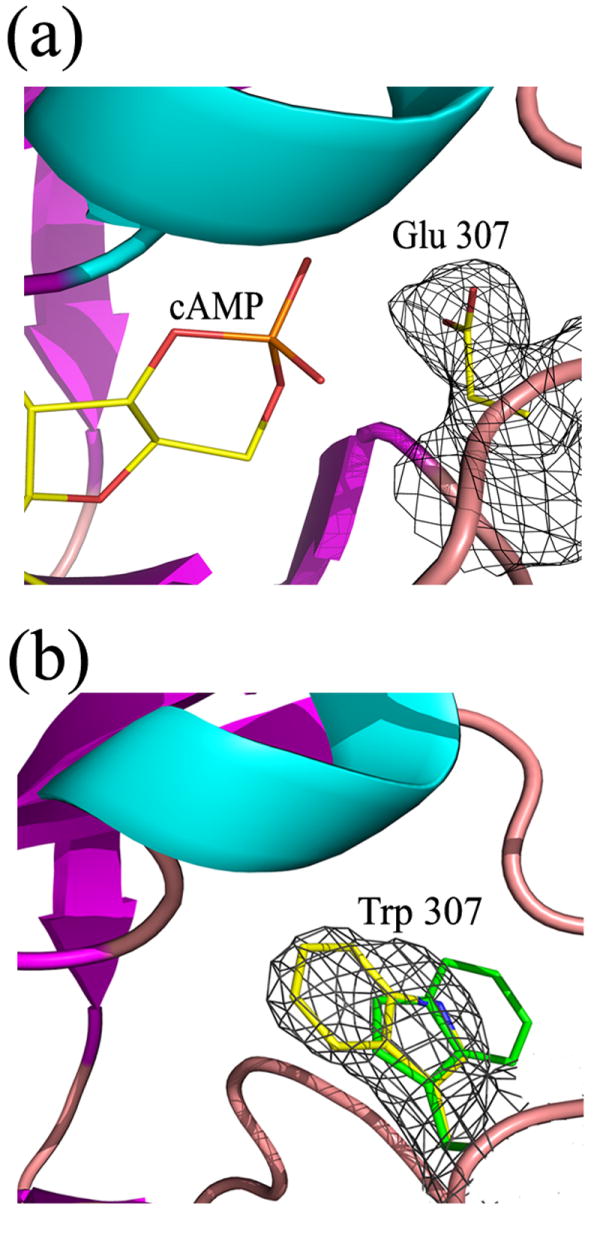
a) Binding pocket of R307E showing the electron density of the glutamate side chain (simulated annealing omit map contoured at 1.0 sigma). b) Binding pocket of R307W with the two tryptophan conformations, as observed in the different molecules of the asymmetric unit, superposed. Electron density (simulated annealing omit map contoured at 1.0 sigma) is shown for just one of the tryptophan side chains. The density map of the other conformer is just as good.
The R307W mutant was crystallized in the absence of cAMP and solved at 2.9 Å resolution by performing a selenomethione multiwavelength anomalous diffraction experiment and then docking and adjusting the R348A mutant structure into the density (Table 1). In these crystals there are 4 molecules per asymmetric unit and, as expected, there is no nucleotide density in the binding pockets. The tryptophan side chain at position 307 adopts two different conformations and appears to reduce ligand affinity by steric hinderance (Figure 5B).
Collection of apo and liganded structures
With the determination of the three new crystal structures we have an extensive collection of structures of the MlotiK1 CNB domain in both the bound and apo states. All of these crystal structures have more than one molecule in the asymmetric unit, each with a unique crystal environment, therefore the molecules are not necessarily identical and can be considered independent determinations. In the bound state we have 6 structures of MlotiK1 CNB domains: 2 structures of the mutant R307E with cAMP, 2 wild type structures with cAMP and 2 more wild type structures with cGMP. We also have 6 different structures for the apo state: the mutants R348A (2 molecules in asymmetric unit) and R307W (4 molecules in the asymmetric unit). This collection of structures provides us with the unique opportunity of analyzing, in the same protein, the differences between states and within states, while controlling for the conformational effects due to crystal contacts and introduction of mutations.
The 12 structures were superimposed over the residue range 250 to 280 and 309 to 323, which spans a large part of the β-roll and does not include the Phosphate Binding Cassette (PBC) region and any of the helices (Figure 6A, 6B and subsequent figures). It is immediately clear that the structures in the bound state (in red) are almost identical, with RMS deviation between different structures varying between 0.15 to 0.3 Å (calculated for the all residues relative to a cAMP-bound wild type structure (subunit A)). In contrast, the structures without nucleotide (in green and gray) are much more diverse, mainly on the αC-helix but also in the disposition of the other helices on the surface of the β-roll and in the position of some of the residues that form the binding pocket. This difference between the two states demonstrates that there is a single, well defined bound state conformation which probably results from the added interactions formed between the ligand and the protein. In the absence of nucleotide, the structure of the domain appears to be much “looser” and capable of adopting different conformations.
Figure 6. Superposition of the entire collection of MlotiK1 CNB domain structures.

Two stereo-views of α-Carbon trace for all structures. All bound structures (R307E, wild type-cAMP and wild type-cGMP) are shown in red while apo structures are in gray (R348A) or green (R307W). Structures were superimposed over the residue range 250 to 280 and 309 to 323. a) View into the mouth of binding pocket. B) Small rotation around vertical axis relative to view in A). Different regions of the molecule are labeled.
The largest differences between the two states are observed in the structural unit formed by αA’-helix/loop/αA-helix, in the αC- and αB-helices, the binding pocket and in the β4-β5 hairpin. In MlotiK1, the αA’-helix/loop/αA-helix unit (αA’-L- αA) is directly connected to the last TM of the channel and is thought to be the major conveyer of the conformational change from the domain to the gate. Direct evidence for the movement of αA’-L- αA in the MlotiK1 CNB domain first came from comparison of the structures of wild type (bound state) and R348A mutant (apo state) domains 10. However, the use of a mutant domain to generate crystals in the apo form raised the possibility that the observed αA’-L- αA movement was actually an artifact of the R348A mutation. The determination of the new apo structures, resulting from a mutation (R307W) in a completely different region of the domain, confirms the shift of the αA’-L- αA unit between apo and bound states (Figure 6A and 7A), and clearly establishes that the rearrangement is induced by ligand binding.
Figure 7. A hydrophobic patch is shielded by the A’- helix/loop/A-helix in the apo structures.

a) Stereo-view of the relative position of the A’-helix/loop/A-helix unit in the bound state (red) and the apo state (green). b) The hydrophobic cluster in the bound state. Residues of the β-roll hydrophobic patch are in yellow. c) The hydrophobic cluster in the apo state. Residues of the β-roll hydrophobic patch are in yellow. The molecules in figures b) and c) were oriented so that residues in yellow superimpose.
Ligand-dependent movement of the αA’-L-αA is associated with remodeling of a hydrophobic cluster formed by residues from the β-roll, the PBC and helices. In the bound state (Figure 7A and 7B), the cluster includes residues on the β-roll (Met272, Phe 274, Phe295, Leu320, Leu322), residues from the αC helix (Ile337 and Phe341) and from the αB helix (Phe327 and Leu330). The cluster reorganizes upon ligand release (Figure 7C): the αC helix shifts away, the residues in the PBC and αB helix relocate and the αA’-L-αA unit moves in, covering the residues on the surface of the β-roll (Figure 6A). αA’-L-αA residues involved in the hydrophobic cluster include Leu229 (αA’-helix), Val233, Leu235, Phe236 and Leu239 (all from the loop), as well as Ile247 (αA-helix) (Figure 7A and 7C). While the αA’-L-αA unit undergoes a large movement (Cα at Phe236 moves 8Å between the wild type and one of the R307W structures), the residues on the β-roll are basically unchanged between the two states (Phe274 Cα shifts just 0.3 Å between one copy of the wild type-cAMP and R307W). In the apo state the hydrophobic cluster also involves two residues that are functionally important and will be discussed in detail below: the PBC residues Leu301, and its αB helix partner Phe327.
The functional importance of the residues in the αA’-L-αA unit, in particular the loop residues, is demonstrated by a 9-fold change in the cAMP-response of the channel (K½ of 0.93 ± 0.02 μM) when Leu235 is mutated to alanine. Moreover, the apolar character of residues in this loop, more specifically Leu235 and Phe236, is conserved in CNB domains.
The importance of the C-terminal helix, αC, in ligand binding and domain function has been well demonstrated in many different CNB domains; in our bound state structures this importance is reflected by a well-defined position. In contrast, in the apo states αC can be in different positions, for example: relative to the wild type liganded position, F341 moves 6 Å in a R348A mutant structure, and 14 Å in a R307W mutant structure (Figure 6A). Interestingly, the αC-helix in some of the apo conformations has partially unwound or became disordered (Figure 8A). The αB-helix, which is directly connected to αC, also moves between the two states (Figure 6B and 8B); for instance, the position of Leu330, a residue in the middle of αB, shifts by 6-8 Å relative to the bound state. However, unlike the C-terminal helix, αB adopts a much better defined position in the apo states (Figure. 8B). This difference between the two tethered helices is most likely due to the extensive and specific contacts established between αB and residues in the β-roll and ligand binding pocket in both states. In contrast, the αC-helix seems to form specific contacts with the rest of the protein only when the ligand is present.
Figure 8. C- and B-helix features in the bound and apo states.
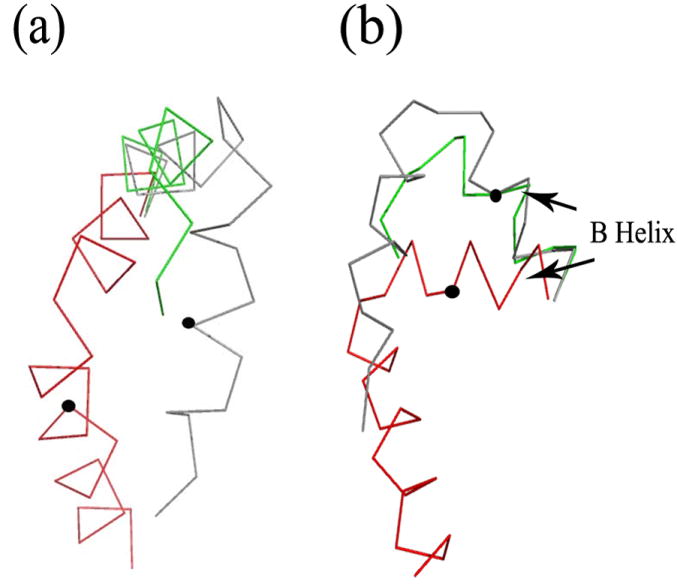
a) Three different αC helix arrangements: cAMP-bound wild type (red), partially unwound (R348A in gray) and disordered (R307W in green). The circle indicates the position of Phe 341, a residue referred in the text (the R307W structure lacks this residue). b) Three examples of the αB helix arrangement emphasizing how it occupies two orientations: bound (wild type in red) and apo (R348A in gray and R307W in green). The circle indicates the position of Leu 330 (see text).
Previous structures show that binding of cyclic nucleotide results in a collapse of the binding pocket around the ligand with the establishment of a network of interactions between ligand and protein. There are very few differences in the positions of the residues of the binding pocket between our bound state structures. As mentioned above, even the mutation of the important arginine (R307E) causes only small distortions in some of the residues in this region. In the absence of ligand, many residues in the binding pocket adopt divergent main chain positions and side-chain conformations (Figure 9A). For example, in the different apo structures the Cα atom of the highly conserved glutamate E298, which hydrogen bonds the sugar ring, is disposed between 1.2 and 2.3 Å away from its position in the bound state. The side chain carboxylic group occupies many different positions, with its carbon atom shifted by as little as 1.2 Å (in one of the R348A mutant domains) or as much as 5.2 Å (in a R307W mutant) relative to its position in the bound state. This extreme position is not the result of the mutation since in another R307W mutant structure the carbon atom of the glutamate carboxylic group is positioned just 1.2 Å relative to the bound state. Clearly, observation of residue movement between bound and apo structures does not necessarily mean that the residue plays a major role in the stability of the complex. For example, we previously described how the side chain of Met299 in the R348A structures has flipped relative to wild type and is accompanied by 1.9 Å shift in the position of the main chain carbon 10. This shift is even more pronounced in the R307W structures, where the main chain distance can be as much as 4.4 Å relative to wild type. However, when we mutated Met299 to Ala in the whole channel there was no change in the activity dependence on cAMP (K½ = 0.109 ± 0.015 μM).
Figure 9. Features of the Binding Pocket.

a) Overlay of the binding pockets (residues 295-312) of two bound: wild type-cAMP (red), R307E (red) and 5 apo: R348A (gray) R307W (green) structures. Glu 298 and Met 299, both highly mobile, are highlighted. The side chains of Leu 301 and position 307 (various mutants) as well as cAMP are shown for reference. b) The positions of Leu 301 and Phe 327 in the bound (tan) and apo structures (green) showing clear differences in the position of the side chains between the two states but not amongst the structures in the individual states. c) The position of the Arg 307 side chain is invariant regardless of state or mutation. Side chain position shown: R307 from cAMP-bound wild type (red), R307 from the R348A apo structure (gray), Glu 307 from the R307E bound structure (cyan) and two different Trp 307s from the R307W apo structure (green). d) Stabilization of position 307 by interactions between its main chain and residues of a parallel loop. Hydrogen bonds between the backbones at Arg 307, Gly 266 and Glu 267 from the cAMP-bound wild type structure are shown.
In the binding pocket region, Leu301 and the conserved arginine Arg307 are exceptions to the structural variability of the apo state. In our structures the side chain of Leu301 occupies fairly well-defined positions in both the apo and bound states (Figure 9B). This binary disposition is also present in Phe327, a residue in the αB helix that is in direct contact with Leu301. The structural and functional importance of this pair of residues was demonstrated in the CNB domain of Epac. The relatively restricted volume occupied by these two residues in both the apo and bound structures is most likely a reflection of their structural and energetic importance as well of their structural connection.
The arginine in the ligand binding pocket (Arg307 in MlotiK1) is conserved across CNB domains. It interacts directly with the ligand by forming an ionic interaction with the cyclic phosphate. Its contribution to the stability of the complex in MlotiK1 CNB domain is manifested by a 3000-fold decrease in affinity when the arginine is mutated to alanine. We were surprised to notice that the position of the side chains at 307 is roughly retained in all structures, independent of state or mutation (Figure 9A and 9C). The two mutations (tryptophan and glutamate) that replace the arginine at position 307 are very different in bulk and charge; it would not have been surprising to see them occupy a different region of the binding pocket. Instead the mutations overlap with the side chain of the arginine in the bound structures. The same happens with Arg307 in the apo state (mutant R348A), the side chain does not move away from the position it occupied when interacting with the ligand.
Analysis of the region surrounding residues at 307 reveals extensive contacts between the side chains at this position and surrounding residues; these contacts are not necessarily the same across the different structures but they probably play a role in restricting the position of the side chains. It is also clear that the conformation and position of the main chain atoms of residue 307 is highly restricted across the different structures (Figure 9A). This imposes strong constraints on the side chain conformation and the region of the binding pocket that it occupies. In all the structures, the main chain carbonyl and amide groups of residue 307 are hydrogen bonded with the amide of Gly266 and carbonyl of Glu267, respectively (Figure 9D). Gly266 and Glu367 are part of the loop between the β2 and β3 strands, which follows a parallel path to the PBC region of the binding pocket; moreover, the glycine at position 266 is conserved in other CNB domains.
Our comparison between all the bound and unbound structures also reveals an interesting conformational change that it was not detected in the original comparison between the wild-type and R348A apo structure. The β4-β5 hairpin is situated at the mouth of the ligand binding pocket and it undergoes a conformational readjustment, closing around the ligand upon binding (Figure 6B and 10A). Residues Val282, Thr284 and Val288 on the hairpin interact with the cAMP molecule, establishing Van der Waal’s contacts with the face of the nucleotide base opposite the Arg348 side-chain (Figure 10B). In the cGMP structures, the flipping of the base to a syn conformation removes the interaction with Val288 (Figure 10C). In the absence of ligand the hairpin is no longer tightly anchored and adopts different conformations (Figure 10A).
Figure 10. Conformational changes in the β4-β5 hairpin.

a) The β4-β5 hairpin is shown in the bound (in red, cAMP-bound wild type and R307E) and apo (R348 in grey and R307W in green) states. b) and c) Van der Waals interactions of the purine base of cAMP (b) or cGMP (c) with Arg 348 and residues in the β4-β5 hairpin.
Discussion
In this work, we explore nucleotide binding and ligand-dependent activation in the MlotiK1 bacterial ion channel using biochemical, functional and crystallographic approaches.
We have shown that MlotiK1 displays the following selectivity among ligands cAMP > cGMP > cIMP > cCMP, for both binding and activation. The selectivity of activation is relatively modest, with a 9-fold difference between cAMP and cGMP and a 21-fold difference between cAMP and cIMP. Eukaryotic cyclic nucleotide-regulated channels show wide variability in their ligand specificity. The HCN channel spIH, for example, is exceptionally selective, preferring cAMP over cGMP by more than 500-fold 19. Other channels such as the CNGA2/CNGA4 native olfactory heterotetramer are largely unselective between cAMP and cGMP 20. MlotiK1 is most reminiscent of the HCN2 channel, in which both cAMP and cGMP modulate channel activity but cAMP is a slightly more effective ligand (10-60 fold difference) 21; 22. Our observation that the pyrimidine nucleotide cCMP activates MlotiK1 channel has precedent in eukaryotic cyclic nucleotide-modulated channels. A recent study indicates that the HCN2 CNB domain is also capable of binding pyrimidines 23, and electrophysiological studies of the native olfactory cyclic nucleotide-dependent channel show that cCMP can act as a agonist 24.
Our structures of wild type MlotiK1 CNB domain in complex with both cGMP and cAMP provide the second case in which both ligands have been crystallized in the same protein 9. As observed for HCN2 CNB domain, the MlotiK1 CNB domain binding pocket has essentially identical configurations when bound to either ligand. Thus, at least for these two ligands, the domain appears to have a single “bound” state and to the extent that ligands have unique effects, it must be through differential stabilization of the bound conformation rather than by inducing unique conformations. Although the protein assumes similar structures, cAMP and cGMP have different conformations: the ligands assume the anti and syn forms, respectively. The difference in geometry means that the nucleotide bases of the two ligands have different interactions with the protein. The purine ring N2 group of cGMP makes a hydrogen bond with the hydroxyl side chain of Ser308. The purine ring of cAMP, in contrast, first does not have an amine group at this position, and second, is located too far to make any direct contact with Ser308. An analogous interaction to that between Ser308 and cGMP is suspected to underlie ligand specificity in other CNB domains 25; 26; the relative importance of this bond in generating cGMP selectivity appears to depend on the precise binding domain 25. In MlotiK1, despite the presence of the hydroxyl-containing side chain Ser308, cAMP activates at lower concentrations than cGMP demonstrating that other factors also affect selectivity. Zagotta, et al. postulate that functional specificity also arises from a hydrogen bond between Arg632 on the αC-helix and the N6 amine of cAMP, but a similar argument cannot be made for MlotK1 where there are no specific hydrogen bonds between the cAMP purine ring and the CNB domain 9.
At least some of the difference in ligand selectivity must reflect the individual energetics of the ligand. Although each nucleotide can physically assume syn and anti configurations, the relative stabilities of the two configurations differ. Computational studies of cyclic nucleotides in solution indicate that cAMP slightly favors the anti conformation (69:31), while cGMP is heavily biased toward the syn orientation (95:5) 27. Structural studies parallel the computational predictions: structures determined through solution NMR show cAMP and cGMP in the anti and syn configurations, respectively 28; 29. The configuration of the nucleotides in our structures exactly mirror their most stable forms in solution and therefore preference to one ligand over the other may depend on the ability of the domain to accommodate the syn or anti conformations. Also, the ligand selectivity observed in MlotiK1 may arise simply because the electronic distribution of cAMP in the anti conformation is better suited to activate the channel than the electronic distribution of cGMP in the syn conformation. If so, future work teasing apart the contributions of various characteristics of the ligands (sugar pucker, anti vs. syn configuration) might ultimately require adding a panel of engineered ligands to the arsenal.
Our parallel studies of binding and activation reveal a particular characteristic of MlotiK1: a large disparity between the Kd of ligand binding (at least to the isolated domain) and the K1/2 of channel activation.
For the R307A mutant, with a weaker binding affinity (205 μM) than K1/2 of activation (30 μM), ligand-dependence is easily described by a simple three state scheme of the sort typically used to model the gating of ligand-dependent ion channels (Scheme I) 30. If we assume the Kd provided by binding experiments reflects the first step of the reaction (ligand binding to domain in the absence of any work required to open the channel), then it together with the K1/2 of activation (reflecting the linked equilibrium of the initial binding and subsequent channel opening steps) can be used to calculate the equilibrium of the second, allosteric, rearrangement. If the final opening transition is heavily biased toward the open state, even sub-Kd concentrations of ligand will drive channel opening; in R307A, the binding and activation data described above are consistent with a closed:open distribution of 1:7.
Scheme 1.

Depiction of ligand-dependent activation mechanism for MlotiK1 channels. In the first step, ligand (cAMP) binds to unbound CNB domain (C) to form liganded but closed channels (C•cAMP). In the second step, the channels undergo an allosteric rearrangement to open (O•cAMP).
Although Scheme I easily accommodates the data from the R307A mutant protein, it cannot account for the data obtained from the wild type MlotiK1, where the Kd for ligand binding is smaller that the K1/2 of channel activation. The paradox presented by the wild type MlotiK1, where high affinity binding is coupled with a comparatively low Kd for activation, requires a different gating scheme. In ligand-dependent proteins, very high ligand affinities are not uncommon when a binding domain is physically uncoupled from its effector domain 31; 32; 33; in theory the energy difference could be harnessed within the effector domain to drive enzymatic catalysis, or in the case of ion channels, within the transmembrane core to promote channel opening. Thus there is hope that, despite the very disparate behaviors of the mutant and wild-type channels, MlotiK1 may turn out to be a tractable system for dissecting the detailed energetics of this type of coupling in future studies, as MlotiK1 clearly displays tight ligand binding and weak ligand activation but is also uniquely amenable to biochemistry, structural and functional approaches.
All of the described crystal forms of the MlotiK1 CNB domain contain more than one molecule in the asymmetric unit: 4 molecules in the R307W mutant and 2 in all the others. Each one of these molecules can be considered as an independent determination and with the three new structures we have created a collection that includes a total of 12 domain structures, 6 in the bound and 6 in the apo state. The dimer interface, relating molecules in the asymmetric unit and originally described in the wild type domain, is retained in all these crystals (in R307W the asymmetric unit contains a dimer of dimers). Therefore, the new structures do not provide any new evidence on the issue of domain organization in the channel. Importantly, our structural analysis at the level of the domain benefits greatly from having a broad panel of structures from the same protein under different conditions: the novel apo state domain structure was attained using a mutation at a site distant from the original R348A mutation, while the two novel liganded structures included both a point mutant as well as a second ligand. These differences provide validation of previous observations as well as new insights into ligand gating. For example, this comparison clearly establishes that the changes within the binding pocket as well as the large movements of the helices on the surface of the MlotiK1 CNB domain result from ligand binding/unbinding.
Another clear conclusion from this analysis is that the bound state has a unique conformation independent of the nature of bound ligand (cAMP versus cGMP) or introduction of a mutation in the binding pocket (R307E). In contrast, in the apo state the domain is looser and explores multiple conformations. For example, the αC helix establishes a single set of interactions with the ligand and the rest of the protein in the bound state while, in the absence of ligand, the helix can move large distances, form interactions with different areas of the domain and even unwind. The comparison also reinforces the importance of the hydrophobic cluster formed by residues from the surface of the β-roll, αC and αB helices, binding pocket and αA’-helix/loop/αA-helix structural unit. Many of the residues in this cluster move upon ligand binding, and consequently the hydrophobic cluster is central in the remodeling of the domain conformation.
The multiplicity of conformations in the apo state is also quite clearly seen in the binding pocket region. However, the functional importance of some of its residues is reflected in a very specific fashion at the structural level. Leu301 (and its partner Phe327 in the αB helix) and Arg307 are exceptional among their immediate neighbors in the binding pocket because they occupy well defined positions in both states. Arg307 directly interacts with the ligand but maintains the same conformation in its absence due to restrictions imposed on its side chain and main chain atoms by neighboring protein regions. The positional restrictions imposed on Arg307 could be important in the interaction between ligand and protein by reducing entropic penalties or favoring the steering of the ligand into the pocket through electrostatic effects. Although Leu301 and Phe327 move upon ligand binding/unbinding their positions are almost fixed within a state and the positional relation between them is retained in both states. This behavior is probably required so that the conformational changes occurring in the binding pocket, and caused by the ligand, can be propagated to the helices on the β-roll surface 8.
Finally, our analysis has revealed the importance of the hairpin formed by the β4 and β5 strands. Previous studies did not notice that the structure of this hairpin is affected by the presence of the ligand. This region, together with the αC helix, forms direct interactions with the nucleotide base, closing over the ligand and shielding it from the solvent.
Materials and Methods
Materials
All materials were reagent grade. The following were obtained from Sigma Aldrich (St. Louis, MO): adenosine 3’:5’ cyclic monophosphate (cAMP), guanosine 3’:5’ cyclic monophosphate (cGMP), inosine 3’:5’ cyclic monophospate (cIMP), cytidine 3’:5’ cyclic monophosphate (cCMP), uridine 3’:5’ cyclic monophosphate (cUMP), 5’ adenosine monophosphate (5’AMP), 3’ adenosine monophosphate (3’AMP) and cAMP-tethered resin (via an 11 atom spacer at position N-6; A7396). Decyl maltoside (DM) was obtained from Anatrace (Maumee, OH). The HitHunter cAMP detection kit (#90-002) was obtained from DiscoverRx (Freemont, CA).
Molecular Biology
The pASK90 vector 34 containing the MlotiK1 gene with a C-terminal His-tag has been described previously 10. The cyclic nucleotide binding domain (residues 216–355) of MlotiK1 was cloned into either pET-15 vector which contains a C-terminal His-tag (Novagen, Madison, WI) for protein used in binding experiments or the GST fusion construct pGEX-2T (Amersham Pharmacia, Piscataway, NJ) for protein used for crystallography. Site-directed mutagenesis was performed using Quick-Change mutagenesis (Stratagene, LaJolla, CA) and confirmed by DNA sequencing.
Purification and Reconstitution
Bacterial expression and Ni2+-affinity purification of full-length MlotiK1 and mutants was performed essentially as described previously 10. Briefly, protein was expressed in BL21 E. coli after a 90 min induction with anhydrotetracycline (ACROS organics, Geel, Belgium) at 37°C with shaking. Membranes were solubilized in 25 mM DM and the protein was bound to Ni2+-matrix in the presence of 25 mM imidizole at pH 7.8. The resin was washed with a buffer containing 5 mM DM, 100 μM cAMP, 5 mM β-mercaptoethanol and 25 mM imidizole, pH 7.8. The protein was eluted from the column by increasing the imidizole concentration to 500 mM and reducing the pH from 7.8 to 7.0. In these studies the protein was subjected to a single round of gel filtration chromatography (Superdex 200 column, Amersham, Piscataway, NJ) in the presence of 100 μM cAMP. Channel protein was then immediately reconstituted into mixed E. coli lipids (Avanti Polar Lipids, Alabaster, AL) as described elsewhere 15 at a density of 5 μg MlotiK1/ mg lipid in 100 μl per sample. Residual cAMP was removed during the reconstitution step, by gel filtration over a small Sephadex G50 (Sigma Aldrich, St. Louis, MO) column and subsequent overnight dialysis.
Cyclic nucleotide binding domain fragment cloned into pGEX-2T was expressed and purified as previously described 10. Briefly, protein was expressed in BL21 E. coli and induced with 0.6 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) overnight at 20°C. Cell lysate was passed over a glutathione-Sepharose column (Amersham, Piscataway, NJ). The protein was cleaved off the column by the addition of thrombin (150 NIH Units/ 4L cell lysate, incubated for 4h at room temperature) and further purified by gel filtration. Constructs in pET-15 were expressed in BL21-DE3 strain overnight at 20°C, induced with 0.6 mM IPTG and purified by Ni2+ matrix and subsequent size exclusion chromatography. All CNB domain protein was dialyzed against ~1000 volumes of appropriate buffer (see below) for at least 12 hour before experiments.
CNB domain affinity matrix competition binding assays
Wild type MlotiK1 CNB domain was incubated with a cAMP-agarose overnight at 4°C in PBS with 2 mM DTT. After extensive washing, protein bound matrix was incubated with sodium salts of the desired ligand (3 mM) at room temperature for 30 min. Eluted protein was separated from the matrix by centrifugation. Purified protein was subjected to 15% SDS PAGE and stained with Commassie blue.
cAMP detection system
All experiments were in PBS with 5 mM β-mercaptoethanol (βME). After purification, CNB domain was dialyzed or run over gel filtration to remove free cAMP and the resultant protein was heat denatured (95°C for 1h) to release bound cAMP. The freed cAMP was separated from the unfolded protein by a series of centrifugations (3 spins at 13, 000 RPM); the supernatant contained cAMP. The HitHunter cAMP detection kit procedure has been described elsewhere 35.We followed the kit instructions exactly, with the following exceptions: Steps 1 and 2 were omitted and the Step 5 incubation was for 1h at room temperature. cAMP from the CNB domains was diluted and aliquoted into Fluorolux black polystyrene 96-well plates (Dynex, Chantilly, VA, product number 3010). A set of cAMP standards was made by serially diluting the stock provided in the kit ([cAMP] ranged from 1.3×10-10 to 2.3×10-5 M) and measured along side MlotiK1 samples. All samples were incubated with an anti-cAMP antibody, a fragment of the enzyme β-galactosidase tethered to cAMP and a mixture of proprietary fluorescent substrate and a second fragment of β-galactosidase for 1h each at room temperature. Measurements were taken with a FluoDia T70 fluorescence plate reader (PTI, London, Ontario, Canada) with an excitation wavelength of 530 nM and an emission wavelength of 610 nM.
Direct binding assays
All binding experiments were performed at room temperature in the following buffer: 100 mM NaCl with 10 mM Hepes, pH 7.5. The reaction volume was 100 μL. Protein was titrated against 2 μM 8-NBD cAMP (Biolog, Bremen, Germany). CNB domain and fluorescent ligand were incubated 1h at room temperature. Fluorescence measurements were performed with a Quantamaster fluorescence spectrometer (Photon Technologies Intl., South Brunswick, NJ) using a 0.3 × 0.3 cm Suprasil quartz cuvette, the Hellma Ultra-Micro Cell (GmbH & Co KG, Müllheim, Germany). The excitation wavelength was 471 nm and emission spectra were collected between 490 and 590 nm. Raw fluorescent measurements were normalized, first by subtracting out the background (no protein samples) and then by dividing by the value obtained for the highest protein tested. The resultant normalized fluorescence was plotted as the dependent variable against the amount of protein concentration titrated in the experiment (independent variable). These data were fit to a hyperbolic function using Microcal Origin (Origin Labs, Northampton, MA). Data shown reflects at least two independent purifications and three independent experiments. Data is presented as mean ± SE, where not visible, error bars are smaller than symbols.
Competition binding assays
For competition experiments, the protein concentration and concentration of 8-NBD cAMP were held constant (for R307A, 50 μM protein with 150 μM 8-NBD cAMP) while the concentration of unlabelled ligand was titrated (0 to 10 mM). Only sodium salts of ligands were used (Sigma Aldrich, St. Louis, MO). Measurements were taken as in the direct binding experiments. Competition fitting was performed as described explicitly elsewhere 36; 37.
86Rb+ Uptake Assay
Flux assays were conducted as previously described 10; 15. For cAMP and cGMP, free acid forms of the ligand were used; for cIMP, cCMP and 5’AMP only the sodium salt forms were available. For these dose-response experiments, NaCl was added from a stock solution into individual samples to ensure that all samples contained the same concentration of sodium. 86Rb+ uptake was quantified after uptake had proceeded for 1.5 hrs. Normalized uptake is the fraction of uptake in comparison to the valinomycin-mediated uptake in an aliquot from the same sample. Data is presented as mean ± SE, performed in triplicate from at least two independent purifications. Where not visible, error bars are smaller than symbols.
Crystallography
All crystals were grown using the hanging drop vapor diffusion method with the protein at 10 mg/mL in 10 mM Tris (pH 7), 0.1 M NaCl, 4 mM dithiotheitol. The cGMP crystal also required 4.4% 1-propanol, as an additive, in the protein drop (Hampton Research, Aliso Viejo, CA). The well solution for this crystal was: 1M ammonium sulfate, 150 mM sodium citrate (pH 5.6) with 1% 1-propanol. Crystallization of the R307E mutant required the addition of 20mM cAMP (sodium salt) to the drop. The pH of the drop was 5.7. The well solution for R307E was: 1M ammonium sulfate, 150 mM sodium citrate (pH 5.6). Both proteins crystallized in the P21 space group. Both crystals were prepared for data collection by fast transfer into cryoprotectant consisting of 0.2 M sodium citrate (pH 5.6), 0.3 M ammonium sulfate, 0.02 M NaCl, 0.08 M Li sulfate, 30% glycerol, and 2 mM Tris (pH 7). Crystals were flash frozen in liquid propane. All data (Table 1) were collected at X25 station, NSLS, Brookhaven National Laboratory. The native data were integrated and scaled using the CCP4 suite set of programs Mosfilm and SCALA 38. The structure was phased using molecular replacement using the CCP4 suite with the cAMP bound structure serving as the search molecule. Refinement was done in CCP4 using Refmac and CNS. Model building was performed in O 39. Figures were made using the program Pymol 40.
Crystals of the R307W mutant were grown and frozen as above, the well solution contained 1.2 M ammonium sulfate, 100 mM sodium citrate (pH 5.6) and 100 mM NaCl. Protein was crystallized in the space group P21212. Data was collected at the X29 beamline at NSLS, Brookhaven National Laboratory. We solved the phases by performing a selenomethionine multiwavelength anomalous diffraction experiment. Selenomethione derivative crystals grew in the same conditions as the native ones. Data was processed in HKL2000 41. The R348A mutant structure was manually docked into an initial map from Resolve 42, altered and refined with CNS and Refmac with TLS refinement. Model building was performed in O 39. Figures were made using the program Pymol 40.
Accession Codes
Structures have been deposited in Protein Data Bank with the following codes: 3CL1, 3CLP and 3CO2.
Acknowledgments
The authors declare they have no competing financial interests. We thank Fred Sigworth for helpful discussions on gating, Ron Albright for help with the figures and the staff of X12C, X25 and X29 beamlines at the National Synchrotron Light source. This work was supported in part by a grant from the American Heart Association (#0555781T) to L.R.T, and grants from the American Heart Association (#0555822T) and National Institutes of Health (GM068585) to J.H.M.-C.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions Author contributions: All authors designed research; G.M.C. and S.L.A. performed crystallography experiments; W.R.S. and S.L.A. performed flux assays, S.L.A. and A.O.O. performed fluorescent binding assays, and S.L.A. performed matrix competition assays; all authors analyzed data; and S.L.A., L.R.T. and J.H.M.-C. wrote the paper.
References
- 1.Rehmann H, Wittinghofer A, Bos JL. Capturing cyclic nucleotides in action: snapshots from crystallographic studies. Nat Rev Mol Cell Biol. 2007;8:63–73. doi: 10.1038/nrm2082. [DOI] [PubMed] [Google Scholar]
- 2.Berman HM, Ten Eyck LF, Goodsell DS, Haste NM, Kornev A, Taylor SS. The cAMP binding domain: an ancient signaling module. Proc Natl Acad Sci U S A. 2005;102:45–50. doi: 10.1073/pnas.0408579102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moore J, Kantorow M, Vanderzwaag D, McKenney K. Escherichia coli cyclic AMP receptor protein mutants provide evidence for ligand contacts important in activation. J Bacteriol. 1992;174:8030–5. doi: 10.1128/jb.174.24.8030-8035.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gronenborn AM, Sandulache R, Gartner S, Clore GM. Mutations in the cyclic AMP binding site of the cyclic AMP receptor protein of Escherichia coli. Biochem J. 1988;253:801–7. doi: 10.1042/bj2530801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang LJ, Taylor SS. Dissecting cAMP binding domain A in the RIalpha subunit of cAMP-dependent protein kinase. Distinct subsites for recognition of cAMP and the catalytic subunit. J Biol Chem. 1998;273:26739–46. doi: 10.1074/jbc.273.41.26739. [DOI] [PubMed] [Google Scholar]
- 6.McKay DB, Steitz TA. Structure of catabolite gene activator protein at 2.9 A resolution suggests binding to left-handed B-DNA. Nature. 1981;290:744–9. doi: 10.1038/290744a0. [DOI] [PubMed] [Google Scholar]
- 7.Su Y, Dostmann WR, Herberg FW, Durick K, Xuong NH, Ten Eyck L, Taylor SS, Varughese KI. Regulatory subunit of protein kinase A: structure of deletion mutant with cAMP binding domains. Science. 1995;269:807–13. doi: 10.1126/science.7638597. [DOI] [PubMed] [Google Scholar]
- 8.Rehmann H, Prakash B, Wolf E, Rueppel A, de Rooij J, Bos JL, Wittinghofer A. Structure and regulation of the cAMP-binding domains of Epac2. Nat Struct Biol. 2003;10:26–32. doi: 10.1038/nsb878. [DOI] [PubMed] [Google Scholar]
- 9.Zagotta WN, Olivier NB, Black KD, Young EC, Olson R, Gouaux E. Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature. 2003;425:200–5. doi: 10.1038/nature01922. [DOI] [PubMed] [Google Scholar]
- 10.Clayton GM, Silverman WR, Heginbotham L, Morais-Cabral JH. Structural basis of ligand activation in a cyclic nucleotide regulated potassium channel. Cell. 2004;119:615–27. doi: 10.1016/j.cell.2004.10.030. [DOI] [PubMed] [Google Scholar]
- 11.Nimigean CM, Shane T, Miller C. A cyclic nucleotide modulated prokaryotic K+ channel. J Gen Physiol. 2004;124:203–10. doi: 10.1085/jgp.200409133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Craven KB, Zagotta WN. CNG and HCN channels: two peas, one pod. Annu Rev Physiol. 2006;68:375–401. doi: 10.1146/annurev.physiol.68.040104.134728. [DOI] [PubMed] [Google Scholar]
- 13.Matulef K, Zagotta WN. Cyclic nucleotide-gated ion channels. Annu Rev Cell Dev Biol. 2003;19:23–44. doi: 10.1146/annurev.cellbio.19.110701.154854. [DOI] [PubMed] [Google Scholar]
- 14.Biel M, Schneider A, Wahl C. Cardiac HCN channels: structure, function, and modulation. Trends Cardiovasc Med. 2002;12:206–12. doi: 10.1016/s1050-1738(02)00162-7. [DOI] [PubMed] [Google Scholar]
- 15.Heginbotham L, Kolmakova-Partensky L, Miller C. Functional reconstitution of a prokaryotic K+ channel. J Gen Physiol. 1998;111:741–9. doi: 10.1085/jgp.111.6.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clayton GM, Altieri S, Heginbotham L, Unger VM, Morais-Cabral JH. Structure of the transmembrane regions of a bacterial cyclic nucleotide-regulated channel. Proc Natl Acad Sci U S A. 2008;105:1511–5. doi: 10.1073/pnas.0711533105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chiu PL, Pagel MD, Evans J, Chou HT, Zeng X, Gipson B, Stahlberg H, Nimigean CM. The structure of the prokaryotic cyclic nucleotide-modulated potassium channel MloK1 at 16 A resolution. Structure. 2007;15:1053–64. doi: 10.1016/j.str.2007.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cukkemane A, Gruter B, Novak K, Gensch T, Bonigk W, Gerharz T, Kaupp UB, Seifert R. Subunits act independently in a cyclic nucleotide-activated K(+) channel. EMBO Rep. 2007;8:749–55. doi: 10.1038/sj.embor.7401025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gauss R, Seifert R, Kaupp UB. Molecular identification of a hyperpolarization-activated channel in sea urchin sperm. Nature. 1998;393:583–7. doi: 10.1038/31248. [DOI] [PubMed] [Google Scholar]
- 20.Nakamura T, Gold GH. A cyclic nucleotide-gated conductance in olfactory receptor cilia. Nature. 1987;325:442–4. doi: 10.1038/325442a0. [DOI] [PubMed] [Google Scholar]
- 21.Ludwig A, Zong X, Jeglitsch M, Hofmann F, Biel M. A family of hyperpolarization-activated mammalian cation channels. Nature. 1998;393:587–91. doi: 10.1038/31255. [DOI] [PubMed] [Google Scholar]
- 22.Zhou L, Siegelbaum SA. Gating of HCN channels by cyclic nucleotides: residue contacts that underlie ligand binding, selectivity, and efficacy. Structure. 2007;15:655–70. doi: 10.1016/j.str.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scott SP, Shea PW, Dryer SE. Mapping ligand interactions with the hyperpolarization activated cyclic nucleotide modulated (HCN) ion channel binding domain using a soluble construct. Biochemistry. 2007;46:9417–31. doi: 10.1021/bi6026049. [DOI] [PubMed] [Google Scholar]
- 24.Dhallan RS, Yau KW, Schrader KA, Reed RR. Primary structure and functional expression of a cyclic nucleotide-activated channel from olfactory neurons. Nature. 1990;347:184–7. doi: 10.1038/347184a0. [DOI] [PubMed] [Google Scholar]
- 25.Flynn GE, Black KD, Islas LD, Sankaran B, Zagotta WN. Structure and rearrangements in the carboxy-terminal region of SpIH channels. Structure. 2007;15:671–82. doi: 10.1016/j.str.2007.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weber IT, Shabb JB, Corbin JD. Predicted structures of the cGMP binding domains of the cGMP-dependent protein kinase: a key alanine/threonine difference in evolutionary divergence of cAMP and cGMP binding sites. Biochemistry. 1989;28:6122–7. doi: 10.1021/bi00440a059. [DOI] [PubMed] [Google Scholar]
- 27.Yathindra N, Sundaralingam M. Conformations of cyclic 3’,5’-nucleotides. Effect of the base on the synanti conformer distribution. Biochem Biophys Res Commun. 1974;56:119–26. doi: 10.1016/s0006-291x(74)80323-2. [DOI] [PubMed] [Google Scholar]
- 28.Chwang AK, Sundaralingam M. Molecular conformation of guanosine 3,’5-cyclic monophosphate. Nat New Biol. 1973;244:136–7. doi: 10.1038/newbio244136a0. [DOI] [PubMed] [Google Scholar]
- 29.Watenpaugh K, Dow J, Jensen LH, Furberg S. Crystal and molecular structure of adenosine 3’,5’-cyclic phosphate. Science. 1968;159:206–7. doi: 10.1126/science.159.3811.206. [DOI] [PubMed] [Google Scholar]
- 30.Varnum MD, Black KD, Zagotta WN. Molecular mechanism for ligand discrimination of cyclic nucleotide-gated channels. Neuron. 1995;15:619–25. doi: 10.1016/0896-6273(95)90150-7. [DOI] [PubMed] [Google Scholar]
- 31.Chen J, Sharma S, Quiocho FA, Davidson AL. Trapping the transition state of an ATP-binding cassette transporter: evidence for a concerted mechanism of maltose transport. Proc Natl Acad Sci U S A. 2001;98:1525–30. doi: 10.1073/pnas.041542498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dao KK, Teigen K, Kopperud R, Hodneland E, Schwede F, Christensen AE, Martinez A, Doskeland SO. Epac1 and cAMP-dependent protein kinase holoenzyme have similar cAMP affinity, but their cAMP domains have distinct structural features and cyclic nucleotide recognition. J Biol Chem. 2006;281:21500–11. doi: 10.1074/jbc.M603116200. [DOI] [PubMed] [Google Scholar]
- 33.Rehmann H, Schwede F, Doskeland SO, Wittinghofer A, Bos JL. Ligand-mediated activation of the cAMP-responsive guanine nucleotide exchange factor Epac. J Biol Chem. 2003;278:38548–56. doi: 10.1074/jbc.M306292200. [DOI] [PubMed] [Google Scholar]
- 34.Schmidt TG, Skerra A. The random peptide library-assisted engineering of a C-terminal affinity peptide, useful for the detection and purification of a functional Ig Fv fragment. Protein Eng. 1993;6:109–22. doi: 10.1093/protein/6.1.109. [DOI] [PubMed] [Google Scholar]
- 35.Golla R, Seethala R. A homogeneous enzyme fragment complementation cyclic AMP screen for GPCR agonists. J Biomol Screen. 2002;7:515–25. doi: 10.1177/1087057102238625. [DOI] [PubMed] [Google Scholar]
- 36.Talavera MA, De La Cruz EM. Equilibrium and kinetic analysis of nucleotide binding to the DEAD-box RNA helicase DbpA. Biochemistry. 2005;44:959–70. doi: 10.1021/bi048253i. [DOI] [PubMed] [Google Scholar]
- 37.Wang ZX. An exact mathematical expression for describing competitive binding of two different ligands to a protein molecule. FEBS Lett. 1995;360:111–4. doi: 10.1016/0014-5793(95)00062-e. [DOI] [PubMed] [Google Scholar]
- 38.Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 39.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 1991;A 47(Pt 2):110–9. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 40.DeLano WL. The PyMOL User’s Manual. DeLano Scientific; Palo Alto, CA, USA: 2002. [Google Scholar]
- 41.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 42.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr. 1999;55:849–61. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]