

Summary
The recent cloning of tumor-associated antigens (TAAs) recognized by CD8 + T lymphocytes (TCD8−) has made it possible to use recombinant and synthetic forms of TAAs to generate TCD8− with anti-tumor activity. To explore new therapeutic strategies in a mouse model, we retrovirally transduced the experimental murine tumor CT26 (H-2d), with the lacZ gene encoding our model TAA, (β-galactosidase (β-gal). The transduced cell line, CT26.CL25, grew as rapidly and as lethally as the parental cell line in normal, immuno-competent animals. In an attempt to elicit TCD8+ directed against our model TAA by using purely recombinant and synthetic forms of our model TAA, we synthesized a nine-amino-acid long immunodominant peptide of (β-gal (TPH-PARIGL), corresponding to amino acid residues 876–884, which was known to be presented by the Ld major histocompatibility complex (MHC) class I molecule, and a recombinant vaccinia virus encoding the full-length β-gal protein (VJS6). Splenocytes obtained from naïve mice and co-cultured with (β-gal peptide could not be expanded in primary ex vivo cultures. However, mice immunized with VJS6, but not with a control recombinant vaccinia virus, yielded splenocytes that were capable of specifically lysing CT26.CL25 in vitro after co-culture with (β-gal peptide. Most significantly, adoptive transfer of these cells could effectively treat mice bearing 3-day-old established pulmonary metastases. These observations show that therapeutic TCD8+ directed against a model TAA could be generated by using purely recombinant and synthetic forms of this antigen. These findings point the way to a potentially useful immunotherapeutic strategy, which has been made possible by the recent cloning of immunogenic TAAs that are expressed by human malignancies.
Keywords: Recombinant vaccinia virus, Peptide, Adoptive immunotherapy, T lymphocyte, MHC class I
Lymphocytes with anti-tumor reactivities can be expanded to large numbers ex vivo and can effect the regression of even large tumor burdens on adoptive transfer in both mouse and human (1–5). The antigens recognized by anti-human melanoma T lymphocytes have been cloned recently, and the exact peptide fragments of these antigens that are presented on the tumor cell surfaces by major histocompatibility complex (MHC) class I molecules and recognized by CD8 + T lymphocytes have also been identified (6–14).
Tumor cells have been used in previous studies as antigenic stimulators of CD8 + T lymphocytes with anti-tumor reactivities (15–18). Knowledge of the identities of tumor-associated antigens (TAAs) has made it possible to develop immunotherapeutic strategies based entirely on recombinant and synthetic materials. Indeed, synthetic peptides have been used to expand T lymphocytes with anti-tumor reactivities from peripheral blood lymphocytes (19–23).
In this study, we employed β-galactosidase (β-gal) as a model TAA to explore alternative strategies to the use of tumor cells in the generation of anti-tumor T lymphocytes for adoptive transfer. Although recombinant vaccinia virus (rVV) encoding this model TAA alone was incapable of treating established tumor, it was capable of priming T lymphocytes, which could be expanded ex vivo for the successful adoptive immunotherapy of tumor-bearing animals.
MATERIALS AND METHODS
Tumors and Animals
CT26 (H-2d) is an N-nitroso-N-methylurethane induced BALB/c undifferentiated colon adenocarcinoma (24). CT26.CL25 is a clone of CT26 that was transduced with a lacZ retrovirus based on the LXSN modification of the Moloney murine leukemia virus and has been described elsewhere (25). EL4.E22 is a clone of EL4 stably transfected with β-gal and was used as a negative control in 51Cr-release assays (kindly provided by Y. Paterson, Philadelphia, PA, U.S.A.). Cell lines were maintained in RPMI 1640, 10% heat-inactivated fetal calf serum (PCS) (Biofluids, Rockville, MD, U.S.A.), 0.03% L-glutamine, 100 μg/ml streptomycin, 100 μg/ml penicillin, and 50 μg/ml gentamycin sulfate (NIH Media Center). CT26.CL25 and EL4.E22 are maintained in the presence of 400 μg/ml G418 sulfate (Gibco, Grand Island, NY, U.S.A.). Female BALB/c mice, 8–12 weeks old, were obtained from the Animal Production Colonies, Frederick Cancer Research Facility, NIH, Frederick, MD, U.S.A.
Viruses
Recombinant vaccinia virus (rVV) stocks were produced using the thymidine kinase-deficient (TK−) human osteosarcoma 143/B cell line (American Type Culture Collection, Rockville, MD, U.S.A.; CRL 8303). rVVs expressing β-gal and influenza virus A/PR/8/34 nucleoprotein (NP) were constructed by previously described methods (26,27). The expression of β-gal was under the control of the early element of the VV natural P7.5 early/late promoter from plasmid pSC65 (S. Chakrabarti, J. Sisler, B. Moss, NIAID, Bethesda, MD, U.S.A.) in the HPV 16-E6Vac (VJS6) (kindly provided by B. Moss, NIAID). The control rVV (designated V69) expressed NP with the natural P7.5 early/late promoter and did not express β-gal (28). In all cases, foreign genes were inserted into the vaccinia virus TK locus by homologous recombination in CV-1 cells (CCL 70). Viruses were plaque purified on 143/B cells in the presence of bromodeoxyuridine. BS-C-1 cells (CCL 26), an African green monkey kidney cell line obtained from the American Type Culture Collection were used to determine virus concentration by plaque titration. Crude 19 is a non-recombinant control vaccinia virus donated by J. Yewdell and J. Bennink (NIAID).
β-Galactosidase Assay
Two × 105 CT26.WT or 1.6 × 105 BS-C-1 were plated in each well of six-well plates and infected in duplicate [multiplicity of infection (MOI), 10:1] with Crude 19, VJS6 or not infected. Cultures were allowed to incubate for 24 h. Cells extracts were prepared, and β-gal activity was measured using the β-galactosidase Enzyme Assay System (Promega, Madison, WI, U.S.A.). Briefly, cell extracts were prepared using the freeze/thaw method. Samples were then incubated with the substrate o-nitrophenyl-β-D-galactopyranoside (ONPG) for 30 min at 37°C, after which the reaction was stopped using 1 M sodium carbonate. β-Galactosidase hydrolizes ONPG to o-nitrophenol, and the absorbance is read at 420 nm with a spectrophotometer. Standard curves were prepared as indicated and β-gal activity was measured and reported as units × 10−4.
Peptides
The synthetic peptide, TPHPARIGL, representing the naturally processed H-2 Ld restricted epitope spanning amino acids 876–884 of β-gal and TYQRTRALV, the H-2 Kd epitope of influenza virus PR8 nucleoprotein corresponding to residues 147–155, were synthesized by Peptide Technologies (Washington, DC, U.S.A.) to a purity of >99%, as assessed by high-pressure liquid chromatography (HPLC) and amino acid analysis.
Effector Cells
TCD8− populations were generated by injecting BALB/c mice intravenously with the designated plaque-forming unit (PFU) of VJS6 or V69. After 21 days, splenocytes were harvested, dispersed into a single-cell suspension, and cultured ex vivo in the presence of 1 μM of the peptide designated (either TPHPARIGL or TYQRTRALV) in a T-75 flask (Nunc, Denmark) at a density of 5.0 × 106 splenocytes/ml. Total volumes of splenocyte cultures were 30 ml. Culture medium that contained 0.1 mM nonessential amino acids, 1.0 mM sodium pyruvate (both from Biofluids) and 5 × 10 −5 M 2-mercaptoethanol (Gibco/BRL, Rockville, MD, U.S.A.) in the absence of IL-2, as described (29). Cultures were harvested on day 7, and splenocytes were washed with fresh culture medium before use in a 51Cr-release assay. Splenocytes were tested for their ability to lyse either CT26 alone, pulsed with TPHPARIGL, or transduced with the lacZ gene (CT26.CL25) in a 6-h 51Cr-release assay.
51Cr-Release Assay
Six-hour 51Cr-release assays were performed as previously described (30). Briefly, 2 × 106 target cells were incubated with 200 mCi Na51CrO4(51Cr) for 90 min. Peptide-pulsed CT26 were incubated with 1 μg/ml of synthetic peptide during labeling. Target cells were then mixed with effector cells for 6 h at the effector-to-target (E/T) ratios indicated. The amount of 51Cr released was determined by γ-counting, and the percentage specific lysis was calculated from triplicate samples as follows: [(experimental cpm − spontaneous cpm/maximal cpm − spontaneous cpm)] × 100.
In Vivo Adoptive Transfer Studies
For primary adoptive transfer experiments, the spleens of mice that had been injected 21 days previously with either 5 × 106 PFUs of VJS6 or V69, and the spleens of unimmunized mice were harvested, dispersed into single-cell suspensions, and immediately reinjected intravenously [2 × 107 cells/0.5 ml Hanks’ Balanced Saline Solution (HBSS)] into mice bearing 3-day pulmonary metastases. One half of the treatment and control groups received 15,000 Cetus units of recombinant IL-2 (rIL-2) intraperitoneally twice a day on days 3–5. On day 12, all mice were killed, tagged, and randomized. Metastatic lung nodules were enumerated in a blinded, coded fashion, as previously described. The adoptive transfer of splenocytes from secondary cultures was carried out as follows (31). The spleens of mice that had been injected 21 days previously with either 5 × 106 PFUs of VJS6 or V69 and the spleens of unimmunized mice were harvested, dispersed into single-cell suspensions, and placed into culture in T-75 flasks (Nunc, Denmark) at a density of 5 × 106 splenocytes/ml with 1 μg/ml of antigenic peptide or control peptide in a total volume of 30 ml of culture medium that also contained 0.1 mM non-essential amino acids, 1.0 mM sodium pyruvate (both from Biofluids) and 5 × 10−5 M 2-mercaptoethanol (Gibco/BRL) in the absence of IL-2. Seven days later, splenocytes were harvested and washed in HBSS and reinjected intravenously (2 × 107 cells/0.5 ml HBSS) into mice bearing established pulmonary metastases. 51Cr-release assays were also done to confirm the specificity and reactive-ness of the secondary splenocyte cultures. On day 12, all mice were killed, tagged, and randomized. Metastatic lung nodules were enumerated in a blinded, coded fashion as previously described (32).
RESULTS
CT26.WT, a murine undifferentiated colon adenocarcinoma, was retrovirally transduced with the lacZ gene and subcloned to generate CT26.CL25 to provide a tumor model with a defined TAA. CT26.CL25 has been previously shown to express β-gal in vivo and grow with equal rate and lethality in normal BALB/c mice (25). Furthermore, it was shown that an intravenous dose of 5 × 105 CT26.WT or CT26.CL25 killed the recipients in 15 days (25).
CT26.WT and BSC-1 cells were infected with VJS6 to assess the function of the rVV in the production of the model TAA, β-gal, as measured by a colorimetric assay. β-Gal activity in lysates of cells infected with VJS6 (MOI, 10:1) and incubated for 24 h at 37°C was quantified, and the results are shown in Fig. 1. Cell extracts from cell lines infected with VJS6 had β-gal activity, whereas uninfected cells and cells infected with a control non-β-gal expressing VV (Crude 19) had no detectable β-gal activity.
FIG. 1.

VJS6-infected cells produce β-galactosidase. Two × 105 CT26.WT or 1.6 × 105 BS-C-1 were plated in each well of six-well plates and infected in duplicate (MO1, 10:1) with Crude 19, VJS6, or not infected. Cultures were allowed to incubate for 24 h. Cell extracts were prepared, and β-gal activity was measured using the β-galactosidase Enzyme Assay System (Promega, Madison, Wl, U.S.A.). (3-Gal activity is reported as units × 10−4. Experiment was repeated with similar results.
Splenocytes from naïve BALB/c mice were cultured in the presence of the nine-amino-acid-long immunodominant peptide of β-gal (TPHPARIGL), corresponding to amino acid residues 876–884, that was known to be presented by the Ld MHC class I molecule. Although these splenocytes did not specifically lyse CT26.CL25 in vitro, cultured splenocytes from mice that were injected intravenously with VJS6 at doses >2 × 105 PFUs were capable of specific recognition of CT26.CL25 or of CT26 pulsed with synthetic peptide (Fig. 2).
FIG. 2.

VJS6 elicits specific anti-β-galactosidase cytotoxic T lymphocytes (CTL). BALB/c mice were intravenously immunized with either 2 × 104, 2 × 105, 2 × 106, 2 × 107 PFUs of VJS6. Twenty-one days later, splenocytes from all of the immunized mice and unimmunized mice (peptide alone) were restimulated with 1 μg/ml of synthetic peptide TPHPARIGL for 7 days and then assayed in a 51Cr-release assay against CT26.WT (closed diamonds), CT26.WT pulsed with TPHPARIGL (closed squares), CT26.CL25 (open triangles), or EL4.E22 (open circles; EL4.E22 is an H-2b tumor that expresses β-gal). Spontaneous release for all reported 51Cr-release assays is <10%. Experiment was repeated with similar results.
To determine the efficacy of anti-β-gal CTLs elicited by VJS6 in the treatment of established pulmonary metastases, BALB/c mice were injected with 5 × 105 CT26.WT or CT26.CL25. Three days later, 2 × 107 fresh splenocytes from animals previously immunized with VJS6 were adoptively transferred intravenously, with or without intraperitoneal administration of 30,000 Cetus units of rIL-2 for 3 days, to determine if primary splenocytes could treat established 3-day pulmonary metastases. There was no reduction of established tumor using primary splenocytes, even with the exogenous administration of rIL-2 (data not shown). Because the administration of these splenocytes was intravenous, this allowed them to travel to the lung; therefore, we hypothesized that either the tumor was incapable of stimulating these splenocytes in vivo, or the number of anti-β-gal-specific CTL precursors was limiting.
Next we evaluated the therapeutic efficacy of splenocytes taken from mice pre-immunized with VJS6 that were grown in culture for 7 days in the presence of TPHPARIGL (Table 1). Splenocyte cultures obtained from either naive mice, or mice previously primed with VJS6, were cultured with peptide and then adoptively transferred to mice bearing 3-day-old pulmonary metastases of CT26 or CT26.CL25. Half of each treatment group was given 15,000 Cetus units of rIL-2 (intraperitoneally) twice daily on days 3–5. In the treatment group bearing CT26.CL25, the adoptive transfer of 2 × 107 splenocytes from mice primed with VJS6 significantly reduced the number of pulmonary metastases from >500 to an average of 1.2 in the presence of exogenously administered rIL-2, or three in the absence of IL-2 (p < 0.001). Mice injected with rIL-2 alone showed minimal response to treatment and had tumor burdens comparable to those of mice receiving only HBSS.
TABLE 1.
Adoptive treatment of tumor-bearing mice with splenocytes elicited with recombinant and synthetic forms of β-gal
| Treatment
|
CT26.WT | CT26.CL25 | |
|---|---|---|---|
| In vivo | In vitro | (Avg no. pulmonary metastases) | |
| Naïve | — | >500 | 425 |
| Naïve | β-gal | ND | >500 |
| VJS6a | β-gal | >500 | 3 |
| VJS6a | β-gal (IL-2)b | 349 | 1.2 |
| Naïve | — (IL-2)b,c | 387 | 303 |
| Naïve | β-gal (IL-2)b | ND | >500 |
BALB/c mice were injected intravenously with 0.5 ml of HBSS containing 5 × 105 cells of either CT26.WT or CT26.CL25. Three days later, they received adoptive transfer of effector cells by single intravenous injection of 0.5 ml of HBSS containing 2 × 107 cells from 7-day cultured splenocytes generated from immunized and unimmunized (naïve) mice after re-stimulation with TPHPARIGL. Lungs were harvested 12 days after tumor inoculation, and pulmonary metastases were enumerated in a coded and blinded fashion.
β-gal, β-galactosidase; IL-2, interleukin-2; ND, not done; HBSS, Hanks’ Balanced Saline Solution; PFU, plaque-forming unit; rIL-2, recombinant IL-2.
Mice were primed 21 days before in vitro culture with 5 × 106 PFU of VJS6 i.v.
Treatment with exogenous rlL-2 (15,000 Cetus units i.p., b.i.d.) was started 6 h after adoptive transfer and continued for 3 days.
This group received no therapeutic splenocytes on day 3.
To assess whether the therapeutic effect could be nonspecifically mediated by any antigen-specific activated CTLs adoptively transferred into a tumor-bearing mouse or that any irrelevant peptide could enhance an anti-β-gal response in vitro, mice were immunized with 5 × 106 PFUs of either VJS6 or V69. After 3 weeks, spleens were harvested, dispersed into single-cell suspensions, and incubated with the antigenic peptides corresponding to a known epitope of β-gal (TPHPARIGL) or of NP (TYQRTRALV). After 7 days in culture, these β-gal- or NP-specific splenocytes were injected intravenously into mice bearing day 3 CT26 or CT26.CL25 pulmonary metastases. The specificity and lytic ability of the splenocytes were confirmed by 51Cr-release assay (Fig. 3). Mice bearing CT26.CL25 that received the adoptive transfer of 2 × 107 splenocytes from mice immunized with VJS6 and stimulated in vitro with the β-gal antigenic peptide TPHPARIGL were able to reduce the average tumor burden of established pulmonary metastases to 10.6/mouse, whereas mice that received splenocytes from unimmunized mice that were identically cultured in vitro carried an average tumor burden of >500 metastatic nodules on day 12 (Fig. 4). All other treatment groups, including the group of mice that received NP-specific CTL and a group that received CTL generated from mice immunized with VJS6 and restimulated with NP peptide (TYQRTRALV), showed no response to treatment. Mice bearing CT26.CL25 were not significantly different from controls if only 2 × 106 anti-β-gal CTLs were injected, which provided evidence that a dose threshold for treatment existed for antigen-specific effector cells generated by an rVV. Mice receiving CT26. WT did not respond to any form of treatment.
FIG. 3.

Mice immunized with recombinant vaccinia viruses mount specific immune responses. BALB/c mice were immunized intravenously with 5 × 106 PFUs of VJS6 (B,C) or V69 (D,E). After 21 days, splenocytes from all immunized mice (B,C,D,E) and unimmunized mice (A) were restimulated with 1 μg/ml of either synthetic peptide, TPHPARIGL (A,C,D) or TYQRTRALV (B,E), for 7 days. These effectors were then assayed in a 51Cr-release assay against CT26.CL25 (closed circles) or CT26.WT pulsed with TYQRTRALV (open circles). Spontaneous release for all reported 51Cr-release assays is <10%. Experiment was repeated with similar results.
FIG. 4.
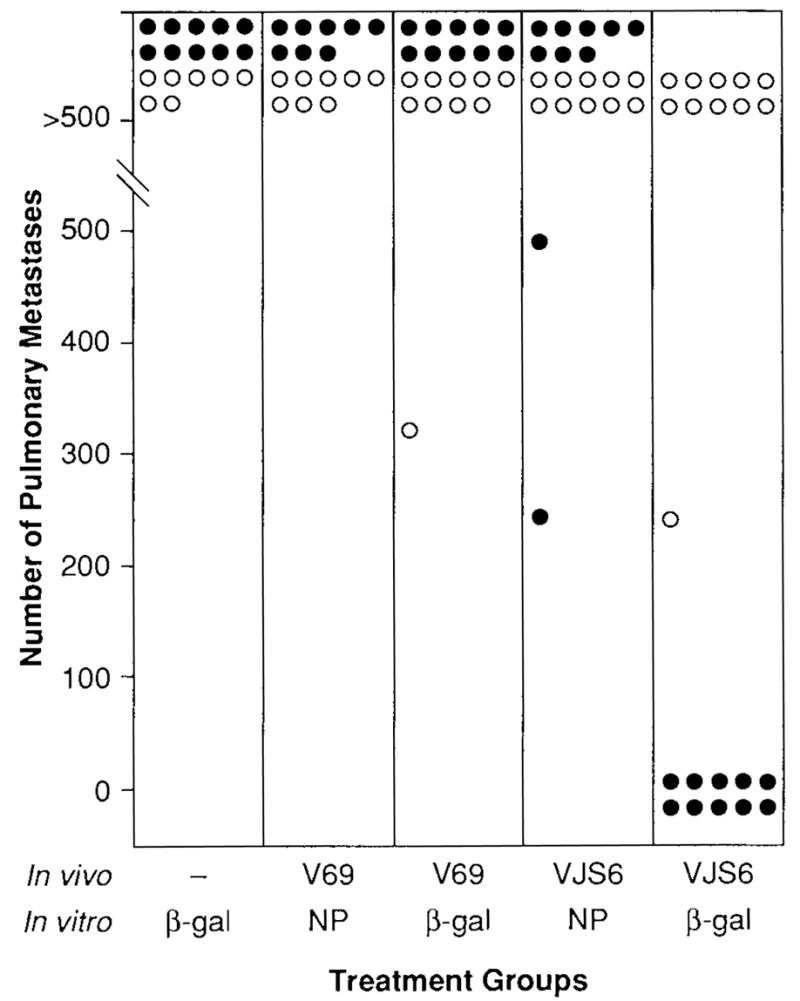
Treatment of CT26.CL25 can be obtained only with the adoptive transfer of anti-β-galactosidase cytotoxic T lymphocytes (CTL). BALB/c mice were injected intravenously with 0.5 ml of Hanks’ Balanced Saline Solution (HBSS) containing either CT26.WT (open circles) or CT26.CL25 (closed circles). Three days later, they received adoptive transfer of 2 × 107 effector cells generated from splenocytes of mice immunized with VJS6 or V69, or unimmunized (naïve) mice, and cultured for 7 days with 1 μg/ml of either synthetic peptide, TPHPARIGL or TYQR-TRALV. Lungs were harvested on day 12 after tumor inoculation, and pulmonary metastases were enumerated in a coded and blinded fashion. No mice received exogenous recombinant in-terleukin-2 (rIL-2). Experiment was repeated with similar results.
DISCUSSION
The immunogenicity of spontaneously arising tumors is usually low despite the presence of one or more TAAs (33). However, even weakly immunogenic tumors in mouse and humans contain T lymphocytes that can be expanded ex vivo and adoptively transferred back to tumor-bearing hosts (together with IL-2), where they can effect the regression of even large tumor burdens (1,34–37). We used a murine tumor model that expresses a model tumor antigen, β-gal, stably expressed by CT26.CL25. Fresh splenocytes harvested from mice bearing subcutaneous CT26.CL25 were unable to lyse (β-gal-expressing targets in primary 51Cr-release assays (data not shown), and untreated mice bearing CT26.CL25 generally had >500 pulmonary metastases 12 days after tumor injection, indicating that an effective immune response was not mounted against the model TAA. A combination of rVV and synthetic peptide, however, could be used to elicit lymphocytes that were therapeutic in adoptive transfer experiments, much like lymphocytes found within tumor deposits (TILs).
There are advantages to immunization with recombinant viruses encoding TAAs. Viruses induce strong cellular immune responses. Poxviruses introduce antigen into the cytoplasm of cells, where they can go through antigen processing and presentation, and then potential recognition by TCD8+. There is a recent report of an rVV encoding the gene for carcinoembryonic antigen (CEA) effectively treating mice with tumors bearing this antigen (38). Current immunization strategies with tumor cells may be largely ineffective because many tumors are not efficient activators of a primary immune response in vivo, even in the context of well-defined model antigens (39). Genes encoding cytokines have been introduced into tumor cells to enhance their immunogenicity, but these methods of generating vaccines are limited because they may address only one source of the possible factors that are deficient in the host anti-tumor response (40,41). Harvesting TILs from tumors or anti-tumor peripheral blood leukocytes of unimmunized patients may also be limited for similar reasons.
Adoptive immunotherapy can cause tumor regression in some patients with established disease (1–5,42). In fact, the first use of anti-melanoma vaccines may not be in the active treatment of disease but in increasing the precursor frequency of tumor-specific TCD8+ that can be specifically expanded in vitro with antigen epitope peptides and given back to the patient. Our findings show that an rVV expressing β-gal can generate specific anti-tumor TCD8+ that can be used to treat an aggressive experimental murine tumor in vivo even in the absence of exogenous rIL-2. Further studies are necessary to establish the therapeutic potential of these effectors in the treatment of tumors growing in sites other than the lung.
We and others have also observed that mice immune to VV cannot mount a primary or secondary response against antigen expressed by an rVV in a second immunization (25). This not only limits the applicability of the use of rVV to people born after 1967 but implies that boosting the immune response through multiple immunizations with rVV will be difficult. Whereas specific boosting of this response may be achieved with other non-cross-reactive viruses (25) or peptides, the efficacy of these methods of secondary boosting of the immune response against a TAA are unknown.
The human melanoma TAA cloned thus far, as well as the mouse antigen P1A are “normal” self proteins. Therefore, a fundamental issue facing the vaccine-based immunotherapy of cancer may be the breaking of immunologic tolerance against self antigens (8). Results obtained in this set of experiments using β-gal as a model TAA may not be reproduced when the antigen is a nonmutated, “self” protein, which will presumably induce tolerance. However, the β-gal model is relevant to other human tumor situations in which TAAs can be targeted that originate from viruses (43,44), fusion proteins resulting from translocations (45), and genetic events that result in totally foreign protein expression, such as mutations resulting in frame-shifts, translation of intronic information, and the loss of stop codons (6,46).
An important consideration in the effective use of therapeutic anti-cancer vaccines is that tumors may escape from immune recognition by a variety of mechanisms, including the functional loss of β2-microglobulin (47–49; Restifo et al., unpublished data), the downregulation of TAP- and MHC-encoded proteasome components (30), the loss of expression of particular MHC alleles (50), and the loss of expression of TAAs (51). Therapies to overcome these problems, such as the transduction of tumors with the gene for γ-interferon and IL-2, have shown success in animal models, but their applicability to the treatment of human cancer is unclear (52).
The use of rVV thus represents one method of immunizing against cancers with well-defined antigens. Evidence presented here supports the efficacy of the adoptive transfer of TCD8+ and shows that the context in which the host recognizes antigen may play an important role in eliciting a TCD8+ response. Our data also suggest that the lack of a primary immune response does not necessarily mean inadequate vaccination, emphasizing the potential importance of epitope-specific synthetic peptides in the in vitro expansion of therapeutic CTL. As the genes for TAAs and their class I MHC-restricted epitopes continue to be revealed, the role of synthetic and recombinant materials in the development of new immunotherapeutic strategies could expand significantly.
Acknowledgments
V. Bronte is a visiting scientist supported by the Italian Association for Cancer Research (A1RC).
References
- 1.Rosenberg SA, Yannelli JR, Yang JC, et al. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J Natl Cancer Inst. 1994;86:1159–66. doi: 10.1093/jnci/86.15.1159. [DOI] [PubMed] [Google Scholar]
- 2.Sabzevari H, Reisfeld RA. Human cytotoxic T-cells suppress the growth of spontaneous melanoma metastases in SCID/hu mice. Cancer Res. 1993;53:4933–7. [PubMed] [Google Scholar]
- 3.Tuttle TM, McCrady CW, Inge TH, Salour M, Bear HD. γ-Interferon plays a key role in T-cell-induced tumor regression. Cancer Res. 1993;53:833–9. [PubMed] [Google Scholar]
- 4.Shu S, Krinock RA, Matsumura T, Sussman JJ, Fox BA, Chang AE, Terman DS. Stimulation of tumor-draining lymph node cells with superantigenic staphylococcal toxins leads to the generation of tumor-specific effector T cells. J Immunol. 1994;152:1277–88. [PubMed] [Google Scholar]
- 5.Chang AE, Yoshizawa H, Sakai K, Cameron MJ, Sondak VK, Shu S. Clinical observations on adoptive immunotherapy with vaccine-primed T-lymphocytes secondarily sensitized to tumor in vitro. Cancer Res. 1993;53:1043–50. [PubMed] [Google Scholar]
- 6.Boon T, Cerottini JC, Van den Eynde B, van der Bruggen P, Van Pel A. Tumor antigens recognized by T lymphocytes. Annu Rev Immunol. 1994;12:337–65. doi: 10.1146/annurev.iy.12.040194.002005. [DOI] [PubMed] [Google Scholar]
- 7.Cox AL, Skipper J, Chen Y, et al. Identification of a peptide recognized by five melanoma-specific human cytotoxic T cell lines. Science. 1994;264:716–9. doi: 10.1126/science.7513441. [DOI] [PubMed] [Google Scholar]
- 8.Houghton AN. Cancer antigens: immune recognition of self and altered self. J Exp Med. 1994;180:1–4. doi: 10.1084/jem.180.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pardoll DM. Tumour antigens. A new look for the 1990s. Nature. 1994;369:357. doi: 10.1038/369357a0. [DOI] [PubMed] [Google Scholar]
- 10.Kawakami Y, Eliyahu S, Delgado CH, et al. Cloning of the gene coding for a shared human melanoma antigen recognized by autologous T cells infiltrating into tumor. Proc Natl Acad Sci USA. 1994;91:3515–9. doi: 10.1073/pnas.91.9.3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawakami Y, Eliyahu S, Delgado CH, et al. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc Natl Acad Sci USA. 1994;91:6458–62. doi: 10.1073/pnas.91.14.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang RF, Robbins PF, Kawakami Y, Kang XQ, Rosenberg SA. Identification of a gene encoding a melanoma tumor antigen recognized by HLA-A31-restricted tumor-infiltrating lymphocytes. J Exp Med. 1995;181:799–804. doi: 10.1084/jem.181.2.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Topalian SL, Rivoltini L, Mancini M, et al. Human CD4+ T cells specifically recognize a shared melanoma-associated antigen encoded by the tyrosinase gene. Proc Natl Acad Sci USA. 1994;91:9461–5. doi: 10.1073/pnas.91.20.9461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawakami Y, Eliyahu S, Sakaguchi K, et al. Identification of the immunodominant peptides of the MART-1 human melanoma antigen recognized by the majority of HLA-A2-restricted tumor infiltrating lymphocytes. J Exp Med. 1994;180:347–52. doi: 10.1084/jem.180.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alexander RB, Rosenberg SA. Adoptively transferred tumor-infiltrating lymphocytes can cure established metastatic tumor in mice and persist long-term in vivo as functional memory T lymphocytes. J Immunother. 1991;10:389–97. doi: 10.1097/00002371-199112000-00001. [DOI] [PubMed] [Google Scholar]
- 16.Matsumura T, Sussman JJ, Krinock RA, Chang AE, Shu S. Characteristics and in vivo homing of long-term T-cell lines and clones derived from tumor-draining lymph nodes. Cancer Res. 1994;54:2744–50. [PubMed] [Google Scholar]
- 17.Barth RJ, Jr, Mule JJ, Spiess PJ, Rosenberg SA. Interferon gamma and tumor necrosis factor have a role in tumor regressions mediated by murine CD8+ tumor-infiltrating lymphocytes. J Exp Med. 1991;173:647–58. doi: 10.1084/jem.173.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jicha DL, Mule JJ, Rosenberg SA. Interleukin 7 generates antitumor cytotoxic T lymphocytes against murine sarcomas with efficacy in cellular adoptive immunotherapy. J Exp Med. 1991;174:1511–5. doi: 10.1084/jem.174.6.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rivoltini L, Kawakami Y, Sakaguchi K, et al. Induction of tumor-reactive CTL from peripheral blood and tumor-infiltrating lymphocytes of melanoma patients by in vitro stimulation with an immunodominant peptide of the human melanoma antigen MART-1. J Immunol. 1995;154:2257–65. [PubMed] [Google Scholar]
- 20.Bellone M, Iezzi G, Manfredi AA, Protti MP, Dellabona P, Casorati G, Rugarli C. In vitro priming of cytotoxic T lymphocytes against poorly immunogenic epitopes by engineered antigen-presenting cells. Eur J Immnnol. 1994;24:2691–8. doi: 10.1002/eji.1830241118. [DOI] [PubMed] [Google Scholar]
- 21.van der Bruggen P, Bastin J, Gajewski T, et al. A peptide encoded by human gene MAGE-3 and presented by HLA-A2 induces cytolytic T lymphocytes that recognize tumor cells expressing MAGE-3. Eur J Immunol. 1994;24:3038–43. doi: 10.1002/eji.1830241218. [DOI] [PubMed] [Google Scholar]
- 22.Yoshino I, Goedegebuure PS, Peoples GE, et al. HER2/neu-derived peptides are shared antigens among human non-small cell lung cancer and ovarian cancer. Cancer Res. 1994;54:3387–90. [PubMed] [Google Scholar]
- 23.Skipper J, Stauss HJ. Identification of two cytotoxic T lymphocyte-recognized epitopes in the ras protein. J Exp Med. 1993;177:1493–8. doi: 10.1084/jem.177.5.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brattain MG, Strobel-Stevens J, Fine D, Webb M, Sarrif AM. Establishment of mouse colonic carcinoma cell lines with different metastatic properties. Cancer Res. 1980;40:2142–6. [PubMed] [Google Scholar]
- 25.Wang M, Bronte V, Chen PW, et al. Active immunotherapy of cancer with a non-replicating recombinant fowlpox virus encoding a model tumor-associated antigen. J Immunol. 1995;154:4685–92. [PMC free article] [PubMed] [Google Scholar]
- 26.Smith GL, Levin JZ, Palese P, Moss B. Synthesis and cellular location of the ten influenza polypeptides individually expressed by recombinant vaccinia viruses. Virology. 1987;160:336–45. doi: 10.1016/0042-6822(87)90004-3. [DOI] [PubMed] [Google Scholar]
- 27.Eisenlohr LC, Bacik I, Bennink JR, Bernstein K, Yewdell JW. Expression of a membrane protease enhances presentation of endogenous antigens to MHC class I-restricted T lymphocytes. Cell. 1992;71:963–72. doi: 10.1016/0092-8674(92)90392-p. [DOI] [PubMed] [Google Scholar]
- 28.Smith GL, Levin JZ, Palese P, Moss B. Synthesis and cellular location of the ten influenza polypeptides individually expressed by recombinant vaccinia viruses [published erratum appears in Virology 1988;163(1):259] Virology. 1987;160:336–45. doi: 10.1016/0042-6822(87)90004-3. [DOI] [PubMed] [Google Scholar]
- 29.Restifo NP, Bacik I, Irvine KR, et al. Antigen processing in vivo and the elicitation of primary CTL responses. J Immunol. 1995;154:4414–22. [PMC free article] [PubMed] [Google Scholar]
- 30.Restifo NP, Esquivel F, Kawakami Y, et al. Identification of human cancers deficient in antigen processing. J Exp Med. 1993;177:265–72. doi: 10.1084/jem.177.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science. 1986;233:1318–21. doi: 10.1126/science.3489291. [DOI] [PubMed] [Google Scholar]
- 32.Mule JJ, Yang JC, Lafreniere R, Shu S, Rosenberg SA. Identification of cellular mechanisms operational in vivo during the regression of established pulmonary metastases by the systemic administration of high-dose recombinant interleukin-2. J Immunol. 1987;139:285–94. [PubMed] [Google Scholar]
- 33.Prehn RT, Main JM. Immunity to methylcholanthrene-l induced sarcomas. J Natl Cancer Inst. 1957;18:769–81. [PubMed] [Google Scholar]
- 34.Rosenberg SA, Lotze MT, Muul LM, et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N Engl J Med. 1985;313:1485–92. doi: 10.1056/NEJM198512053132327. [DOI] [PubMed] [Google Scholar]
- 35.Rosenberg SA, Lotze MT, Muul LM, et al. A progress report on the treatment of 157 patients with advanced cancer using lymphokine activated killer cells and interleukin-2 or high dose interleukin-2 alone. N Engl J Med. 1987;316:889–97. doi: 10.1056/NEJM198704093161501. [DOI] [PubMed] [Google Scholar]
- 36.Rosenberg SA, Packard BS, Aebersold PM, et al. Use of tumor infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. Preliminary report. N Engl J Med. 1988;319:1676–80. doi: 10.1056/NEJM198812223192527. [DOI] [PubMed] [Google Scholar]
- 37.Rosenberg SA, Aebersold PM, Cornetta K, et al. Gene transfer into humans: immunotherapy of patients with advanced melanoma using tumor infiltrating lymphocytes modified by retroviral gene transduction. N Engl J Med. 1990;323:570–8. doi: 10.1056/NEJM199008303230904. [DOI] [PubMed] [Google Scholar]
- 38.Kantor J, Abrams S, Irvine K, Snoy P, Kaufman H, Schlom J. Specific immunotherapy using a recombinant vaccinia virus expressing human carcinoembryonic antigen. Ann NY AcadSci. 1993;690:370–3. doi: 10.1111/j.1749-6632.1993.tb44034.x. [DOI] [PubMed] [Google Scholar]
- 39.Kundig TM, Bachmann MF, Lefrancois L, Puddington L, Hengartner H, Zinkernagel RM. Nonimmunogenic tumor cells may efficiently restimulate tumor antigen-specific cytotoxic T cells. J Immunol. 1993;150:4450–6. [PubMed] [Google Scholar]
- 40.Golumbek PT, Lazenby AJ, Levitsky HI, et al. Treatment of established renal cancer by tumor cells engineered to secrete interleukin-4. Science. 1991;254:713–6. doi: 10.1126/science.1948050. [DOI] [PubMed] [Google Scholar]
- 41.Connor J, Bannerji R, Saito S, Heston W, Fair W, Gilboa E. Regression of bladder tumors in mice treated with interleukin 2 gene-modified tumor cells [published erratum appears in J Exp Med 1993;177(6):1831] J Exp Med. 1993;177:1127–34. doi: 10.1084/jem.177.4.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Greenberg PD. Adoptive T cell therapy of tumors: mechanisms operative in the recognition and elimination of tumor cells. Adv Immunol. 1991;49:281–355. doi: 10.1016/s0065-2776(08)60778-6. [DOI] [PubMed] [Google Scholar]
- 43.Ressing ME, Sette A, Brandt RM, et al. Human CTL epitopes encoded by human papillomavirus type 16 E6 and E7 identified through in vivo and in vitro immunogenicity studies of HLA-A*0201-binding peptides. J Immunol. 1995;154:5934–43. [PubMed] [Google Scholar]
- 44.Trivedi P, Hu LF, Chen F, et al. Epstein–Barr virus (EBV)-encoded membrane protein LMPI from a nasopharyngeal carcinoma is non-immunogenic in a murine model system, in contrast to a B cell-derived homologue. Eur J Cancer. 1994;30A:84–8. doi: 10.1016/s0959-8049(05)80024-3. [DOI] [PubMed] [Google Scholar]
- 45.Chen W, Peace DJ, Rovira DK, You SG, Cheever MA. T-cell immunity to the joining region of p210BCR-ABL protein. Proc Natl Acad Sci USA. 1992;89:1468–72. doi: 10.1073/pnas.89.4.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Townsend A, Ohlen C, Rogers M, Edwards J, Mukherjee S, Bastin J. Source of unique tumour antigens. Nature. 1994;371:662. doi: 10.1038/371662a0. [DOI] [PubMed] [Google Scholar]
- 47.Bicknell DC, Rowan A, Bodmer WF. Beta 2-microglobulin gene mutations: a study of established colorectal cell lines and fresh tumors. Proc Natl Acad Sci USA. 1994;91:4751–6. doi: 10.1073/pnas.91.11.4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.D’Urso CM, Wang ZG, Cao Y, Tatake R, Zeff RA, Ferrone S. Lack of HLA class I antigen expression by cultured melanoma cells FO-1 due to a defect in B2m gene expression. J Clin Invest. 1991;87:284–92. doi: 10.1172/JCI114984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maio M, Altomonte M, Tatake R, Zeff RA, Ferrone S. Reduction in susceptibility to natural killer cell-mediated lysis of human FO-1 melanoma cells after induction of HLA class I antigen expression by transfection with B2m gene. J Clin Invest. 1991;88:282–9. doi: 10.1172/JCI115289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marincola FM, Shamamian P, Alexander RB, et al. Loss of HLA haplotype and B locus down-regulation in melanoma cell lines. J Immunol. 1994;153:1225–37. [PubMed] [Google Scholar]
- 51.Lehmann F, Marchand M, Hainaut P, et al. Differences in the antigens recognized by cytolytic T cells on two successive metastases of a melanoma patient are consistent with immune selection. Eur J Immunol. 1995;25:340–7. doi: 10.1002/eji.1830250206. [DOI] [PubMed] [Google Scholar]
- 52.Pardoll DM. Cancer vaccines. Immunol Today. 1993;14:310–6. doi: 10.1016/0167-5699(93)90051-L. [DOI] [PubMed] [Google Scholar]