

Abstract
The roles of the Pseudomonas aeruginosa-derived pigment pyocyanin
(PYO) as an oxidant and activator of the proinflammatory transcription factor
NF-κB were tested in a cystic fibrosis (CF) airway epithelial cell line,
CF15. 100 μm PYO on its own had no effect or only small effects
to activate NF-κB (<1.5-fold), but PYO synergized with the TLR5
agonist flagellin. Flagellin activated NF-κB 4–20-fold, and PYO
increased these activations >2.5-fold. PYO could have synergized with
flagellin to activate NF-κB by redox cycling with NADPH, generating
superoxide
(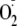 ),
hydrogen peroxide (H2O2), and hydroxyl radical
(HO.). Cytosol-targeted, redox-sensitive roGFP1 and imaging
microscopy showed that 1–100 μm PYO oxidized CF15 cytosol
redox potential (Ψcyto) from -325 mV (control) to -285 mV.
),
hydrogen peroxide (H2O2), and hydroxyl radical
(HO.). Cytosol-targeted, redox-sensitive roGFP1 and imaging
microscopy showed that 1–100 μm PYO oxidized CF15 cytosol
redox potential (Ψcyto) from -325 mV (control) to -285 mV.
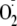 (derived from
(derived from
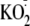 .
or xanthine + xanthine oxidase) or H2O2 oxidized
Ψcyto dose-dependently but did not activate NF-κB, even
in the presence of flagellin, and 400 μm
H2O2 inhibited NF-κB. Overexpressing intracellular
catalase decreased effects of PYO and H2O2 on
Ψcyto but did not affect flagellin + PYO-activated NF-κB.
Catalase also reversed the inhibitory effects of H2O2 on
NF-κB. The HO. scavenger DMSO did not alter the effects of
PYO on Ψcyto and NF-κB. The synergistic NF-κB
activation was calcium-independent. Thus, in the presence of flagellin, PYO
activated NF-κB through a redox- and calcium-independent effect.
.
or xanthine + xanthine oxidase) or H2O2 oxidized
Ψcyto dose-dependently but did not activate NF-κB, even
in the presence of flagellin, and 400 μm
H2O2 inhibited NF-κB. Overexpressing intracellular
catalase decreased effects of PYO and H2O2 on
Ψcyto but did not affect flagellin + PYO-activated NF-κB.
Catalase also reversed the inhibitory effects of H2O2 on
NF-κB. The HO. scavenger DMSO did not alter the effects of
PYO on Ψcyto and NF-κB. The synergistic NF-κB
activation was calcium-independent. Thus, in the presence of flagellin, PYO
activated NF-κB through a redox- and calcium-independent effect.
Pseudomonas aeruginosa is commonly present in lungs of cystic fibrosis (CF)2 and immunocompromised patients (1, 2). The bacterium secretes a large number of products that contribute to virulence. These include type III-secreted exotoxins that disrupt the cytoskeleton and lyse the cells, as well as proteases, phospholipase, rhamnolipids, and hemolysin (3–8). In addition, P. aeruginosa produce and secrete the blue pigment pyocyanin (PYO), which is found in the sputum of patients with CF and bronchiectasis at concentrations up to 100 μm; this is responsible for the blue-green color often observed in CF sputum (4, 5, 9, 10). PYO-deficient P. aeruginosa elicits less mortality in P. aeruginosa-mediated burn-sepsis model in mice, and PYO appears to be important for persistence of P. aeruginosa in lungs of CF patients (3, 4, 11). PYO has a multitude of effects on the physiology of epithelial cells, including inhibition or alteration of antioxidant enzymes (12, 13), ciliary function (14), cellular metabolism, and organelle H+ v-ATPase (2, 15). A key aspect of PYO pathology may result from its ability to trigger inflammation leading to the influx of neutrophils to the P. aeruginosa-infected region; PYO stimulates ICAM-1 and IL8 production on its own and also synergizes with IL1β and TNFα in stimulating IL8 production (16–18). The resulting IL8 production triggers polymorphonuclear leukocyte infiltration. The polymorphonuclear leukocytes are critical for fighting infections through production of reactive oxygen species (ROS) and proteases, but these products also contribute to the tissue destruction characteristic of CF.
PYO activation of IL8 production may occur through effects on cellular
signaling leading to activation of the transcription factors AP-1,
NF-IL6/C-EBP, and/or NF-κB, which control IL8 production
(19). It is widely assumed
that the effects of PYO on signaling are mediated at least in part through its
ability to redox cycle with cellular NADPH and/or GSH leading to the
production of ROS (9,
13,
20,
21) and oxidation of the
cytosol and/or mitochondria
(21,
22). Experiments using the
electron spin resonance method showed that PYO caused the production of
superoxide
(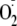 )
but not hydroxyl radical (HO.), indicating that
)
but not hydroxyl radical (HO.), indicating that
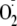 and, through the action of superoxide dismutase, H2O2
might contribute to proinflammatory signaling by PYO
(16). In addition, the
antioxidant N-acetylcysteine and the HO. scavenger DMSO
reduced the proinflammatory effects of PYO, consistent with PYO triggering
inflammatory processes through its pro-oxidant effects
(18). However, none of these
experiments made direct, quantitative measurements on the proposed effect of
PYO to produce ROS and oxidize the cytosol. In addition, there were no direct
tests of the role of this hypothesized oxidation on the activation of
inflammatory signaling in airway cells. Furthermore, because PYO synergizes
with IL1β in triggering IL8 production and IL1β and flagellin
trigger similar signaling
(23), it was predicted that
PYO would similarly synergize with P. aeruginosa flagellin in
activation of inflammatory signaling and production of IL8.
and, through the action of superoxide dismutase, H2O2
might contribute to proinflammatory signaling by PYO
(16). In addition, the
antioxidant N-acetylcysteine and the HO. scavenger DMSO
reduced the proinflammatory effects of PYO, consistent with PYO triggering
inflammatory processes through its pro-oxidant effects
(18). However, none of these
experiments made direct, quantitative measurements on the proposed effect of
PYO to produce ROS and oxidize the cytosol. In addition, there were no direct
tests of the role of this hypothesized oxidation on the activation of
inflammatory signaling in airway cells. Furthermore, because PYO synergizes
with IL1β in triggering IL8 production and IL1β and flagellin
trigger similar signaling
(23), it was predicted that
PYO would similarly synergize with P. aeruginosa flagellin in
activation of inflammatory signaling and production of IL8.
The present experiments were therefore designed to test the hypothesis that
PYO activates inflammatory signaling by triggering production of the reactive
oxygen species
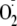 ,
H2O2, and HO. that then oxidize the cytosol
and activate NF-κB. The experiments also tested whether this potentially
proinflammatory effect was synergized in the presence of flagellin, the key
product required for P. aeruginosa activation of inflammatory
responses in airway epithelial cells
(24,
25). The general approach was
to measure redox potentials in the cytosol (Ψcyto) of the CF
nasal cell line CF15 using genetically targeted, redox-sensitive GFP (roGFP1)
(26–28)
and ratiometric imaging microscopy during treatment with PYO,
H2O2, and
,
H2O2, and HO. that then oxidize the cytosol
and activate NF-κB. The experiments also tested whether this potentially
proinflammatory effect was synergized in the presence of flagellin, the key
product required for P. aeruginosa activation of inflammatory
responses in airway epithelial cells
(24,
25). The general approach was
to measure redox potentials in the cytosol (Ψcyto) of the CF
nasal cell line CF15 using genetically targeted, redox-sensitive GFP (roGFP1)
(26–28)
and ratiometric imaging microscopy during treatment with PYO,
H2O2, and
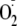 to compare changes in redox potential to NF-κB activation measured under
the same conditions using both Western blot analyses of NF-κB (p65), IKK
and IκB, and NF-κB promotor-driven luciferase assays. The role of
H2O2 in these processes was tested by overexpressing
catalase (catalyzes hydrolysis of H2O2) and the role of
HO. by adding the HO. scavenger DMSO. Finally, because
PYO activation of NF-κB appeared to be independent of redox changes and
previous experiments showed a potential role for cytosolic [Ca2+],
we tested whether increases in cytosolic [Ca2+] were involved.
to compare changes in redox potential to NF-κB activation measured under
the same conditions using both Western blot analyses of NF-κB (p65), IKK
and IκB, and NF-κB promotor-driven luciferase assays. The role of
H2O2 in these processes was tested by overexpressing
catalase (catalyzes hydrolysis of H2O2) and the role of
HO. by adding the HO. scavenger DMSO. Finally, because
PYO activation of NF-κB appeared to be independent of redox changes and
previous experiments showed a potential role for cytosolic [Ca2+],
we tested whether increases in cytosolic [Ca2+] were involved.
EXPERIMENTAL PROCEDURES
Reagents—Unless otherwise specified, reagents and chemicals were obtained from Sigma. Thapsigargin was dissolved in DMSO at 1.0 mm and then dissolved into solutions at 1–10 μm; these concentrations yielded similar effects on cellular functions.
Pyocyanin—PYO was purchased from Color Your Enzyme (Bath, Ontario, Canada). PYO was dissolved in PBS, pH 7.4, at 10 mm and diluted into medium or Ringer's solution as mentioned in the text. To ensure complete solubility, we also dissolved PYO in DMSO, which was then added to the cells at the appropriate concentrations. Total [DMSO] in these experiments was 0.5%, which did not affect cellular responses. We observed no differences in responses to PYO that had been dissolved initially in PBS, Ringer's, or DMSO.
Flagellin—P. aeruginosa flagellin (10-3 g/ml in PBS, pH 7.4; Inotek, Beverly, MA) was stored at -20 °C and diluted from the stock into the incubation media at concentrations stated in the text. This solution was vortexed vigorously and heated to 37 °C before adding to the solutions to ensure dispersal as monomers. As described by Inotek, recombinant flagellin is expressed with tags in Escherichia coli and purified to >95% homogeneity by SDS-PAGE. Previous experiments showed that lipopolysaccharide contamination of this preparation is small and cannot account for effects of flagellin to activate NF-κB (29). Flagellin isolated from Salmonella typhimurium (InvivoGen, San Diego) gave similar results. Flagellin was sensitive to freeze-thaw cycles, so comparisons among different treatment regimes were always performed with one batch of flagellin.
Solutions—In experiments to measure cytosolic redox potentials and Ca2+i, epithelial cells were incubated in Ringer's solutions containing (in mm) the following: 145 NaCl, 1.2 MgSO4, 2 CaCl2, 2.4 K2HPO4, 0.6 KH2PO4, 10 HEPES, and 10 glucose, pH 7.4.
Tissue Culture—Cystic fibrosis airway cells JME/CF15, termed CF15 throughout this paper (30), a continuous SV40 large T antigen-transformed human nasal epithelial cell line homozygous for ΔF508 cystic fibrosis transmembrane regulator, were cultured in Dulbecco's modified Eagle's medium/F-12 media supplemented with 10% fetal bovine serum, 2 mm l-glutamine, 200 milliunits/ml penicillin, 200 μg/ml streptomycin, 10 ng/ml epidermal growth factor, 1 μm hydrocortisone, 5 μg/ml insulin, 5 μg/ml transferrin, 30 nm triiodothyronine, 180 μm adenine, and 5.5 μm epinephrine. Cells were passaged at a 1:5–1:10 dilution, and the remaining cell suspension was seeded directly onto 18-mm diameter cover glasses or onto 48-well or 24-well tissue culture plates (BD Biosciences).
Measurement of Cai—High concentrations of PYO have been shown to elicit small increases in cytosolic [Ca2+], Cai, through effects to release Ca2+ from internal stores (presumably the endoplasmic reticulum) (20). Because increases in Cai potently synergize with flagellin in stimulating NF-κB in airway epithelial cells (19), this Cai-stimulating effect of PYO could be particularly important when the bacteria also produce and release flagellin into the airway surface liquid. We therefore tested whether PYO or H2O2 triggered changes in Cai in CF15 cells. Cells grown on cover glasses were incubated with growth media containing 2 μm fura-2/AM for 40–60 min at room temperature and then washed three times with Ringer's solution to remove the extra dye. Fura-2-loaded cells were mounted onto a chamber on the stage of the imaging microscope and maintained at room temperature. Treatments with agonists were made by diluting stock solutions into Ringer's solution at the concentrations stated in the text. Fluorescence ratio imaging measurements of cytosolic Cai were performed using methods that have been reported previously (19, 31). Briefly, a Nikon Diaphot inverted microscope was used with a 40× Neofluar objective (1.4 NA). A CCD camera collected emission (>510 nm) images during alternate excitation at 350 ± 5 and 380 ± 5 nm using a filter wheel (Lambda-10, Sutter Instruments, Novato, CA). Axon Imaging Workbench 4.0 (Axon Instruments, Foster City, CA) controlled both filters and collection of data. Calibration of fura-2 signals was performed as described by Grynkiewicz et al. (32). All images were corrected for background (region without cells).
Confocal Microscopy—Expression of roGFP1 in transiently transfected CF15 cells was analyzed on a Solamere spinning disk confocal microscope with excitation at 488 nm. Cells were bathed in Ringer's solution containing 500 μm DTT to increase roGFP1 fluorescence intensity for excitation at 488 nm. Images were obtained using a 515 nm long pass emission filter and ×40 objective. Differential interference contrast images were also recorded to correlate cell morphology and roGFP1 fluorescence. Images were overlaid using Adobe Photoshop.
Redox Potential Measurements Using roGFP1 and Imaging Microscopy—Measurements of cytosolic redox potentials in CF15 cells were performed as described recently (28). Briefly, CF15 cells grown on cover glasses were transiently transfected with plasmids coding for a redox-sensitive GFP mutant roGFP1 (26, 27) using the Effectene transfection reagent according to the manufacturer's protocol (Qiagen, Valencia, CA). roGFP1-expressing cells were bathed in Ringer's solution and mounted in a chamber on the stage of a Nikon Diaphot microscope with a ×40 Neofluar objective (1.4 NA). Ratiometric imaging was performed using a CCD camera, filter wheel (Lambda-10, Sutter Instruments, Novato, CA), and Axon Imaging Workbench 4 (Axon Instruments, Foster City, CA) to collect emission (>510 nm) images during alternate excitation at 385 ± 5 and 474 ± 5 nm. Cells were exposed to the various treatments in Ringer's solution, and roGFP1 ratios were recorded over time. Alternatively, ratios from multiple regions each containing ≥10 cells were recorded at the beginning of experiments and after 150 min of incubation with Ringer's solution, 100 μm PYO, or 400 μm H2O2. At this time the ratios reached a steady state, i.e. no further change in roGFP1 ratio.
Calibration of the roGFP1 ratios in terms of cytosolic redox potentials was performed using a protocol that has been described previously (27, 28). Briefly, at the end of each experiment roGFP1 385:474 ratios were recorded during maximal oxidation by treatment with 10 mm H2O2 and then during maximal reduction by treatment with 10 mm DTT. Images were background-subtracted, and normalized roGFP1 385:474 ratios were averaged and converted to redox potentials (mV) using an in situ calibration curve that has been published previously (28). The calibration curve was generated by first preparing the standard solutions consisting of trans-4,5-dihydroxy-1,2-dithiane and dl-DTT under nitrogen atmosphere to exclude oxidation by air. Then roGFP1-expressing cells were permeabilized by adding 1–5 μg/ml digitonin for 5–10 min. The permeabilized cells were then perfused with different DTT standard solutions covering redox potentials from -330 to -195 mV at pH 7 (calculated using the Nernst equation (27)), and 385:474 nm excitation ratios were recorded. The roGFP1 excitation ratios were normalized to the values measured using 10 mm DTTred as 0% oxidation and 10 mm H2O2 as 100% oxidation, and the normalized ratios were plotted against the calculated redox potentials of the DTT standard solutions to generate the curve (28) that was used for calibrating the experiments.
Phosphorylation of NF-κB p65, IκBα, and IKK—Immunoblot analysis was used to assay activity of NF-κB p65, IκBα, and IKKα and phosphorylated NFκB p65, IκBα, and IKKα/β. Cells were treated with pyocyanin, H2O2, and/or flagellin for 20 or 40 min and then lysed in M-PER mammalian protein extraction reagent (Pierce) containing 5 μg/ml leupeptin, 5 μg/ml pepstatin, 1 mm phenylmethylsulfonyl fluoride, and 50 nm calyculin A. Protein concentrations were determined using Bradford reagent (Bio-Rad). Immunoblot analysis was performed by first separating protein (10–50 μg/lane) electrophoretically using SDS-PAGE and subsequently transferring to polyvinylidene difluoride membranes. Membranes were blocked (5% nonfat dried milk) in 20 mm Tris-HCl, pH 7.5, 150 mm NaCl, and 0.05% Tween 20 followed by incubation with specific antibodies. Primary antibodies (diluted 1:1000) for NF-κB p65, phospho-p65 (serine 536), IκBα, phospho-IκBα (serine 32), IKKα, and phospho-IKKα/β (serine 176/180) were purchased from Cell Signaling (Danvers, MA). Blots were first probed for phosphorylated proteins. The membranes were then stripped and probed with antibodies for nonphosphorylated proteins. Immunostaining of β-actin was performed to control for the amount of protein in each sample. Binding of primary antibodies was visualized by enhanced chemiluminescence using horseradish peroxidase-conjugated secondary antibodies (1:2000) and Renaissance Chemiluminescence Reagent Plus (PerkinElmer Life Sciences).
NF-κB-Luciferase Adenovirus and NF-κB Activation Assays— A recombinant adenoviral vector expressing a luciferase reporter gene driven by NF-κB transcriptional activation (Ad5HSVNF-κB luciferase, termed adv-NF-κB-luc) was used for studies to determine the effects of flagellin, PYO, and/or various other potential oxidants. This vector contained the luciferase gene driven by four tandem copies of the NF-κB consensus sequence (33). Recombinant adenoviral stocks (6 × 1010 plaque-forming units) were stored in 10 mm Tris with 20% glycerol at -80 °C. The virus was added to CF15 cells at 100 multiplicities of infection and returned to the incubator for 24 h, followed by washing of the adenovirus and further growth for 24 h. Control experiments with a β-galactosidase- or enhanced GFP-expressing adenovirus showed that this infection protocol generated expression in 75–100% of the cells (28). Adenoviral constructs were obtained from Gene Transfer Vector Core (University of Iowa, Iowa City). Cells were washed with fresh medium and exposed to the various agonists for 4 h. Cells were then washed and processed using the luciferase assay system with Reporter Lysis Buffer (Promega, Madison, WI) to measure NF-κB-mediated transcriptional induction according to the manufacturer's protocol. Luciferase activity (relative light units) was analyzed with a TD-20/20 luminometer (Turner Designs, Sunnyvale, CA) in at least triplicate for each sample and normalized to the protein concentration (Bradford assay, Bio-Rad). These averages were then expressed relative to the average control value in the epithelial cells, which was set equal to 1.0.
Effects of Catalase on Redox and NF-κB—Overexpression of human catalase was obtained by overnight incubation of CF15 cells with an adenovirus coding for human catalase (100 multiplicities of infection). To control for adenoviral infection cells were infected with an adenovirus coding for β-galactosidase (100 multiplicities of infection). Cytosolic redox potentials were analyzed in CF15 cells after transfection with roGFP1-coding plasmids (see above), and transcriptional activation of NF-κB was studied by co-infection of cells with adv-catalase adv-NF-κB-luc in the presence of medium, [H2O2], or PYO with or without flagellin.
Statistics—Data have been presented as original values or as means ± S.D.; n refers to the number of averaged experiments. Significance was tested using t test for paired or unpaired samples as appropriate. Calculated p values <0.05 were considered significant.
RESULTS
Effects of PYO and Flagellin on NF-κB—Inflammatory signaling leading to the activation of NF-κB was assayed in CF15 airway epithelia by measuring phosphorylation/degradation of involved mediators in the presence or absence of flagellin (0.1 μg/ml, a submaximal dose; see Ref. 19) and PYO (100 μm, concentration found in sputum of CF patients; see Ref. 10). NF-κB signaling in many cells is characterized by kinase cascade-induced phosphorylations of IKK, p65, and IκB, subsequent degradation of IκB, and migration of p65 into the nucleus to activate production of proinflammatory cytokines and chemokines. Fig. 1A shows Western blots characterizing the phosphorylation of IKKα/β, p65, and IκBα upon treatment with flagellin, PYO, and with both bacterial products for 20 and 40 min. These times were chosen because preliminary experiments using immunofluorescence imaging microscopy showed that NF-κB (p65) migration into the nucleus during flagellin treatment occurred maximally after 45 min, so that activation/phosphorylation of IKKα/β, p65, and IκBα was expected to occur before this time. Treatment of cells with flagellin increased phosphorylation of IKKα/β, p65, and IκB. After 40 min of treatment, loss/degradation of IκB was observed. Treatment with PYO also increased phosphorylation of IKKα/β, p65, and IκB, although effects were less pronounced compared with those of flagellin. PYO + flagellin increased phosphorylation of p65 and IKKα/β compared with either PYO or flagellin alone, but other effects of PYO + flagellin were similar to those elicited by flagellin. The amount of nonphosphorylated p65 was not affected by either PYO or flagellin. Similar signals for β-actin for every experiment suggested that equal amounts of cell lysates were analyzed for each treatment. These data were consistent with flagellin and PYO both activating NF-κB signaling, and the effects of PYO + flagellin to phosphorylate IKKα/β and p65 were larger than either stimulant alone.
FIGURE 1.

Synergistic activation of NF-κB by PYO + flagellin in CF15 cells. A, CF15 cells grown to confluency in wells were exposed to medium, 100 μm PYO, 10-7 g/ml flagellin, or PYO + flagellin (Flag). Activation of IKKα/β, p65, and IκBα was analyzed after 20 and 40 min by observing protein phosphorylation and degradation using specific antibodies as indicated next to each panel. B, CF15 cells grown in wells were infected with NF-κB-luciferase adenovirus and then exposed to different [PYO] or to [PYO] + 10-7 g/ml flagellin for 4 h, followed by measurements of luciferase activity, expressed relative to controls (=1.0). Data are averages ± S.D. (n ≥ 5 experiments for each point). *, p < 0.05 compared with control; **, p < 0.05 compared with flagellin. C, CF15 cells grown to confluency on permeable filter inserts were infected with NF-κB-luciferase adenovirus and then exposed to either apical or basal 100 μm PYO + 10-7 g/ml flagellin or to apical flagellin and basal PYO for 4 h, followed by measurements of luciferase activity, expressed relative to controls (=1.0). Data are averages ± S.D. (n ≥ 3 experiments for each point). *, p < 0.05 compared with control; **, p < 0.05 compared with flagellin. Effect of apical flagellin + basal PYO was compared with apical flagellin.
Further experiments utilized NF-κB-regulated luciferase to test the effects of PYO and flagellin on NF-κB signaling. CF15 airway epithelia cells expressing the luciferase reporter gene driven by NF-κB transcriptional activation were exposed to increasing concentrations of PYO in the absence and presence of flagellin (0.1 μg/ml) (Fig. 1B). PYO on its own had a small stimulatory effect on NF-κB compared with untreated controls only at the highest concentration tested, 100 μm. Flagellin increased NF-κB-luciferase 5.5-fold over control, and this increase was further amplified when cells were treated with both flagellin + PYO. Physiologically relevant concentrations of PYO of 50 and 100 μm (as described for sputum of CF patients; see Ref. 10) synergized with flagellin in activating NF-κB 10- and 15.7-fold, respectively. Experiments were also performed to test the sidedness of these responses. Addition of flagellin and PYO to the apical side of cells grown to confluency on filter inserts activated NF-κB with similar synergism compared with basal addition, although the magnitude of activation by flagellin and PYO was less when added to the basal side, perhaps because of restricted access to the membrane (Fig. 1C). Apical addition of flagellin and basal addition of PYO also yielded a synergistic activation of NF-κB (13.5-fold increase) compared with flagellin (5.5-fold) and PYO (1.2-fold). These results showed that flagellin and PYO synergized in activating NF-κB when they were exposed to either apical or basal side or even when flagellin and PYO were on opposite sides of the epithelium.
Effects of PYO, H2O2, and
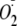 on
NF-κB and Cytosolic Redox Potential—As proposed previously
(34), PYO is expected to react
with cytosolic reducing agents like NADPH to generate
on
NF-κB and Cytosolic Redox Potential—As proposed previously
(34), PYO is expected to react
with cytosolic reducing agents like NADPH to generate
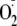 ,
which would be expected to dismutate to H2O2 by cellular
superoxide dismutase. Both H2O2 and
,
which would be expected to dismutate to H2O2 by cellular
superoxide dismutase. Both H2O2 and
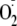 would be expected to oxidize the cytosolic redox potential. Expression of a
redox-sensitive GFP mutant roGFP1 allowed the cytosol-specific analysis of the
redox potential (Ψcyto) in response to PYO. Typical images of
roGFP1 expressed in CF15 cells showed that the sensor was localized throughout
the cytosol and also in the nucleus (Fig.
2A). We detected no differences in the redox properties
of the cytosol and nucleus of CF15 cells, consistent with previous results
(26,
28). Changes in the 385:474
excitation ratio of roGFP1 fluorescence were converted to Ψcyto
(in mV) by applying a recently published calibration curve
(28). As shown in
Fig. 2B, PYO oxidized
the cytosol of CF15 cells in a dose- and time-dependent way. 10 and 100
mm PYO both caused Ψcyto to oxidize slowly over the
course of 30–45 min to new steady values. These effects of PYO were
relatively irreversible, as Ψcyto remained oxidized even after
60 min following removal of PYO from the bathing solutions for 60 min. On
average 10 and 100 μm PYO oxidized Ψcyto from
-325 to -306 mV and -285 mV, respectively
(Fig. 2C). Flagellin
did not affect Ψcyto in the absence (data not shown) or
presence of PYO (filled circle Fig.
2C).
would be expected to oxidize the cytosolic redox potential. Expression of a
redox-sensitive GFP mutant roGFP1 allowed the cytosol-specific analysis of the
redox potential (Ψcyto) in response to PYO. Typical images of
roGFP1 expressed in CF15 cells showed that the sensor was localized throughout
the cytosol and also in the nucleus (Fig.
2A). We detected no differences in the redox properties
of the cytosol and nucleus of CF15 cells, consistent with previous results
(26,
28). Changes in the 385:474
excitation ratio of roGFP1 fluorescence were converted to Ψcyto
(in mV) by applying a recently published calibration curve
(28). As shown in
Fig. 2B, PYO oxidized
the cytosol of CF15 cells in a dose- and time-dependent way. 10 and 100
mm PYO both caused Ψcyto to oxidize slowly over the
course of 30–45 min to new steady values. These effects of PYO were
relatively irreversible, as Ψcyto remained oxidized even after
60 min following removal of PYO from the bathing solutions for 60 min. On
average 10 and 100 μm PYO oxidized Ψcyto from
-325 to -306 mV and -285 mV, respectively
(Fig. 2C). Flagellin
did not affect Ψcyto in the absence (data not shown) or
presence of PYO (filled circle Fig.
2C).
FIGURE 2.

Expression of roGFP1 and PYO-induced oxidation of the cytosol and activation of NF-κB in CF15 cells. A, CF15 cells were transiently transfected with plasmids coding for cytosolic roGFP1. Confocal fluorescence image (excitation: 488 nm; emission >510 nm) overlaid on the bright field image was taken 48 h post-transfection on living cells in PBS containing 0.5 mm DTT to increase brightness at 488 nm. B, redox responses of cytosolic roGFP1 were measured during treatment of cells with 10 and 100 μm PYO in Ringer's solution. Raw data were background-subtracted and calibrated into redox potentials. Experiment was typical of five similar experiments. C, summary of Ψcyto versus [PYO] measured in the steady state in experiments like that in B. Data are averages ± S.D. (n ≥ 5 for each point). Cells that were also exposed to 10-7 g/ml flagellin (Flag) (closed circle) exhibited similar PYO-induced oxidation as cells that were not exposed to flagellin (open circles). *, p < 0.05 compared with control.
If PYO were activating NF-κB through its effect to generate
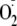 and H2O2, it was expected that exogenous addition of
these ROS would also oxidize the cytosol and activate NF-κB. Effects of
and H2O2, it was expected that exogenous addition of
these ROS would also oxidize the cytosol and activate NF-κB. Effects of
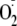 were investigated by treating CF15 cells with either xanthine + xanthine
oxidase or
were investigated by treating CF15 cells with either xanthine + xanthine
oxidase or
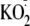 .
Xanthine (X) alone or xanthine oxidase (XO) alone (not shown) had no effect on
Ψcyto, but addition of both X + XO caused rapid oxidation of
Ψcyto by up to 50 mV (Fig.
3A). These results were consistent with the idea that
extracellular X + XO generated
.
Xanthine (X) alone or xanthine oxidase (XO) alone (not shown) had no effect on
Ψcyto, but addition of both X + XO caused rapid oxidation of
Ψcyto by up to 50 mV (Fig.
3A). These results were consistent with the idea that
extracellular X + XO generated
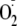 ,
which entered the cell and oxidized the cytosol. Similar oxidation occurred
during addition of
,
which entered the cell and oxidized the cytosol. Similar oxidation occurred
during addition of
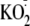 (Fig. 3B). Although
(Fig. 3B). Although
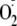 (either from X + XO or from
(either from X + XO or from
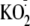 )
effectively oxidized the cytosol, it did not activate NF-κB either on
its own or when added with flagellin (Fig.
3, C and D).
)
effectively oxidized the cytosol, it did not activate NF-κB either on
its own or when added with flagellin (Fig.
3, C and D).
FIGURE 3.

Effects of superoxide
(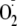 )
on cytosolic redox and NF-κB activity in CF15 cells. A,
Ψcyto was measured during treatment of cells with xanthine
(X, 200 μm) and then xanthine oxidase (XO, 30
milliunits/ml) in Ringer's solution. Experiment is typical of three similar
experiments. B, Ψcyto was measured during treatment of
cells with
)
on cytosolic redox and NF-κB activity in CF15 cells. A,
Ψcyto was measured during treatment of cells with xanthine
(X, 200 μm) and then xanthine oxidase (XO, 30
milliunits/ml) in Ringer's solution. Experiment is typical of three similar
experiments. B, Ψcyto was measured during treatment of
cells with
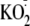 (10, 100, 1000 μm). Experiment is typical of three similar
experiments. C and D, NF-κB-expressing CF15 cells were
exposed to X + XO (C) or to
(10, 100, 1000 μm). Experiment is typical of three similar
experiments. C and D, NF-κB-expressing CF15 cells were
exposed to X + XO (C) or to
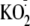 (D) ± 10-7 g/ml flagellin (Flag) for 4 h,
followed by measurements of luciferase activities, expressed relative to
controls (=1.0). Data are averages ± S.D. (n ≥ 3
experiments). **, p < 0.05 compared with flagellin.
(D) ± 10-7 g/ml flagellin (Flag) for 4 h,
followed by measurements of luciferase activities, expressed relative to
controls (=1.0). Data are averages ± S.D. (n ≥ 3
experiments). **, p < 0.05 compared with flagellin.
Similarly, if PYO were activating NF-κB through its effect to generate H2O2, exogenous addition of H2O2 should oxidize the cytosol and activate NF-κB. H2O2 dose-dependently (threshold effect at 1 μm, highest concentration applied 400 μm) oxidized Ψcyto from -325 to -250 mV (Fig. 4, A and B). Although such single additions of H2O2 effectively oxidized the cytosol, H2O2 (added either alone or in combination with flagellin) did not affect NF-κB-luciferase activity (Fig. 5A).
FIGURE 4.

Effects of H2O2 and catalase on cytosolic redox in CF15 cells. A, Ψcyto was measured during short term treatment of roGFP1-expressing cells with 1, 10, and 100 μm H2O2 in Ringer's solution. Experiment is typical of 13 similar experiments. B, summary change in Ψcyto (compared with controls) as function of [H2O2]. Average ± S.D. (n = 13 experiments); *, p < 0.05 compared with control. C, Ψcyto was measured during 2 h of treatment with 25 or 100 μm H2O2. There was a rapid increase during the first 15 min followed by slower decrease toward control levels over the course of about 100 min. D, Ψcyto was measured during repeated treatment of cells with 25 μm H2O2. Experiment is typical of three similar experiments. E, dose-dependent effects of H2O2 on Ψcyto were tested in CF15 cells infected with catalase (Ad-Cat, solid line) or β-galactosidase (Ad-lacZ, as control, dotted line) adenovirus. Data are typical of three similar experiments in each case. F, CF15 cells expressing catalase or β-galactosidase were treated with a single dose of 100 μm PYO or with repeated 30-min additions of 100 or 400 μm H2O2. Ψcyto were recorded before and after 2 h in five different regions of the cover glass each containing ≥10 different roGFP1-expressing cells. Averages ± S.D. (n = 3 experiments) for changes in Ψcyto in response to PYO or [H2O2] are shown. *, p < 0.05 compared with no catalase expression.
FIGURE 5.

Effects of flagellin, H2O2, and PYO on NF-κB activation in catalase and lacZ-expressing CF15 cells. CF15 cells infected with adenoviruses coding for catalase (cat) or β-galactosidase along with NF-κB-regulated luciferase were treated with different [H2O2] (A) or [PYO] (B) alone and in combination with flagellin (Flag) (10-7 g/ml). A, H2O2 did not affect NF-κB, except at 400 μm, which inhibited NF-κB in both controls and in the presence of flagellin. Catalase expression eliminated the inhibitory effect of 400 μm H2O2. B, 50 and 100 μm PYO slightly activated NF-κB, and these concentrations synergized with flagellin in activating NF-κB (expressed relative to control = 1.0). Data are averages ± S.D. (n = 3 experiments); *, p < 0.05 compared with control; **, p < 0.05 compared with flagellin.
Previous experiments (35) showed that H2O2 can be metabolized by cells and that repeated additions of H2O2 to the media bathing the cells were required to maintain extracellular concentrations. We therefore tested the possibility that the lack of effect of H2O2 on NF-κB was caused by transient oxidation of Ψcyto resulting from single additions of H2O2. As shown in Fig. 4C, 25 and 100 μm H2O2 both oxidized Ψcyto over the course of 15–20 min followed by a slow reduction of Ψcyto over time back toward control levels. In contrast, sequential additions of H2O2 caused Ψcyto to remain oxidized to a roughly constant level of -310 mV (10 μm H2O2), -290 mV (25 μm H2O2), and -270 mV (100 μm H2O2) (Fig. 4D).
Armed with this information on the time and concentration dependence of effects of H2O2 on redox potentials, we tested for effects on activation of NF-κB in CF15 cells. Although 0.1 μg/ml flagellin induced typical phosphorylation of IKKα/β, p65, and IκBα after 30 and 40 min (Fig. 1A), 100 μm H2O2 elicited no changes in phosphorylation of IKKα/β, p65, and IκB in the absence or presence of 0.1 μg/ml flagellin (not shown). Further tests were performed using the sequential-addition protocol (Fig. 4D) to obtain relatively constant cellular oxidative redox potentials over the 4 h required for the NF-κB-luciferase experiments. Using this protocol, H2O2 had no effect (at 25, 50, and 100 μm) or only inhibitory effects (at 400 μm) on NF-κB activity when added alone or in the presence of flagellin (Fig. 5B, open circles). 1 and 10 μm H2O2 also had small oxidizing effect on Ψcyto (Fig. 4A), but there were no effects of these concentrations of H2O2 (added either alone or in the presence of flagellin) on NF-κB (not shown).
A second approach to testing a role for H2O2 in oxidizing Ψcyto and activating NF-κB was to use an adenovirus to overexpress intracellular catalase (converts H2O2 to H2O and O2) in CF15 cells. β-Galactosidase (lacZ) adenovirus was used as a control. These cells were also infected with NF-κB-luciferase adenovirus to measure NF-κB activity or transfected with roGFP1 to measure Ψcyto. lacZ-infected (Fig. 4E) and uninfected control cells (Fig. 4A) exhibited similar changes in Ψcyto in response to H2O2. In contrast, catalase-expressing cells exhibited much smaller oxidations of Ψcyto in response to H2O2 (Fig. 4E). Similar experiments were performed using repeated additions of H2O2 over 2 h to allow comparison with the NF-κB measurements. CF15 cells expressing either LacZ or catalase and transfected with roGFP1 were treated with repeated additions of 100 or 400 μm H2O2. Catalase expression almost abolished the 50–60 mV oxidation of Ψcyto in response to 100 and 400 μm H2O2 (Fig. 4F).
As summarized in Fig. 5B, catalase expression had no significant effect on NF-κB activation of control cells and did not significantly alter responses to the following: (i) 25, 50, or 100 μm H2O2; (ii) flagellin; or (iii) flagellin plus 25, 50, or 100 μm H2O2. Importantly, catalase expression reversed the inhibitory effects of 400 μm H2O2 on NF-κB activation in the presence or absence of flagellin.
Similar experiments were performed testing PYO. CF15 cells expressing either LacZ or catalase and transfected with roGFP1 were exposed to 100 μm PYO. Catalase expression almost abolished PYO-induced oxidation of Ψcyto (Fig. 4F). These data indicated that the oxidative effect of PYO resulted from cytosolic production of H2O2 and that overexpression of catalase prevented oxidation. However, the synergistic stimulation of NF-κB in the presence of flagellin plus PYO was unaffected by intracellular catalase expression (Fig. 5C), indicating that changes in oxidation played no role in the responses.
We investigated whether PYO was activating NF-κB through effects to produce HO. by treating cells with the HO. scavenger DMSO (36). CF15 cells expressing either roGFP1 or NF-κB-luciferase were treated with 100 μm PYO or 100 μm PYO + ≥50 mm DMSO. There were no effects of DMSO on the effects of PYO to oxidize Ψcyto (Fig. 6A) or to activate NF-κB in the presence of flagellin (Fig. 6B).
FIGURE 6.

Effects of DMSO on Ψcyto and NF-κB activity in cells treated with PYO and/or flagellin. A, CF15 cells expressing roGFP1 were treated with 50 mm DMSO, 100 μm PYO, and 100 μm PYO + 50 mm DMSO. Ψcyto were recorded before and after 2 h in five different regions of the cover glass each containing ≥10 different roGFP1-expressing cells. Averages ± S.D. for changes in Ψcyto in response to PYO/DMSO are shown; *, p < 0.05 compared with nontreated control. B, summary of effects of 100 μm PYO ± 70 mm DMSO in the absence or presence of flagellin (Flag) (10-7 g/ml) on NF-κB activity (expressed relative to controls = 1.0). Averages ± S.D. *, p < 0.05 compared with control; **, p < 0.05 compared with flagellin.
Effects of PYO and Flagellin on NF-κB: Role for Cai?—Previous research showed 80–300 μm PYO triggered increases in Cai in human bronchial epithelial cells and A549 cells (20) and that increases in Cai synergized markedly with flagellin in activating NF-κB and IL8 secretion (19). We therefore tested whether the synergism noted between flagellin and PYO in activating NF-κB involved similar changes in Cai in CF15 cells. As shown in Fig. 7, 100 μm of PYO had no effect on Cai (initial Cai, 48 ± 12 nm versus Cai; after PYO treatment, 64 ± 11 nm), although a typical increase in Cai was recorded upon stimulation with the known Cai-regulating agents ATP (activates purinergic receptors) and thapsigargin (blocks Ca2+ pump in the endoplasmic reticulum, leading to loss of Ca2+ into the cytosol) (see Ref. 19). Previous experiments also showed flagellin did not alter Cai in CF15 cells (19). Therefore, the synergistic activation of NF-κB by PYO and flagellin in CF15 cells did not require changes in Cai.
FIGURE 7.

Effects of PYO, ATP, and thapsigargin on Cai in CF15 cells. CF15 cells grown to confluency on cover glasses were loaded with fura-2 and mounted in the imaging microscope for measurements of Cai. Calibrations of 350:385 ratio in terms of Cai (in nm, shown on y axis) were performed at the end of the experiment (data not shown). Bold line shows average response of ∼30 cells in the field, and lighter lines show responses of five single cells. 500-s time bar is also shown. Addition of 100 μm of PYO had no effects on Cai, whereas subsequent addition of 100 μm ATP caused a characteristically rapid spike in Cai followed by a secondary slower response. Further addition of 1 μm thapsigargin (TG) also caused a large increase in Cai. Experiments are typical of three similar experiments.
DISCUSSION
PYO Synergizes with Flagellin in Activating NF-κB—A major conclusion from these studies was that during the initial stages of exposure (0.5–4 h) PYO elicited no activation or only small activation of NF-κB on its own, but large (>2.5-fold) synergistic stimulation when flagellin was also present. Unpublished experiments have shown similar synergism between PYO and TNFα in activating NF-κB-luciferase (data not shown).3 Previous experiments showed that 24 h of exposure to PYO elicited similar activation of NF-κB in the presence of TNFα or IL1β (18). Thus, PYO elicited synergistic activation of NF-κBinthe presence of agonists that activated NF-κB. The synergy between flagellin and PYO occurred during additions to the apical or basolateral side of the monolayers and also when flagellin was added apically and PYO basally. It therefore seems likely that the synergistic interactions between PYO and flagellin occurred through interactions among cytosolic signaling pathways and not through interactions at the cell surface. Because NF-κB is a key regulator of IL8 production and secretion (19) and [PYO] in the sputum of CF patients can reach 100 μm (4, 10, 15), these and previous (4, 11) data show that PYO may be an important modulator of innate immune responses to P. aeruginosa in vivo. During chronic infections in CF, P. aeruginosa lose their flagella and become immotile, so the concentration of flagellin in the sputum will likely decrease. However, even in this condition PYO may be an important modulator of innate immune responses of airway epithelial cells because both neutrophils and macrophages release IL1β and TNFα and other proinflammatory cytokines that activate NF-κB into the ASL, and PYO may then synergize with these to augment responses of the airway epithelia (17, 18).
PYO Triggers Production of Cellular H2O2 That
Oxidizes the Cytosol—A second major conclusion was that PYO induced
the predicted oxidation of the cytosol, likely through redox cycling with
cytosolic reductants and generation of
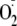 that is converted in the cells to H2O2. As determined
from measurements of Ψcyto, [PYO] as low as 1 μm
caused small oxidation of Ψcyto, and 10 and 100
μm PYO caused large and sustained oxidation of
Ψcyto by 20 and 40 mV. These quantitative measurements of
PYO-induced oxidation of the cytosol therefore confirm previous qualitative
measurements based on the use of oxidation-dependent fluorescein derivatives
(21,
22) or electron spin resonance
(16) that provide estimates of
redox of the entire cell, including cytosol, mitochondria, and endoplasmic
reticulum, all of which have unique redox potentials
(28). Cytosol-targeted roGFP1
was advantageous compared with these other methods in providing specific,
quantitative estimates of Ψcyto.
that is converted in the cells to H2O2. As determined
from measurements of Ψcyto, [PYO] as low as 1 μm
caused small oxidation of Ψcyto, and 10 and 100
μm PYO caused large and sustained oxidation of
Ψcyto by 20 and 40 mV. These quantitative measurements of
PYO-induced oxidation of the cytosol therefore confirm previous qualitative
measurements based on the use of oxidation-dependent fluorescein derivatives
(21,
22) or electron spin resonance
(16) that provide estimates of
redox of the entire cell, including cytosol, mitochondria, and endoplasmic
reticulum, all of which have unique redox potentials
(28). Cytosol-targeted roGFP1
was advantageous compared with these other methods in providing specific,
quantitative estimates of Ψcyto.
Even though PYO is membrane-permeant, its relatively slow effect to oxidize
the cytosol (e.g. compared with H2O2) may
result from the slow redox cycling that occurs in interactions of PYO with
cytosolic glutathione or NAD(P)H
(13); these reactions will
generate
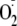 which dismutates on its own and may also be catalyzed by cytosolic superoxide
dismutase to H2O2. Based on the lack of effect of PYO on
Ψcyto in catalase-overexpressing cells, it seems likely that
the PYO-produced H2O2 caused the observed cytosolic
oxidation. H2O2 elicited rapid oxidation of
Ψcyto, likely resulting from rapid penetration of nonpolar,
membrane-permeant H2O2 through the membrane bilayer. The
rapid oxidation of Ψcyto in response to increased extracellular
which dismutates on its own and may also be catalyzed by cytosolic superoxide
dismutase to H2O2. Based on the lack of effect of PYO on
Ψcyto in catalase-overexpressing cells, it seems likely that
the PYO-produced H2O2 caused the observed cytosolic
oxidation. H2O2 elicited rapid oxidation of
Ψcyto, likely resulting from rapid penetration of nonpolar,
membrane-permeant H2O2 through the membrane bilayer. The
rapid oxidation of Ψcyto in response to increased extracellular
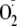 likely also resulted from its rapid penetration across the membrane, cytosolic
conversion to H2O2, and subsequent oxidation of
Ψcyto.
likely also resulted from its rapid penetration across the membrane, cytosolic
conversion to H2O2, and subsequent oxidation of
Ψcyto.
Activation of NF-κB by PYO Occurs through a Nonredox-mediated
Effect—Several observations indicated that the PYO-induced
generation of ROS and oxidation of Ψcyto played only a minor
role in the ability of PYO to synergize with flagellin in activating
NF-κB during a 4-h exposure. (i) Exogenous additions of
H2O2 or
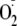 at concentrations that oxidized the cytosol similar to that exhibited by 100
μm PYO (i.e. ∼40 mV, from control -325 mV to about
-285 mV in the presence of PYO, H2O2, or
at concentrations that oxidized the cytosol similar to that exhibited by 100
μm PYO (i.e. ∼40 mV, from control -325 mV to about
-285 mV in the presence of PYO, H2O2, or
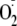 )
had no effect on NF-κB alone or in the presence of flagellin. (ii) The
oxidizing effect of PYO was reduced in cells overexpressing catalase, but
there was no significant effect of catalase expression on the synergistic
stimulation of NF-κBby PYO + flagellin. (iii) The HO.
scavenger DMSO had no effect on PYO-induced oxidation of Ψcyto
or activation of NF-κBby flagellin + PYO.
)
had no effect on NF-κB alone or in the presence of flagellin. (ii) The
oxidizing effect of PYO was reduced in cells overexpressing catalase, but
there was no significant effect of catalase expression on the synergistic
stimulation of NF-κBby PYO + flagellin. (iii) The HO.
scavenger DMSO had no effect on PYO-induced oxidation of Ψcyto
or activation of NF-κBby flagellin + PYO.
In contrast to the present work showing PYO elicited its stimulatory effects on NF-κB through nonredox mechanisms, previous work (18) showed there were important roles for H2O2, HO., and nitric oxide in the activation of IL8 and ICAM production over 1–2 days of exposure to PYO and one of its precursors, phenazine 1-carboxylic acid. A possible explanation for the differences between these previous (18) and present results could be that the redox-dependent proinflammatory effects of PYO and phenazine 1-carboxylic acid on IL8 and ICAM1 may be mediated through signaling pathways that lead to other transcription factors besides NF-κB that are important for overall gene regulation, e.g. NF-IL6 and/or AP-1 in the case of the IL8 promoter (19). Another possibility is that the redox-dependent proinflammatory effects of PYO on IL8 and ICAM1 resulted from the longer time course of the previous experiments. Thus, the proinflammatory effects of PYO could result from nonredox activation of NF-κB during 4-h incubations but by generating cellular ROS that activate proinflammatory signaling during 1–2-day incubations.
Because PYO oxidation of Ψcyto appeared not to be involved in activating NF-κB, how does PYO work? Western analysis showed that PYO and flagellin each increased phosphorylation of IKK and p65, and there were increased phosphorylations in the presence of both PYO and flagellin. This may indicate that PYO activated one of the kinases in the MyD88/IRAK/TRAF/TAK pathway (23) leading to NF-κB.
Previous experiments have shown that high [PYO] (80–300 μm) stimulated inositol 1,4,5-trisphosphate production, apparent release of Ca2+ from internal stores, and small increases (50–300 nm) in Cai in human bronchial epithelial cells and A549 cells (20). However, we were unable to detect any changes in Cai during treatments of either CF15 (Fig. 7) or Calu-34 cells with 100 μm PYO. Thus, although changes in Cai may occur in some cells during exposure to high [PYO], PYO synergism with flagellin in activation of NF-κB does not require increases in Cai in CF15 and Calu-3 cells.
We considered the possibility that PYO elicited its stimulatory effects by altering redox properties in other cellular compartments besides the cytosol. Altering the redox properties of the endoplasmic reticulum could trigger an unfolded protein response that leads to activation of NF-κB (37), but PYO did not activate the unfolded protein response (analyzed by IRE1α-dependent splicing of XBP-1),3 indicating that this was an unlikely explanation for the mechanism by which PYO synergizes with NF-κB-activating agonists in stimulating NF-κB. Previous work has indicated that PYO may oxidize mitochondria selectively (22), and such oxidation could couple to cytosolic signaling (38) to activate NF-κB. By using mitochondria-targeted roGFP, we found that PYO oxidized mitochondrial redox potential (Ψmito), although about 20% less than Ψcyto,3 indicating that PYO-induced oxidation of Ψmito could contribute to activation of NF-κB. Further studies will be required to determine the role of mitochondrial redox in controlling proinflammatory signaling in the cytosol.
Too Much Oxidation Inhibits NF-κB—Flagellin-activated
NF-κB was slightly inhibited by extracellular treatments with
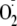 ,
which oxidized Ψcyto from control -325 mV to about -270 mV
(Fig. 3), and NF-κB was
nearly completely inhibited by 400 μm
H2O2, which oxidized Ψcyto to about -260
mV (Fig. 4). That this
inhibitory effect on NF-κB resulted directly from oxidizing effects of
H2O2 was shown by the reversal of the inhibition by
overexpressing catalase. In contrast, overexpressing catalase prevented
PYO-induced oxidation of Ψcyto but did not affect activation of
NF-κB. Taken together, these results indicated that the threshold for
the inhibitory effect of excessive oxidation of Ψcyto on
NF-κB in CF15 cells may occur near -270 mV.
,
which oxidized Ψcyto from control -325 mV to about -270 mV
(Fig. 3), and NF-κB was
nearly completely inhibited by 400 μm
H2O2, which oxidized Ψcyto to about -260
mV (Fig. 4). That this
inhibitory effect on NF-κB resulted directly from oxidizing effects of
H2O2 was shown by the reversal of the inhibition by
overexpressing catalase. In contrast, overexpressing catalase prevented
PYO-induced oxidation of Ψcyto but did not affect activation of
NF-κB. Taken together, these results indicated that the threshold for
the inhibitory effect of excessive oxidation of Ψcyto on
NF-κB in CF15 cells may occur near -270 mV.
Controversial Role of Redox Regulation of Inflammation— Previous data have shown that cytosolic oxidation on its own or in combination with cytokines activates NF-κB (35, 39, 40) and that antioxidants may reduce proinflammatory signaling (18). In contrast, others have provided evidence showing that oxidation can mediate anti-inflammatory effects (41, 42). The present data showed that NF-κB activity was insensitive to oxidation of Ψcyto by up to 40–50 mV, but further oxidations past this threshold had large inhibitory effects. These contrasting results indicate that responses to oxidation are likely to depend on subtle, cell-specific effects that may alter the magnitudes and localizations of oxidation. Furthermore, our data as well as that of others (43, 44) emphasized that at least some redox-active molecules can alter proinflammatory NF-κB signaling through redox-independent mechanisms. Future quantitative investigations of the role of Ψcyto in controlling proinflammatory signaling may yield insights into why oxidants can either stimulate, inhibit, or have no effect on inflammatory signaling in different cells.
Acknowledgments
We thank William Reenstra, Dianne Newman (California Institute of Technology), and Linda Thomashow (Washington State University) for suggestions and Adam Schindler for help with measurements of endoplasmic reticulum stress using IREα. Adenovirus to express catalase was kindly provided by Dwight Look and Gerene Denning (University of Iowa).
This work was supported, in whole or in part, by National Institutes of Health Grant R01 DK51799. This work was also supported by Cystic Fibrosis Foundation Grants MACH03, MACH07, FISCHE07, and SCHWA01, Cystic Fibrosis Research Inc., and the Hawn Fund (University of California, Berkeley). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: CF, cystic fibrosis;
Cai, cytosolic or cell [Ca2+];
Ψcyto, redox potential in the cytosol; PA, Pseudomonas
aeruginosa; PYO, pyocyanin; IL1β, interleukin 1β; IL8,
interleukin 8; TNFα, tumor necrosis factor α; ICAM1, intercellular
adhesion molecule 1;
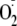 ,
superoxide; DTT, dithiothreitol; DMSO, dimethyl sulfoxide; X, xanthine; XO,
xanthine oxidase;
,
superoxide; DTT, dithiothreitol; DMSO, dimethyl sulfoxide; X, xanthine; XO,
xanthine oxidase;
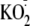 ,
potassium superoxide; ROS, reactive oxygen species; GFP, green fluorescent
protein; roGFP1, redox-sensitive GFP; DTT, dithiothreitol.
,
potassium superoxide; ROS, reactive oxygen species; GFP, green fluorescent
protein; roGFP1, redox-sensitive GFP; DTT, dithiothreitol.
C. Schwarzer and T. E. Machen, unpublished observations.
Z. Fu and T. Machen, unpublished results.
References
- 1.Lyczak, J. B., Cannon, C. L., and Pier, G. B. (2002) Clin. Microbiol. Rev. 15 194-222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kong, F., Young, L., Chen, Y., Ran, H., Meyers, M., Joseph, P., Cho, Y. H., Hassett, D. J., and Lau, G. W. (2006) Cell. Microbiol. 8 1121-1133 [DOI] [PubMed] [Google Scholar]
- 3.Cao, H., Krishnan, G., Goumnerov, B., Tsongalis, J., Tompkins, R., and Rahme, L. G. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 14613-14618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lau, G. W., Ran, H., Kong, F., Hassett, D. J., and Mavrodi, D. (2004) Infect. Immun. 72 4275-4278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mavrodi, D. V., Bonsall, R. F., Delaney, S. M., Soule, M. J., Phillips, G., and Thomashow, L. S. (2001) J. Bacteriol. 183 6454-6465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olson, J. C., Fraylick, J. E., McGuffie, E. M., Dolan, K. M., Yahr, T. L., Frank, D. W., and Vincent, T. S. (1999) Infect. Immun. 67 2847-2854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pederson, K. J., Vallis, A. J., Aktories, K., Frank, D. W., and Barbieri, J. T. (1999) Mol. Microbiol. 32 393-401 [DOI] [PubMed] [Google Scholar]
- 8.Sato, H., and Frank, D. W. (2004) Mol. Microbiol. 53 1279-1290 [DOI] [PubMed] [Google Scholar]
- 9.Muller, M. (2002) Free Radic. Biol. Med. 33 1527-1533 [DOI] [PubMed] [Google Scholar]
- 10.Wilson, R., Sykes, D. A., Watson, D., Rutman, A., Taylor, G. W., and Cole, P. J. (1988) Infect. Immun. 56 2515-2517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahajan-Miklos, S., Tan, M. W., Rahme, L. G., and Ausubel, F. M. (1999) Cell 96 47-56 [DOI] [PubMed] [Google Scholar]
- 12.O'Malley, Y. Q., Reszka, K. J., Rasmussen, G. T., Abdalla, M. Y., Denning, G. M., and Britigan, B. E. (2003) Am. J. Physiol. 285 L1077-L1086 [DOI] [PubMed] [Google Scholar]
- 13.O'Malley, Y. Q., Reszka, K. J., Spitz, D. R., Denning, G. M., and Britigan, B. E. (2004) Am. J. Physiol. 287 L94-L103 [DOI] [PubMed] [Google Scholar]
- 14.Wilson, R., Pitt, T., Taylor, G., Watson, D., MacDermot, J., Sykes, D., Roberts, D., and Cole, P. (1987) J. Clin. Investig. 79 221-229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ran, H., Hassett, D. J., and Lau, G. W. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 14315-14320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Denning, G. M., Wollenweber, L. A., Railsback, M. A., Cox, C. D., Stoll, L. L., and Britigan, B. E. (1998) Infect. Immun. 66 5777-5784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pan, N. Y., Hui, W. S., Tipoe, G. L., Taylor, G. W., Leung, R. Y., Lam, W. K., Tsang, K. W., and Mak, J. C. (2006) Respir. Med. 100 1614-1622 [DOI] [PubMed] [Google Scholar]
- 18.Look, D. C., Stoll, L. L., Romig, S. A., Humlicek, A., Britigan, B. E., and Denning, G. M. (2005) J. Immunol. 175 4017-4023 [DOI] [PubMed] [Google Scholar]
- 19.Fu, Z., Bettega, K., Carroll, S., Buchholz, K. R., and Machen, T. E. (2007) Am. J. Physiol. 292 L353-L364 [DOI] [PubMed] [Google Scholar]
- 20.Denning, G. M., Railsback, M. A., Rasmussen, G. T., Cox, C. D., and Britigan, B. E. (1998) Am. J. Physiol. 274 L893-L900 [DOI] [PubMed] [Google Scholar]
- 21.O'Malley, Y. Q., Reszka, K. J., and Britigan, B. E. (2004) Free Radic. Biol. Med. 36 90-100 [DOI] [PubMed] [Google Scholar]
- 22.O'Malley, Y. Q., Abdalla, M. Y., McCormick, M. L., Reszka, K. J., Denning, G. M., and Britigan, B. E. (2003) Am. J. Physiol. 284 L420-L430 [DOI] [PubMed] [Google Scholar]
- 23.Akira, S. (2001) Adv. Immunol. 78 1-56 [DOI] [PubMed] [Google Scholar]
- 24.Zhang, Z., Louboutin, J. P., Weiner, D. J., Goldberg, J. B., and Wilson, J. M. (2005) Infect. Immun. 73 7151-7160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hybiske, K., Ichikawa, J. K., Huang, V., Lory, S. J., and Machen, T. E. (2004) Cell. Microbiol. 6 49-63 [DOI] [PubMed] [Google Scholar]
- 26.Dooley, C. T., Dore, T. M., Hanson, G. T., Jackson, W. C., Remington, S. J., and Tsien, R. Y. (2004) J. Biol. Chem. 279 22284-22293 [DOI] [PubMed] [Google Scholar]
- 27.Hanson, G. T., Aggeler, R., Oglesbee, D., Cannon, M., Capaldi, R. A., Tsien, R. Y., and Remington, S. J. (2004) J. Biol. Chem. 279 13044-13053 [DOI] [PubMed] [Google Scholar]
- 28.Schwarzer, C., Illek, B., Remington, S. J., Fischer, H., and Machen, T. E. (2007) Free Radic. Biol. Med. 43 300-316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tseng, J., Do, J., Widdicombe, J. H., and Machen, T. E. (2006) Am. J. Physiol. 290 C678-C690 [DOI] [PubMed] [Google Scholar]
- 30.Jefferson, D. M., Valentich, J. D., Marini, F. C., Grubman, S. A., Iannuzzi, M. C., Dorkin, H. L., Li, M., Klinger, K. W., and Welsh, M. J. (1990) Am. J. Physiol. 259 L496-L505 [DOI] [PubMed] [Google Scholar]
- 31.Jacob, T., Lee, R. J., Engel, J., and Machen, T. E. (2002) Infect. Immun. 70 6399-6408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grynkiewicz, G., Poenie, M., and Tsien, R. Y. (1985) J. Biol. Chem. 260 3440-3450 [PubMed] [Google Scholar]
- 33.Sanlioglu, S., Williams, C. M., Samavati, L., Butler, N. S., Wang, G., McCray, P. B., Jr., Ritchie, T. C., Hunninghake, G. W., Zandi, E., and Engelhardt, J. F. (2001) J. Biol. Chem. 276 30188-30198 [DOI] [PubMed] [Google Scholar]
- 34.Reszka, K., O'Malley, Y., McCormick, M., Denning, G., and Britigan, B. (2004) Free Radic. Biol. Med. 36 1448-1459 [DOI] [PubMed] [Google Scholar]
- 35.de Oliveira-Marques, V., Cyrne, L., Marinho, H. S., and Antunes, F. (2007) J. Immunol. 178 3893-3902 [DOI] [PubMed] [Google Scholar]
- 36.Wasil, M., Halliwell, B., Grootveld, M., Moorhouse, C. P., Hutchison, D. C. S., and Baum, H. (1987) Biochem. J. 243 867-870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pahl, H. L., and Baeuerle, P. A. (1997) Trends Biochem. Sci. 22 63-67 [DOI] [PubMed] [Google Scholar]
- 38.Hughes, G., Murphy, M. P., and Ledgerwood, E. C. (2005) Biochem. J. 389 83-89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schreck, R., Rieber, P., and Baeuerle, P. A. (1991) EMBO J. 10 2247-2258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Flohe, L., Brigelius-Flohe, R., Saliou, C., Traber, M. G., and Packer, L. (1997) Free Radic. Biol. Med. 22 1115-1126 [DOI] [PubMed] [Google Scholar]
- 41.Kaileh, M., Vanden Berghe, W., Heyerick, A., Horion, J., Piette, J., Libert, C., De Keukeleire, D., Essawi, T., and Haegeman, G. (2007) J. Biol. Chem. 282 4253-4264 [DOI] [PubMed] [Google Scholar]
- 42.Na, H. J., Lee, G., Oh, H. Y., Jeon, K. S., Kwon, H. J., Ha, K. S., Lee, H., Kwon, Y. G., and Kim, Y. M. (2006) Int. Immunopharmacol. 6 1597-1608 [DOI] [PubMed] [Google Scholar]
- 43.Hayakawa, M., Miyashita, H., Sakamoto, I., Kitagawa, M., Tanaka, H., Yasuda, H., Karin, M., and Kikugawa, K. (2003) EMBO J. 22 3356-3366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sandur, S. K., Pandey, M. K., Sung, B., Ahn, K. S., Murakami, A., Sethi, G., Limtrakul, P., Badmaev, V., and Aggarwal, B. B. (2007) Carcinogenesis 28 1765-1773 [DOI] [PubMed] [Google Scholar]