

Abstract
In the vasculature, nitric oxide (NO) is generated by endothelial NO
synthase (eNOS) in a calcium/calmodulin-dependent reaction. With oxidative
stress, the critical cofactor BH4 is depleted, and NADPH oxidation
is uncoupled from NO generation, leading to production of
(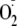 ).
Although phosphorylation of eNOS regulates in vivo NO generation, the
effects of phosphorylation on eNOS coupling and
).
Although phosphorylation of eNOS regulates in vivo NO generation, the
effects of phosphorylation on eNOS coupling and
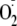 generation are unknown. Therefore, we phosphorylated recombinant
BH4-free eNOS in vitro using native kinases and determined
generation are unknown. Therefore, we phosphorylated recombinant
BH4-free eNOS in vitro using native kinases and determined
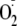 generation using EPR spin trapping. Phosphorylation of Ser-1177 by Akt led to
an increase (>50%) in maximal
generation using EPR spin trapping. Phosphorylation of Ser-1177 by Akt led to
an increase (>50%) in maximal
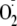 generation from eNOS. Moreover, Ser-1177 phosphorylation greatly altered the
Ca2+ sensitivity of eNOS, such that
generation from eNOS. Moreover, Ser-1177 phosphorylation greatly altered the
Ca2+ sensitivity of eNOS, such that
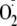 generation became largely Ca2+-independent. In contrast,
phosphorylation of eNOS at Thr-495 by protein kinase Cα (PKCα) had
no effect on maximum activity or calcium sensitivity but decreased calmodulin
binding and increased association with caveolin. In endothelial cells,
eNOS-dependent
generation became largely Ca2+-independent. In contrast,
phosphorylation of eNOS at Thr-495 by protein kinase Cα (PKCα) had
no effect on maximum activity or calcium sensitivity but decreased calmodulin
binding and increased association with caveolin. In endothelial cells,
eNOS-dependent
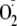 generation was stimulated by vascular endothelial growth factor that induced
phosphorylation of Ser-1177. With PKC activation that led to phosphorylation
of Thr-495, no inhibition of
generation was stimulated by vascular endothelial growth factor that induced
phosphorylation of Ser-1177. With PKC activation that led to phosphorylation
of Thr-495, no inhibition of
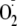 generation occurred. As such, phosphorylation of eNOS at Ser-1177 is pivotal
in the direct regulation of
generation occurred. As such, phosphorylation of eNOS at Ser-1177 is pivotal
in the direct regulation of
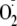 and NO generation, altering both the Ca2+ sensitivity of the enzyme
and rate of product formation, whereas phosphorylation of Thr-495 indirectly
affects this process through regulation of the calmodulin and caveolin
interaction. Thus, Akt-mediated phosphorylation modulates eNOS uncoupling and
greatly increases
and NO generation, altering both the Ca2+ sensitivity of the enzyme
and rate of product formation, whereas phosphorylation of Thr-495 indirectly
affects this process through regulation of the calmodulin and caveolin
interaction. Thus, Akt-mediated phosphorylation modulates eNOS uncoupling and
greatly increases
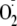 generation from the enzyme at low Ca2+ concentrations, and
PKCα-mediated phosphorylation alters the sensitivity of the enzyme to
other negative regulatory signals.
generation from the enzyme at low Ca2+ concentrations, and
PKCα-mediated phosphorylation alters the sensitivity of the enzyme to
other negative regulatory signals.
Nitric-oxide synthase
(NOS)2 is a critical
enzyme that converts l-arginine (l-Arg) to
l-citrulline and nitric oxide (NO) with the consumption of NADPH.
NO is a signaling molecule that promotes vascular smooth muscle relaxation and
functions as an endogenous mediator of a wide range of effects in different
tissues (1,
2). After oxidant stress, as
occurs in postischemic tissues, production of
 and its derived oxidants, including peroxynitrite (ONOO-), hydrogen
peroxide (H2O2), and hydroxyl radical (·OH),
induce NOS dysfunction with uncoupling of the enzyme leading to the production
of NOS-derived
and its derived oxidants, including peroxynitrite (ONOO-), hydrogen
peroxide (H2O2), and hydroxyl radical (·OH),
induce NOS dysfunction with uncoupling of the enzyme leading to the production
of NOS-derived
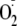 instead of NO (3,
4). It has been reported that
an imbalance between NO and
instead of NO (3,
4). It has been reported that
an imbalance between NO and
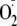 can contribute to the onset of a variety of cardiovascular diseases, including
hypertension, atherosclerosis, and heart failure
(5). Therefore, tight coupling
of the enzyme is important for normal cardiovascular function and prevention
of disease.
can contribute to the onset of a variety of cardiovascular diseases, including
hypertension, atherosclerosis, and heart failure
(5). Therefore, tight coupling
of the enzyme is important for normal cardiovascular function and prevention
of disease.
The catalytic domains of NOS include a flavin-containing NADPH binding
reductase and a heme-binding oxygenase that also contains the binding sites
for the redox labile cofactor tetrahydrobiopterin (BH4) and the
substrate l-Arg. In the presence of Ca2+ and calmodulin
(CaM), electrons flow from NADPH through the reductase domain to the oxygenase
domain resulting in the activation of oxygen at the heme center followed by
substrate monooxygenation. This process requires the presence of the fully
reduced BH4. Our laboratory and several others have demonstrated
that besides synthesizing NO, all three isoforms of NOS can also generate
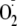 ,
depending on substrate and cofactor availability
(3,
6–9).
One of the primary mechanisms implicated in the oxidant-induced switch of NOS
from the production of NO to the generation of
,
depending on substrate and cofactor availability
(3,
6–9).
One of the primary mechanisms implicated in the oxidant-induced switch of NOS
from the production of NO to the generation of
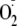 is the oxidation of the enzyme bound BH4
(10,
11).
is the oxidation of the enzyme bound BH4
(10,
11).
Various extracellular signals, including shear stress and additional stimuli such as vascular endothelial growth factor (VEGF), estrogen, sphingosine 1-phosphate, bradykinin, and aldosterone, modulate eNOS NO generation through several signal transduction pathways (12–16). Cellular studies have demonstrated that phosphorylation of eNOS at specific amino acids regulates enzyme-mediated NO production (17). The majority of previous work has focused on two residues, serine 1177 and threonine 495. It has been shown that Akt specifically induces phosphorylation of Ser-1177 (18, 19) and that PKC specifically phosphorylates Thr-495 (20). Although phosphorylation of Ser-1177 has been shown to increase NO production from eNOS (21), in contrast, phosphorylation of Thr-495 has been reported to down-regulate NO generation (18, 22, 23).
Although there is strong evidence indicating that phosphorylation of eNOS
is involved in directly modulating eNOS-mediated NO generation, the definitive
mechanisms involved remain unclear. Moreover, there is a lack of prior
investigation directed toward understanding how phosphorylation alters
 generation from the uncoupled enzyme. Determination of the effects of
phosphorylation on eNOS-derived
generation from the uncoupled enzyme. Determination of the effects of
phosphorylation on eNOS-derived
 generation is of particular importance, because of the implications of this
regulation in cardiovascular disease and other physiological settings in which
eNOS is uncoupled (3,
4). Delineation of the
mechanisms involved in the phosphorylation-dependent regulation of uncoupled
eNOS will provide critical insights regarding the pathophysiology of eNOS
dysfunction.
generation is of particular importance, because of the implications of this
regulation in cardiovascular disease and other physiological settings in which
eNOS is uncoupled (3,
4). Delineation of the
mechanisms involved in the phosphorylation-dependent regulation of uncoupled
eNOS will provide critical insights regarding the pathophysiology of eNOS
dysfunction.
Therefore, studies were performed to investigate how phosphorylation by the
critical signaling kinases Akt or PKCα modulates eNOS uncoupling and the
production of
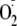 from the enzyme. Studies were performed with in vitro phosphorylation
of recombinant human eNOS and also with intact endothelial cells. Our results
demonstrated that phosphorylation of eNOS at Ser-1177 is pivotal in the
regulation of
from the enzyme. Studies were performed with in vitro phosphorylation
of recombinant human eNOS and also with intact endothelial cells. Our results
demonstrated that phosphorylation of eNOS at Ser-1177 is pivotal in the
regulation of
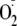 generation, altering both the Ca2+ sensitivity of the enzyme and
maximal rate of product generation, whereas phosphorylation of Thr-495
indirectly affects
generation, altering both the Ca2+ sensitivity of the enzyme and
maximal rate of product generation, whereas phosphorylation of Thr-495
indirectly affects
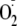 generation by modulating the binding of proteins known to regulate the
activity of the enzyme.
generation by modulating the binding of proteins known to regulate the
activity of the enzyme.
EXPERIMENTAL PROCEDURES
Materials—Akt1 kinase, PKCα kinase, anti-eNOS antibody, anti-phospho-Ser-1177, and anti-phospho-Thr-495 eNOS antibodies were purchased from Cell Signaling Technology, Inc. (Danvers, MA). Terrific broth, LB broth base, EZQ phosphoprotein quantification kit, dithiothreitol (DTT), and carbenicillin were obtained from Invitrogen. Protease inhibitor mixture tablets were purchased from Roche. NADPH, l-Arg, and hemoglobin were purchased from Sigma-Aldrich. Isopropyl β-d-1-thiogalactopyranoside was purchased from Anatrace, Inc. (Maumee, OH). Chloramphenicol was obtained from Fluka (St. Louis, MO). 5-Diethoxyphosphoryl-5-methyl-1-pyrroline N-oxide (DEPMPO) was purchased from Alexis Biochemicals (San Diego, CA). Caveolin peptide (Cav-P) was custom-synthesized by Bio-Synthesis Inc. (Lewisville, TX). The sequence of Cav-P is DGIWKASFTTFTVTKYWFYR (24).
Protein Expression and Purification—The bacterial expression plasmid of pCWheNOS was a gift from Dr. Ortiz de Montellano from University of California at San Franscisco. Overexpression of active human eNOS in Escherichia coli was greatly enhanced by coexpression with calmodulin (pCaM). Typically, a 10-ml LB overnight culture of pCWheNOS and pCaM in BL21(DE3) E. coli was used to inoculate 1 liter of terrific broth in a 4-liter flask containing 125 μg/ml carbenicillin and 50 μg/ml chloramphenicol. The culture was grown at 37 °C until the cell density reached an A600 of 0.8, then δ-aminolevulinic acid was added to a final concentration of 0.5 mm, and the cells were induced by the addition of isopropyl β-d-1-thiogalactopyranoside (1 mm final concentration). The cultures were then grown at 22 °C at 200 rpm for 20 h before harvest by centrifugation at 4 °C. The cell pellet was stored at -80 °C until purification (25, 26).
Typically, 2 liters of human eNOS (heNOS)-expressing cells were pelleted
and resuspended in 50 ml of lysis buffer containing 40 mm HEPES, pH
7.6, 10% glycerol, 500 mm NaCl, 40 mm imidazole, 1
mm DTT, 10 μm BH4, and 5 tablets of
protease inhibitor mixture, EDTA free. The cells were lysed in the presence of
20 mg/ml lysozyme on ice for 30 min with stirring by pulse sonication 10 s
each cycle with a total of ∼15 cycles. During the sonication the solution
was always kept below 4 °C to prevent any degradation or inactivation of
the enzyme. The cell debris was cleared by centrifugation at 48,000 ×
g for 1 h at 4 °C. The supernatant was applied to a 5-ml HisTrap
nickel-nitrilotriacetic acid column (GE Healthcare) equilibrated with lysis
buffer using an AKTA fast protein liquid chromatography system (GE
Healthcare). The column was then washed with 10 column volumes of buffer A (40
mm HEPES, pH 7.6, 10% glycerol, 40 mm imidazole, 1
mm DTT, 10 μm BH4). Finally the heNOS was
eluted with buffer B (40 mm HEPES, pH 7.6, 10% glycerol, 500
mm NaCl, 250 mm imidazole, 1 mm DTT, 10
μm BH4). The colored fractions were pooled, and 20
mm l-Arg, 20 μm BH4, and 5 mm
DTT were added and incubated on ice for 4 h. The purified protein was
subsequently loaded into Hi-Load Superdex 200 size exclusion column
equilibrated with buffer C (40 mm HEPES, pH 7.6, 10% glycerol, 150
mm NaCl, 1 mm DTT). The colored fractions were pooled
and concentrated to greater than 1 mg/ml by ultrafiltration. The concentrated
protein was separated into aliquots and quickly frozen in liquid nitrogen
before storage at -80 °C. When heNOS was coexpressed with CaM, 2
mm CaCl2 was included in all purification buffers
(26). Because of the
instability of purified heNOS, the entire purification process was completed
within 1 day. For the BH4-free heNOS, used for
 determinations, l-Arg and BH4 were excluded during the
purification process. To remove CaM from the heNOS purification, 2
mm EGTA was included in Buffer C.
determinations, l-Arg and BH4 were excluded during the
purification process. To remove CaM from the heNOS purification, 2
mm EGTA was included in Buffer C.
Phosphorylation of heNOS by Akt and PKCα— heNOS (5 μm, 0.675 μg/μl) was phosphorylated at RT for 20 min with Akt or PKCα (100 ng) in a total volume of 20 μl in 1× kinase buffer containing 25 mm Tris-HCl, pH 7.5, 10 mm MgCl2, 5 mm β-glycerophosphate, 0.1 mm Na3VO4, 2 mm DTT, 200 μm ATP according to manufacturer's protocol. After phosphorylation, the phosphorylated heNOS was subjected to immunoblotting analysis or EZQ phosphoprotein determination. The Invitrogen EZQ phosphoprotein quantification kit was used to determine the phosphorylation efficiency of heNOS by Akt and PKCα. According to the procedures in the manufacture's manual, the phosphorylated heNOS by Akt, PKCα, or both was spotted onto specially prepared assay paper, fixed onto the paper with methanol, and then stained with EZQ phosphoprotein quantification reagent. The dried assay paper was inserted onto EZQ 96-well microplate cassette, and the stained protein spots were analyzed in a fluorescence-based reader (Spectra-Max GEMINIXS, Molecular Devices, Sunnyvale, CA) using excitation/emission wavelengths of 550/580 nm. Relative phosphate content of phosphorylated heNOS was determined from a standard curve of ovalbumin, which contains two phosphate groups per molecule. The control experiments with kinase alone were always performed, and the fluorescent intensity of control experiments was subtracted from all experiments.
Protein and Heme Content Determination—Protein concentration of purified heNOS was determined by the Bradford assay from Bio-Rad using a bovine serum albumin standard. The heme content of the purified heNOS was determined by pyridine hemochromogen assay. 50 μg of heNOS was added to a solution containing 0.15 m NaOH and 1.8 m pyridine, and the difference spectrum (reduced minus oxidized bispyridine heme) was recorded using Δε = 24 mm-1 cm-1 at 556–538 nm (27). The reduced bispyridine heme was generated by the addition of a few grains of dithionite.
SDS-PAGE and Immunoblotting—The reaction mixture was mixed with the sample loading buffer at a ratio 3:1 (v/v), incubated at 80 °C for 10 min, and then immediately loaded onto a 4–20% Tris-glycine polyacrylamide gradient gel. Samples were run at room temperature for 1.5 h at 125 V. Protein bands were electrophoretically transferred to a nitrocellulose membrane in 12 mm Tris, 96 mm glycine, and 20% methanol with a Xcell II Blot Module from Invitrogen with 25 V constant for 90 min. Membranes were blocked for 1 h at room temperature in Tris-buffered saline (TBS) containing 0.05% Tween 20 (TBST) with 5% dry milk (Bio-Rad). Membranes were then incubated overnight with anti-eNOS, anti-phospho-Thr-495, or anti-phospho-Ser-1177 eNOS polyclonal antibodies at 4 °C. Membranes were then washed 3 times in TBST and incubated for 1 h with horseradish peroxidase-conjugated anti-rabbit IgG in TBST at room temperature. Membranes were again washed three times in TBST and then visualized using ECL immunoblotting detection reagents (Amersham Biosciences). The signal intensity of blotting was digitized and quantified using an AlphaImager™ high performance gel documentation and image analysis system, model 3300 (Alpha Innotech Co. San Leandro, CA).
Measurement of
 Generation by EPR Spin Trapping—Spin-trapping measurements of
oxygen radicals were performed in 50 mm Tris-HCl buffer, pH 7.4,
containing 0.5 mm NADPH, 0.5 mm Ca2+, 10
μg/ml CaM, 15 μg/ml purified heNOS, and 25 mm spin trap
DEPMPO. EPR spectra were recorded in a 50-μl capillary at room temperature
with a Bruker EMX spectrometer operating at 9.86 GHz with 100 kHz modulation
frequency as described (28,
29). The sample was scanned
using the following parameters: center field, 3510 G; sweep width, 140 G;
power, 20 milliwatts; receiver gain, 2 × 105; modulation
amplitude, 1 G; time of conversion, 41 ms; time constant, 328 ms.
Generation by EPR Spin Trapping—Spin-trapping measurements of
oxygen radicals were performed in 50 mm Tris-HCl buffer, pH 7.4,
containing 0.5 mm NADPH, 0.5 mm Ca2+, 10
μg/ml CaM, 15 μg/ml purified heNOS, and 25 mm spin trap
DEPMPO. EPR spectra were recorded in a 50-μl capillary at room temperature
with a Bruker EMX spectrometer operating at 9.86 GHz with 100 kHz modulation
frequency as described (28,
29). The sample was scanned
using the following parameters: center field, 3510 G; sweep width, 140 G;
power, 20 milliwatts; receiver gain, 2 × 105; modulation
amplitude, 1 G; time of conversion, 41 ms; time constant, 328 ms.
Ca2+ Dependence of
 Generation from heNOS—100 μl of reaction volume was typically
used for EPR measurements containing 100 μm EGTA. The reaction
was initiated by the addition of 0.5 mm NADPH. The
Generation from heNOS—100 μl of reaction volume was typically
used for EPR measurements containing 100 μm EGTA. The reaction
was initiated by the addition of 0.5 mm NADPH. The
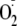 measurements were performed by EPR spin-trapping as described above. The free
Ca2+ concentration in the Ca2+ dependence studies was
calculated using winmaxc32 program version 2.5 (Stanford University)
(30,
31) accounting for pH and
buffer components of phosphorylation reactions and 100 μm
EGTA.
measurements were performed by EPR spin-trapping as described above. The free
Ca2+ concentration in the Ca2+ dependence studies was
calculated using winmaxc32 program version 2.5 (Stanford University)
(30,
31) accounting for pH and
buffer components of phosphorylation reactions and 100 μm
EGTA.
Cav-P Inhibition of
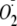 Generation from heNOS—A 100-μl reaction volume was typically
used for EPR measurements, identical to that described above except that Cav-P
(400 μm final concentration) was added to each reaction. The
reaction was initiated by the addition of 0.5 mm NADPH. The
Generation from heNOS—A 100-μl reaction volume was typically
used for EPR measurements, identical to that described above except that Cav-P
(400 μm final concentration) was added to each reaction. The
reaction was initiated by the addition of 0.5 mm NADPH. The
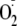 measurements were performed and quantified by EPR spin-trapping as described
above.
measurements were performed and quantified by EPR spin-trapping as described
above.
Measurement of
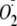 from
Uncoupled eNOS in Endothelial Cells—Bovine aortic endothelial cells
(BAECs) cultured on sterile coverslips (Harvard Apparatus, 22 mm2)
in 35-mm sterile dishes at a density of 104 cells/dish were
subjected to BH4 depletion. To deplete BH4, BAECs were
treated with 5 mm 2,4-diamino-6-hydroxypyrimidine, an inhibitor of
GTPCH1 (GTP-cyclohydrolase I) involved in BH4 biosynthesis, for 18
h (32). For VEGF treatment
(22), a final concentration of
50 ng/ml VEGF was added to BAECs or 2,4-diamino-6-hydroxypyrimidine-treated
BAECs for 10 min to activate Ser-1179 phosphorylation of bovine eNOS (beNOS).
Ionomycin (calcium ionophore (CaI))
(22) was added to a final
concentration of 1 μm in control BAECs or BAECs with
2,4-diamino-6-hydroxypyrimidine treatment for 10 min. The phosphorylation of
beNOS at Thr-497 was achieved by the addition of phorbol 12-myristate
13-acetate (PMA) to a final concentration of 0.1 μm for 10 min
(22). For negative control
experiments, l-NG-nitroarginine methyl ester
(l-NAME), a NOS inhibitor was added to a final concentration of 1
mm, 15 min before VEGF, PMA, or CaI treatments. Cells were then
incubated with the
from
Uncoupled eNOS in Endothelial Cells—Bovine aortic endothelial cells
(BAECs) cultured on sterile coverslips (Harvard Apparatus, 22 mm2)
in 35-mm sterile dishes at a density of 104 cells/dish were
subjected to BH4 depletion. To deplete BH4, BAECs were
treated with 5 mm 2,4-diamino-6-hydroxypyrimidine, an inhibitor of
GTPCH1 (GTP-cyclohydrolase I) involved in BH4 biosynthesis, for 18
h (32). For VEGF treatment
(22), a final concentration of
50 ng/ml VEGF was added to BAECs or 2,4-diamino-6-hydroxypyrimidine-treated
BAECs for 10 min to activate Ser-1179 phosphorylation of bovine eNOS (beNOS).
Ionomycin (calcium ionophore (CaI))
(22) was added to a final
concentration of 1 μm in control BAECs or BAECs with
2,4-diamino-6-hydroxypyrimidine treatment for 10 min. The phosphorylation of
beNOS at Thr-497 was achieved by the addition of phorbol 12-myristate
13-acetate (PMA) to a final concentration of 0.1 μm for 10 min
(22). For negative control
experiments, l-NG-nitroarginine methyl ester
(l-NAME), a NOS inhibitor was added to a final concentration of 1
mm, 15 min before VEGF, PMA, or CaI treatments. Cells were then
incubated with the
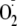 indicator 10 μm dihydroethidine (DHE) to detect
indicator 10 μm dihydroethidine (DHE) to detect
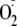 in live cells. DHE fluoresces when oxidized by
in live cells. DHE fluoresces when oxidized by
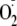 .
Nuclei were stained with blue fluorescent DAPI (1 μm) for 10 min
in the incubator. After the incubation, cells were washed with 1×
phosphate-buffered saline and mounted using a mounting medium Fluoromount-G,
and images were captured and analyzed at a magnification of 20× for DHE
and DAPI by confocal fluorescence microscopy (LSM 510; Zeiss Inc., Peabody,
MA) and overlaid using LSM software.
.
Nuclei were stained with blue fluorescent DAPI (1 μm) for 10 min
in the incubator. After the incubation, cells were washed with 1×
phosphate-buffered saline and mounted using a mounting medium Fluoromount-G,
and images were captured and analyzed at a magnification of 20× for DHE
and DAPI by confocal fluorescence microscopy (LSM 510; Zeiss Inc., Peabody,
MA) and overlaid using LSM software.
Immunofluorescence Microscopy—BAECs cultured on sterile coverslips (Harvard Apparatus, 22 mm2) in 35-mm sterile dishes at a density of 104 cells/dish were subjected to BH4 depletion and Ser-1179 and Thr-497 phosphorylation, as described in the previous section. At the end of the experiment, cells attached to coverslips were washed with 1× phosphate-buffered saline and fixed with 3.7% paraformaldehyde for 10 min, permeabilized with 0.25% Triton X-100 in TBST containing 0.01% Tween 20 for 5 min, and blocked for 30 min with 1% bovine serum albumin in 0.01% TBST. For visualization of beNOS Ser-1179 and Thr-497 phosphorylation, the fixed and permeabilized cells were incubated with rabbit primary anti-phospho-Ser-1179 and anti-phospho-Thr-497 eNOS antibodies, respectively, at a dilution of 1:2000 in 0.01% TBST containing 1% bovine serum albumin for 1 h at room temperature followed by an anti-rabbit AlexaFluor 488-conjugated antibody (1:1000 dilution) for 1 h at room temperature. The coverslips with cells were then mounted on a glass slide with the antifade mounting medium and viewed with a Zeiss confocal microscope at a magnification of 60×, and images were captured digitally.
RESULTS
Protein Expression, Purification, and Characterization—To mitigate problems with low yield and instability of heNOS when expressed in an E. coli system, recombinant human eNOS was coexpressed with CaM as reported previously (26) and isolated using nickel-nitrilotriacetic acid affinity and size exclusion chromatography. The coexpression of heNOS with CaM increased the yield 3-fold compared with expression of heNOS alone (3 mg/liter versus 1 mg/liter), consistent with previous reports (26). The purified protein preparations exhibited one prominent band (>95% pure) on 4–20% SDS-PAGE with Coomassie staining. The molecular weight of the purified protein, 135 kDa, is in accordance with the molecular mass for native heNOS monomer (3, 10, 33), and the identity of this band was confirmed by immunoblotting using an anti-eNOS antibody (Fig. 1), and mass spectrometry (data not shown). The NO generation, as determined by monitoring the conversion of oxyhemoglobin to ferric methemoglobin from the recombinant protein, was consistent with reported values (100–140 nmol·min-1·mg-1) (3, 26), and as expected the catalytic activity was dependent upon the addition of Ca2+ and CaM and could be totally blocked by the NOS inhibitor, l-NAME (1 mm).
FIGURE 1.

Immunoblotting of phosphorylated heNOS at Thr-495 and Ser-1177. Upper panel, a loading control, blotted against eNOS antibody. Middle panel, heNOS was phosphorylated in vitro by PKCα, Akt, or Akt and PKCα at RT for 20 min and blotted against anti-phospho-Thr-495 eNOS polyclonal antibody. Lower panel, heNOS was phosphorylated in vitro by PKCα, Akt, or Akt and PKCα at RT for 20 min and blotted against anti-phospho-Ser-1177 eNOS polyclonal antibody.
Phosphorylation of heNOS by Akt and PKCα—Purified recombinant heNOS was phosphorylated at Ser-1177, Thr-495, or at both sites using the kinases Akt and PKCα in the presence of 1× kinase buffer at RT. The reactions reached completion by 20 min for both Akt and PKCα based on Akt phosphorylation time course immunoblotting. The phosphorylation events were confirmed by immunoblotting with anti-phospho-Ser-1177 and anti-phospho-Thr-495 antibodies (Fig. 1). These results indicated that Akt specifically phosphorylated heNOS at Ser-1177, whereas PKCα phosphorylation was specific for Thr-495. Additionally, equivalent phosphorylation of both sites occurs when using both Akt and PKCα. The extent of phosphorylation of heNOS at each site was determined by EZQ phosphoprotein quantitation kit. The phosphorylation of heNOS at Ser-1177 by Akt was determined to be ∼80%, and the phosphorylation of Thr-495 by PKCα was determined to be close to 100% (34).
Effect of Phosphorylation of heNOS by Akt and PKCα on
Its 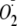 Generation—Under certain conditions NOS-dependent NADPH oxidation
is uncoupled from NO generation. This uncoupling occurs when either the
substrate l-Arg or the redox active cofactor BH4 is not
present. EPR spin-trapping was used to directly measure the magnitude of
Generation—Under certain conditions NOS-dependent NADPH oxidation
is uncoupled from NO generation. This uncoupling occurs when either the
substrate l-Arg or the redox active cofactor BH4 is not
present. EPR spin-trapping was used to directly measure the magnitude of
 generation from heNOS. Purified BH4-free heNOS (5 μg) was
incubated with 1× kinase buffer in the presence or absence of any kinase
in a volume of 10 μl at RT for 20 min before DEPMPO spin-trapping. For the
measurement of
generation from heNOS. Purified BH4-free heNOS (5 μg) was
incubated with 1× kinase buffer in the presence or absence of any kinase
in a volume of 10 μl at RT for 20 min before DEPMPO spin-trapping. For the
measurement of
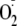 production, this 10-μl reaction mixture was diluted into a total volume of
100 μl containing only Ca2+/CaM, and the reaction was initiated
by the addition of NADPH to a final concentration of 0.5 mm. EPR
measurements were carried out, as described under “Experimental
Procedures,” with the nitrone spin trap DEPMPO, which forms a stable
production, this 10-μl reaction mixture was diluted into a total volume of
100 μl containing only Ca2+/CaM, and the reaction was initiated
by the addition of NADPH to a final concentration of 0.5 mm. EPR
measurements were carried out, as described under “Experimental
Procedures,” with the nitrone spin trap DEPMPO, which forms a stable
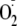 adduct with a half-life of ∼16 min
(28). Kinetics of
adduct with a half-life of ∼16 min
(28). Kinetics of
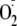 generation from heNOS were determined by setting the static field at
∼3501.1 G, on the maximum of the fourth peak of the EPR spectrum of the
DEPMPO/·OOH spin trap adduct (Fig.
2A). The reaction rate of
generation from heNOS were determined by setting the static field at
∼3501.1 G, on the maximum of the fourth peak of the EPR spectrum of the
DEPMPO/·OOH spin trap adduct (Fig.
2A). The reaction rate of
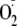 generation was determined by a linear fitting of this signal amplitude change
versus time during the first 5 min of reaction
(Fig. 2B). The
relative activities were further calculated by comparison with control
experiments of unmodified heNOS.
generation was determined by a linear fitting of this signal amplitude change
versus time during the first 5 min of reaction
(Fig. 2B). The
relative activities were further calculated by comparison with control
experiments of unmodified heNOS.
FIGURE 2.

EPR spin-trapping of the kinetics of
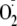 generation from control heNOS and phospho-Ser-1177 heNOS. A,
incremental scanning EPR spectra of oxygen free radical generation. The
reaction system contains 50 mm Tris-HCl buffer, pH 7.4, 0.5
mm NADPH, 0.5 mm Ca2+, 10 μg/ml CaM, 15
μg/ml purified heNOS, and 25 mm spin trap DEPMPO. Spectra were
recorded at room temperature with a microwave frequency of 9.863 GHz, 20
milliwatts of microwave power, and 1.0 G modulation amplitude. Center field
was 3510 G with 140-G sweep width. Time constant was 328 ms; each spectrum was
an 84-s acquisition. B, the continuous time course of
generation from control heNOS and phospho-Ser-1177 heNOS. A,
incremental scanning EPR spectra of oxygen free radical generation. The
reaction system contains 50 mm Tris-HCl buffer, pH 7.4, 0.5
mm NADPH, 0.5 mm Ca2+, 10 μg/ml CaM, 15
μg/ml purified heNOS, and 25 mm spin trap DEPMPO. Spectra were
recorded at room temperature with a microwave frequency of 9.863 GHz, 20
milliwatts of microwave power, and 1.0 G modulation amplitude. Center field
was 3510 G with 140-G sweep width. Time constant was 328 ms; each spectrum was
an 84-s acquisition. B, the continuous time course of
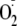 generation from control and phospho-Ser-1177 heNOS was measured by fixing the
static field at 3501.1 G on the maximum of the 4th peak of the DEPMPO-OOH
adduct, as marked by the asterisk. The time constant was 5243 ms. The reaction
system and acquisition parameters were otherwise the same as in
A.
generation from control and phospho-Ser-1177 heNOS was measured by fixing the
static field at 3501.1 G on the maximum of the 4th peak of the DEPMPO-OOH
adduct, as marked by the asterisk. The time constant was 5243 ms. The reaction
system and acquisition parameters were otherwise the same as in
A.
Compared with the control enzyme, phosphorylation of heNOS at Ser-1177
increased
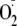 generation by 54% ± 3% (Fig.
3). However, there was no significant effect on
generation by 54% ± 3% (Fig.
3). However, there was no significant effect on
 generation produced by phosphorylation of Thr-495. Furthermore,
phosphorylation of heNOS by both Akt and PKCα increased its
generation produced by phosphorylation of Thr-495. Furthermore,
phosphorylation of heNOS by both Akt and PKCα increased its
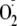 generation by a similar magnitude, 50% ± 4%, as seen with
phosphorylation by Akt alone.
generation by a similar magnitude, 50% ± 4%, as seen with
phosphorylation by Akt alone.
FIGURE 3.

Effects of phosphorylation on the magnitude of
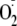 from heNOS.
from heNOS.
 generation rates from control and phosphorylated heNOS were determined from
preparations of enzyme by EPR spin-trapping with DEPMPO as described in
Fig. 2. The phosphorylation of
heNOS by Akt or Akt and PKCα increased its
generation rates from control and phosphorylated heNOS were determined from
preparations of enzyme by EPR spin-trapping with DEPMPO as described in
Fig. 2. The phosphorylation of
heNOS by Akt or Akt and PKCα increased its
 generation by greater than 50%; however, there was no significant effect from
the phosphorylation of heNOS by PKCα. Data were expressed as the mean
± S.E., n = 3 (*, p < 0.001
versus control).
generation by greater than 50%; however, there was no significant effect from
the phosphorylation of heNOS by PKCα. Data were expressed as the mean
± S.E., n = 3 (*, p < 0.001
versus control).
Ca2+ Dependence of Phosphorylation of
heNOS on
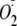 Generation—The generation of
Generation—The generation of
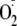 from eNOS is regulated by Ca2+/CaM, and as noted above,
phosphorylation of heNOS at Ser-1177 caused a marked increase in
from eNOS is regulated by Ca2+/CaM, and as noted above,
phosphorylation of heNOS at Ser-1177 caused a marked increase in
 production. Therefore, studies were performed to examine how phosphorylation
of this residue alters the Ca2+ sensitivity of the uncoupled
BH4-free enzyme. The desired free Ca2+ concentration was
achieved by including 100 μm EGTA in the reaction mixture, with
subsequent addition of CaCl2 stock solution. The free
Ca2+ concentration was then calculated as described under
“Experimental Procedures.” Plotting free [Ca2+]
versus activity and fitting the data with the Hill equation, we found
that phosphorylation at Ser-1177 decreased the EC50 of
Ca2+ for
production. Therefore, studies were performed to examine how phosphorylation
of this residue alters the Ca2+ sensitivity of the uncoupled
BH4-free enzyme. The desired free Ca2+ concentration was
achieved by including 100 μm EGTA in the reaction mixture, with
subsequent addition of CaCl2 stock solution. The free
Ca2+ concentration was then calculated as described under
“Experimental Procedures.” Plotting free [Ca2+]
versus activity and fitting the data with the Hill equation, we found
that phosphorylation at Ser-1177 decreased the EC50 of
Ca2+ for
 generation to a much lower concentration, 4.5 μm for the
phospho-Ser-1177 eNOS compared with 22.1 μm for native eNOS
(Fig. 4;
Table 1). Thus, phosphorylation
of heNOS at Ser-1177 triggered
generation to a much lower concentration, 4.5 μm for the
phospho-Ser-1177 eNOS compared with 22.1 μm for native eNOS
(Fig. 4;
Table 1). Thus, phosphorylation
of heNOS at Ser-1177 triggered
 generation at much lower Ca2+ concentrations compared with
non-phosphorylated heNOS, demonstrating ∼45% of maximal
generation at much lower Ca2+ concentrations compared with
non-phosphorylated heNOS, demonstrating ∼45% of maximal
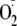 generation even in the absence of free Ca2+. The NOS inhibitor
l-NAME, which is known to inhibit both NO and
generation even in the absence of free Ca2+. The NOS inhibitor
l-NAME, which is known to inhibit both NO and
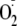 generation from eNOS, was able to almost totally block
generation from eNOS, was able to almost totally block
 production from the phosphorylated enzyme over the full range of
Ca2+ concentrations studied. PKCα phosphorylation, however,
only slightly altered the Ca2+-dependent activation of
production from the phosphorylated enzyme over the full range of
Ca2+ concentrations studied. PKCα phosphorylation, however,
only slightly altered the Ca2+-dependent activation of
 generation compared with that of non-phosphorylated heNOS (23.2
versus 22.1 μm)
(Fig. 4;
Table 1).
generation compared with that of non-phosphorylated heNOS (23.2
versus 22.1 μm)
(Fig. 4;
Table 1).
FIGURE 4.

Ca2+ dependence of
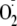 generation from control, phospho-Ser-1177, and phospho-Thr-495 heNOS.
generation from control, phospho-Ser-1177, and phospho-Thr-495 heNOS.
 generation rates from control, phospho-Ser-1177, and phospho-Thr-495 heNOS
were determined by EPR spin-trapping with DEPMPO as described in
Fig. 2. In the reaction 100
μm EGTA was included in the assay buffer. The desired
Ca2+ concentration was achieved by the addition of a concentrated
CaCl2 stock solution. The free [Ca2+] was calculated as
described under “Experimental Procedures.”
generation rates from control, phospho-Ser-1177, and phospho-Thr-495 heNOS
were determined by EPR spin-trapping with DEPMPO as described in
Fig. 2. In the reaction 100
μm EGTA was included in the assay buffer. The desired
Ca2+ concentration was achieved by the addition of a concentrated
CaCl2 stock solution. The free [Ca2+] was calculated as
described under “Experimental Procedures.”
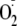 generation from eNOS was almost totally blocked by 1 mm l-NAME over
the full range of Ca2+ concentrations studied. All data points show
the relative magnitude of
generation from eNOS was almost totally blocked by 1 mm l-NAME over
the full range of Ca2+ concentrations studied. All data points show
the relative magnitude of
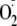 generation compared with the maximal values measured from control heNOS and
correspond to the mean ± S.E. from triplicate experiments.
generation compared with the maximal values measured from control heNOS and
correspond to the mean ± S.E. from triplicate experiments.
TABLE 1.
Effects of heNOS phosphorylation on Ca2+,
CaM activation, or Cav-P inhibition of
 generation
generation
 generation rates from control and Akt or PKCα-phosphorylated heNOS
(BH4-free) were measured by EPR spin-trapping as described in
Fig. 2 with EC50
measured as in Figs. 4 and
6. The effect of
phosphorylation of heNOS on the interaction with Cav-P was determined with
Cav-P inhibition expressed as the percent inhibition produced by the addition
of Cav-P (400 μm) to a reaction containing 500 μm
Ca2+, 600 nm CaM.
generation rates from control and Akt or PKCα-phosphorylated heNOS
(BH4-free) were measured by EPR spin-trapping as described in
Fig. 2 with EC50
measured as in Figs. 4 and
6. The effect of
phosphorylation of heNOS on the interaction with Cav-P was determined with
Cav-P inhibition expressed as the percent inhibition produced by the addition
of Cav-P (400 μm) to a reaction containing 500 μm
Ca2+, 600 nm CaM.
| EC50 of [calcium] | EC50 of [CaM] | Cav-P inhibition of superoxide generation | |
|---|---|---|---|
| μm | nm | % | |
| Control | 22.1 | 208 | 35.7 ± 3.9 |
| Akt | 4.5 | 149 | 50.6 ± 2.6a |
| PKCα | 23.2 | 496 | 67.8 ± 1.6b |
p < 0.02 versus control
p < 0.01 versus control. Data are expressed as the mean ± S.E., n = 3
EGTA Inactivation of
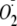 Generation from heNOS—In these experiments the active
eNOS·CaM·Ca2+ complex was allowed to form, and then
increasing concentrations of EGTA were added to chelate the Ca2+
and initiate the dissociation of the complex. After 1 min, the rate of
Generation from heNOS—In these experiments the active
eNOS·CaM·Ca2+ complex was allowed to form, and then
increasing concentrations of EGTA were added to chelate the Ca2+
and initiate the dissociation of the complex. After 1 min, the rate of
 generation was determined using EPR spin-trapping, as described above. At an
EGTA concentration of 800 μm, no
generation was determined using EPR spin-trapping, as described above. At an
EGTA concentration of 800 μm, no
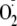 signal was detected from the nonphosphorylated enzyme; however, the
phospho-Ser-1177 eNOS still retained ∼40% of its maximal
signal was detected from the nonphosphorylated enzyme; however, the
phospho-Ser-1177 eNOS still retained ∼40% of its maximal
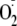 generation capacity (Fig. 5).
Even when EGTA was added to a final concentration of 1000 μm,
the phosphorylated enzyme still retained ∼25% of its maximal
generation capacity (Fig. 5).
Even when EGTA was added to a final concentration of 1000 μm,
the phosphorylated enzyme still retained ∼25% of its maximal
 generation capacity. Thus, it is clear that phosphorylation of heNOS at
Ser-1177 increased resistance to EGTA inactivation of
generation capacity. Thus, it is clear that phosphorylation of heNOS at
Ser-1177 increased resistance to EGTA inactivation of
 generation.
generation.
FIGURE 5.

EGTA inactivation of
 generation by control heNOS and phospho-Ser-1177 heNOS.
generation by control heNOS and phospho-Ser-1177 heNOS.
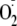 generation rates from control and phosphoSer-1177 heNOS were determined by EPR
spin-trapping with DEPMPO as described in
Fig. 2. The effect of
phosphorylation of heNOS on the EGTA inactivation of
generation rates from control and phosphoSer-1177 heNOS were determined by EPR
spin-trapping with DEPMPO as described in
Fig. 2. The effect of
phosphorylation of heNOS on the EGTA inactivation of
 generation was determined. 200 μm Ca2+ was included
in the reaction. In control experiments, when EGTA was added to a final
concentration of 800 μm, there is no measurable
generation was determined. 200 μm Ca2+ was included
in the reaction. In control experiments, when EGTA was added to a final
concentration of 800 μm, there is no measurable
 generation, although with Akt phosphorylation, even with 1000 μm
EGTA, still ∼25% of the
generation, although with Akt phosphorylation, even with 1000 μm
EGTA, still ∼25% of the
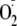 generation remained. All data points show the relative magnitude of
generation remained. All data points show the relative magnitude of
 generation compared with the maximal values measured from control heNOS and
correspond to the mean ± S.E. from triplicate experiments. The line
fitting of the experimental points for each curve was performed using a
sigmoidal function.
generation compared with the maximal values measured from control heNOS and
correspond to the mean ± S.E. from triplicate experiments. The line
fitting of the experimental points for each curve was performed using a
sigmoidal function.
CaM Dependence of Phosphorylation of heNOS on
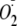 Generation—The
Generation—The
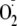 generation rate of heNOS and phosphorylated heNOS at Ser-1177 was determined
by EPR spin-trapping with DEPMPO, as a function of increasing concentrations
of CaM. The calculated EC50 of control heNOS was 208 nm,
and the calculated EC50 of phosphorylated heNOS at Ser-1177 was 149
nm (Fig. 6;
Table 1); this result is
consistent with the reported CaM dependence of NO production
(21,
35). Furthermore,
phosphorylation of heNOS by PKCα at Thr-495 greatly lowered its CaM
binding affinity, increasing the calculated EC50 of Thr-495
phosphorylated heNOS to 496 nm
(Fig. 6;
Table 1).
generation rate of heNOS and phosphorylated heNOS at Ser-1177 was determined
by EPR spin-trapping with DEPMPO, as a function of increasing concentrations
of CaM. The calculated EC50 of control heNOS was 208 nm,
and the calculated EC50 of phosphorylated heNOS at Ser-1177 was 149
nm (Fig. 6;
Table 1); this result is
consistent with the reported CaM dependence of NO production
(21,
35). Furthermore,
phosphorylation of heNOS by PKCα at Thr-495 greatly lowered its CaM
binding affinity, increasing the calculated EC50 of Thr-495
phosphorylated heNOS to 496 nm
(Fig. 6;
Table 1).
FIGURE 6.

CaM dependence of
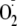 generation from control, phospho-Ser-1177, and phospho-Thr-495 heNOS.
generation from control, phospho-Ser-1177, and phospho-Thr-495 heNOS.
 generation rate from control, phospho-Ser-1177, and phospho-Thr-495 heNOS was
determined from preparations of enzyme by EPR spin-trapping with DEPMPO as
described in Fig. 2. The effect
of phosphorylation of heNOS on the interaction with CaM under uncoupled
(BH4-free) conditions was determined. In the presence of 0.5
mm CaCl2, CaM was added to the desired concentration as
indicated. The concentration of CaM required for 50% maximal activity of
generation rate from control, phospho-Ser-1177, and phospho-Thr-495 heNOS was
determined from preparations of enzyme by EPR spin-trapping with DEPMPO as
described in Fig. 2. The effect
of phosphorylation of heNOS on the interaction with CaM under uncoupled
(BH4-free) conditions was determined. In the presence of 0.5
mm CaCl2, CaM was added to the desired concentration as
indicated. The concentration of CaM required for 50% maximal activity of
 generation (EC50) was determined from CaM dependence of heNOS
generation (EC50) was determined from CaM dependence of heNOS
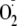 generation. The EC50 of control heNOS was 208 nm, the
EC50 of phospho-Ser-1177 heNOS was 149 nm, and the
EC50 of phospho-Thr-495 heNOS was 496 nm. Data were
expressed as mean ± S.E., n = 3.
generation. The EC50 of control heNOS was 208 nm, the
EC50 of phospho-Ser-1177 heNOS was 149 nm, and the
EC50 of phospho-Thr-495 heNOS was 496 nm. Data were
expressed as mean ± S.E., n = 3.
Effects of Phosphorylation of heNOS on the Cav-P Inhibition of
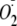 Generation from Uncoupled heNOS—The association with caveolin is
known to negatively regulate eNOS
(36,
37). To investigate the
effects of Cav-P on the
Generation from Uncoupled heNOS—The association with caveolin is
known to negatively regulate eNOS
(36,
37). To investigate the
effects of Cav-P on the
 generation from control or phosphorylated heNOS, Cav-P was added to a final
concentration of 400 μm. The percentage inhibition of
generation from control or phosphorylated heNOS, Cav-P was added to a final
concentration of 400 μm. The percentage inhibition of
 generation under each condition was used to determine how phosphorylation
affects the association of heNOS with Cav-P. In control experiments using
native enzyme, a 35.7% inhibition of
generation under each condition was used to determine how phosphorylation
affects the association of heNOS with Cav-P. In control experiments using
native enzyme, a 35.7% inhibition of
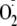 generation was produced by incubation with Cav-P. Phosphorylation in general
produced an increase in Cav-P inhibition. When Ser-1177 was phosphorylated,
Cav-P induced a 50.6% inhibition of
generation was produced by incubation with Cav-P. Phosphorylation in general
produced an increase in Cav-P inhibition. When Ser-1177 was phosphorylated,
Cav-P induced a 50.6% inhibition of
 .
production, whereas with Thr-495 phosphorylation a 67.8% decrease of
.
production, whereas with Thr-495 phosphorylation a 67.8% decrease of
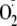 was seen (Table 1).
was seen (Table 1).
Phosphorylation of eNOS Alters
 Generation from Uncoupled eNOS in BAECs—To test how phosphorylation
of eNOS affects the activity of uncoupled eNOS in endothelial cells, we
treated BAECs with stimuli known to induce eNOS phosphorylation. We used
confocal microscopy with DHE for the detection of
Generation from Uncoupled eNOS in BAECs—To test how phosphorylation
of eNOS affects the activity of uncoupled eNOS in endothelial cells, we
treated BAECs with stimuli known to induce eNOS phosphorylation. We used
confocal microscopy with DHE for the detection of
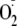 and phospho-specific eNOS antibodies to confirm the eNOS phosphorylation state
in cells. Treatment of BAECs with VEGF and PMA is known to lead to the
phosphorylation of eNOS on Ser-1179 and Thr-497 (equivalent to Ser-1177 and
Thr-495 in the human eNOS), respectively
(22). In control experiments,
untreated BAECs, or untreated BAECs stimulated with VEGF, PMA, or CaI, there
was no detectable DHE fluorescence signal. Thus, as expected, endothelial
cells containing BH4-repleted eNOS did not produce
and phospho-specific eNOS antibodies to confirm the eNOS phosphorylation state
in cells. Treatment of BAECs with VEGF and PMA is known to lead to the
phosphorylation of eNOS on Ser-1179 and Thr-497 (equivalent to Ser-1177 and
Thr-495 in the human eNOS), respectively
(22). In control experiments,
untreated BAECs, or untreated BAECs stimulated with VEGF, PMA, or CaI, there
was no detectable DHE fluorescence signal. Thus, as expected, endothelial
cells containing BH4-repleted eNOS did not produce
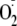 ,
and phosphorylation of the enzyme did not lead to uncoupling.
,
and phosphorylation of the enzyme did not lead to uncoupling.
To uncouple eNOS, BAECs were first treated with 5 mm
2,4-diamino-6-hydroxypyrimidine for 18 h to deplete BH4
(32). In control
BH4-depleted cells, there was no detectable red HE fluorescence
(Fig. 7A). Note, DAPI
staining was used to counter stain the nuclei (blue). However, when
the BH4-depleted BAECs were treated with either CaI, commonly used
to induce Ca2+ influx, or VEGF to induce Akt activation with
phosphorylation of Ser-1179, strong red HE fluorescence was detected, and this
stimulated
 generation was completely inhibited by 1 mm l-NAME. This HE
fluorescence was also completely quenched by the superoxide dismutase mimetic
MnTBAP (manganese (III) tetrakis (4-benzoic acid) porphyrin), 1 mm,
confirming that it was derived from
generation was completely inhibited by 1 mm l-NAME. This HE
fluorescence was also completely quenched by the superoxide dismutase mimetic
MnTBAP (manganese (III) tetrakis (4-benzoic acid) porphyrin), 1 mm,
confirming that it was derived from
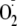 (data not shown). Treatment of BH4-depleted BAECs with the PKC
activator PMA, which increases intracellular Ca2+ and induces
phosphorylation of Thr-497
(38), was performed, and these
cells also exhibited red HE fluorescence similar to that induced by CaI. The
phosphorylation of beNOS produced by each stimulus was determined by
immunofluorescence microscopy using phospho-specific eNOS antibodies
(Fig. 7B).
Phosphorylation of uncoupled beNOS at Ser-1179 was seen when cells were
treated with either VEGF or CaI, whereas treatment of BH4-depleted
cells with PMA led to the phosphorylation of eNOS mainly on Thr-497.
Therefore, the formation of
(data not shown). Treatment of BH4-depleted BAECs with the PKC
activator PMA, which increases intracellular Ca2+ and induces
phosphorylation of Thr-497
(38), was performed, and these
cells also exhibited red HE fluorescence similar to that induced by CaI. The
phosphorylation of beNOS produced by each stimulus was determined by
immunofluorescence microscopy using phospho-specific eNOS antibodies
(Fig. 7B).
Phosphorylation of uncoupled beNOS at Ser-1179 was seen when cells were
treated with either VEGF or CaI, whereas treatment of BH4-depleted
cells with PMA led to the phosphorylation of eNOS mainly on Thr-497.
Therefore, the formation of
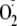 from uncoupled beNOS in endothelial cells is stimulated by phosphorylation of
the uncoupled enzyme at Ser-1179 or Thr-497. Moreover, just as in our isolated
enzyme experiments, phosphorylation of Thr-497 did not inhibit the formation
of eNOS-derived
from uncoupled beNOS in endothelial cells is stimulated by phosphorylation of
the uncoupled enzyme at Ser-1179 or Thr-497. Moreover, just as in our isolated
enzyme experiments, phosphorylation of Thr-497 did not inhibit the formation
of eNOS-derived
 .
.
FIGURE 7.

Imaging of
 generation and immunostaining in BAECs. A, confocal microscopy
measurements of
generation and immunostaining in BAECs. A, confocal microscopy
measurements of
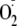 generation from uncoupled eNOS in endothelial cells. BH4 depletion
was achieved by incubation of BAECs with 5 mm
2,4-diamino-6-hydroxypyrimidine (DAHP) 18 h at 37 °C. The
generation from uncoupled eNOS in endothelial cells. BH4 depletion
was achieved by incubation of BAECs with 5 mm
2,4-diamino-6-hydroxypyrimidine (DAHP) 18 h at 37 °C. The
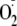 generation was visualized from BH4-depleted BAECs loaded with DHE
that reacts to form HE, that exhibits red fluorescence. The blue
color corresponds to the nuclei stained with DAPI. Measurable
generation was visualized from BH4-depleted BAECs loaded with DHE
that reacts to form HE, that exhibits red fluorescence. The blue
color corresponds to the nuclei stained with DAPI. Measurable
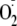 fluorescence was seen only when the BH4-depleted cells were treated
with CaI, VEGF, or PMA, and this was inhibited by l-NAME.
B, immunostaining using phosphorylation specific anti-eNOS antibodies
in BH4-depleted BAECs. The left column shows the results
using the anti-phospho-Ser-1179 antibody. Ser-1179 phosphorylation was
detected in BH4-depleted BAECs when the cells were treated with CaI
or VEGF but not with PMA. The right column shows the results using
the anti-phospho-Thr-497 antibody, demonstrating that PMA treatment produced
strong phosphorylation of Thr-497. These two immunostaining experiments were
done in two sets of cells, both under the same conditions.
fluorescence was seen only when the BH4-depleted cells were treated
with CaI, VEGF, or PMA, and this was inhibited by l-NAME.
B, immunostaining using phosphorylation specific anti-eNOS antibodies
in BH4-depleted BAECs. The left column shows the results
using the anti-phospho-Ser-1179 antibody. Ser-1179 phosphorylation was
detected in BH4-depleted BAECs when the cells were treated with CaI
or VEGF but not with PMA. The right column shows the results using
the anti-phospho-Thr-497 antibody, demonstrating that PMA treatment produced
strong phosphorylation of Thr-497. These two immunostaining experiments were
done in two sets of cells, both under the same conditions.
DISCUSSION
Nitric-oxide synthase requires BH4 to produce NO from
l-Arg in a reaction involving the NADPH-dependent formation of a
heme-bound two-electron reduced molecular oxygen moiety. In the absence of
BH4, NOS is unable to timely donate the second electron, resulting
in
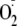 release from the heme center, thus uncoupling NADPH oxidation from NO
formation. The switch from NO to
release from the heme center, thus uncoupling NADPH oxidation from NO
formation. The switch from NO to
 production that is triggered by oxidative depletion of BH4 has been
implicated in the pathophysiology of numerous diseases
(11,
39–43).
Therefore, it is of critical importance to understand how the production of
both of these eNOS-derived products is regulated.
production that is triggered by oxidative depletion of BH4 has been
implicated in the pathophysiology of numerous diseases
(11,
39–43).
Therefore, it is of critical importance to understand how the production of
both of these eNOS-derived products is regulated.
Several sites have been identified in eNOS with the potential for
phosphorylation and posttranslational regulation, including Ser-1177, Thr-495,
and serines 114, 615, and 633 (using the human amino acid numbering). However,
prior work has shown that modifications of Ser-1177 and Thr-495 are of
particular importance. It is commonly accepted that eNOS NO production is
inhibited by phosphorylation at Thr-495 and enhanced by phosphorylation at
Ser-1177 (20,
22,
44,
45). However, questions remain
regarding the effect that phosphorylation of these two key residues has on the
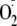 production of the uncoupled enzyme. Therefore, in this work we have used EPR
spin-trapping to determine how phosphorylation of recombinant human eNOS by
native kinases affects the
production of the uncoupled enzyme. Therefore, in this work we have used EPR
spin-trapping to determine how phosphorylation of recombinant human eNOS by
native kinases affects the
 production of the BH4-free enzyme. We show that phosphorylation of
Ser-1177 increased the rate of
production of the BH4-free enzyme. We show that phosphorylation of
Ser-1177 increased the rate of
 generation by >50% in the presence of excess Ca2+/CaM, whereas
phosphorylation of Thr-495 had no effect under these conditions.
generation by >50% in the presence of excess Ca2+/CaM, whereas
phosphorylation of Thr-495 had no effect under these conditions.
Phosphorylation of eNOS at Ser-1177 for human or Ser-1179 for bovine was first demonstrated to regulate its NO production in 1999 by Fulton et al. (18) and separately by Dimmeler et al. (19). It was shown that phosphorylation of this residue via the kinase Akt in cells leads to an increase in NOS-derived NO. Since then, isolated enzyme studies have shown that the S1177D eNOS mutant (mimicking the Ser-1177 phosphorylated eNOS) leads to an increase in the electron flow through the reductase domain, and it was hypothesized that this increase is the mechanism responsible for the phosphorylation-dependent increase in eNOS-derived NO (21).
In our current study, we directly demonstrate that under conditions where
eNOS is uncoupled, phosphorylation of Ser-1177 increases the rate of
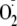 generation. With 80% phosphorylation of Ser-1177 we observe a 54% increase
similar to that previously demonstrated for the phosphorylation-dependent
increase in NO production. If one corrects for the incomplete phosphorylation,
approximately a 68% increase in
generation. With 80% phosphorylation of Ser-1177 we observe a 54% increase
similar to that previously demonstrated for the phosphorylation-dependent
increase in NO production. If one corrects for the incomplete phosphorylation,
approximately a 68% increase in
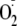 generation would be expected with 100% phosphorylation of the enzyme.
Moreover, we demonstrated in endothelial cells that this increase in
generation would be expected with 100% phosphorylation of the enzyme.
Moreover, we demonstrated in endothelial cells that this increase in
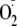 is also produced by Akt activation with phosphorylation of the critical
serine. This increased
is also produced by Akt activation with phosphorylation of the critical
serine. This increased
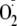 generation of phospho-Ser-1177 heNOS was inhibited by the addition of
l-NAME, a NOS-specific inhibitor that blocks
generation of phospho-Ser-1177 heNOS was inhibited by the addition of
l-NAME, a NOS-specific inhibitor that blocks
 formation by preventing electron transfer to the heme of the oxygenase domain.
There has been a report that eNOS-dependent
formation by preventing electron transfer to the heme of the oxygenase domain.
There has been a report that eNOS-dependent
 generation from BAECs is not dependent upon Ser-1177 phosphorylation
(46). Our results agree that
phosphorylation is not required for the generation of
generation from BAECs is not dependent upon Ser-1177 phosphorylation
(46). Our results agree that
phosphorylation is not required for the generation of
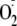 from eNOS. However, it is clear that Ser-1177 phosphorylation significantly
increases
from eNOS. However, it is clear that Ser-1177 phosphorylation significantly
increases
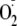 generation from eNOS and, perhaps more importantly, shifts the calcium
requirement to much lower levels.
generation from eNOS and, perhaps more importantly, shifts the calcium
requirement to much lower levels.
There are two potential mechanisms for
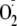 generation in eNOS, direct electron transfer from the FMN to molecular oxygen
(commonly termed electron leakage) and electron transfer from the reductase
domain to the oxygenase domain forming the ferrous heme, followed by oxygen
binding and then regeneration of the ferric heme by the release of
generation in eNOS, direct electron transfer from the FMN to molecular oxygen
(commonly termed electron leakage) and electron transfer from the reductase
domain to the oxygenase domain forming the ferrous heme, followed by oxygen
binding and then regeneration of the ferric heme by the release of
 .
Because l-NAME blocked the observed increase in
.
Because l-NAME blocked the observed increase in
 in the phosopho-Ser-1177 eNOS, we conclude that Akt phosphorylation alters
in the phosopho-Ser-1177 eNOS, we conclude that Akt phosphorylation alters
 production from the heme. The rate-limiting step for the generation of
production from the heme. The rate-limiting step for the generation of
 from the heme is the transfer of electrons from the reductase domain to the
oxygenase domain (47,
48), which in the global
kinetic model of NOS activity proposed by Stuehr et al.
(49) is the same rate-limiting
step for NO production. Thus, it would be expected that any modification that
alters eNOS NO production will similarly alter the activity of the uncoupled
enzyme. Indeed, Ser-1177 phosphorylation increased the
from the heme is the transfer of electrons from the reductase domain to the
oxygenase domain (47,
48), which in the global
kinetic model of NOS activity proposed by Stuehr et al.
(49) is the same rate-limiting
step for NO production. Thus, it would be expected that any modification that
alters eNOS NO production will similarly alter the activity of the uncoupled
enzyme. Indeed, Ser-1177 phosphorylation increased the
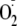 production from the enzyme switching eNOS from Ca2+-dependent to
more Ca2+-independent
production from the enzyme switching eNOS from Ca2+-dependent to
more Ca2+-independent
 generation, more like NOS-2. The EC50 for CaM activation of
generation, more like NOS-2. The EC50 for CaM activation of
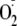 production was also altered. These observations are in good agreement with
reports measuring NO output from the S1177D eNOS and from an eNOS in which the
C-terminal 27 amino acids (containing Ser-1177) were deleted (Δ27)
(21,
50).
production was also altered. These observations are in good agreement with
reports measuring NO output from the S1177D eNOS and from an eNOS in which the
C-terminal 27 amino acids (containing Ser-1177) were deleted (Δ27)
(21,
50).
Both the C-terminal region of eNOS and an autoinhibitory region located in the FMN binding domain regulate the flow of electrons within the reductase domain, modulate CaM activation, and are thought to act in concert to regulate NOS function (51). Ser-1177 lies within the C-terminal region, and its phosphorylation is thought to regulate NO production from eNOS by altering the interaction between the C-terminal and autoinhibitory region (AR), releasing the AR-dependent inhibition. Along with the prior work using S1177D and Δ27, our data support this hypothesis. However, although Ser-1177 phosphorylation enhances the eNOS-CaM interaction, this phosphorylation event also would lead to an increase in the absolute rate of electron transfer from the reductase domain to the heme with increased flavin reduction rate, a hallmark of AR inhibition release (47).
Thr-495 lies within the CaM binding domain of eNOS, and the currently
accepted dogma is that phosphorylation of eNOS at Thr-495 (human) or Thr-497
(bovine) inhibits NO production by interfering with CaM binding
(27). However, more recent
studies have questioned this. It has been shown that treatment of BAECs with
okadaic acid and PMA, which increased Thr-497 phosphorylation, enhanced NO
production compared with control
(44). Moreover, although the
T495A eNOS mutant, which mimics dephosphorylation, did increase NO production,
mutagenesis of this residue to aspartic acid to mimic phosphorylation had no
effect on eNOS NO activity
(22). The dephosphorylation of
Thr-495 has been linked to increasing eNOS-dependent
 generation in endothelial cells via the inhibition of PKCα
(52). Conversely it has been
reported that PKCα overexpression activates eNOS and increases arterial
blood flow in vivo
(53), but it was hypothesized
that this occurred due to an increase in Ser-1177 phosphorylation, not
phosphorylation or dephosphorylation of Thr-495.
generation in endothelial cells via the inhibition of PKCα
(52). Conversely it has been
reported that PKCα overexpression activates eNOS and increases arterial
blood flow in vivo
(53), but it was hypothesized
that this occurred due to an increase in Ser-1177 phosphorylation, not
phosphorylation or dephosphorylation of Thr-495.
Our work demonstrated that phosphorylation of Thr-495 using PKCα did
indeed greatly decrease CaM binding affinity; however, under conditions where
CaM was saturating there was no effect on the maximal rate of
 generation. Our cellular findings show strong heme-dependent
generation. Our cellular findings show strong heme-dependent
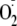 generation from BH4-depleted BAECs even with the phosphorylation of
Thr-495, clearly demonstrating that Thr-495 phosphorylation is not strictly an
“off-switch.” It has been shown that the cellular concentration of
CaM is greater than 10 μm
(54), which is higher than the
saturating concentration of CaM used in our in vitro study. As such,
although phosphorylation of Thr-495 does decrease CaM binding affinity for
eNOS and thereby potentially leads to a decrease in eNOS activity, any
potential negative regulation in cells would require alterations of other
cellular factors that influence the eNOS-CaM interaction.
generation from BH4-depleted BAECs even with the phosphorylation of
Thr-495, clearly demonstrating that Thr-495 phosphorylation is not strictly an
“off-switch.” It has been shown that the cellular concentration of
CaM is greater than 10 μm
(54), which is higher than the
saturating concentration of CaM used in our in vitro study. As such,
although phosphorylation of Thr-495 does decrease CaM binding affinity for
eNOS and thereby potentially leads to a decrease in eNOS activity, any
potential negative regulation in cells would require alterations of other
cellular factors that influence the eNOS-CaM interaction.
Previously it has been shown that the association of eNOS with caveolin
inhibits eNOS activity (36,
37). Our results showed that
the caveolin-dependent inhibition of eNOS
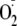 generation was enhanced by phosphorylation of eNOS, with the greatest
inhibition seen when Thr-495 was phosphorylated. This indicates that
phosphorylation of eNOS increases the association between eNOS and caveolin.
As such, our in vitro data are consistent with the hypothesis that
Thr-495 phosphorylation negatively regulates eNOS activity
(13,
20,
22,
44,
52,
55,
56). Previous reports have
indicated that phosphorylation of eNOS at Ser-1177 leads to a dissociation of
eNOS and caveolin; however, this dissociation was dependent upon the induction
of caveolin-dependent endocytosis
(57,
58). We conclude that
phosphorylation of heNOS at Thr-495 plays a role in the regulation of eNOS
activity indirectly through the alteration of CaM binding and the association
with caveolin. However, the precise in vivo regulation induced by
Thr-495 phosphorylation will clearly be dependent upon processes that regulate
local CaM and caveolin concentrations.
generation was enhanced by phosphorylation of eNOS, with the greatest
inhibition seen when Thr-495 was phosphorylated. This indicates that
phosphorylation of eNOS increases the association between eNOS and caveolin.
As such, our in vitro data are consistent with the hypothesis that
Thr-495 phosphorylation negatively regulates eNOS activity
(13,
20,
22,
44,
52,
55,
56). Previous reports have
indicated that phosphorylation of eNOS at Ser-1177 leads to a dissociation of
eNOS and caveolin; however, this dissociation was dependent upon the induction
of caveolin-dependent endocytosis
(57,
58). We conclude that
phosphorylation of heNOS at Thr-495 plays a role in the regulation of eNOS
activity indirectly through the alteration of CaM binding and the association
with caveolin. However, the precise in vivo regulation induced by
Thr-495 phosphorylation will clearly be dependent upon processes that regulate
local CaM and caveolin concentrations.
In the process of ischemia-reperfusion injury, the oxidative stress in
tissues can lead to depletion of BH4
(3,
4,
59). Recently we have reported
that hearts subjected to various durations of ischemia show a time-dependent
decrease in BH4 levels that trigger increased NOS-derived
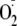 production (60). Our current
work demonstrates that with Akt-mediated phosphorylation, which would normally
enhance eNOS-derived NO, increased eNOS-derived
production (60). Our current
work demonstrates that with Akt-mediated phosphorylation, which would normally
enhance eNOS-derived NO, increased eNOS-derived
 production is also triggered from the uncoupled enzyme. Thus, when designing
treatment strategies to ameliorate oxidative stress induced diseases by
altering posttranslational modification of eNOS, one must consider that the
activity of both coupled and uncoupled eNOS will be modified. For example, a
strategy to enhance eNOS-derived NO by treatment with VEGF or other stimulus
that induces Ser-1177 phosphorylation may exacerbate eNOS dysfunction in the
post-ischemic heart. Moreover, although it was previously predicted that
treatments to phosphorylate Thr-495 would decrease eNOS output, our data
indicate that these would not necessarily decrease eNOS-derived
production is also triggered from the uncoupled enzyme. Thus, when designing
treatment strategies to ameliorate oxidative stress induced diseases by
altering posttranslational modification of eNOS, one must consider that the
activity of both coupled and uncoupled eNOS will be modified. For example, a
strategy to enhance eNOS-derived NO by treatment with VEGF or other stimulus
that induces Ser-1177 phosphorylation may exacerbate eNOS dysfunction in the
post-ischemic heart. Moreover, although it was previously predicted that
treatments to phosphorylate Thr-495 would decrease eNOS output, our data
indicate that these would not necessarily decrease eNOS-derived
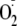 generation.
generation.
Our results demonstrate that phosphorylation can regulate
 in addition to NO generation from eNOS. Peroxynitrite (ONOO-) is
formed by the diffusion-limited reaction of
in addition to NO generation from eNOS. Peroxynitrite (ONOO-) is
formed by the diffusion-limited reaction of
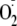 and NO. Thus, ONOO- is a third potential eNOS-derived effector
molecule regulated by the posttranslational modification of eNOS. Any
modification that would partially uncouple eNOS, with generation of both
and NO. Thus, ONOO- is a third potential eNOS-derived effector
molecule regulated by the posttranslational modification of eNOS. Any
modification that would partially uncouple eNOS, with generation of both
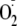 and NO, would lead to ONOO- generation. Additionally, any
modification changing the flux of either
and NO, would lead to ONOO- generation. Additionally, any
modification changing the flux of either
 or NO from eNOS would alter the relative amount of ONOO- formed.
ONOO- is known to be cytotoxic, functioning by both apoptotic and
necrotic pathways (61).
Additionally, ONOO- has been found to be a non-toxic signaling
molecule, altering a number of cell signal transduction pathways
(62). Thus, with partial
BH4 depletion, differential phosphorylation of eNOS could modulate
ONOO- generation, leading to altered cell signaling or cell
death.
or NO from eNOS would alter the relative amount of ONOO- formed.
ONOO- is known to be cytotoxic, functioning by both apoptotic and
necrotic pathways (61).
Additionally, ONOO- has been found to be a non-toxic signaling
molecule, altering a number of cell signal transduction pathways
(62). Thus, with partial
BH4 depletion, differential phosphorylation of eNOS could modulate
ONOO- generation, leading to altered cell signaling or cell
death.
In conclusion, PKCα-mediated phosphorylation of heNOS can decrease
 production from the enzyme through alterations in its interactions with CaM
and caveolin. Thus, Thr-495 phosphorylation indirectly regulates eNOS via
modulation of protein-protein interactions. In contrast, Akt-mediated
phosphorylation markedly enhances this
production from the enzyme through alterations in its interactions with CaM
and caveolin. Thus, Thr-495 phosphorylation indirectly regulates eNOS via
modulation of protein-protein interactions. In contrast, Akt-mediated
phosphorylation markedly enhances this
 production directly by altering the kinetics of electron transfer within the
enzyme. Additionally, this phosphorylation of Ser-1177 greatly increases
production directly by altering the kinetics of electron transfer within the
enzyme. Additionally, this phosphorylation of Ser-1177 greatly increases
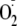 production at low levels of Ca2+ such that eNOS-dependent
production at low levels of Ca2+ such that eNOS-dependent
 generation becomes largely Ca2+-independent. Thus, phosphorylation
is of key importance in regulating the overall function, activation, and
potentially the coupling, of eNOS, modulating both the production of NO and
generation becomes largely Ca2+-independent. Thus, phosphorylation
is of key importance in regulating the overall function, activation, and
potentially the coupling, of eNOS, modulating both the production of NO and
 from the enzyme.
from the enzyme.
Acknowledgments
We greatly appreciate the gift of pCWheNOS bacterial expression plasmid from Dr. Ortiz de Montellano (University of California San Francisco).
This work was supported, in whole or in part, by National Institutes of Health Grants HL63744, HL65608, and HL38324 (to J. L. Z.) and HL83237 (Y.-R. C.). This work was also supported by an American Heart Association postdoctoral fellowship (to C.-A. C.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: NOS, nitric-oxide synthase; eNOS, endothelial NOS; heNOS, human eNOS; beNOS, bovine eNOS; PKC, protein kinase C; BAECs, bovine aortic endothelial cells; CaI, calcium ionophore; CaM, calmodulin; Cav-P, caveolin peptide; DAPI, 4′,6-diamidino-2-phenylindole dihydrochloride; DEPMPO, 5-diethoxyphosphoryl-5-methyl-1-pyrroline N-oxide; DHE, dihydroethidine; DTT, dithiothreitol; EPR, electron paramagnetic resonance; HE, hydroethidine; l-NAME, l-NG-nitroarginine methyl ester hydrochloride; BH4, tetrahydrobiopterin; TBST, Tris-buffered saline (TBS) and Tween; VEGF, vascular endothelial growth factor; RT, room temperature; PMA, phorbol 12-myristate 13-acetate.
References
- 1.Sessa, W. C. (2004) J. Cell Sci. 117 2427-2429 [DOI] [PubMed] [Google Scholar]
- 2.Kone, B. C., Kuncewicz, T., Zhang, W., and Yu, Z. Y. (2003) Am. J. Physiol. Renal Physiol. 285 178-190 [DOI] [PubMed] [Google Scholar]
- 3.Xia, Y., Tsai, A. L., Berka, V., and Zweier, J. L. (1998) J. Biol. Chem. 273 25804-25808 [DOI] [PubMed] [Google Scholar]
- 4.Vasquez-Vivar, J., Kalyanaraman, B., Martasek, P., Hogg, N., Masters, B. S., Karoui, H., Tordo, P., and Pritchard, K. A., Jr. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 9220-9225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muscoli, C., Cuzzocrea, S., Riley, D. P., Zweier, J. L., Thiemermann, C., Wang, Z. Q., and Salvemini, D. (2003) Br. J. Pharmacol. 140 445-460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pou, S., Pou, W. S., Bredt, D. S., Snyder, S. H., and Rosen, G. M. (1992) J. Biol. Chem. 267 24173-24176 [PubMed] [Google Scholar]
- 7.Xia, Y., Dawson, V. L., Dawson, T. M., Snyder, S. H., and Zweier, J. L. (1996) Proc. Natl. Acad. Sci. U. S. A. 93 6770-6774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xia, Y., and Zweier, J. L. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 6954-6958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xia, Y., Roman, L. J., Masters, B. S., and Zweier, J. L. (1998) J. Biol. Chem. 273 22635-22639 [DOI] [PubMed] [Google Scholar]
- 10.Griffith, O. W., and Stuehr, D. J. (1995) Annu. Rev. Physiol. 57 707-736 [DOI] [PubMed] [Google Scholar]
- 11.Sun, J., Druhan, L. J., and Zweier, J. L. (2008) Arch. Biochem. Biophys. 471 126-133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simoncini, T., Hafezi-Moghadam, A., Brazil, D. P., Ley, K., Chin, W. W., and Liao, J. K. (2000) Nature 407 538-541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris, M. B., Ju, H., Venema, V. J., Liang, H., Zou, R., Michell, B. J., Chen, Z.-P., Kemp, B. E., and Venema, R. C. (2001) J. Biol. Chem. 276 16587-16591 [DOI] [PubMed] [Google Scholar]
- 14.Igarashi, J., and Michel, T. (2001) J. Biol. Chem. 276 36281-36288 [DOI] [PubMed] [Google Scholar]
- 15.Morales-Ruiz, M., Lee, M. J., Zollner, S., Gratton, J. P., Scotland, R., Shiojima, I., Walsh, K., Hla, T., and Sessa, W. C. (2001) J. Biol. Chem. 276 19672-19677 [DOI] [PubMed] [Google Scholar]
- 16.Nagata, D., Takahashi, M., Sawai, K., Tagami, T., Usui, T., Shimatsu, A., Hirata, Y., and Naruse, M. (2006) Hypertension 48 165-171 [DOI] [PubMed] [Google Scholar]
- 17.Fulton, D., Gratton, J. P., and Sessa, W. C. (2001) J. Pharmacol. Exp. Ther. 299 818-824 [PubMed] [Google Scholar]
- 18.Fulton, D., Gratton, J.-P., McCabe, T. J., Fontana, J., Fujiot, Y., Walsh, K., Franke, T. F., Papapetropoulos, A., and Sessa, W. C. (1999) Nature 399 597-601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dimmeler, S., Fleming, I., Fisslthaler, B., Hermann, C., Busse, R., and Zeiher, A. M. (1999) Nature 399 601-605 [DOI] [PubMed] [Google Scholar]
- 20.Fleming, I., Fisslthaler, B., Dimmeler, S., Kemp, B. E., and Busse, R. (2001) Circ. Res. 88 68-75 [DOI] [PubMed] [Google Scholar]
- 21.McCabe, T. J., Fulton, D., Roman, L. J., and Sessa, W. C. (2000) J. Biol. Chem. 275 6123-6128 [DOI] [PubMed] [Google Scholar]
- 22.Lin, M. I., Fulton, D., Babbitt, R., Fleming, I., Busse, R., Pritchard, K. A., Jr., and Sessa, W. C. (2003) J. Biol. Chem. 278 44719-44726 [DOI] [PubMed] [Google Scholar]
- 23.Michell, B. J., Harris, M. B., Chen, Z. P., Ju, H., Venema, V. J., Blackstone, M. A., Huang, W., Venema, R. C., and Kemp, B. E. (2002) J. Biol. Chem. 277 42344-42351 [DOI] [PubMed] [Google Scholar]
- 24.Bucci, M., Gratton, J. P., Rudic, R. D., Acevedo, L., Roviezzo, F., Cirino, G., and Sessa, W. C. (2000) Nat. Med. 6 1362-1367 [DOI] [PubMed] [Google Scholar]
- 25.Gerber, N. C., and Ortiz de Montellano, P. R. (1995) J. Biol. Chem. 270 17791-17796 [DOI] [PubMed] [Google Scholar]
- 26.Rodriguez-Crespo, I., and Ortiz de Montellano, P. R. (1996) Arch. Biochem. Biophys. 336 151-156 [DOI] [PubMed] [Google Scholar]
- 27.Berka, V., Palmer, G., Chen, P. F., and Tsai, A. L. (1998) Biochemistry 37 6136-6144 [DOI] [PubMed] [Google Scholar]
- 28.Cardounel, A. J., Xia, Y., and Zweier, J. L. (2005) J. Biol. Chem. 280 7540-7549 [DOI] [PubMed] [Google Scholar]
- 29.Chen, Y. R., Chen, C. L., Yeh, A., Liu, X., and Zweier, J. L. (2006) J. Biol. Chem. 281 13159-13168 [DOI] [PubMed] [Google Scholar]
- 30.Bers, D. M., Patton, C. W., and Nuccitelli, R. (1994) Methods Cell Biol. 40 3-29 [DOI] [PubMed] [Google Scholar]
- 31.Patton, C., Thompson, S., and Epel, D. (2004) Cell Calcium 35 427-431 [DOI] [PubMed] [Google Scholar]
- 32.Delgado-Esteban, M., Almeida, A., and Medina, J. M. (2002) J. Neurochem. 82 1148-1159 [DOI] [PubMed] [Google Scholar]
- 33.Nathan, C., and Xie, Q. W. (1994) Cell 78 915-918 [DOI] [PubMed] [Google Scholar]
- 34.Gallis, B., Corthals, G. L., Goodlett, D. R., Ueba, H., Kim, F., Presnell, S. R., Figeys, D., Harrison, D. G., Berk, B. C., Aebersold, R., and Corson, M. A. (1999) J. Biol. Chem. 274 30101-30108 [DOI] [PubMed] [Google Scholar]
- 35.Michell, B. J., Griffiths, J. E., Mitchelhill, K. I., Rodriguez-Crespo, I., Tiganis, T., Bozinovski, S., de Montellano, P. R., Kemp, B. E., and Pearson, R. B. (1999) Curr. Biol. 9 845-848 [DOI] [PubMed] [Google Scholar]
- 36.Michel, J. B., Feron, O., Sacks, D., and Michel, T. (1997) J. Biol. Chem. 272 15583-15586 [DOI] [PubMed] [Google Scholar]
- 37.Ju, H., Zou, R., Venema, V. J., and Venema, R. C. (1997) J. Biol. Chem. 272 18522-18525 [DOI] [PubMed] [Google Scholar]
- 38.Andrews, D. A., Yang, L., and Low, P. S. (2002) Blood 100 3392-3399 [DOI] [PubMed] [Google Scholar]
- 39.Zweier, J. L., Flaherty, J. T., and Weisfeldt, M. L. (1987) Proc. Natl. Acad. Sci. U. S. A. 84 1404-1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zweier, J. L., Kuppusamy, P., and Lutty, G. A. (1988) Proc. Natl. Acad. Sci. U. S. A. 85 4046-4050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zweier, J. L. (1988) J. Biol. Chem. 263 1353-1357 [PubMed] [Google Scholar]
- 42.Zweier, J. L., Kuppusamy, P., Williams, R., Rayburn, B. K., Smith, D., Weisfeldt, M. L., and Flaherty, J. T. (1989) J. Biol. Chem. 264 18890-18895 [PubMed] [Google Scholar]
- 43.Zweier, J. L. (1998) Transplant. Proc. 30 4228-4232 [DOI] [PubMed] [Google Scholar]
- 44.Michell, B. J., Chen, Z., Tiganis, T., Stapleton, D., Katsis, F., Power, D. A., Sim, A. T., and Kemp, B. E. (2001) J. Biol. Chem. 276 17625-17628 [DOI] [PubMed] [Google Scholar]
- 45.Greif, D. M., Kou, R., and Michel, T. (2002) Biochemistry 41 15845-15853 [DOI] [PubMed] [Google Scholar]
- 46.Whitsett, J., Martasek, P., Zhao, H., Schauer, D. W., Hatakeyama, K., Kalyanaraman, B., and Vasquez-Vivar, J. (2006) Free Radic. Biol. Med. 40 2056-2068 [DOI] [PubMed] [Google Scholar]
- 47.Haque, M. M., Panda, K., Tejero, J., Aulak, K. S., Fadlalla, M. A., Mustovich, A. T., and Stuehr, D. J. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 9254-9259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berka, V., Wu, G., Yeh, H. C., Palmer, G., and Tsai, A. L. (2004) J. Biol. Chem. 279 32243-32251 [DOI] [PubMed] [Google Scholar]
- 49.Stuehr, D. J., Santolini, J., Wang, Z. Q., Wei, C. C., and Adak, S. (2004) J. Biol. Chem. 279 36167-36170 [DOI] [PubMed] [Google Scholar]
- 50.Lane, P., and Gross, S. S. (2002) J. Biol. Chem. 277 19087-19094 [DOI] [PubMed] [Google Scholar]
- 51.Nishida, C. R., and Ortiz de Montellano, P. R. (1999) J. Biol. Chem. 274 14692-14698 [DOI] [PubMed] [Google Scholar]
- 52.Fleming, I., Mohamed, A., Galle, J., Turchanowa, L., Brandes, R. P., Fisslthaler, B., and Busse, R. (2005) Cardiovasc. Res. 65 897-906 [DOI] [PubMed] [Google Scholar]
- 53.Partovian, C., Zhuang, Z., Moodie, K., Lin, M., Ouchi, N., Sessa, W. C., Walsh, K., and Simons, M. (2005) Circ. Res. 97 482-487 [DOI] [PubMed] [Google Scholar]
- 54.Niki, I., Yokokura, H., Sudo, T., Kato, M., and Hidaka, H. (1996) J. Biochem. (Tokyo) 120 685-698 [DOI] [PubMed] [Google Scholar]
- 55.Chen, P. F., and Wu, K. K. (2000) J. Biol. Chem. 275 13155-13163 [DOI] [PubMed] [Google Scholar]
- 56.Chen, P. F., and Wu, K. K. (2003) J. Biol. Chem. 278 52392-52400 [DOI] [PubMed] [Google Scholar]
- 57.Maniatis, N. A., Brovkovych, V., Allen, S. E., John, T. A., Shajahan, A. N., Tiruppathi, C., Vogel, S. M., Skidgel, R. A., Malik, A. B., and Minshall, R. D. (2006) Circ. Res. 99 870-877 [DOI] [PubMed] [Google Scholar]
- 58.Lim, E. J., Smart, E. J., Toborek, M., and Hennig, B. (2007) Am. J. Physiol. Heart Circ. Physiol. 293 3340-3347 [DOI] [PubMed] [Google Scholar]
- 59.Xia, Y., Biondi, R., Liebgott, T., Paolocci, N., Cardounel, A., Ambrosio, G., and Zweier, J. L. (2004) J. Am. Coll. Cardiol. 43 294 [Google Scholar]
- 60.Dumitrescu, C., Biondi, R., Xia, Y., Cardounel, A. J., Druhan, L. J., Ambrosio, G., and Zweier, J. L. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 15081-15086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Szabo, C. (2003) Toxicol. Lett. 140–141, 105-112 [DOI] [PubMed] [Google Scholar]
- 62.Pacher, P., Beckman, J. S., and Liaudet, L. (2007) Physiol. Rev. 87 315-424 [DOI] [PMC free article] [PubMed] [Google Scholar]