

Abstract
Fhit, a member of the Histidine Triad superfamily of nucleotide-binding proteins, binds and cleaves diadenosine polyphosphates and functions as a tumor suppressor in human epithelial cancers. Function of Fhit in tumor suppression does not require diadenosine polyphosphate cleavage but correlates with the ability to form substrate complexes. As diadenosine polyphosphates are at lower cellular concentrations than mononucleotides, we sought to quantify interactions between Fhit and competitive inhibitors with the use of diadenosine polyphosphate analogs containing fluorophores in place of one nucleoside. ApppAMC, ApppBODIPY and GpppBODIPY, synthesized in high yield, are effective Fhit substrates, producing AMP or GMP plus fluorophore diphosphates. GpppBODIPY cleavage is accompanied by a 5.4-fold increase in flourescence because BODIPY fluorescence is quenched by stacking with guanine. Titration of unlabeled diadenosine polyphosphates, inorganic pyrophosphate, mononucleotides, and inorganic phosphate into fluorescent assays provided values of Km and KI as competitive inhibitors. The data indicate that Fhit discriminates between good substrates via kcat and against cellular competitors in equilibrium binding terms. Surprisingly, pyrophosphate competes better than purine mononucleotides.
Encoded at 3p14.2 (1), the most fragile site in the human genome (2), Fhit is a dimeric protein of 147 amino acids with diadenosine triphosphate1 (ApppA) hydrolase activity (3). Because of the fragility of the FHIT chromosomal location, allele loss at FHIT, the earliest and most frequent known event in lung carcinogenesis (4,5), could have been considered either a cause or a consequence of cancer. Suppression of tumor formation by reexpression of Fhit protein in kidney, gastric and lung cancer cell lines with FHIT deletions demonstrated that Fhit is an authentic tumor suppressor (6). In lung cancer suppressor systems, suppression of tumorigenesis is accompanied by induction of apoptosis (7,8). In mice, FHIT inactivation induces stomach and sebaceous tumors that resemble human Muir-Torre syndrome.2
As anticipated from structural similarity between histidine triad (HIT) protein dimers and galactose-1-phosphate uridylyltransferase (GalT) (9), Fhit proceeds through a GalT-like (10), covalent His96-adenylate intermediate that is hydrolyzed with retention of configuration (11). The His96Asn allele of Fhit, which is more than a million-fold reduced in kcat and intermediate formation (12), is nonetheless functional in tumor suppression (6). Because this mutant protein retains ApppA binding in the low micromolar range, it was hypothesized that the tumor-suppressing function of Fhit consists of the ability to form complexes with substrates rather than cleavage of diadenosine polyphosphates (ApnA) (12). Much as G-proteins are receptors for regulated GTP binding and hydrolysis that transmit a signal as enzyme-substrate complexes, Fhit protein has been hypothesized to signal for apoptosis as an enzyme-substrate complex (13,14). An alternative but not exclusive model suggests that the tumor-suppressing function of Fhit may depend on promoting microtubule assembly (15).
To determine the structural consequences of ApnA substrate-binding, stable complexes of wild-type and mutant Fhit proteins bound to nonhydrolyzable ApppA analogs were prepared (16,17). Co-crystal structures indicated that the Fhit dimer binds two ApnA molecules in a manner that fills a large, positively charged groove with substrate phosphates, potentially presenting an altered surface for protein-protein interactions to a proapoptotic effector (12). Because this model of Fhit function depends on forming complexes with inabundant substrates, and recognition (and stabilization) of these complexes by a Fhit effector, it was important to assay the more abundant cellular mononucleotides as competitive inhibitors of Fhit. To a first approximation, the cellular abundance of a Fhit-ApnA complex is expected to be a function of the equilibrium binding constant of Fhit for that ApnA, the binding constants of Fhit for competing compounds, the abundance of Fhit and each compound, and the lifetimes of these complexes, potentially as modulated by cellular proteins.
In the past, determination of the Km values for competing substrates would have required assaying products from each substrate. Determination of KI values for competitive inhibitors would have required measuring rates of 3H-AMP and 3H-ADP formation from 3H-ApppA as a function of each inhibitor (18). A recently reported method of visualizing ApppA reaction products by staining them with SYBR GREEN II is unsuitable for kinetic measurements (19). Dietheno-ApppA has also been synthesized and shown to be a Fhit substrate with a 2 μM Km (20). Here, we synthesized novel fluorescent analogs of ApppA by joining thiol-reactive derivatives of 7-diethylamino-4-methylcoumarin (AMC) and 4-4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacine-3-yl (BODIPY FL) to ATP-γS and GTP-γS. We characterized the resulting ApppAMC, ApppBODIPY and GpppBODIPY compounds biochemically and found them to be sensitive probes of Fhit activity. Despite a lack of any fluorescence acceptor function on guanine, GpppBODIPY had fluorescence that was more than 5-fold quenched relative to parental BODIPY compounds and with respect to the ppBODIPY enzymatic product. Proton NMR measurements provided evidence for the proximity of the guanine to BODIPY, suggesting that BODIPY fluorescence is quenched by a stacked or collisional mechanism (21).
Having developed fluorescent and fluorogenic assays for the Fhit active site, we titrated unlabeled competitive inhibitors into these assay to determine the Km of Fhit for ApppA, AppppA, ApppppA, ATP-αS and GTP-αS, and the KI of Fhit for AMP, inorganic pyrophosphate, and monophosphate. Surprisingly, pyrophosphate competes 10-fold better for the Fhit active site than do purine mononucleotides. The results indicate that the ability of Fhit to function as an ApnA-dependent signaling protein in epithelial tumor suppression may depend on a hierarchy of competing compounds: ApnA competing out pyrophosphate, and pyrophosphate competing out purine mononucleotides.
EXPERIMENTAL PROCEDURES
Synthesis of ApppAMC, ApppBODIPY and GpppBODIPY
ATP-γS or GTP-γS tetralithium salts (25 mg, Sigma 85% pure, 0.05 mmol) were dissolved in 3 ml water, buffered to pH 9.0 with sodium bicarbonate. Equimolar amounts of thiol-reactive fluorescent dyes (7-diethylamino-3-(4′-maleimidylphenyl)-4-methylcoumarin (22) or N-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacine-3-yl)methyl)iodoacetamide (23), Molecular Probes) were added in 3.0 ml dioxane. Reactions were stirred and monitored by silica gel TLC, using dioxane:2-propanol:water:ammonium hydroxide (40:20:35:35) as the mobile phase. ATP-γS, GTP-γS and the unreacted dyes migrated with Rf values of 0.25, 0.20 and 0.95, respectively, while fluorescent adenosine nucleotide products had an Rf of 0.7 and GpppBODIPY had an Rf of 0.65. When, after several hours, reactions were judged complete by TLC, the resulting solutions were concentrated via rotary evaporation to 2 ml. Crude products were applied to an aqueous 2×30 cm column of Sephadex LH-20 (Pharmacia). Pooling and lyophilization of pure product fractions afforded nucleotide-dye conjugates as their sodium salts with 76-86% yield. For ApppAMC, 1H NMR (D2O) peaks were 8.51 (s, 1H, adenosine H2); 8.10 (s, 1H, adenosine H8); 7.55 (m, 2H, dye CH-3 and CH-5); 7.38 (m, 4H, dye phenyl); 6.81 (m, 1H, dye CH-6); 6.59 (d, 1H, dye CH-8); 6.10 (d, 1H, C1); 4.6 (m, 1H, -C(O)CHS-); 4.3 (m, 3H, C2,C3,C4); 3.50 (m, 6H, C5 + NCH2CH3); 2.17 (s, 2H, C(O)-CH2-CH); 1.22 (t, 6H, NCH2CH3); and 31P (D2O) peaks were 6.5 ppm (d, 1P); -9.3 (m, 1P); -21.4 (m, 1P). For ApppBODIPY, the proton peaks were 8.31 (s, 1H, adenosine H2); 8.03 (s, 1H, adenosine H8); 7.29 (s, 1H, dye CH); 6.83 (d, 1H, dye CH); 6.40 (d, 1H, dye CH); 6.29 (s, 1H, dye CH); 5.98 (d, 1H, C1); 4.59 (s, 2H, -C(O)CH2S-); 4.51 (d, 2H, C5); 4.3 (m, 3H, C2,C3,C4); 3.75 (d, 2H, dye CH2NH-); 2.47 (s, 3H, dye CH3); 2.28 (s, 3H, dye CH3); and phosphorous peaks were 8.6 ppm (d, 1P); -9.4 (d, 1P); -21.45 (dd, 1P). For GpppBODIPY, the proton peaks were 7.90 (s, 1H, guanosine C8); 7.83 (br s, 1H, NH); 7.77 (br m, 4H, NH2 and OH); 7.29 (s, 1H, dye CH); 6.92 (s, 1H, dye CH); 6.39 (s, 1H, dye CH); 6.25 (s, 1H, dye CH); 5.73 (m, 1H, C1); 4.57 (s, 2H, SCH2); 4.49 (t, 1H, C3); 4.41 (br s, 1H, C2); 4.28 (br s, 1H, C4); 4.21 (dd, 2H, C5); 3.79 (dq, 2H, NCH2); 2.43 (s, 3H, dye CH3); and 2.25 (s, 3H, dye CH3). The covalent structures of ApppA, ApppAMC, ApppBODIPY and GpppBODIPY are represented in Fig. 1.
FIG. 1. Covalent structures of ApppA, ApppAMC, ApppBODIPY, and GpppBODIPY.

Fluorescent ApppA analogs were synthesized from ATP-γS, GTP-γS, and thiol reactive fluorescent dyes as described in Experimental Procedures.
Fluorescent thin layer enzyme assays
Fluorescent substrates at 1 - 30 μM were incubated with purified Fhit protein (16) in reactions at 37 °C containing 20 mM Na HEPES, pH 7.0, 0.5 mM MnCl2, and 0.2 mg/ml bovine serum albumin. Competitive compounds, when indicated, were mixed with fluorescent substrate at eight to ten concentrations, surrounding the Km or KI values of the competitor. Reaction samples (≤ 5 μl) were spotted on silica TLC plates (E. Merck) at 60 - 120 s intervals. Plates were air-dried and developed in 2-propanol:NH4OH:1,4-dioxane:H2O (50:35:8:7 for ApppAMC and ApppBODIPY; 50:33:6:11 for GpppBODIPY). Developed plates were imaged by epi-UV illumination and quantitated on a BioRad Fluor-S instrument using Multi•Analyst 1.0.2 software. Three types of thin-layer assays were performed. First-order decay assays, reactions in which time-courses of complete consumption of low concentrations of fluorescent substrate were measured, were used to determine the specificity constant, kcat/Km. Initial rate assays, reactions that measured the rates of appearance of the first few percent of products formed as a function of substrate concentration, were used to determine kcat and Km. Competitive inhibition assays, first-order assays with titration of nonlabeled competitive inhibitors, were used to determine binding constants of competitors from their ability to reduce kcat/Km apparent. For first-order decay assays, substrate fluorescence minus background fluorescence was obtained in arbitary units. Plots of log[remaining substrate] against time yielded experimental slopes, having units of s-1, which were multiplied by -1/[enzyme], to obtain values of kcat/Km or kcat/Km apparent. For initial rate assays, ppBODIPY fluorescence was converted to pmol with a standard product curve and kcat and Km values were derived from Lineweaver-Burk plots (24). For calculation of KI (or Km) in competitive inhibition assays, kcat/Km apparent values were plotted against [I] and values of kcat/Km and KI (or Km) were determined using the equation in Scheme 1.
In these experiments, the value for kcat/Km, though treated as an independent variable, was always within 15% of the experimentally determined value.
Fluorogenic enzyme assays
Reactions in black 96-well plates containing GpppBODIPY at 1.25 to 40 μM in 60 μl of 20 mM Na HEPES, pH 7.0, 0.5 mM MnCl2, and 0.2 mg/ml bovine serum albumin were initiated with addition of 167 fmole of Fhit and incubated for 300 – 360 s at 37 °C. Duplicate reactions were stopped by addition of 60 μl 200 mM Na Citrate pH 3.0 and read with a Wallach Victor2 Multilabel Counter 1420 with a 485 nm excitation filer and a 535 nm emission filter. At each substrate concentration, mock reactions were created by mixing 0%, 5% and 10% volumes of acid-stopped complete hydrolysates with acid-stopped mock reactions (100%, 95% and 90%). Fluorescent emissions of experimental and mock reactions were counted and plotted to calculate ppBODIPY production per active site per second at each concentration of GpppBODIPY. To prove that ApppA, pyrophosphate and AMP are competitive inhibitors, these compounds were titrated into 96-well GpppBODIPY assays at four concentrations at each of five concentrations of GpppBODIPY (2 μM, 4 μM, 8 μM, 16 μM and 32 μM). Initial rates as a function of GpppBODIPY and inhibitor concentration were plotted by the method of Eadie and Hofstee (24).
RESULTS
Design criteria for fluorescent Fhit substrates
HIT proteins are a superfamily of homodimeric nucleotide-binding proteins with a unique mode of nucleotide recognition (14). Each monomer contributes 5 strands to a 10-stranded anti-parallel β sheet that forms two identical purine nucleotide-binding sites (9,12,25). In Hint dimers, the two nucleotide-binding sites accommodate two equivalents of AMP or GMP with the α phosphates proximal to the catalytic histidines (9). In Fhit dimers bound to ApnA, one pair of AMP moieties are buried proximal to the catalytic histidines with polyphosphate chains extending from the tightly bound AMP groups to expose the second pair of adenosines in a solvent-accessible site near Thr 79 (12). Because Fhit tolerates substitutions of the second adenosine (3,11), we expected that Fhit would hydrolyze ApppX-type substrates to produce AMP plus ppX. Thiol-reactive fluorescent dyes, 7-diethylamino-3-(4’-maleimidylphenyl)-4-methylcoumarin (22) and BODIPY FL C1 -iodoacetamide (23), which were initially synthesized to label protein thiols, were reacted with ATP-γS to generate first generation fluorescent compounds, ApppAMC and ApppBODIPY. The second generation substrate, GpppBODIPY, was made by conjugating BODIPY FL C1-iodoacetamide to GTP-γS. Fluorescent substrates are depicted in Fig. 1.
Fluorescent ApppA analogs are good Fhit substrates
At substrate concentrations below Km, reactions proceed with first-order kinetics such that the negative slope of a plot of log[remaining substrate] against time equals kcat/Km times the enzyme concentration (24). To test whether Fhit would tolerate substitution of one adenosine in a fluorescent ApppA analog, we incubated 0.5 to 2.5 nM Fhit with 1.5 to 5 μM ApppAMC and ApppBODIPY. Time courses of such reactions, analyzed by TLC and digitally imaged upon UV illumination, made it clear that Fhit cleaves each substrate, generating one fluorescent product with reduced chromatographic mobility (Figs. 2A and B). One thousand times more concentrated substrates were required to visualize the nonfluorescent product by UV-shadowing on TLC plates with fluorescent indicator (12) and, as expected, that product was indistinguishable from AMP (data not shown). Thus, Fhit cleaves ApppAMC and ApppBODIPY to produce AMP + ppAMC and AMP + ppBODIPY, respectively.
FIG. 2. Time courses of cleavage of fluorescent substrates under kcat /Km conditions.

Digital fluorescent images of ApppAMC (A), ApppBODIPY (B), and GpppBODIPY (C) reactions separated by TLC.
At an initial substrate concentration of 3 μM ApppAMC ([enzyme] = 2.5 nM) or 1.5 μM ApppBODIPY ([enzyme] = 1.2 nM), the first-order decay kinetics of ApppAMC and ApppBODIPY were examined. As shown in Figs. 3A and B and Table 1, calculated kcat/Km values for each substrate were 1.2 × 106 s-1 M-1 and 2.3 × 106 s-1 M-1, respectively.
FIG. 3. kcat/Km determination of fluorescent substrates.

Plots of log remaining substrate fluorescence against time: ApppAMC (A); ApppBODIPY (B); GpppBODIPY (C).
Table 1.
Kinetic parameters with Fhit
| Compound |
Kcat1 (s-1) |
Km1 (μM) |
Kcat /Km1 (s-1 M-1) |
Kcat /Km2 (S-1 M-1) |
Km3 (μM) |
Kr3 (μM) |
|---|---|---|---|---|---|---|
| ApppA | 73 ±4.3 | 1.9 ±0.2 | 3.8 E7 | 2.0 ±0.2 | ||
| ApppAMC | 1.2 E6 | |||||
| ApppBODIPY | 7.3 | 3.0 | 2.4 E6 | 2.3 E6 | ||
| GpppBODIPY | 0.58 | 1.4 | 4.1 E5 | 5.8 E5 | ||
| AppppA | 2.6 ±0.4 | |||||
| ApppppA | 13.9 ±2.3 | |||||
| PPi | 19.9 ±3.8 | |||||
| AMP | 196 ±25 | |||||
| ATP-αS | 163 ±19 | |||||
| GTP-αS | 257 ±34 | |||||
| Pi | 2900 ±430 |
From initial rate assays. ApppA data are from Pace et al. (1998).
From substrate decay assays.
By titration into ApppBODIPY decay assays.
The BODIPY substitution affects kcat
ApppBODIPY is an effective and useful substrate for Fhit with a quantitative disadvantage in cleavage (kcat/Km for ApppBODIPY = 2.3 × 106 s-1 M-1 versus 3.8 × 107 s-1 M-1 for ApppA (12)). Initial rates as a function of ApppBODIPY concentration were measured and revealed that the basis for the catalytic disadvantage is in the kcat term. While the Km for ApppBODIPY, 3.0 μM, is less than 60% higher than that for ApppA, the kcat for ApppBODIPY, 7.3 s-1, is 10-fold lower than that for ApppA (Table 1). Considering that ApppBODIPY is an ApppX-type compound, containing an unaltered adenylating functionality (11), it is interesting to consider why the BODIPY substitution affects kcat rather than Km. Retention of a low Km at the expense of reduced kcat could indicate that ApppBODIPY has multiple binding modes, only some of which are productive, or that ppBODIPY is not as good a leaving group as ADP. Thus, the 10-fold reduction in kcat might correspond to the reciprocal fraction of time ApppBODIPY is in a productive, ApppA-type conformation or a tendency of the ppBODIPY product to re-attack His96-AMP rather than exiting the active site.
GpppBODIPY is a stacked and quenched fluorogenic substrate
Cleavage of ApppG by Fhit yielded a two-thirds mixture of AMP + GDP with a one-third mixture of GMP + ADP (3). Thus, the guanine in GpppBODIPY was expected to be an acceptable substitution for adenine in ApppBODIPY. Upon synthesis, GpppBODIPY was found to have only 11% of the fluorescence in aqueous solution of the parental dye. Because guanine does not absorb photons in the visible range, fluorescent resonance energy transfer was ruled out as the mechanism of quenching. In methanol, the yield of GpppBODIPY fluorescence rose to 60% that of BODIPY fluorescence, suggesting that a solvent-sensitive binding interaction between guanine and BODIPY is responsible for quenching. The NMR signal in D2O for the guanine C8 proton was shifted from 8.2 ppm (GTP) to 7.9 ppm (GpppBODIPY) indicating that guanine is bound in the electron-rich BODIPY environment. No proton NMR shift or alteration of fluorescence was observed with ApppBODIPY. By analogy to fluorescent uridine nucleotide analogs, we conclude that GpppBODIPY is quenched by stacking (21).
The TLC assay of 2 μM GpppBODIPY ([enzyme] = 2.4 nM) in Figs. 2C and 3C shows that GpppBODIPY is within 4-fold as good a substrate as ApppBODIPY (kcat/Km = 5.8 × 105 s-1 M-1 versus 2.3 × 106 s-1 M-1) and that cleavage to ppBODIPY produces a 5.4-fold increase in fluorescence. The fluorogenic nature of GpppBODIPY allowed us to measure hydrolysis continuously or as stopped endpoints in a multi-well fluorescent plate reader. As shown in Table 1, substitution of guanine for adenine reduces kcat 10-fold while reducing Km two-fold. In the crystal structure of Hint-GMP, the carbonyl oxygen of His 42 makes a buried hydrogen bond to the N2 nitrogen of GMP (9). A similar interaction in Fhit could be positioning the GMP moiety of GpppBODIPY nonoptimally for catalysis and/or changing the rate-limiting step of the reaction.
Nucleotides and nucleotide analogs as competitive inhibitors of Fhit-ApppBODIPY and GpppBODIPY kinetics
The readout of first-order ApppBODIPY assays, kcat/Km times enzyme concentration, is well suited for assays of competitive inhibitors. Competitive inhibitors rob from the pool of available enzyme, decreasing kcat/Km(apparent). The decrease in kcat/Km(apparent) is a function of the equilibrium binding constant and the concentration of the competitor. When the competing ligand is not a substrate, the equilibrium binding constant represents KI. When the competing ligand is itself a Fhit substrate, the equilibrium binding constant is Km, the apparent dissociation constant for all bound forms with enzyme.
As shown in Fig. 4 and summarized in Table 1, ApnA compounds, purine mononucleotides, inorganic pyrophosphate and monophosphate were titrated into ApppBODIPY assays as competitive inhibitors. ApppA and AppppA displayed the lowest Km values. The Km for ApppA measured as a competing substrate was 2.0 ± 0.2 μM, consistent with the Km value measured for ApppA by 3H product formation, 1.9 ± 0.2 μM (12). The additional phosphate group and two phosphate groups in AppppA and ApppppA increased Km by 30% and 600%, respectively. The next best competitor was inorganic pyrophosphate with a KI of 19.9 ± 3.8 μM. ATP and GTP, probably the most abundant cellular mononucleotides (26), were difficult to fit to Scheme 1 (see Methods), potentially because of formation of pyrophosphate. To estimate the Km values of ATP and GTP, the slowly hydrolyzed αS forms of these nucleotides (11) were examined. All purine mononucleotides tested had KI and Km values approximately 10-fold higher than that of pyrophosphate. Monophosphate, on the other hand, inhibited with a KI of 2.9 ± 0.4 mM.
FIG. 4. Titration of ApppA as an inhibitor of ApppBODIPY.
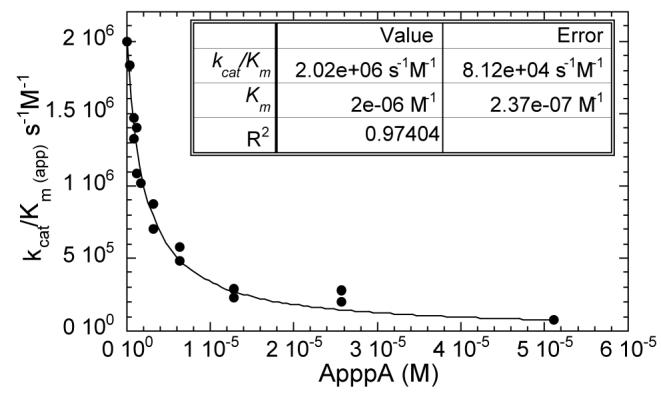
Increasing concentrations of ligands reduce kcat /Km apparent and allow determination of K values (Table 1) using Scheme 1.
To prove that the compounds inhibit competitively, rather than by a noncompetitive or uncompetitive mechanism, compounds were titrated into fluorogenic GpppBODIPY initial-rate assays over a range of substrate and inhibitor concentrations. As shown in Fig. 5, ApppA, pyrophosphate and AMP all increase the apparent Km of GpppBODIPY without affecting the kcat value, displaying the classical signature of competitive inhibitors in Eadie-Hofstee plots. These experiments also demonstrated that, much as the BODIPY for adenosine substitution was responsible for a 10-fold decrease in kcat with minimal effect on Km, the guanine for adenine substitution is responsible for a 10-fold decrease in kcat with minimal effect on Km.
FIG. 5. Nucleotides and related compounds are competitive with substrate.

Increasing amounts of ApppA (A), pyrophosphate (B) and AMP (C) were titrated into initial rate assays of GpppBODIPY. For each concentration of substrate and inhibitor, rates were plotted by the method of Eadie and Hofstee.
DISCUSSION
The new compounds described herein can be considered analogs of ATP and GTP as well as ApnA and have potential uses with a wide variety of enzymes including ATPases and GTPases. The compounds have been used to purify and characterize the Fhit active site of NitFhit,3 the Fhit ortholog in C. elegans which is fused to Nit (27), a tetrameric protein that binds two Fhit dimers and confers a novel protein-association activity to the complex.3
In addition to basic science applications, by combining the fluorogenic Fhit substrate with newly developed Fhit-specific inhibitors (28), a Fhit diagnostic kit can be developed for use in clinical laboratories. The GpppBODIPY assay for Fhit, as a 96-well fluorogenic assay, is also suitable for a high-throughput screen for inhibitors. Among Fhit inhibitors, uncompetitive compounds (those that bind and retard the enzyme-substrate complex) might include compounds that are competitive with a Fhit effector (12,13).
The present study provides valuable information about properties of substrates and related compounds that determine binding to Fhit. Fhit-ApnA complexes may be required for signaling (12) and formation of these complexes would have to occur in the face of competition with mononucleotides, monophosphate, pyrophosphate and other compounds. The finding that reversible binding constants for ApnA, pyrophosphate and purine mononucleotides are 2 E-6 M, 2 E-5 M and 2 E-4 M, respectively, suggests that Fhit may have evolved a hierarchical system to avail itself of ApnA in the presence of competing compounds. Prior to our measurements of pyrophosphate and purine mononucleotides as competitive inhibitors of Fhit, one might have expected that the ground state of Fhit would be ATP-bound. Indeed, if purine mononucleotides are at low millimolar levels (26), ten times their equilibrium-binding constants, ∼90% of Fhit would be occupied by mononucleotides in the absence of other competitors. The reported amount of inorganic pyrophosphate (1.33 × 10-16 mol, ref 29) per lymphocyte (volume = 1.37 × 10-13 l, ref 30) suggests that the concentration of pyrophosphate (∼1 mM) exceeds the KI for pyrophosphate by 50-fold. Thus, Fhit may be primarily bound to pyrophosphate rather than ATP under low ApnA conditions. From the competition binding data presented in Table 1, we calculate that a spike of ∼100 μM ApnA would convert half the Fhit-pyrophosphate to Fhit-ApnA. Conditions that lead to dramatic increases in ApnA levels include interferon (31) and etoposide (32) treatment of promyelocytes, and administration of glucose to pancreatic β cells (33).
The data presented herein illustrate a surprising degree of kcat discrimination among chemically similar substrates. Though the kinetics of Fhit-dependent cleavage of ApppA are robust, the function of Fhit appears to depend not on cleavage but binding ApnA substrates (5,6,12). We have argued that important Fhit substrates are those with low Km values but not necessarily the highest kcat values. Less optimal substrates, such as AppppA, may be more important than ApppA if they are more abundant and if their residence time on Fhit is longer than that of ApppA (12). In the cell, the lifetime of Fhit-substrate complexes may be stabilized by effector interactions. The observation that substrates with fundamentally unaltered chemical lability such as ApppBODIPY and GpppBODIPY are cleaved with reduced kcat values suggests that Fhit has binding modes that are at least 100-fold stabilized with respect to optimized in vitro kinetics.
The stereochemical course of the Fhit reaction (11) dictates that ADP (in case of ApppA), ATP (in case of AppppA) or ppBODIPY (in case of fluorescent substrates) must leave before the hydrolytic water can be bound by the enzyme. Thus, any Fhit or Fhit-effector conformation that prevents leaving-group exit or blocks water entry would be expected to retard substrate hydrolysis. The 20 μM pyrophosphate-binding site that is competitive with substrate is a likely facilitator of leaving group-reattack. A Fhit effector may stabilize Fhit-ApnA by pinning the leaving group down on this site.
Many intracellular and extracellular receptors are enzymatically inert while others, such as GTPases, are slow enzymes with intrinsically long-lived enzyme-substrate complexes. Fhit appears to be a receptor for ApnA with rapid kinetics but with features that allow it to be stabilized in vivo. The kinetics and structural biology of Fhit associated proteins (15,27) are expected to shed more light on this matter.
Acknowledgements
We thank Kay Huebner, Mike Blackburn and Perry Frey for helpful discussions, and Dieter Klaubert of Molecular Probes for suggesting synthesis of GpppBODIPY. Synthesis was performed at Molecular Probes.
Footnotes
This work was supported by National Cancer Institute research grant CA75954 (C.B.), an institutional National Cancer Institute training grant (A.D.), and new investigator awards from the March of Dimes Birth Defects Foundation, the Burroughs Wellcome Fund, and the Arnold and Mabel Beckman Foundation (C.B.).
- ApppA
- diadenosine triphosphate
- HIT
- histidine triad
- GalT
- galactose-1-phosphate uridylyltransferase
- ApnA
- diadenosine polyphosphate
- AMC
- 7-diethylamino-4-methylcoumarin
- BODIPY FL
- 4-4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacine-3-yl
L.Y.Y. Fong, V. Fidanza, N. Zanesi, L.F. Lock, L.D. Siracusa, R. Mancini, Z. Siprashvili, M. Ottey, S.E. Martin, R. Dolsky, T. Druck, P.A. McCue, C.M. Croce, and K. Huebner, in preparation.
H.C. Pace, J. Huang, A. Dragansescu, S.C. Hodawadekar, Y. Pekarsky, C.M. Croce, and C. Brenner, in preparation.
REFERENCES
- 1.Ohta M, Inoue H, Cotticelli MG, Kastury K, Baffa R, Palazzo J, Siprashvili Z, Mori M, McCue P, Druck T, Croce CM, Huebner K. Cell. 1996;84:587–597. doi: 10.1016/s0092-8674(00)81034-x. [DOI] [PubMed] [Google Scholar]
- 2.Huebner K, Garrison PN, Barnes LD, Croce CM. Ann. Rev. Genet. 1998;32:7–31. doi: 10.1146/annurev.genet.32.1.7. [DOI] [PubMed] [Google Scholar]
- 3.Barnes LD, Garrison PN, Siprashvili Z, Guranowski A, Robinson AK, Ingram SW, Croce CM, Ohta M, Huebner K. Biochem. 1996;35:11529–11535. doi: 10.1021/bi961415t. [DOI] [PubMed] [Google Scholar]
- 4.Sozzi G, Pastorino U, Moiraghi L, Tagliabue E, Pezzella F, Ghirelli C, Tornielli S, Sard L, Huebner K, Pierotti MA, Croce CM, Pilotti S. Cancer Res. 1998;58:5032–5037. [PubMed] [Google Scholar]
- 5.Huebner K, Sozzi G, Brenner C, Pierotti MA, Croce CM. Advances in Oncology. 1999;15:3–10. [Google Scholar]
- 6.Siprashvili Z, Sozzi G, Barnes LD, McCue P, Robinson AK, Eryomin V, Sard L, Tagliabue E, Greco A, Fusetti L, Schwartz G, Pierotti MA, Croce CM, Huebner K. Proc. Natl. Acad. Sci. USA. 1997;94:13771–13776. doi: 10.1073/pnas.94.25.13771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ji L, Fang B, Yeh N, Fong K, Minna JD, Roth JA. Cancer Res. 1999;59:3333–3339. [PubMed] [Google Scholar]
- 8.Sard L, Accornero P, Tornielli S, Delia D, Bunone G, Campiglio M, Colombo MP, Gramegna M, Croce CM, Pierotti MA, Sozzi G. Proc. Natl. Acad. Sci. USA. 1999;96:8489–8492. doi: 10.1073/pnas.96.15.8489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brenner C, Garrison P, Gilmour J, Peisach D, Ringe D, Petsko GA, Lowenstein JM. Nat. Struct. Biol. 1997;4:231–238. doi: 10.1038/nsb0397-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geeganage S, Frey PA. Biochem. 1998;37:14500–14507. doi: 10.1021/bi9815546. [DOI] [PubMed] [Google Scholar]
- 11.Abend A, Garrison PN, Barnes LD, Frey PA. Biochem. 1999;38:3668–3676. doi: 10.1021/bi981895j. [DOI] [PubMed] [Google Scholar]
- 12.Pace HC, Garrison PN, Robinson AK, Barnes LD, Draganescu A, A R, Blackburn GM, Siprashvili Z, Croce CM, Huebner K, Brenner C. Proc. Natl. Acad. Sci. USA. 1998;95:5484–9. doi: 10.1073/pnas.95.10.5484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brenner C. Phosphorous, Sulfur and Silicon. 1999;144:745–748. [Google Scholar]
- 14.Brenner C, Bieganowski P, Pace HC, Huebner K. J. Cell. Physiol. 1999;181:179–187. doi: 10.1002/(SICI)1097-4652(199911)181:2<179::AID-JCP1>3.0.CO;2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chaudhuri AR, Khan IA, Prasad V, Robinson AK, Ludeuna RF, Barnes LD. J. Biol. Chem. 1999;274:24378–24382. doi: 10.1074/jbc.274.34.24378. [DOI] [PubMed] [Google Scholar]
- 16.Brenner C, Pace HC, Garrison PN, Robinson AK, Rosler A, Liu XH, Blackburn GM, Croce CM, Huebner K, Barnes LD. Protein Eng. 1997;10:1461–1463. doi: 10.1093/protein/10.12.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blackburn GM, Liu XH, Rosler A, Brenner C. Nucleosides & Nucleotides. 1998;17:301–308. doi: 10.1080/07328319808005178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barnes LD, Robinson AK, Mumford CH, Garrison PN. Anal. Biochem. 1985;144:296–304. doi: 10.1016/0003-2697(85)90120-4. [DOI] [PubMed] [Google Scholar]
- 19.Ji L, Fang B, Roth JA. Anal. Biochem. 1999;271:114–116. doi: 10.1006/abio.1999.4131. [DOI] [PubMed] [Google Scholar]
- 20.Asensio AC, Oaknin S, Rotllan P. Biochim. Biophys. Acta. 1999;1432:396–400. doi: 10.1016/s0167-4838(99)00102-8. [DOI] [PubMed] [Google Scholar]
- 21.Dhar G, Bhaduri A. J. Biol. Chem. 1999;274:14568–72. doi: 10.1074/jbc.274.21.14568. [DOI] [PubMed] [Google Scholar]
- 22.Khalfan H, Abuknesha R, Rand-Weaver M, Price RG, Robinson D. Histochem. J. 1986;18:497–9. doi: 10.1007/BF01675617. [DOI] [PubMed] [Google Scholar]
- 23.Karolin J, Johansson LB-A, Strandberg L, Ny T. J. Am. Chem. Soc. 1994;116:7801–7806. [Google Scholar]
- 24.Jencks WP. Catalysis in Chemistry and Enzymology. 1987 [Google Scholar]
- 25.Lima CD, Damico KL, Naday I, Rosenbaum G, Westbrook EM, Hendrickson WA. Structure. 1997;5:763–774. doi: 10.1016/s0969-2126(97)00231-1. [DOI] [PubMed] [Google Scholar]
- 26.Kornberg A, Baker TA. DNA Replication, Second Edition. 1992 [Google Scholar]
- 27.Pekarsky Y, Campiglio M, Siprashvili Z, Druck T, Sedkov Y, Tillib S, Draganescu A, Wermuth P, Rothman JH, Huebner K, Buchberg AM, Mazo A, Brenner C, Croce CM. Proc. Natl. Acad. Sci. USA. 1998;95:8744–8749. doi: 10.1073/pnas.95.15.8744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu X, Brenner C, Guranowski A, Starzynska E, Blackburn GM. Angew. Chem. Int. Ed. 1999;38:1245–1247. doi: 10.1002/(SICI)1521-3773(19990503)38:9<1244::AID-ANIE1244>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 29.Barshop BA, Adamson DT, Vellom DC, Rosen F, Epstein BL, Seegmiller JE. Anal. Biochem. 1991;197:266–72. doi: 10.1016/0003-2697(91)90387-9. [DOI] [PubMed] [Google Scholar]
- 30.Thompson CB, Scher I, Schaefer ME, Lindsten T, Finkelman FD, Mond JJ. J. Immunol. 1984;133:2333–42. [PubMed] [Google Scholar]
- 31.Vartanian A, Narovlyansky A, Amchenkova A, Turpaev K, Kisselev L. FEBS Lett. 1996;381:32–4. doi: 10.1016/0014-5793(96)00073-7. [DOI] [PubMed] [Google Scholar]
- 32.Vartanian A, Prudovsky I, Suzuki H, Dal Pra, I., Kisselev L. FEBS Lett. 1997;415:160–2. doi: 10.1016/s0014-5793(97)01086-7. [DOI] [PubMed] [Google Scholar]
- 33.Ripoll C, Martin F, Manuel Rovira J, Pintor J, Miras-Portugal MT, Soria B. Diabetes. 1996;45:1431–4. doi: 10.2337/diab.45.10.1431. [DOI] [PubMed] [Google Scholar]