

Abstract
Fragile histidine triad protein (Fhit) is a diadenosine triphosphate (ApppA) hydrolase encoded at the human chromosome 3 fragile site which is frequently disrupted in tumors. Reintroduction of FHIT coding sequences to cancer cell lines with FHIT deletions suppressed the ability of these cell lines to form tumors in nude mice even when the reintroduced FHIT gene had been mutated to allow ApppA binding but not hydrolysis. Because this suggested that the tumor suppressor activity of Fhit protein depends on substrate-dependent signaling rather than ApppA catabolism, we prepared two crystalline forms of Fhit protein that are expected to model its biologically active, substrate bound state. Wild-type and the His96Asn forms of Fhit were overexpressed in Escherichia coli, purified to homogeneity and crystallized in the presence and absence of ApppA and an ApppA analog. Single crystals obtained by vapor diffusion against ammonium sulfate diffracted Xrays to beyond 2.75 Å resolution. High quality native synchrotron X-ray data were collected for an orthorhombic and a hexagonal crystal form.
Keywords: Fhit, nucleotide-binding, tumor-suppressor, ApppA
Introduction
The FHIT gene, located at the human chromosome 3 fragile site (Ohta et al., 1996; Zimonjic et al., 1997), encodes a dimeric polypeptide of 147 amino acids which cleaves diadenosine triphosphate (ApppA) to yield AMP plus ADP (Barnes et al., 1996). Lesions in the FHIT gene are extremely common in tumors (Mao et al., 1996; Mau et al., 1996; Negrini et al., 1996; Ohta et al., 1996; Panagopoulos et al., 1996; Shridhar et al., 1996; Sozzi et al., 1996a,b; Virgilio et al., 1996; Yanagisawa et al., 1996; Druck et al., 1997; Gartenhaus, 1997; Geurts et al., 1997; Shridhar et al., 1997) and occur as early events in carcinogen exposed tissues (Sozzi et al., 1996b). In a family predisposed to early-onset, multifocal renal carcinoma, a translocation that disrupts FHIT is transmitted to affected children; tumors from these individuals have lost both copies of FHIT (Ohta et al., 1996). Strikingly, in gastric, lung and kidney cancer cell lines that contain deletions of the FHIT gene, stable re-expression of Fhit protein suppressed the ability of the cell lines to form tumors in nude mice (Siprashvili et al., 1997). The tumor suppressor activity of transfected FHIT genes was not lost when the codon for His96 was mutated to Asn (Siprashvili et al., 1997) even though this mutation results in Fhit protein at least 1000-fold reduced for ApppA hydrolase activity (Barnes et al., 1996). These observations suggest that FHIT is a tumor suppressor gene whose loss contributes to malignant transformation in multiple tissues. The ability to separate tumor suppressor function from hydrolysis suggests, as it had been hypothesized (Brenner et al., 1997), that a substrate-bound form of the enzyme may be the biologically active state whose cellular function suppresses carcinogenesis. Fhit-related proteins have been found in animals (Ohta et al., 1996) and yeasts (Huang et al., 1995; Brenner et al., 1997) and constitute one branch of the histidine triad (HIT) superfamily (Seraphin, 1992) of nucleotide-binding proteins (Brenner et al., 1997). An older branch of the HIT superfamily, the Histidine triad nucleotide-binding protein (Hint) branch, apparently predates the separation of bacteria, archaea and eukarya, having been found in all organisms (Brenner et al., 1997). Crystal structures of nucleotide-bound forms of rabbit Hint demonstrated that the most conserved amino acids in the superfamily form an AMP or GMP binding site with the HIT motif, His-ϕ-His-ϕ-His-ϕ-ϕ (ϕ is a hydrophobic amino acid), constituting part of the α-phosphate binding site (Brenner et al., 1997). Despite no overall sequence similarity, a monomer of E.coli galactose-1-phosphate uridylyltransferase (GalT) with its associated pyrimidine nucleotide (Wedekind et al., 1995) can be superimposed upon the rabbit Hint dimer with its two bound purine nucleotides (Brenner et al., 1997).
Recently, Lima and co-workers reported high resolution crystal structures of Fhit bound to adenosine (Lima et al., 1997a), adenosine α/β-methylene diphosphate (AMP-CP), without adenosine bound, and in a form exposed to a mixture of adenosine and tungstate (Lima et al., 1997b). These studies (Lima et al., 1997a,b) demonstrated that Fhit possesses a GalThalf- barrel closely related to that of Hint with unique amino and C-terminal elaborations. Two identical adenosine nucleotide binding sites, as predicted from nucleotide-bound Hint structures (Brenner et al., 1997), were described in the Fhit structure determinations (Lima et al., 1997a,b). Anticipating that the active, signaling state of Fhit protein may be bound to ApppA or AppppA substrates (Brenner et al., 1997), and encouraged by the activity of the FHIT His96Asn allele as a tumor suppressor (Siprashvili et al., 1997), we have undertaken a program to synthesize new hydrolysis-resistant analogs of this class of compounds (Blackburn et al., 1998) for crystallographic analysis with wild-type enzyme. We have also undertaken a program to mutagenize active-site residues of Fhit (Barnes et al., 1996) for crystallographic analysis with authentic ApppA. Here we report purification and crystallization of Fhit—diadenosine nucleotide complexes.
Materials and methods
Overexpression, purification and crystallization
The FHIT cDNA was cloned into pSGA02, transformed into E.coli strain SG100, and induced for 6 h at 30°C with 1 mM isopropyl-β-D-thiogalactoside (Ghosh and Lowenstein, 1997). A 52 ml crude supernatant was prepared from 14 g wet bacterial cell pellet by sonication in 42 ml buffer A [50 mM Na HEPES pH 6.8, 10% (vol/vol) glycerol] containing 0.5 mM phenylmethylsulfonyl fluoride, 1.1 μM leupeptin and 0.7 μM pepstatin. This material, dialyzed against buffer B [25 mM Na HEPES pH 6.8, 10% (vol/vol) glycerol] with 0.5 mM phenylmethylsulfonyl fluoride, was subjected to chromatography on a 210 ml column of DEAE-Sephacel (Pharmacia Biotech, Sweden) and eluted with an 800 ml gradient to 0.3 M NaCl in buffer B. Fractions containing ApppA hydrolase activity, assayed as described (Barnes et al., 1996), were concentrated with a PM10 membrane (Amicon Corporation, Beverly, MA) and applied to a 180 ml column of Sephadex G75 superfine gel (Pharmacia) equilibrated with buffer A. Though the gel filtration step had only a one-third yield and resulted in no observable increase in specific activity, it was necessary to remove a peak of aggregated ApppA hydrolase activity from the major peak of activity which ran as expected of a Fhit dimer. The Fhit dimer peak was concentrated as above and chromatographed on a 21 ml column of QAE-Sepharose (Pharmacia). The homogeneous Fhit eluate from a 200 ml gradient to 0.2 M NaCl in buffer A was concentrated to 13.4 mg/ml (0.40 mM of dimer) in buffer A for crystallization. Purification data are summarized in Table I. Fhit protein was mixed with an equal volume (2.5-4.0 μl) of 2 M ammonium sulfate, 4% polyethylene glycol 400, 0.1 M Na HEPES pH 7.5 on a siliconized cover slip which was sealed over a 0.8 ml volume of the same solution. Crystals grew at room temperature over the course of 1-4 weeks. Inhibited Fhit crystals were prepared by preincubating Fhit with 2.5 molar equivalents of IB2 (Blackburn et al., 1997) (Figure 1) prior to vapor diffusion. Crystals grown in the absence of IB2 were typically orthorhombic prisms with dimensions as large as ∼250 μm × ∼250 μm × 1000 μm (Figure 2A). Inhibited crystals were usually hexagonal needles, ∼150 μm wide by ∼1000 μm in length (Figure 2B). Occasionally, crystals with pentagonal habits formed (Figure 2C) which diffracted X-rays to 2 Å resolution but did not appear to be single (data not shown). Fhit His96Asn protein (Barnes et al., 1996) was expressed, purified and crystallized as above except that hexagonal needles were obtained with IB2 and with authentic ApppA (Sigma).
Table I.
Purification of Fhit
| Fraction | Protein (mg) |
Activity (unitsa) |
Specific activity (units/mg) |
Yield (%) |
|---|---|---|---|---|
| Dialyzed crude supernatant | 993 | 8750 | 8.8 | 100 |
| DEAE-Sephacel | 129 | 9550 | 74.0 | 110 |
| Sephadex G75 | 62 | 3300 | 53.0 | 38 |
| QAE-Sepharose | 44 | 4700 | 107.0 | 54 |
ApppA hydrolase activity of Fhit was assayed at 37°C with 100 μM [3H]ApppA in 50 mM HEPES, pH 6.8, 0.5 MnCl2.
One unit of activity was defined as the amount of enzyme that releases 1 μmol AMP per min under linear conditions.
Fig. 1.
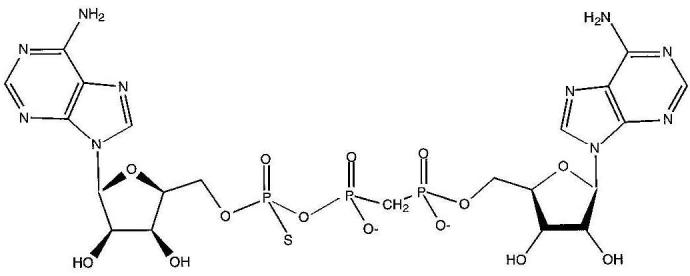
Hydrolysis-resistant ApppA analog, IB2. The compound is a diadenosine triphosphate with P1, P2-methylene and P3-thio modifications (Blackburn et al., 1997). Only one stereoisomer at the phosphorothioate center is shown.
Fig. 2.
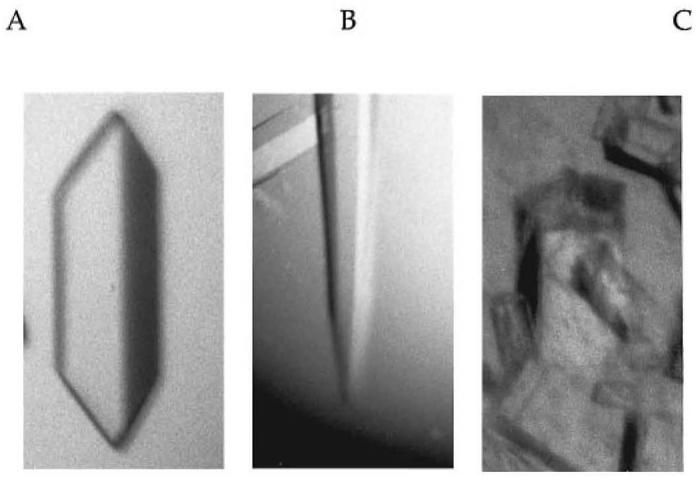
Fhit crystals. Human Fhit protein, salted out with ammonium sulfate as described in Materials and methods, forms single orthorhombic prisms (A), single hexagonal needles (B), and pentagonal columns (C).
Synchrotron X-ray data collection
Orthorhombic crystals were mounted in quartz capillaries and transported to beamline A1 (wavelength 0.908 Å, collimated to 150 μm) of the Cornell High Energy Synchrotron Source (CHESS). X-Ray diffraction data were collected on an ADSC charge coupled device (CCD) camera (Walter et al., 1995), 185 mm from the crystal at 5 s per 1° oscillation. At CHESS beamline F1 (wavelength 0.918 Å, collimated to 150 μm), hexagonal crystals were washed in 2 M ammonium sulfate with 4% (vol/vol) polyethylene glycol 400 and 12% (vol/vol) glycerol, removed on nylon loops (Hampton Research, Laguna Hills, CA), and rapidly centered onto a goniometer head chilled by a liquid nitrogen cold stream. Data were collected on a Princeton 1k CCD, 225 mm from the crystal at 30 s per 1° oscillation. Data were indexed, integrated and averaged using the HKL package (HKL Research Inc., Charlottesville, VA).
Results and discussion
Active, dimeric human Fhit protein was purified from E.coli with a high yield (Table I) and with a substantially higher specific activity than an affinity-purified Fhit fusion protein (Barnes et al., 1996). Whereas the glutathione S-transferase—Fhit fusion protein had an apparent kcat value of ∼3 s-1 with ApppA as a substrate, the form of the protein purified in this study had a kcat value of at least 60 s-1 and a KM value of 1.9 μM (kcat/KM. 3.2 3 107 M-1 s-1). Moving from the affinity-purified fusion protein system to the current system has either increased the fraction of enzyme that is active, released an inhibitory fusion protein domain, or both. The preparation of Lima and co-workers (Lima et al., 1997b) was only 2% as active as this preparation for reasons that are not apparent. Purified Fhit crystallized in at least three discrete forms, two of which were suitable for X-ray diffraction analysis.
Surprisingly, the orthorhombic form (Figure 2A) exhibited little decay in diffraction intensities in the synchrotron beam over the course of ∼100° data collection sweeps. The hexagonal form (Figure 2B) was radiation sensitive at room temperature but, despite the presence of 2 M ammonium sulfate in the mother liquor, could be frozen in the cold stream simply by brief equilibration in 2 M ammonium sulfate, 4% (vol/vol) polyethylene glycol 400, 12% (vol/vol) glycerol. An additional crystal form had a pentagonal habit (Figure 2C) and diffracted well but could not be indexed as a single crystal. Nearly complete data were collected for the wild-type orthorhombic form and the wild-type—IB2 complexed hexagonal form to 2.75 and 3.1 Å respectively (Table II). Because these crystals contain long cell lengths of ∼220 and ∼270 Å, data collection at home radiation sources is difficult. In order to separate reflections along the long axes of these crystal forms, X-ray detectors must be positioned at least 180 or 220 mm, respectively, from the crystals. The fine focus and high intensity of CHESS beamlines and the precise Cartesian resolution of CCD detectors were required for reliable diffraction intensities to be measured. The orthorhombic form is of space group P2(1)2(1)2(1) and has a unit cell volume of 1 700 000 Å3, consistent with a noncrystallographic multiplicity of 12 (six dimers) and a 42% solvent content (Matthews, 1968). The hexagonal form obeys the symmetry of space group P6(1)22 and its enantiomorph P6(5)22 and appears to be isomorphous with the P6(1)22 crystals of selenomethionyl Fhit (Lima et al., 1997a). Crystal structures of the orthorhombic form of Fhit and the two diadenosine complexed forms modeling the active state of Fhit protein are in progress.
Table II.
Crystallographic data
| Ligand | Space group | Temperature (°C) |
Unit Cell lengths | Resolution (Å) | Reflectiotls measured/unique |
Completeness (%) |
Rsyma (%) |
||
|---|---|---|---|---|---|---|---|---|---|
| a | b | c | |||||||
| NONE | P2(1)2(1)2(1) | 20 | 51.96 | 149.56 | 219.20 | 50.0-2.75 | 177 630 / 39 117 | 85.8 | 5.8 |
| IB2 | P6(1)22 | -175 | 50.73 | 50.73 | 268.51 | 70.0-3.35 | 6201 / 2584 | 74.1 | 9.8 |
| 70.0-3.10 | 6659 / 2965 | 68.3 | 9.7 | ||||||
Σ|Iobs - Iavg| ÷ ΣI. I = intensity.
Acknowledgments
This work was supported by NIH/NCI Grants CA75954 (to C.B.) and CA56336 (to C.M.C. for Kimmel Cancer Center core facilities), NSF Grant MCB-9604124 (to L.D.B.) and BBSRC Grant CHM.RA73861 (to G.M.B.). H.C.P. was supported by NIH/NCI Training Grant T32CA09662. CHESS is supported by NSF Grant DMR-9311772 and NIH Grant RR-01646. We thank Dan Thiel, the staff at CHESS, and Xiaofeng Zheng for assistance with data collection.
References
- Barnes LD, Garrison PN, Siprashvili Z, Guranowski A, Robinson AK, Ingram SW, Croce CM, Ohta M, Huebner K. Biochemistry. 1996;35:11529–11535. doi: 10.1021/bi961415t. [DOI] [PubMed] [Google Scholar]
- Blackburn GM, Liu X, Rosler A, Brenner C.Nucleosides Nucleotides 199817 in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner C, Garrison P, Gilmour J, Peisach D, Ringe D, Petsko GA, Lowenstein JM. Nature Struct. Biol. 1997;4:231–238. doi: 10.1038/nsb0397-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druck T, Hadaczek P, Fu TB, Ohta M, Siprashvili Z, Baffa R, Negrini M, Kastury K, Veronese ML, Rosen D, Rothstein J, McCue P, Cotticelli MG, Inoue H, Croce CM, Huebner K. Cancer Res. 1997;57:504–512. [PubMed] [Google Scholar]
- Gartenhaus RB. Oncogene. 1997;14:375–378. doi: 10.1038/sj.onc.1200867. [DOI] [PubMed] [Google Scholar]
- Geurts JMW, Schoenmakers EFPM, Roijer E, Stenman G, Van de Ven WJM. Cancer Res. 1997;57:13–17. [PubMed] [Google Scholar]
- Ghosh S, Lowenstein JM. Gene. 1997;176:249–255. doi: 10.1016/0378-1119(96)00260-0. [DOI] [PubMed] [Google Scholar]
- Huang Y, Garrison PN, Barnes LD. Biochem. J. 1995;312:925–932. doi: 10.1042/bj3120925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima CD, Dı’ Amico KL, Naday I, Rosenbaum G, Westbrook EM, Hendrickson WA. Structure. 1997a;5:763–774. doi: 10.1016/s0969-2126(97)00231-1. [DOI] [PubMed] [Google Scholar]
- Lima CD, Klein MG, Hendrickson WA. Science. 1997b;278:286–290. doi: 10.1126/science.278.5336.286. [DOI] [PubMed] [Google Scholar]
- Mao L, Fan YH, Lotan R, Hong WK. Cancer Res. 1996;56:5128–5131. [PubMed] [Google Scholar]
- Matthews BW. J. Mol. Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- Mau S, Ellis IO, Sibbering M, Blamey RW, Brook JD. Cancer Res. 1996;56:5484–5489. [PubMed] [Google Scholar]
- Negrini M, Monaco C, Vorechovsky I, Ohta M, Druck T, Baffa R, Huebner K, Croce CM. Cancer Res. 1996;56:3173–3179. [PubMed] [Google Scholar]
- Ohta M, Inoue H, Cotticelli MG, Kastury K, Baffa R, Palazzo J, Siprashvili Z, Mori M, McCue P, Druck T, Croce CM, Huebner K. Cell. 1996;84:587–597. doi: 10.1016/s0092-8674(00)81034-x. [DOI] [PubMed] [Google Scholar]
- Panagopoulos I, Pandis N, Thelin S, Petersson C, Mertens F, Borg A, Kristoffersson U, Mitelman F, Aman P. Cancer Res. 1996;56:4871–4875. [PubMed] [Google Scholar]
- Seraphin B. DNA Sequence. 1992;3:177–179. doi: 10.3109/10425179209034013. [DOI] [PubMed] [Google Scholar]
- Shridhar R, Shridhar V, Wang XH, Paradee W, Dugan M, Sarkar F, Wilke C, Glover TW, Vaitkevicius VK, Smith DI. Cancer Res. 1996;56:4347–4350. [PubMed] [Google Scholar]
- Shridhar V, Wang L, Rosati R, Paradee W, Shridhar R, Mullins C, Sakr W, Grignon D, Miller OJ, Sun QC, Petros J, Smith DI. Oncogene. 1997;14:1269–1277. doi: 10.1038/sj.onc.1201100. [DOI] [PubMed] [Google Scholar]
- Siprashvili Z, Sozzi G, Barnes LD, McCue P, Robinson AK, Eryomin V, Sard L, Tagliabue E, Greco A, Fusetti L, Schwartz G, Pierotti MA, Croce CM, Huebner K. Proc. Natl. Acad. Sci. USA. 1997;94:13771–13776. doi: 10.1073/pnas.94.25.13771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sozzi G, Alder H, Tornielli S, Corletto V, Baffa R, Veronese ML, Negrini M, Pilotti S, Pierotti MA, Huebner K, Croce CM. Cancer Res. 1996a;56:2472–2474. [PubMed] [Google Scholar]
- Sozzi G, Veronese ML, Negrini M, Baffa R, Cotticelli MG, Inoue H, Tornielli S, Pilotti S, De Gregorio L, Pastorini U, Pierotti MA, Ohta M, Huebner K, Croce CM. Cell. 1996b;85:17–26. doi: 10.1016/s0092-8674(00)81078-8. [DOI] [PubMed] [Google Scholar]
- Virgilio L, Schuster M, Gollin SM, Veronese ML, Ohta M, Huebner K, Croce CM. Proc. Natl Acad. Sci. USA. 1996;93:9770–9775. doi: 10.1073/pnas.93.18.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter RL, Thiel DJ, Barna SL, Tate MW, Wall ME, Eikenberry EF, Gruner SM, Ealick SE. Structure. 1995;3:835–844. doi: 10.1016/s0969-2126(01)00218-0. [DOI] [PubMed] [Google Scholar]
- Wedekind JE, Frey PA, Rayment I. Biochemistry. 1995;34:11049–11061. doi: 10.1021/bi00035a010. [DOI] [PubMed] [Google Scholar]
- Yanagisawa K, Kondo M, Osada H, Uchida K, Takagi K, Masuda A, Takahashi T, Takahashi T. Cancer Res. 1996;56:5579–5582. [PubMed] [Google Scholar]
- Zimonjic DB, Druck T, Ohta M, Kastury K, Croce CM, Popescu NC, Huebner K. Cancer Res. 1997;57:1166–1170. [PubMed] [Google Scholar]