

Abstract
We report energy-transferring organically modified silica nanoparticles for two-photon photodynamic therapy. These nanoparticles co-encapsulate two-photon fluorescent dye nanoaggregates as an energy up-converting donor and a photosensitizing PDT drug as an acceptor. They combine two features: i) aggregation-enhanced two-photon absorption and emission properties of a novel two-photon dye, and ii) nanoscopic fluorescence resonance energy transfer between this nanoaggregate and a photosensitizer, 2-devinyl-2-(1-hexyloxyethyl)pyropheophorbide. Stable aqueous dispersions of the co-encapsulating nanoparticles (diameter≤30 nm) have been prepared in the nonpolar interior of micelles by co-precipitating an organically modified silica sol with the photosensitizer and an excess amount of the two-photon dye which forms fluorescent aggregates by phase separation from the particle matrix. Using a multidisciplinary nanophotonic approach, we show: i) indirect excitation of the photosensitizer through efficient two-photon excited intraparticle energy transfer from the dye aggregates in the intracellular environment of tumor cells, and ii) generation of singlet oxygen and in-vitro cytotoxic effect in tumor cells by photosensitization under two-photon irradiation.
Introduction
Photodynamic therapy (PDT), a light-activated treatment for cancer and other diseases, has emerged as one of the important areas in biophotonics, a new field dealing with biomedical applications of light-matter interactions.1,2 PDT utilizes light-sensitive drugs or photosensitizers that can be preferentially localized in malignant tissues. Its therapeutic effect is activated by photoexcitation of the localized photosensitizer to generate a cytotoxic species, e.g., singlet oxygen (1O2), free radicals, or peroxides. This leads to selective and irreversible destruction of diseased tissues, without damaging adjacent healthy ones.
Despite PDT's advantages over current treatments (e.g. surgery, radiation therapy and chemotherapy), PDT still has yet to gain general clinical acceptance. First, currently approved PDT photosensitizers absorb in the visible spectral regions below 700 nm, where light penetration into the skin is only a few millimeters, thus clinically limiting PDT to treating topical lesions. Two-photon absorption (TPA)-induced excitation of photosensitizers is one of the promising approaches to increase light penetration. It enables the use of light in the tissue transparent window (750-1000 nm). Furthermore, due to a quadratic dependence on laser intensity of TPA, it provides a tool for improving treatment of deeper tumors with enhanced spatial resolution.
Approaches to obtain large intrinsic TPA cross-sections in the near-infrared region, include either design of new photosensitizers or chemical modification of existing ones to produce efficient two-photon sensitization.3 Another approach is to exploit the energy-transferring combination of existing photosensitizers with TPA dyes. Here, the photosensitizing unit (energy acceptor) is indirectly excited through fluorescence resonance energy transfer (FRET) from the two-photon absorbing dye unit (energy donor).4 The FRET approach was realized by chemically assembling the TPA donors into dendrimer arms with a photosensitizer as the central core.4b,c Then, energy harvesting by the TPA donors strongly enhances the efficiency of two-photon excitation of the photosensitizer, while preserving the photophysical properties for PDT due to insignificant chromophore modification.
Second, the preparation of pharmaceutical formulations of photosensitizers for parenteral administration is another major challenge in PDT. Most existing photosensitizers are hydrophobic with an inherent poor water solubility and aggregate easily under physiological condition. Thus they cannot be simply injected intravenously. Moreover, even with water-soluble photosensitizers, the accumulation selectivity to diseased tissues is not high enough for clinical use. In this respect, colloidal carriers for photosensitizers,5 such as oil-dispersions, liposomes, low-density lipoproteins, polymeric micelles, and nanoparticles, offer benefits of hydrophilicity, appropriate size for passive targeting to tumor tissues by the enhanced permeability and retention (EPR) effect,6 and the possibility of active targeting by using surface modification.
To produce efficient two-photon PDT effect by addressing the above issues together, we introduce a nanomedicine approach which utilizes nanophotonics7 through the fusion of nanotechnology with biomedical technology. This approach involves energy-transferring nanoscopic co-assembly of photosensitizers and fluorescent TPA dyes produced by physical entrapment within a single nanocarrier. It provides several advantages: (i) Co-encapsulation offers ease of preparation as opposed to chemical conjugation, together with the flexibility to choose a wide range of donor-acceptor pairs. (ii) Surface functionalization provides opportunity to improve biodistribution and to incorporate active targeting. (iii) A nanoscopic volume offers the opportunity for high level of loading of a TPA donor to efficiently harvest IR photons.
To accomplish efficient two-photon excitation using this concept, the loading density of the energy-donating TPA dye needs to be much higher than that of the energy-accepting photosensitizer. In addition, the nanoparticle matrix needs to be compatible with the PDT application, as well as rigid enough to preserve the initially loaded energy-transferring composition without undesirable release. In this regard, organically modified silica (ORMOSIL) nanoparticles have proven to be biocompatible,8-10 stable without releasing encapsulated hydrophobic molecules,9 and suitable for PDT drug delivery because their porous matrix is permeable to oxygen molecules.10
With high loading density of a TPA dye within an individual nanoparticle, additional nonradiative decay which leads to fluorescence quenching at high concentrations or in the aggregated state, typically observed for most organic dyes, needs to be overcome so as to maintain the ability for energy transfer. Recently, a special class of fluorescent molecules has been reported, showing intense fluorescence, instead of quenching in the aggregated state.11 As a highly two-photon active version of such molecules, we have developed a novel 9,10-bis[4′-(4″-aminostyryl)styryl]anthracene dye with a severely distorted geometry (BDSA, see Figure 1), which forms nanoaggregates with intense and red-shifted fluorescence as well as enhanced two-photon absorptivity due to partial planarization of the geometry upon aggregation and the resultant loose packing of molecules in the aggregated state.12 The value of TPA cross-section in the aggregated state, measured at 775 nm by the nonlinear transmission method, is 217 GM. This unusual fluorescence behavior of BDSA offers an opportunity to prepare TPA dye-concentrated nanoparticles, which do not exhibit concentration quenching at high loading.
Figure 1.

Chemical structure of BDSA (top) and optimized geometry of a model structure with dimethylamino endgroups by semiempirical calculation (pm3 formalism, bottom), where the torsion angles (φ), indicated by the solid arrows, are calculated as ca. 74° in the isolated state due to the internal steric hindrance arising from the proximate hydrogens indicated by the dotted arrows.12
Based on these considerations, we prepared organically modified silica nanoparticles co-encapsulating a known photosensitizer and an excess amount of BDSA as a two-photon energy donor. The photosensitizing energy acceptor used in this work is 2-devinyl-2-(1-hexyloxyethyl)pyropheophorbide (HPPH) which is an effective photosensitizer in phase I/II clinical trials at the Rosewell Park Cancer Institute, Buffalo, NY.13 Nanophotonics is used here to control optical interactions and dynamics in two ways: (i) aggregation enhancement of two-photon absorption and emission of BDSA, and (ii) efficient nanoscopic FRET to HPPH. We report two-photon sensitization of singlet oxygen in an aqueous dispersion of the co-encapsulating ORMOSIL nanoparticles, activated by indirect excitation of HPPH via efficient intraparticle FRET. Also presented is two-photon laser scanning fluorescence microscopy imaging of live tumor cells uptaking the co-encapsulating nanoparticles, as well as cytotoxic effect under two-photon irradiation, to demonstrate their potential as a drug-carrier nanoassembly for two-photon activated PDT.
Experimental Section
Materials
Surfactant, Aerosol OT (AOT, sodium bis(2-ethylhexyl)sulfosuccinate, 98%), triethoxyvinylsilane (VTES, 97%), and N-methyl-2-pyrrolidinone (NMP) were purchased from Aldrich. Cosurfactant, 1-butanol and NH4OH (28.0∼30.0%), are products of J. T. Baker. Deuterium oxide (99.9 atom % D) was obtained from Isotec Inc. The drug, HPPH, was provided by the Roswell Park Cancer Institute, Buffalo, NY. 9,10-Anthracenedipropionic acid, disodium salt (ADPA) was purchased from Molecular Probes. All the above chemicals were used as received without further purification. The details on BDSA, including preparation, semiempirical calculation, and characterizations, are described in Ref. 12.
Preparation of Dye-Encapsulating ORMOSIL Nanoparticles
The nanoparticles, entrapping either one or both of HPPH and BDSA, were synthesized by co-precipitating the dyes with polymeric organically modified silica sol in the nonpolar core of AOT/1-butanol micelles in deionized water or D2O. NMP was used as a hydrophilic solvent which has unlimited water miscibility as well as suitable solubility for both HPPH and BDSA. To obtain a clear solution of prepolymerized silica sol, 0.2 g of VTES in 2 mL NMP was hydrolyzed and condensed in the presence of 40 μL NH4OH at room temperature for 12 h to 1 day, until adding one drop of the resulting solution into excess pure water made white bulk precipitate without liquid phase of unreacted VTES or oligomers. After syringe filtering by membrane filter (0.2 μm pore size), 0.15 mL of the sol solution was homogeneously mixed with 0.57 mL NMP solutions containing either or both HPPH (0.15 mg; 1.1 wt% loading with respect to the added VTES amount) and BDSA (1.35, 2.7, or 4.05 mg; 10, 20, or 30 wt% loading amount, respectively). The micelles were prepared by dissolving 0.22 g of AOT and 0.4 mL of 1-butanol in 10 mL of water or D2O. Nanoprecipitation was induced by one-shot syringe injection of the above NMP solutions (0.6 mL) into the prepared micelle dispersions under vigorous magnetic stirring. The resulting mixtures were further stirred at room temperature, to ensure completion of sol-gel condensation within the co-precipitated nanoparticles. After 2 days of stirring, AOT and 1-butanol were removed either by dialyzing the water dispersion against water in a 12∼14 kDa cutoff cellulose membrane for 48 h, or by thoroughly washing the D2O dispersion with n-hexane.
Characterization
Transmission electron microscopy (TEM) was performed to determine the size and shape of the prepared nanoparticles, using a JEOL JEM-100cx microscope at an accelerating voltage of 80 kV. Absorption and fluorescence spectra were acquired using a Shimadzu UV-3101 PC spectrophotometer and a Jobin-Yvon Fluorog FL-3.11 spectrofluorometer, respectively. Fluorescence quantum yields (Φf) of the BDSA-entrapping nanoparticles were determined using methanol solution of rhodamine B as a reference for which the absorbance was kept below 0.1 at the excitation wavelength (480 nm). Two-photon excitation for the fluorescence and the singlet oxygen generation experiments, as well as for two-photon laser scanning fluorescence microscopy was performed at 850 nm using a Ti-sapphire laser (Tsunami, Spectra-Physics) pumped by a frequency-doubled diode-pumped solid-state laser (Millennia, Spectra-Physics). It provided 90 fs pulses at 82 MHz repetition rate. A spectrum analyzer (IST-REES, Germany) was used to monitor the wavelength and the bandwidth of excitation light.
Fluorescence lifetime measurements were performed using the Time correlated Single Photon Counting (TCSPC) technique. For this measurement, a Ti:Sapphire laser providing 800 nm femtosecond pulses was used as the excitation source and a SPC-830 TCSPC module (Becker & Hickl) with an H7422 (Hamamatsu) detector having a response time in the range of 250 ps was used for the measurements. For measuring the lifetime of the donor molecules (BDSA) in presence and absence of the acceptor molecules (HPPH), a band pass filter (HQ540/80-2p from Chroma) was used to cut off the acceptor emission. For an estimation of the instrument response function (IRF), a dye with a known fluorescence lifetime (from Streak camera measurements), less than 50 ps was used. By deconvoluting this IRF with the obtained lifetime, a lifetime much shorter than the detector response can be estimated.14 In our case, we used the SPCImage software (Becker & Hickl) for deconvolution and data fitting to estimate lifetime values.
For quantitative comparison, the concentrations of the nanoparticle water dispersions were kept the same for all the sample sets. The dispersions of all examined HPPH-loaded nanoparticles had the absorbance of 0.07 at 665 nm.
Detection of Singlet Oxygen
One- and two-photon induced generations of singlet oxygen were monitored by singlet oxygen luminescence and chemical oxidation methods, respectively. Singlet oxygen luminescence at 1270 nm was recorded for the surfactant-removed D2O dispersions, using a SPEX 270M spectrometer (Jobin Yvon) equipped with an InGaAs photodetector (Electro-Optical Systems Inc.). A diode-pumped solid-state laser (Millenia, Spectra-Physics) at 532 nm was used as the excitation source. Chemical oxidation of ADPA in the nanoparticle water dispersions was used as a chemical method to detect singlet oxygen generation.10 It was monitored by a decrease in the absorbance of the added ADPA at 400 nm under excitation with 850 nm. The mixture solution was prepared by combining 1 mL of the stock dispersions of nanoparticles in water with 0.1 mL of ADPA stock solution in water (0.5 mM). Two-photon excitation at 850 nm was performed by focusing the laser beam through a cuvette (1 cm path length and 2 mm width) containing 0.5 cm3 solution.
In-Vitro Studies with Tumor Cells: Nanoparticle Uptake and Two-Photon Excited Fluorescence Imaging
HeLa (human cervix epitheloid carcinoma) cells (American Type Culture Collection, Manassas, VA) were cultured in a Minimum Essential Medium (MEM) supplemented with 10% fetal bovine serum (FBS), following the established protocol. The detailed procedures are described elsewhere.9b,10 For nanoparticle uptake, cells in a 60-mm suspension in the medium was combined with nanoparticle dispersion and incubated at 37 °C (5% CO2) for 3 h. Two-photon laser scanning fluorescence microscopy was performed using confocal laser scanning microscopy (Bio-Rad, model MRC-1024), with an upright microscope (Nikon, model Eclipse E800). A water immersion objective lens (Nikon, Fluor-60X, NA 1.0) was used for cell imaging. A long-pass filter (585 LP) was used as the emission filters for imaging. For localized spectrofluorometry, the fluorescence signal was collected without filtering, from the upper port of the confocal microscope, using a multimode optical fiber of core diameter 1 mm, and was delivered to a spectrometer (Holospec from Kaiser Optical Systems, Ann Arbor, MI) equipped with a cooled charge coupled device (CCD) camera (Princeton Instruments, Monmouth Junction, NJ) as a detector.
Results and Discussion
Dye encapsulation in ORMOSIL nanoparticles was accomplished in the nonpolar interior of aqueous AOT micelle, by transient emulsification and spontaneous co-precipitation via a ‘solvent displacement’ process, i.e., diffusive depletion of hydrophilic solvent into the aqueous exterior.15Figure 2 shows representative TEM images of nanoparticles entrapping 1.1 wt% HPPH (a) and 1.1 wt% HPPH/20 wt% BDSA (b), where the sizes are estimated as 25±7 and 30±6 nm, respectively.
Figure 2.

TEM images of silica nanoparticles entrapping (a) 1.1 wt% HPPH and (b) 1.1 wt% HPPH/20 wt% BDSA.
To produce fluorescent domains of BDSA aggregates in the particle matrix, the following amounts were loaded to induce phase separation: 10, 20, and 30 wt% with respect to the total VTES amount. All the prepared nanoparticles entrapping BDSA exhibited almost the same fluorescence spectra peaking at ca. 610 nm. A representative spectrum of the nanoparticles entrapping 20 wt% BDSA is shown in Figure 3, along with typical state-dependent spectra of BDSA in PMMA films (monomer state at 0.5 wt% and aggregated state at 30 wt%). Typically, at lower concentrations in the polymer matrix, BDSA emits blue-shifted, greenish fluorescence, because it exists in the molecularly dispersed frozen monomer state with π-conjugation limited by a distorted geometry.12 At concentrations high enough to induce self-aggregation, the BDSA emission is red-shifted to orange without quenching. This shift is a consequence of molecular stacking by aggregation which results in partial planarization due to severe intramolecular steric hindrance, which not only extends the π-conjugation, but also diminishes intermolecular quenching effects, derived from loose packing of molecules retaining a certain amount of distortion. Accordingly, the intense orange fluorescence from the prepared nanoparticles suggests that the loaded BDSA exists in the aggregated state at the studied loading concentrations of 10∼30 wt%. Importantly, as opposed to concentration quenching generally observed for most dye aggregates, fluorescence quantum yields of the BDSA-loaded nanoparticles are rather increased by raising the extent of aggregation with higher loading (the inset of Figure 3).
Figure 3.

Normalized fluorescence spectra of BDSA, entrapped in PMMA films (a: 0.5 wt%, b: 30 wt%) and in water-dispersed silica nanoparticles (c: 20 wt%). Inset: dependence of fluorescence quantum yields (Φf) of BDSA-loaded silica nanoparticles, on the loading amount.
The spectral matching for FRET between HPPH and the BDSA aggregate, both entrapped separately in nanoparticles, is shown in Figure 4. The aqueous dispersion of HPPH-loaded (1.1 wt%) nanoparticles exhibits typical HPPH fluorescence with peak at ∼667 nm, indicating that by co-precipitation with polymeric VTES sol, the hydrophobic HPPH molecules have successfully been embedded in the particle matrix, without self-aggregation or significant interaction with water. Note that the HPPH fluorescence is completely quenched in water dispersions by self-aggregation.10 Moreover, the HPPH absorption in nanoparticles has significant spectral overlap with the fluorescence of BDSA aggregates, which enables an energy transfer between them. This spectral matching, together with the aggregation-enhanced fluorescence of BDSA, allows the approach based on the use of BDSA aggregates to overcome the issue of fluorescence quenching in the preparation of donor-concentrated FRET nanoparticles.
Figure 4.

Normalized absorption (dashed) and fluorescence (solid) spectra of HPPH (1.1 wt%) and BDSA (20 wt%) entrapped separately in water-dispersed silica nanoparticles. The excitation wavelengths are 600 and 480 nm for HPPH and BDSA, respectively.
Figure 5 shows the one-photon excited fluorescence spectra of the water-dispersed nanoparticles co-encapsulating HPPH (1.1 wt%) and BDSA (20 wt%). When excited at 425 nm, the obtained fluorescence spectrum of the co-encapsulating nanoparticles is a composite of the emission contributions from the donor BDSA aggregates and the acceptor HPPH. Compared with the fluorescence intensities of the nanoparticles containing the corresponding amount of each dye separately, BDSA emission is quenched by ca. 70 % and HPPH emission is amplified ca. 5 times, indicating the occurrence of FRET. This energy harvesting provides evidence that HPPH and the BDSA aggregates have successfully been co-encapsulated in the same nanospace by co-precipitation with polymeric VTES sol.
Figure 5.

One-photon excited fluorescence spectra of the same amount of water-dispersed silica nanoparticles entrapping (a) only 20 wt% BDSA, (b) only 1.1 wt% HPPH, and (c) 1.1 wt% HPPH/20 wt% BDSA together. Because of significant overlapping of BDSA and HPPH emission spectra, the composite fluorescence spectra in (c) were deconvoluted. Curves (d), (e) present the deconvoluted BDSA and HPPH fluorescence spectra, respectively. The excitation wavelength was 425 nm.
Indirect two-photon excitation of HPPH was examined with the HPPH-loaded nanoparticles with different amounts of BDSA. For quantitative comparison, only the BDSA amount was varied under the same conditions, concentration of the nanoparticles in water dispersion was same for every sample set. Equality in the amounts of the loaded HPPH was controlled spectrophotometrically, as described in the Experimental section. Figure 6 shows the fluorescence spectra of the nanoparticle dispersions, obtained by excitation at 850 nm. Under two-photon excitation, the water dispersion of the HPPH-only silica nanoparticles demonstrates weak fluorescence (Figure 6, (a)). However, the HPPH component of the emission from the HPPH/BDSA co-encapsulating nanoparticles is amplified by factors of ∼10 and ∼30 for the BDSA loading of 10 and 20 wt%, respectively, which is much greater than the one-photon FRET amplification obtained by the excitation at 425 nm (Figure 5). More pronounced energy harvesting by two-photon excitation is unambiguously attributed to a large difference in the two-photon absorptivities between HPPH and BDSA at the wavelength of 850 nm. Due to low two-photon absorptivity of HPPH itself, direct excitation of HPPH would have a minimal contribution. Therefore, the intense two-photon fluorescence of HPPH is mainly originating from the indirect excitation through the intraparticle FRET, indicating that the energy of the near-IR light is efficiently up-converted by BDSA aggregates to excite HPPH.
Figure 6.
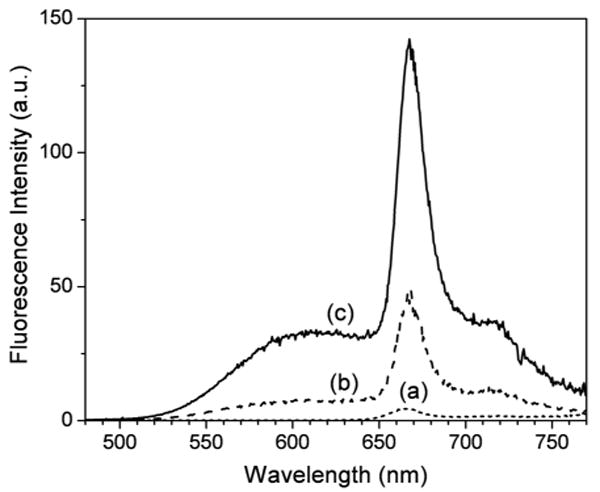
Two-photon excited fluorescence spectra of the same amount of water-dispersed silica nanoparticles, entrapping (a) 1.1 wt% HPPH, (b) 1.1 wt% HPPH/10 wt% BDSA, and (c) 1.1 wt% HPPH/20 wt% BDSA. The excitation wavelength is 850 nm.
To confirm the occurrence of FRET, we also used fluorescence lifetime measurements. Figure 7 shows the fluorescence decay curves with a biexponential fit for nanoparticles doped with (1) 20 wt% BDSA and (2) 1.1 wt% HPPH/20 wt% BDSA. In the case of nanopartilcles encapsulating BDSA alone, the fluorescence decay was found to be biexponential in nature, with an average lifetime (τm) of 636 ps. In case of nanoparticles co-encapsulating BDSA and HPPH, the average lifetime of BDSA was found to be shortened to 173 ps, using a biexponential fitting. FRET efficiency of this donor-acceptor pair can be estimated using the equation, 1-τDA/τD, where τDA is the lifetime of the donor in the presence of acceptor and τD is the lifetime of the donor alone. From this data, the estimated FRET efficiency of the BDSA-HPPH pair, when co-doped in silica particles, was found to be 73%, which is in close agreement with the FRET efficiency estimated from the fluorescence quenching of the donor (BDSA) in Figure 5.
Figure 7.

BDSA fluorescence decay for nanoparticles doped with (a) 20 wt% BDSA and (b) 1.1 wt% HPPH/20 wt% BDSA, each plotted with the best-fitted kinetic functions (solid line). Instrument response function (IRF) is shown as unlabeled dashed line.
We assessed the capability of the encapsulating nanoparticles to generate singlet oxygen, by monitoring the oxygen luminescence under the excitation at 532 nm. Instead of water, D2O was used as a dispersion solvent, because it extends the lifetime of singlet oxygen. As shown in Figure 8, the characteristic singlet oxygen emission with a peak at 1270 nm was clearly observed under the photoexcitation of the dispersion of nanoparticles co-encapsulating HPPH (1.1 wt%) and BDSA (20 wt%), indicating the generation of singlet oxygen (1O2) by sensitizing with HPPH. These data confirm the previous report10 that the polymerized VTES matrix is suitable for PDT due to its porosity, which allows molecular oxygen to diffuse in and out of the particle matrix.
Figure 8.
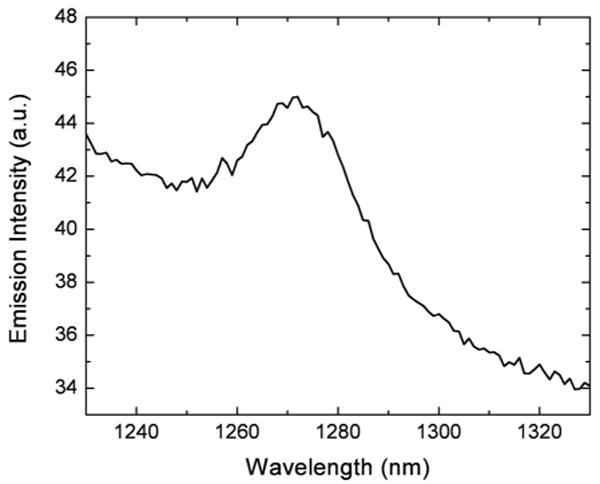
Emission of singlet oxygen generated in the D2O dispersion of nanoparticles co-encapsulating 1.1 wt% HPPH/20 wt% BDSA. The excitation wavelength was 532 nm.
It is difficult to observe singlet oxygen emission in water dispersions under the two-photon excitation condition, due to short lifetime of singlet oxygen in water, the relatively low inherent efficiency of two-photon processes and small volume of excitation due to quadratic intensity dependence of two-photon excitation. Thus, to evaluate the ability for the two-photon sensitization of singlet oxygen in water, we employed a chemical method based on the oxidation of disodium salt of 9,10-anthracenedipropionic acid (ADPA).16 In this approach, photobleaching of the ADPA absorbance was used to monitor singlet oxygen generation.4c,10Figure 9 shows the bleaching of ADPA in water in the presence of nanoparticles co-encapsulating HPPH (1.1 wt%) and BDSA (20 wt%), under two-photon irradiation at 850 nm, where ADPA has no linear absorption. As one can see, in the presence of the HPPH/BDSA co-encapsulating nanoparticles, the ADPA absorption below ∼450 nm was decreased continuously over the course of irradiation, while HPPH and BDSA showed no noticeable changes in their absorptions above 450 nm, suggesting that both compounds are stable under these irradiation conditions. It should be stressed that the observed photobleaching of ADPA is not caused by its photoreaction in the excited state, but by the oxidation by singlet oxygen liberated from the particles after the generation by two-photon excited photosensitizers entrapped inside. In the case of nanoparticles containing only HPPH, only a minimal photobleaching of ADPA was observed under the same irradiation conditions, as shown in the inset of Figure 9. This confirms that the intraparticle FRET, based on the use of fluorescent BDSA aggregates, is truly manifested in the two-photon sensitized generation of singlet oxygen in water.
Figure 9.

Photobleaching of ADPA by singlet oxygen generated upon two-photon excitation of the water-dispersed nanoparticles co-encapsulating 1.1 wt% HPPH/20 wt% BDSA at 850 nm. Inset: ADPA remaining in water dispersions of the nanoparticles encapsulating 1.1 wt% HPPH (square) and 1.1 wt% HPPH/20 wt% BDSA (circle), as a function of time of irradiation with 850 nm.
The potential of the HPPH/BDSA co-encapsulating nanoparticles as a drug-carrier nanoassembly was evaluated in vitro by fluorescence imaging of live tumor cells under two-photon excitation at 850 nm. Figure 10 shows the two-photon laser scanning microscopic image of HeLa cells treated with nanoparticles co-encapsulating HPPH (1.1 wt%) and BDSA (20 wt%). An intense fluorescence signal is observed from the cells, indicating active uptake of the co-encapsulating nanoparticles by tumor cells. Localized spectrum in the inset of Figure 10 shows that the two-photon fluorescence from the cytoplasm exhibits the characteristic HPPH fluorescence. This affirms that the indirect two-photon excitation of HPPH through intraparticle FRET is still operative in the cellular environment, suggesting the intracellular stability of the ORMOSIL nanoparticles co-encapsulating HPPH and BDSA. The intracellular FRET efficiency was estimated up to 36% by using ‘acceptor bleaching’ method,17 without correcting for donor bleaching due to the generated singlet oxygen (see Supporting Information).
Figure 10.

Merged transmission (blue) and two-photon excited fluorescence (red) images of HeLa cells, stained with nanoparticles co-encapsulating 1.1 wt% HPPH/20 wt% BDSA. Inset: Localized two-photon fluorescence spectrum from the cytoplasm of the stained cell. The excitation wavelength is 850 nm.
Two-photon induced cytotoxic effect with nanoparticles co-encapsulating 1.1 wt% HPPH/20 wt% BDSA is shown in Figure 11. Two photon irradiation of HeLa cells treated overnight with nanoparticles containing only BDSA (20 wt%) did not cause any significant change in cell morphology (Figure 11, upper row). On the contrary, drastic changes in the morphology of HeLa cells, which were treated overnight with nanoparticles containing 1.1 wt% HPPH/20 wt% BDSA, have been observed after irradiation under the same conditions (Figure 11, lower row). These changes are associated with cell necrosis and are apparently induced by the reactive oxygen species generated by HPPH which is excited as a result of two-photon excitation of BDSA, followed by energy transfer to HPPH.
Figure 11.

Transmission images of HeLa cells treated overnight with nanoparticles containing 20 wt% BDSA (a, b) and 1.1 wt% HPPH/20 wt% BDSA (d, c). Left column (a, c): before irradiation. Right column (b, d): 15 min after 90 s of irradiation with femtosecond pulsed laser at 850 nm (30 scans of 3 s each with 10 s interval).
Conclusions
We have prepared stable aqueous dispersions of organically modified silica nanoparticles (≤30 nm), co-encapsulating a photosensitizing anticancer drug, HPPH, and fluorescent aggregates of a two-photon absorbing dye, BDSA. It has been shown by two-photon spectroscopic measurements that the BDSA aggregates, formed by phase separation from the particle matrix, can efficiently up-convert the energy of near-IR light and transfer it to the co-encapsulated HPPH molecules through the intraparticle FRET, which results in enhanced two-photon generation of singlet oxygen in water. Using two-photon laser scanning fluorescence microscopy, we demonstrated that the co-encapsulating nanoparticles are actively uptaken by tumor cells in vitro, and that the excitation efficiency of HPPH through energy transfer from two-photon excited BDSA is preserved in the intracellular environment. Cytotoxic effect induced by the co-encapsulating nanoparticles under two-photon irradiation was demonstrated, as compared to the control cells treated with nanoparticles encapsulating BDSA only and irradiated under the same conditions. It can be concluded that our approach of two-photon induced intraparticle FRET, based on the use of two-photon fluorescent aggregates as a donor and a photosensitizing drug as an acceptor, offers a simple and proper methodology for developing formulations of drug-carrier nanoassemblies applicable in two-photon activated PDT.
Supplementary Material
Fluorescence imaging of a live cell treated with co-encapsulating nanoparticles before and after photobleaching of HPPH.
Acknowledgments
This work was supported in part by a grant from the John R. Oishei Foundation and in part by a grant from the National Institute of Health (No. R01-CA119358-1). Partial support from the Center of Excellence in Bioinformatics and Life Sciences at the University at Buffalo is also acknowledged.
References
- 1.Prasad PN. Introduction to Biophotonics. John Wiley & Sons; Hoboken, New Jersey: 2003. [Google Scholar]
- 2.(a) Dougherty TJ. Photochem Photobiol. 1987;45:879. doi: 10.1111/j.1751-1097.1987.tb07898.x. [DOI] [PubMed] [Google Scholar]; (b) Henderson BW, Dougherty TJ. Photochem Photobiol. 1992;55:145. doi: 10.1111/j.1751-1097.1992.tb04222.x. [DOI] [PubMed] [Google Scholar]; (c) Levy JG, Obochi M. Photochem Photobiol. 1996;64:737. doi: 10.1111/j.1751-1097.1996.tb01828.x. [DOI] [PubMed] [Google Scholar]
- 3.(a) Fisher WG, Partridge WP, Jr, Dees C, Wachter EA. Photochem Photobiol. 1997;66:141. doi: 10.1111/j.1751-1097.1997.tb08636.x. [DOI] [PubMed] [Google Scholar]; (b) Karotki A, Kruk M, Drobizhev M, Rebane A, Nickel E, Spangler CW. IEEE J Sel Top Quantum Electron. 2001;7:971. [Google Scholar]; (c) Frederiksen PK, Jørgensen M, Ogilby PR. J Am Chem Soc. 2001;123:1215. doi: 10.1021/ja003468a. [DOI] [PubMed] [Google Scholar]; (d) Karotki A, Drobizhev M, Kruk M, Spangler CW, Nickel E, Mamardashvili N, Rebane A. J Opt Soc Am B. 2003;20:321. [Google Scholar]; (e) Frederiksen PK, Mcllroy SP, Nielsen CB, Nikolajsen L, Skovsen E, Jørgensen M, Mikkelsen KV, Ogilby PR. J Am Chem Soc. 2005;127:255. doi: 10.1021/ja0452020. [DOI] [PubMed] [Google Scholar]; (f) Mcllroy SP, Cló E, Nikolajsen L, Frederiksen PK, Nielsen CB, Mikkelsen KV, Gothelf KV, Ogilby PR. J Org Chem. 2005;70:1134. doi: 10.1021/jo0482099. [DOI] [PubMed] [Google Scholar]
- 4.(a) Bhawalkar JD, Kumar ND, Zhao CF, Prasad PN. J Clin Laser Med Surg. 1997;15:201. doi: 10.1089/clm.1997.15.201. [DOI] [PubMed] [Google Scholar]; (b) Dichtel WR, Serin JM, Edder C, Fréchet JMJ, Matuszewski M, Tan LS, Ohulchanskyy TY, Prasad PN. J Am Chem Soc. 2004;126:5380. doi: 10.1021/ja031647x. [DOI] [PubMed] [Google Scholar]; (c) Oar MA, Serin JM, Dichtel WR, Fréchet JMJ, Ohulchanskyy TY, Prasad PN. Chem Mater. 2005;17:2267. [Google Scholar]
- 5.(a) Konan YN, Gruny R, Allemann E. J Photochem Photobiol, B. 2002;66:89. doi: 10.1016/s1011-1344(01)00267-6. [DOI] [PubMed] [Google Scholar]; (b) van Nostrum CF. Adv Drug Deliv Rev. 2004;56:9. doi: 10.1016/j.addr.2003.07.013. [DOI] [PubMed] [Google Scholar]; (c) Wang S, Gao R, Zhou F, Selke M. J Mater Chem. 2004;14:487. [Google Scholar]
- 6.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. J Control Release. 2000;65:271. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 7.Prasad PN. Nanophotonics. John Wiley & Sons; Hoboken, New Jersey: 2004. [Google Scholar]
- 8.(a) Shimada M, Shoji N, Takahashi A.Anticancer Res 1995151097537484 [Google Scholar]; (b) Lal M, Levy L, Kim KS, He GS, Wang X, Min YH, Pakatchi S, Prasad PN. Chem Mater. 2000;12:2632. [Google Scholar]; (c) Zhao X, Bagwe RP, Tan W. Adv Mater. 2004;16:173. [Google Scholar]
- 9.(a) Jain TK, Roy I, De TK, Maitra AN. J Am Chem Soc. 1998;120:11092. [Google Scholar]; (b) Roy I, Ohulchanskyy TY, Bharali DJ, Pudavar HE, Mistretta RA, Kaur N, Prasad PN. P Natl Acad Sci USA. 2005;102:279. doi: 10.1073/pnas.0408039101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roy I, Ohulchanskyy TY, Pudavar HE, Bergey EJ, Oseroff AR, Morgan J, Dougherty TJ, Prasad PN. J Am Chem Soc. 2003;125:7860. doi: 10.1021/ja0343095. [DOI] [PubMed] [Google Scholar]
- 11.(a) Levitus M, Schmieder K, Ricks H, Shimizu KD, Bunz UHF, Garcia-Garibay MA. J Am Chem Soc. 2001;123:4259. doi: 10.1021/ja003959v. [DOI] [PubMed] [Google Scholar]; (b) Belton C, O'Brien DF, Blau WJ, Cadby AJ, Lane PA, Bradley DDC, Byrne HJ, Stockmann R, Hörhold HH. Appl Phys Lett. 2001;78:1059. [Google Scholar]; (c) Luo J, Xie Z, Lam JWY, Cheng L, Chen H, Qiu C, Kwok HS, Zhan X, Liu Y, Zhu D, Tang BZ. Chem Commun. 2001:1740. doi: 10.1039/b105159h. [DOI] [PubMed] [Google Scholar]; (d) An BK, Kwon SK, Jung SD, Park SY. J Am Chem Soc. 2002;124:14410. doi: 10.1021/ja0269082. [DOI] [PubMed] [Google Scholar]; (e) Xiao D, Xi L, Yang W, Fu H, Shuai Z, Fang Y, Yao J. J Am Chem Soc. 2003;125:6740. doi: 10.1021/ja028674s. [DOI] [PubMed] [Google Scholar]; (f) Ryu SY, Kim S, Seo J, Kim YW, Kwon OH, Jang DJ, Park SY. Chem Commun. 2004:70. doi: 10.1039/b311648d. [DOI] [PubMed] [Google Scholar]; (g) Lim SJ, An BK, Jung SD, Chung MA, Park SY. Angew Chem Int Ed. 2004;43:6346. doi: 10.1002/anie.200461172. [DOI] [PubMed] [Google Scholar]
- 12.Kim S, Zheng Q, He GS, Bharali DJ, Pudavar HE, Baev A, Prasad PN. Adv Funct Mater. 2006;16:2317. [Google Scholar]
- 13.Henderson BW, Bellnier DA, Graco WR, Sharma A, Pandey RK, Vaughan L, Weishaupt K, Dougherty TJ. Cancer Res. 1997;57:4000. [PubMed] [Google Scholar]
- 14.(a) Becker W, Bergmann A, Hink MA, Konig K, Benndorf K, Biskup C. Microscopy Research and Technique. 2004;63:58. doi: 10.1002/jemt.10421. [DOI] [PubMed] [Google Scholar]; (b) Becker W, Bergmann A, Biscotti G, Rück A. Proceedings of SPIE-The International Society for Optical Engineering. 2004:5340. [Google Scholar]
- 15.Horn D, Rieger J. Angew Chem Int Ed. 2001;40:4330. doi: 10.1002/1521-3773(20011203)40:23<4330::aid-anie4330>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 16.Lindig BA, Rodgers MAJ, Schaap AP. J Am Chem Soc. 1980;102:5590. [Google Scholar]
- 17.(a) Rizzo MA, Springer G, Segawa K, Zipfel WR, Piston DW. Microsc Microanal. 2006;12:238. doi: 10.1017/S1431927606060235. [DOI] [PubMed] [Google Scholar]; (b) Mekler VM, Averbakh AZ, Sudarikov AB, Kharitonova OV. J Photochem Photobiol, B: Biology. 1997;40:278. doi: 10.1016/s1011-1344(97)00070-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fluorescence imaging of a live cell treated with co-encapsulating nanoparticles before and after photobleaching of HPPH.