

Abstract
This study tests the feasibility of large porous particles as long-acting carriers for pulmonary delivery of low molecular weight heparin (LMWH). Microspheres were prepared with a biodegradable polymer, poly(lactic-co-glycolic acid) (PLGA), by a double-emulsion–solvent-evaporation technique. The drug entrapment efficiencies of the microspheres were increased by modifying them with three different additives—polyethyleneimine (PEI), Span 60 and stearylamine. The resulting microspheres were evaluated for morphology, size, zeta potential, density, in vitro drug-release properties, cytotoxicity, and for pulmonary absorption in vivo. Scanning electron microscopic examination suggests that the porosity of the particles increased with the increase in aqueous volume fraction. The amount of aqueous volume fraction and the type of core-modifying agent added to the aqueous interior had varying degrees of effect on the size, density and aerodynamic diameter of the particles. When PEI was incorporated in the internal aqueous phase, the entrapment efficiency was increased from 16.22±1.32% to 54.82±2.79%. The amount of drug released in the initial burst phase and the release-rate constant for the core-modified microspheres were greater than those for the plain microspheres. After pulmonary administration, the half-life of the drug from the PEI- and stearylamine-modified microspheres was increased by 5- to 6-fold compared to the drug entrapped in plain microspheres. The viability of Calu-3 cells was not adversely affected when incubated with the microspheres. Overall, the data presented here suggest that the newly developed porous microspheres of LMWH have the potential to be used in a form deliverable by dry-powder inhaler as an alternative to multiple parenteral administrations of LMWH.
Keywords: Large porous particles, microspheres, Low molecular weight heparin, pulmonary delivery
Introduction
The pulmonary route has recently emerged as a viable alternative to the needle-based route of administration for an expanding array of biotechnology-derived drugs with superior therapeutic activity. However, the vast majority of currently available pulmonary drug delivery systems, including recently approved inhaled insulin—Exubera, which was later withdrawn from the market—are designed as immediate-release formulations to produce local and systemic effects for a short period of time [1]. Little has been done in the development of viable, inhaled formulations of therapeutic agents, especially biopharmaceuticals that can be administered via the lungs to release drugs for a prolonged period. In fact, inhaled long-acting formulations remain elusive because of the lack of efficient delivery devices and optimal drug carriers. The factors that have been major barriers to the development of long-acting pulmonary formulations are (i) suboptimal size and shape of the drug substance or the encapsulating carriers within the respirable fraction, (ii) short residence time of the inhaled drug particles or formulations in the respiratory tract, and (iii) poor loading of drugs into the particulate carriers frequently used to prepare inhaled formulations [2-4]. The recent advances in the dry-powder inhalation (DPI) technology have addressed some of the limitations associated with inhaled formulations, including unwanted loss of drug due to oropharyngeal deposition [5, 6]. However, the limitations associated with poor deposition of particles larger than 5 μm have yet to be overcome. In a seminal paper, Edwards et al. first proposed that particles with mass densities <0.4 g/cm3 and geometric diameter >5 μm could be used to ease respirability as well as to enhance residence time in the lungs [3]. Indeed, the report of Edwards et al. spurred significant growth in studies on PLGA-based large porous particles for delivery of biopharmaceuticals. However, suboptimal pore size of the microspheres and poor encapsulation efficiency of many drugs remain major impediments to the widespread use of large porous PLGA microspheres as carriers for inhaled long-acting formulations [7]. In order to achieve sustained release properties and increase drug load, attempts have been made to modify particulate carriers by using polymer blends [8], porosity-altering or surface-active agents [9], and by means of the cyclodextrin-mediated double-entrapment technique [10-13].
Over the past few years, the large porous particle technology has been used for a number of biopharmaceuticals and conventional therapeutic agents, including insulin, testosterone, estradiol, deslorelin, tobramycin, ciprofloxacin and para-aminosalicylic acid [3, 13-17]. However, currently it is not known if this technology can be used for pulmonary delivery of hydrophilic macromolecules carrying a negative surface charge. Low molecular weight heparins (LMWHs) are negatively charged oligosaccharides used in the treatment of deep vein thrombosis and pulmonary embolism [18]. Because of their relatively high molecular weight, negative surface charge and short half-lives, LMWHs are administered by subcutaneous injection multiple times a day. Recent studies in our laboratory showed that pulmonary administration of LMWH is feasible [19, 20]. But the proposed formulation of the drug with absorption enhancers and dendrimers failed to produce a prolonged release of the drug. In this study, we propose that long-acting pulmonary formulations of LMWH can address the limitations imposed by the short duration of action of the drug.
Very recently, PLGA-based microspheres and nanoparticles have been used for oral and nasal delivery of unfractionated heparin and LMWH, although not for pulmonary delivery of LMWH. Moreover, the drug load and release profiles of the proposed formulations were not that encouraging [21-24]. We believe that the poor entrapment efficiency of the formulations was because of the excessive hydrophilicity of the drug, and that the poor release profiles were the result of the use of nonporous particulate carriers. Thus, it is reasonable to assume that the entrapment efficiency of the carriers can be increased by complexing the drug with cationic polymers or surfactants, and that a refined controlled release can be achieved by using microspheres with optimal porosity. This study, therefore, tests the hypothesis that core-modified PLGA-based large porous microspheres can enhance the entrapment efficiency of LMWH and facilitate release of the drug for an extended period after pulmonary administration.
Materials and Methods
Materials
Poly(DL-lactide-co-glycolide) (50:50) PLGA (inherent viscosity 0.15-0.25 dl/g; weight average molecular weight = 10.6 kDa) was purchased from Boehringer Ingelheim (Lactel Absorbable Polymers, Pelham, AL). LMWH (average molecular weight and anti-factor Xa activity, 4493 Da and 61 U/mg, respectively) was obtained from Celsus laboratories (Cincinnati, OH). Poly vinyl alcohol (PVA), Span 60 (SP), polyethyleneimine (PEI) and stearylamine (SA) were purchased from Sigma (Sigma-Aldrich Inc., St. Louis, MO).
Preparation of LMWH-Loaded Porous Microspheres
The LMWH-loaded PLGA microspheres were prepared by the water-in-oil-in-water (w/o/w) emulsion and evaporation method [3]. Briefly, an aqueous solution of LMWH (internal aqueous phase, IAP) was first emulsified in 5.0 ml of dichloromethane (organic phase, OP) containing (0.25 g) PLGA polymer by homogenization (Ultra-Turrex T25 basic, IKA, Wilmington, DE) at 15,000 rpm for 3 min. The volume of the IAP was varied from 0.25 to 1 ml to obtain microspheres of varying porosity (Table 1). To prepare core-modified microspheres, three different core modifying agents—Span 60, stearylamine and polyethyleneimine—were added to the IAP. As both Span 60 and stearylamine are hydrophobic in nature, they were dispersed in water by heating at 85°C for 15 min. The concentrations of the core-modifying agents used were 1.25 and 2.5%. The resulting water-in-oil (w/o) emulsion was then poured into 25 ml of 1.0% w/v PVA aqueous solution (external aqueous phase, EAP) and emulsified by homogenization at 8,000 rpm for 5 min. The w/o/w emulsion thus obtained, the secondary emulsion, was stirred overnight at room temperature for evaporation of dichloromethane. The polymeric particles were then washed thrice and lyophilized to get free-flowing powder. A blank microsphere formulation without LMWH was also prepared according to the same procedure. Each batch was prepared in triplicate.
Table 1.
Composition and entrapment efficiency of the microspheric formulations
| Formulations | IAP:OP: EAP (v/v/v) | IAP composition (w/v) | Entrapment efficiency of LMWH (%) |
|---|---|---|---|
| PM-1 | 0.25: 5: 25 | - | 17.87±1.79 |
| PM-2 | 0.5: 5: 25 | - | 16.22±1.32 |
| PM-3 | 1: 5: 25 | - | 9.39±1.21 |
| PM-SP-1 | 0.5: 5: 25 | 1.25% Span 60 | 36.7±2.08 |
| PM-SP-2 | 0.5: 5: 25 | 2.5% Span 60 | 47.34±1.98 |
| PM-SA-1 | 0.5: 5: 25 | 1.25% SA | 40.2±2.77 |
| PM-SA-2 | 0.5: 5: 25 | 2.5% SA | 54.44±2.02 |
| PM-PEI-1 | 0.5: 5: 25 | 1.25% PEI | 43.5±1.32 |
| PM-PEI-2 | 0.5: 5: 25 | 2.5% PEI | 54.82±2.79 |
SP: Span 60; SA: Stearylamine; PEI: Polyethyleneimine
Particle Characterization
Microspheres were characterized for their morphology, size, zeta potential, tapped density, and aerodynamic diameter. The morphology of the formulations was studied under a scanning electron microscope. The samples for SEM were prepared on a conductive, double-sided adhesive tape and then sputter-coated with gold under argon (Emitech K550X, Kent, UK). The microphotographs were taken using a Hitachi S-3400N (Freehold, NJ) scanning electron microscope. The mean volume-based diameter and size distribution of the particles were determined by a Microtrac® S3500 (North Largo, FL) particle size analyzer after dispersing the freeze-dried microspheres in a 0.2% w/v aqueous solution of PVA. The polydispersity indices of all formulations were calculated as the ratio of volume-averaged mean particle size to number-averaged mean particle size [25]. The zeta potential was measured by Zeta potential analyzer V. 3.40 (Brookhaven Instruments Corp. Holtsville, NY) after dispersing the formulations in PBS buffer. The density of the particles was estimated from the tapped density as described previously [4, 12]. An aliquot of 100 mg microspheres was transferred to a 10 (±0.05) ml graduated cylinder and the initial volume was recorded. Tapped density of the particles (ρ) was calculated as the ratio between sample weight (g) and the volume (ml) occupied after 200 tappings. The aerodynamic diameter of the dry-powder formulations was determined in an eight-stage Andersen cascade impactor (Westech Instruments Inc., Marietta, Georgia). The formulations were fired 5-6 times into the cascade impactor at a flow rate of 28.3 L/min. The deposited formulation at each stage was collected by rinsing the impactor and the samples were analyzed for LMWH content. The studies were performed in triplicate. In addition to actual aerodynamic diameter, the theoretical mass mean aerodynamic diameter (MMAD) of the particles was also calculated using the following equation [26]:
Where d is the geometric mean diameter obtained from particle size analysis, ρ is the tapped density, ρ0 is the reference density of 1 g/cm3 and X is the shape factor, which is 1 for a sphere.
Encapsulation Efficiency
The amount of heparin entrapped within microspheres was determined using an azure A colorimetric assay by measuring the amount of unentrapped drug in the external aqueous solution recovered after centrifugation and washing of the microspheres [27]. Typically, aliquots (50 μl) of aqueous samples were reacted with 150 μl of the azure A solution (0.01 mg/ml) and assayed in triplicate at 595 nm. The drug entrapment efficiency was expressed as the percentage of calculated heparin load in the microspheres with the actual amount of heparin added during the preparation.
In vitro Release Experiments
Freeze-dried microspheres (50 mg) were suspended in 10 ml of PBS buffer (pH 7.4) in a flask containing 0.1% Tween 80 and incubated in a water bath at 37±1°C under gentle magnetic stirring (200 rpm). At various time intervals, 200 μl samples were withdrawn and centrifuged for 15 minutes at 16,000×g. The supernatant was removed and assayed for heparin content according to a previously described colorimetric method [27].
In vivo Studies
Male Sprague-Dawley rats (Charles River Laboratories, Charlotte, NC) weighing between 250 and 350 g were used for the in vivo absorption experiments (n=5-6). Absorption was monitored by measuring plasma anti-factor Xa activity (4). Prior to the experiment, the animals were anesthetized by an intramuscular injection of an anesthetic cocktail containing xylazine (20 mg/kg) and ketamine (100 mg/kg). Anesthesia was maintained with additional intramuscular injections of anesthetic mixture as needed throughout the experiments. Freeze-dried microspheres containing LMWH were administered intratracheally (50 U/kg) by using a specially designed dry-powder insufflator for aerosol inhalation in small animals (Penn Century, Philadelphia). The total amount of polymeric formulation administered was about 5 mg per animal carrying a 50 U/kg dose of LMWH. For subcutaneous administration, 50 U/kg LMWH in normal saline was administered as a single 100 μl injection under the back skin. Blood samples were collected from the tip of the tail at 0, 0.5, 1, 2, 4, 6, 8, 12 and 24 h in citrated microcentrifuge tubes and placed on ice. Subsequently, the plasma was separated by centrifugation (1600×g for 5 min) and stored at −20 °C until further analysis. All animal studies were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals under an approved protocol (AM-02004).
Anti-factor Xa activity Assay
Anti-factor Xa activity present in blood samples was determined by colorimetric assay using a Chromogenix Coatest Heparin Kit® (Diapharma Group Inc., West Chester, OH). The assay was performed according to the protocol supplied by the manufacturer. Standard curves for determination of anti-factor Xa activity in plasma or saline were prepared by diluting the drug with pooled plasma or saline. Plasma obtained from untreated rats was used as a negative control to account for the effect of rat endogenous anti-factor Xa that otherwise could give a false positive increase in anti-factor Xa activity.
Cytotoxicity Studies
For the cytotoxicity studies, Calu-3 cells were seeded at a density of 50,000 cells/well in flat-bottom, 96-well micro-titer tissue culture plates. Immediately prior to the start of the experiment, the medium was removed from the wells and the cells were washed with normal saline. Subsequently, the cells were incubated with 20 μl of formulations for 4 h, separately. The test samples contained 0.25 mg/ml, 0.025 mg/ml or 0.0025 mg/ml of plain core-modifying surfactants or microspheres containing equivalent amounts of core-modifying agents. Drug-loaded or blank microspheres with no core-modifying agents were also tested for cytotoxicity. Sodium dodecyl sulfate (SDS) was used at 0.1% as a positive cytotoxic control. Cell viability was measured by the MTT assay as previously described [28]. Briefly, after 4 h, MTT (5 mg/ml) solution was added to each well and the cells were incubated at 37°C for 4 h. Next, the solution in each well was removed and acidified isopropyl alcohol (100 μl of 1% v/v, concentrated hydrochloric acid in isopropyl alcohol) was added. Finally, the plates were incubated at 37°C for 1 h and absorbance was measured on a microtiter-plate reader (TECAN U.S. Inc, Research Triangle Park, NC) at 570 nm. Each assay was performed on eight samples and cell viability was expressed as the percentage of MTT released by cells exposed to core-modifying agents and microspheric formulation or SDS compared to cells incubated with saline alone.
Data Analysis
Standard noncompartmental analysis (Kinetica®, Version 4.0, Innaphase Corp., Philadelphia, PA) was performed to calculate the area under the plasma concentration versus time curve (AUC0→24h), relative and absolute bioavailability. Pharmacokinetic parameters of different formulations were compared by ANOVA. When the differences in the means were significant, post-hoc pair wise comparisons were conducted using Newman–Keuls multiple comparison (GraphPad Prism, version 3.03, GraphPad Software, San Diego, CA). Differences in p-values less than 0.05 were considered statistically significant.
Results and Discussion
Particle Characterization
In this set of experiments we have used scanning electron microscopy to investigate how particle morphology was affected by varying the fraction of the IAP volume and by the presence of additives in the internal phase (Fig. 1 and Table 1). The photomicrographs of unmodified (in absence of core modifying agents) microspheres were spherical in shape and the pore size of the particles increased with increasing volume of the IAP from 0.25 ml to 1 ml. Also, empty and fragmented cores were observed when the IAP volume was ≥1 ml (data not shown). This observation agrees with previously published studies showing that polymeric microspheres prepared by the emulsion solvent-evaporation technique become porous owing to the presence of the IAP and that the degree of porosity increases as the fraction of the aqueous phase volume increases [29].
Figure 1.
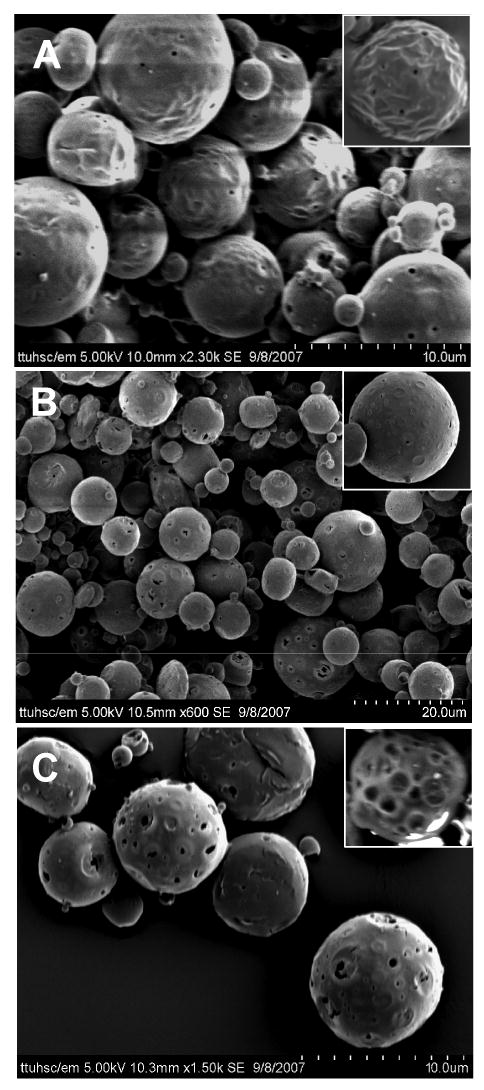
SEM of LMWH-loaded PLGA microspheres (A) PM-SP-2, (B) PM-SA-2 and (C) PM-PEI-2. See Table 1 for composition of the different formulations.
Incorporation of Span 60, stearylamine or PEI into the IAP affected surface morphology, porosity and particle–particle interaction of the microspheres. Span 60 and stearylamine produced particles with rough surfaces containing sporadic pores (Figs. 1A and 1B). Particles with sporadic pores may result from the tendency of Span 60 and stearylamine to migrate to the oil-water interface and the polymeric phase due to their lipophilic character. However, no significant differences were observed between the morphologies of the Span 60- and stearylamine-modified particles despite the two surfactants being electrostatically different. Uniform sized particles with rough surfaces have previously been observed when neutral or cationic surfactants were incorporated in the internal aqueous or OP of the primary emulsion [10, 11, 30]. The rough surface of the particles was caused by shrinkage of the polymer as water was being removed during drying. Furthermore, the morphology of particles prepared using either of the surfactants was very similar, perhaps because the hydrophilic–lipophilic balance (HLB) values for two surfactants are very close—the HLB of Span 60 is 4.7 and that for stearylamine is 7.9 [31].
When PEI, a polycation, was present in the IAP, the microspheres were more porous compared to those formulated without PEI, i.e., PM-2 (Fig. 1C). A similar concentration-dependent increase in porosity and pore diameter was observed when PEI was complexed with oligonucleotides in the IAP to prepare PLGA-based microspheres of the complex [32]. LMWH, like oligonucleotides, is a negatively charged molecule, so that PEI formed an electrostatic complex with LMWH, helping to retain more of the drug in the microspheres. As a result, the space occupied by the dispersed drug may have increased along with the amount of bound water. Alternatively, differences between the osmotic pressure of the internal and external aqueous phases may have played a role in the increase in porosity. In fact, it has been reported that increased osmotic pressure in the aqueous phase renders the particles more porous [9, 33]. As PEI is an osmotically active polycation, its presence in the IAP may lead to an increased influx of water, resulting in larger aqueous droplets during emulsification. Removal of water during lyophilization leaves void space in the particles, making them more porous and with relatively larger-diameter pores.
Particle size, Zeta Potential, Tapped Density and Aerodynamic Diameter
Aerodynamic diameter and particle density play an important role in successful deposition of inhaled particles in the lungs. As discussed in the Introduction, for traditional dry-powder inhalers, a geometric diameter of 1-5 μm is required for efficient deposition of particles in the respiratory tract. Recently, it has been proposed that lighter particles with a mass density <0.4 g/ml and geometric diameter >5 μm could be used to enhance the residence time in the lungs and provide a prolonged release effect. To investigate if the particles meet the above criteria for pulmonary delivery, the volume-based diameter, tapped density, and aerodynamic diameter of the microspheres were determined (Figs. 2 and 3). The data presented in Fig. 2A show that the particles were moderately polydispersed, with a polydispersity index ranging from 1.58 to 2.31. The mean volume-based diameter of the microspheres suggests that the size of the microspheres increases as the aqueous phase volume fraction increases; the highest size increase was observed with an IAP to OP ratio of 1:5. At a ratio of 2:1 a significant increase in the size of the microspheres occurred due to the formation of a relatively unstable secondary emulsion, as observed in the SEM photographs. Incorporation of Span 60 in the IAP did not affect the particle size of the microspheres compared to those without Span 60. When PEI or stearylamine was incorporated in the aqueous phase, an increase in particle size was observed, although no further increase was observed when the concentration of the agents was increased from 1.25% to 2.5% (Fig. 2A). The basis for the increase in particle size could be that these agents may leak out to the external aqueous phase and remain on the surface of the negatively charged PLGA. Such a relocation of cationic core-modifying agents onto the surface of microspheres can increase the particle–particle interactions during the secondary emulsification and this may lead to an increase in particle size. Furthermore, complexation of PEI or stearylamine with the negatively charged drug may have contributed to the increase in size of the particles.
Figure 2.

Volume-based mean diameter (A), the data in parentheses show the polydispersity index, PI, and Zeta potential (B) of LMWH-loaded PLGA microspheres. Data represent mean ± standard error of the mean (n=3).
Figure 3.

Tapped density (A) and Theoretical (MMADt)/Actual mass median aerodynamic (B) diameter (MMADa) of LMWH-loaded PLGA microspheres. Data represent mean ± standard error of the mean (n=3).
In addition to particle size, zeta potential and tapped density of the particles were also determined. The tapped density provides important information about the flowability of the particles from the inhaler device, the porosity of the particles, the particle size distribution, the true density, and interparticulate forces—cohesive and adhesive. The tapped density is also affected by surface properties such as wettability and surface charge. The data presented in Fig. 2B show that plain microspheres were negatively charged but the particles became positively charged upon addition of cationic core-modifying agents, suggesting that the negative surface charge of the drug and PLGA polymer was partially neutralized by the cationic core-modifying agents. The tapped densities decreased with the increase in the internal aqueous volume fraction (Fig. 3A). When the internal aqueous volume was increased from 0.25 ml to 1 ml, a twofold decrease in the tapped density was observed, suggesting that the particles became lighter because of the increase in porosity of the microspheres. These data agree with the porous nature of the microspheres as observed in SEM photomicrographs. However, when Span 60 was used as a core-modifying agent, a reduction in tapped density was observed compared to plain microspheres and the magnitude of reduction was dependent on the concentration of Span 60 used. The presence of Span 60 may have made the particles more hydrophobic and increased the interparticle repulsive forces. As a result, the interparticular distances increased and the particles became fluffier and lighter compared to the plain microspheres. However, when stearylamine or PEI was added to the aqueous internal core, an increase in the density was observed compared to microspheres without stearylamine or PEI, and the tapped density increased slightly with the increase in concentration of the additives. No difference between the tapped densities of the PEI- and stearylamine-based microspheres was observed. Increased tapped density suggests a compact and dense internal structure of the particles. The increase in bulk density may be the result of reduced interparticular distances because of the hydrophilic surface of the microspheres conferred by PEI or stearylamine. It is also likely that the presence of cationic core-modifying agents rendered the microparticles' internal environment hydrophilic, causing the particles to retain residual water and making them heavier than the plain microspheres, as observed by Pistel and Kissel [33].
The mass median diameter (MMAD), a parameter important for the deposition and distribution of particles in the respiratory tract, was determined. The theoretical mass median diameter (MMADt) was determined from the volume-based diameter and tapped density, and the actual mass median diameter (MMADa) was calculated using an eight-stage Anderson cascade impactor, which is a widely used method to characterize the aerodynamic particle size distribution emitted from therapeutic inhalation devices [34]. In terms of size distribution, the MMADt and MMADa data presented in Fig 3B show a similar pattern. For example, for plain microspheres, an increase in MMADt and MMADa was observed when the internal aqueous phase volume was increased. Similarly, for core-modified microspheres, the MMADa of PM-PEI-2 was greater than those of the other two core-modified microspheres. Importantly the MMADt and MMADa of the formulations were <6 μm, which is below the maximum value for aerosolized particles for efficient deposition in the lungs, as described by Edwards et al. and Van Campen and Venthoye [35]. On the whole, the data presented in Figs. 2 and 3 suggest that the presence of additives in the IAP plays an important role in modifying the properties of particles. Moreover, the aerodynamic diameter of the particles was determined to be within the respirable range, indicating their suitability for administering as a dry powder into the respiratory tract.
Entrapment Efficiency
To prepare microspheres containing a single dose of the drug that can be administered in a single insufflation to the lungs, 50 mg LMWH was encapsulated in 250 mg of PLGA microspheres. As indicated above, a fixed amount of the drug was dissolved in varying aqueous phase volume fractions in order to obtain microspheres of different porosity. The data in Table 1 suggest that entrapment efficiency decreases with increasing water volume in the internal phase. When the IAP volume was increased from 0.25 ml to 1 ml, the entrapment efficiency was reduced from 17.87±1.79% to 9.39±1.21%. However, the entrapment efficiency of the formulations containing 0.5 ml of internal phase was close to that obtained with 0.25 ml of internal phase. The reduction in entrapment efficiency observed with the increase in IAP likely occurred because of the formation of particles with larger pores, allowing the encapsulated drug to leach out through the pores during the washing process. The large amount of IAP can also lead to destabilization of the primary emulsion and compromise the stability of the final particles. In fact, because of such instability of the primary emulsion, a large number of empty cores were observed in microspheres prepared with 1 ml of IAP. To enhance the entrapment efficiency and achieve extended-release properties, the drug was complexed with a polycation, PEI, or cationic surfactant, stearylamine, in the IAP. The effect of cationic agents on increasing the entrapment efficiency of anionic LMWH was further compared with the effect of the nonionic surfactant, Span 60. For the preparation of core-modified microspheres, we chose to use 0.5 ml IAP because plain microspheres prepared with this amount of internal phase volume showed an optimal in vitro release pattern. Incorporation of each of the three additives into the internal phase significantly increased the entrapment of LMWH in the microspheres (Table 1). When Span 60, stearylamine or PEI was used at a concentration of 1.25% or 2.5%, the LMWH entrapment efficiency was increased compared to that of particles prepared without any of these additives. The increase in entrapment efficiency was dependent on the concentration of the additives used in the internal phase. For example, when 1.25% PEI was used, a threefold increase in LMWH entrapment efficiency was observed (43.5±1.32%) compared to particles prepared in the absence of PEI (16.22±1.32%). When the concentration of PEI was increased to 2.5% of the IAP, a further increase in entrapment efficiency was observed.
The higher entrapment efficiency of LMWH in the microspheres was because of the enhanced stability of the primary emulsion due to the presence of any one of the three core-modifying agents: Span 60, stearylamine or PEI. However, as these additives are chemically different, they may have increased the stability of the primary emulsion and the entrapment efficiency by different mechanisms. Indeed, it has been previously shown that a critical parameter for high drug-entrapment efficiency in the final particle is the homogeneous dispersion of the aqueous phase in the OP of the emulsion [36]. The entrapment efficiency in the stearylamine-based formulation was higher compared to the Span 60-based formulation because stearylamine acts as both a complexing and emulsifying agent. Stearylamine's positive surface charge may have helped the drug to complex with the surfactant and increase the total amount of the drug in the emulsion. Furthermore, the increase in entrapment efficiency was the highest with PEI, perhaps because a different mechanism is involved. It is hypothesized that positively charged PEI forms an electrostatic complex with the negatively charged LMWH. This hypothesis agrees with a previous study that showed greater entrapment efficiency of heparin in PLGA microspheres prepared with Eudragit®, a quaternary ammonium group-carrying polymer, as compared to microspheres without Eudragit® [22]. We believe that PEI-LMWH complex formed in the IAP becomes relatively hydrophobic compared to the drug alone. The resulting complex may migrate to the water–oil interface and facilitate the formation of a stable primary emulsion. A similar increase in entrapment efficiency was observed when PEI was complexed with oligonucleotides in the internal phase. Overall, the ability of the three agents to increase the entrapment efficiency can be ranked as PEI > stearylamine > Span 60.
In vitro Release Profiles
The amount of heparin released from all formulations was estimated and the release kinetics calculated. The amount of LMWH released at the zero time point was considered as surface-associated drug, and that released during the first 30 min was considered as the initial burst release. The in vitro release profile presented in Fig. 4 and Table 2 suggests a biphasic release pattern for all formulations: an initial burst release followed by near zero-order release kinetics. The phenomenon of burst release occurs because of the release of heparin adsorbed on the surface of the particles and development of a high concentration gradient across the particle surface. For plain PLGA microspheres containing no core-modifying agents, the amount of drug released during the initial burst phase was related to the porosity of the particles. As the porosity of the formulations increased, the amount of drug released also increased (Fig. 4A). As such, the amounts of drug released from formulations prepared with 0.25, 0.5 and 1 ml IAP were 3.33±0.69, 5.79±0.75, and 10.36±1.21%, respectively (Table 2). Because of the presence of larger pores in the latter two formulations (PM-2 and PM-3), a larger amount of medium is likely to come in contact with the particles and facilitate the release of the drug from the microspheres.
Figure 4.

In vitro performances of the formulations. Data represent mean ± standard error of the mean. (A) Release profiles of LMWH from plain microspheres (n=3) (B) Release profiles of LMWH from core-modified particles (n=3).
Table 2.
The microsphere surface-associated LMWH, initial burst effect and release rate of LMWH
| Formulation | Surface-associated LMWH (%± SEM) | Burst Release (%± SEM) | Release constant (%± SEM) |
|---|---|---|---|
| PM-1 | 2.0±0.5 | 3.33±0.69 | 0.259±0.012 |
| PM-2 | 4.6±0.6 | 5.79±0.75 | 0.232±0.018 |
| PM-3 | 8.2±1.2 | 10.36±1.21 | 0.102±0.01 |
| PM-SP-2 | 7.5±1.4 | 10.3±1.23 | 0.687±0.021 |
| PM-SA-2 | 13.1±1.5 | 21.3±1.85 | 1.121±0.036 |
| PM-PEI-2 | 17.7±2.1 | 25.01±1.62 | 1.247±0.030 |
SEM: Standard error of the mean
For core-modified microspheres, the amount of drug released in the initial burst phase was dependent on both the entrapment efficiency and the amount of drug associated with the surface of the particles. Because more drug was available in the core-modified microspheres, more drug was released. For example, because the entrapment efficiency of the PEI-based formulation was greater than that of the Span 60-modified microspheres, the amount of drug released both at the initial burst and during the constant-release phase was also higher for the PEI-modified microspheres (Fig. 4B). In addition to the entrapment efficiency, the amount of drug released was also affected by the core-modifying agent used in the IAP. The presence of either of the surfactants, Span 60 or stearylamine, produced an increase in the amount of drug released during the initial and burst phases. However, the amount of drug released from the Span-modified microspheres was higher than for the stearylamine-modified microspheres (P<0.05) (Fig. 4B). These slight differences could be because stearylamine is more hydrophilic than Span 60, as suggested by their HLB values [37]. In agreement with these data, it was previously shown that surfactants can affect the release of entrapped heparin from PLGA microspheres [23]. Span 40 was shown to decrease the release of heparin from PLGA particles; however, both Span 80 and 85 have been shown to increase the release considerably [23]. Similarly, for PEI, which is more hydrophilic than either Span 60 or stearylamine, the rate and extent of release of the drug from PEI-based formulations was higher than that from the two other formulations. Hydrophilic PEI facilitates interaction of the aqueous medium with PLGA, which results in faster release, as observed by De Rosa et al. [32, 38]. Furthermore, an excess of PEI-LMWH complex may have adsorbed onto the surface of the microspheres and been released soon after the particles came in contact with water.
Overall, in terms of the amount of drug released in the burst phase, the formulations can be ranked in the following order: PM-PEI-2 > PM-SA-2 > PM-SP-2. Furthermore, the rate of release was independent of the entrapment efficiency, suggesting a constant rate of release after the burst phase (Table 2). The data also suggest that core-modified microspheres can release LMWH for 24 h, and that the amount of drug released during 24 h is greater than that released from previously reported PLGA microspheres of LMWH [21, 23, 24]. A larger amount of drug was released from this formulation because the PLGA used was of low molecular weight, and low molecular weight PLGA undergoes faster degradation in dissolution medium at 37±1°C due to its relatively low glass transition temperature of 28-37°C [39]. However, caution should be exercised in correlating the in vitro release data with the release of the drug in the lungs because the composition of the medium used for in vitro release studies is significantly different from that of the lung fluid.
Pulmonary Absorption of LMWH Entrapped in Microspheres
Pulmonary absorption of the encapsulated drug was studied in a rodent model and the amount of drug absorbed was monitored by measuring plasma anti-factor Xa activity. To confirm that pulmonary administration of blank PLGA microspheres has no effect on anti-factor Xa activity, microspheres without LMWH was administered and anti-factor Xa levels were measured. No change in anti-factor Xa levels was observed, indicating that PLGA had no effect on endogenous anti-factor Xa levels (data not shown). When plain LMWH was administered via the pulmonary route, a rapid onset of absorption was observed, with a maximum increase in blood anti-factor Xa activity (Cmax = 0.20±0.01 U/ml) at 2.2±0.44 h (Tmax) and a drug half-life of 4.28±0.31 h (Table 3). Administration of plain PLGA microspheres containing an equivalent dose of the drug resulted in a considerable prolongation of anti-factor Xa activity (T1/2 = 12.13±0.93 h) in the blood, although the maximum increase in anti-factor Xa activity was below the level required for an anti-thrombotic effect in a rodent model [40].
Table 3.
Pharmacokinetic parameters of LMWH-loaded PLGA microspheric formulations. Data represent mean ± SEM (n=5-6)
| Formulations | Pharmacokinetic parameters | ||||||
|---|---|---|---|---|---|---|---|
| Cmax (U/ml) | Tmax (h) | AUC0-24 (U.h/ml) | AUMC0-24 | T1/2 (h) | MRT (h) | Frelative (%) | |
| Plain LMWH Subcutaneous | 0.33±0.03 | 4.4±0.31 | 3.98±0.22 | 36.6±1.72 | 4.35±0.33 | 7.79±0.60 | ---- |
| Plain LMWH pulmonary | 0.20±0.01 | 2.2±0.44 | 1.58±0.12 | 9.63±0.77 | 4.28±0.31 | 6.63±0.47 | 39.7±3.0 |
| PM-2 | 0.07±0.04 | 0.8±0.4 | 1.05±0.11 | 10.57±2.04 | 12.13±0.93 | 18.43±1.42 | 26.38±2.8 |
| PM-SP-2 | 0.13±0.03 | 1.7±0.3 | 1.98±0.29 | 21.22±1.86 | 23.75±2.97 | 34.33±4.29 | 49.74±7.31 |
| PM-SA-2 | 0.20±0.04 | 6.9±0.89 | 3.64±0.36 | 43.14±3.7 | 25.28±2.81 | 39.09±4.34 | 91.45±9.01 |
| PM-PEI-2 | 0.21±0.03 | 7.6±0.70 | 3.37±0.40 | 40.8±2.38 | 20.13±1.68 | 32.39±2.70 | 84.67±10.11 |
SEM: Standard error of the mean
In agreement with in vitro release data presented in Figs. 4A and 4B, the levels of anti-factor Xa produced by the core-modified microspheres and the time required to achieve the Cmax was dependent on the entrapment efficiency of the formulations (Fig. 5A). The Span 60-based formulation, PM-SP-2, showed an initial increase in anti-factor Xa activity in the blood and produced a maximum increase (Cmax = 0.13±0.03 U/ml) at 1.7±0.3 h). Although the Cmax produced by this formulation was lower than that produced by plain pulmonary LMWH, the blood concentration of anti-factor Xa activity was at a steady state for 24 h. The stearylamine- and PEI-based microspheres also showed a slower absorption phase compared to plain LMWH. However, the anti-factor Xa levels produced by these two formulations were similar to that produced by plain LMWH administered by the pulmonary route. For both formulations, the time required to achieve maximum anti-factor Xa levels was longer than the Tmax for plain LWMH. The stearylamine-based formulation reached its Cmax (0.20±0.04 U/ml) at 6.9±0.89 h, and the PEI-based formulation reached its Cmax (0.21±0.03 U/ml) at 7.6±0.7 h. The elimination phase of both formulations was longer than those for subcutaneous and pulmonary administration of plain LMWH. Furthermore, a high concentration of anti-factor Xa in the blood was observed for 24 h, underscoring the prolonged release properties of the formulations. The data presented in Table 3 also clearly demonstrate that the half-life of the drug released from the core-modified microspheres was increased by 5- to 6-fold compared to subcutaneous LMWH. Similar to the half-life, the relative bioavailabilities of the stearylamine- and PEI-based formulations were increased by 2-fold compared to plain LMWH administered by the pulmonary route.
Figure 5.

(A) In vivo performances of the formulations. Changes in plasma anti-factor Xa activity after pulmonary administration of LMWH (50 U/kg) in plain and core-modified PLGA microspheres (n=5-6). (B) Effects of plain core-modifying agents and microspheres containing core-modifying agents on the viability of Calu-3 cells. The test samples contained 0.25 mg/ml, 0.025 mg/ml or 0.0025 mg/ml of plain core-modifying surfactants or microspheres containing an equivalent amount of core-modifying agents. Data represent the mean ± standard error of the mean (n=8).
Several factors may have contributed to the increase in half-life and bioavailability of the formulations. First, slower release of the drug from the formulations should increase the drug's half-life, and a higher magnitude of drug release should enhance its bioavailability. Second, prolongation of the drug's effect can be explained by the fact that the particles were too big to be phagocytosed by alveolar macrophages [3] so that the drug remained in the lung for a longer period of time. In addition to particle size, the rate of release of a drug from the microspheres may also depend on the molecular weight of the PLGA used and the porosity of the microspheres. As discussed above, the biodegradability of PLGA microspheres depends on the molecular weight and glass transition temperature of the polymer. The PLGA used in this formulation has a glass transition temperature of 28-37°C, and hence can easily degrade at physiological temperature and release the drug from the formulations for absorption via the respiratory epithelium [39]. However, the fate of PLGA after release of the drug is unknown. It is likely that some residue of PLGA remains in the lung after the drug has been released. Further investigations are required to determine the fate of PLGA after pulmonary ingestion. The porosity of the microspheres also plays a major role in enhancing the rate of biodegradation by exposing more surface area of the polymer matrix to the lung fluid [41]. The presence of hydrophilic molecules such as PEI and surfactant may also facilitate degradation of polymers by increasing the wettability and reducing the contact angle of the polymer with the physiological fluid, as suggested previously [42, 43]. Another important factor that may contribute to the enhanced bioavailability is the property of the core-modifying agent to increase the permeability of the respiratory epithelium. It is hypothesized that core-modifying agents used to prepare microspheres may separate out from the microspheres and act as absorption promoters [37].
MTT Cytotoxicity Study
To evaluate the cytotoxicity of the core-modifying agents to Calu-3 cells, the viability of the cells was assessed with the MTT assay. MTT, a tetrazolium salt, is cleaved by mitochondrial dehydrogenase in living cells to form a measurable, dark blue product called formazan. Damaged or dead cells have reduced dehydrogenase activity and diminished formazan production. Results of the MTT test showed high levels of viability of Calu-3 cells after incubation with saline; in contrast, only 21% of Calu-3 cells were viable following treatment with 0.1% SDS (p<0.05) (Fig. 5B). A concentration-dependent increase in cytotoxicity was observed for both plain core-modifying agents and microspheres containing core-modifying agents. Of the three core-modifying agents tested, the effect of PEI in cell death was more pronounced than that of the other two agents. However, when PEI was used as a core-modifying agent to prepare LMWH-entrapped microspheres, a reduction in cytotoxicity was observed compared to plain PEI. This reduced cytotoxicity was because of partial neutralization of the positive surface charge of PEI in the presence of negatively charged LMWH. Overall, the cytotoxicity produced by all core-modified microspheres was comparable to that produced by blank or plain LMWH-loaded microspheres and that of the saline control.
Taken together, the entrapment efficiency of LMWH in PLGA microspheres can be increased by incorporating Span 60, stearylamine and PEI in the IAP. In terms of particle size, porosity, aerodynamic diameter, in vitro release behavior and cytotoxicity, the core-modified microspheres were optimal for pulmonary administration. The drug was released, both in vitro and in vivo, for 24 h and the maximum increase in anti-factor Xa levels was observed for the PEI- and stearylamine-modified microspheres. The half-life of the drug from the core-modified microspheres was considerably higher than that of plain LMWH administered via the pulmonary route. The data presented here demonstrate that PLGA-based large porous microspheres could be a viable option for delivering LMWH via the pulmonary route for a prolonged release effect.
Acknowledgments
This work was supported by NIH grant R 15 HL7713302 (FA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lenzer J. Inhaled insulin is approved in Europe and United States. Bmj. 2006;332:321. doi: 10.1136/bmj.332.7537.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agu RU, Ugwoke MI, Armand M, Kinget R, Verbeke N. The lung as a route for systemic delivery of therapeutic proteins and peptides. Respir Res. 2001;2:198–209. doi: 10.1186/rr58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Edwards DA, Hanes J, Caponetti G, Hrkach J, Ben-Jebria A, Eskew ML, Mintzes J, Deaver D, Lotan N, Langer R. Large porous particles for pulmonary drug delivery. Science. 1997;276:1868–1871. doi: 10.1126/science.276.5320.1868. [DOI] [PubMed] [Google Scholar]
- 4.Vanbever R, Mintzes JD, Wang J, Nice J, Chen D, Batycky R, Langer R, Edwards DA. Formulation and physical characterization of large porous particles for inhalation. Pharm Res. 1999;16:1735–1742. doi: 10.1023/a:1018910200420. [DOI] [PubMed] [Google Scholar]
- 5.Bell J, Newman S. The rejuvenated pressurised metered dose inhaler. Expert Opin Drug Deliv. 2007;4:215–234. doi: 10.1517/17425247.4.3.215. [DOI] [PubMed] [Google Scholar]
- 6.Heyder J, Gebhart J, Rudolph G, Schiller CF, Stahlhofen W. Deposition of particles in the human respiratory tract in the size range 0.005–15 microns. J Aerosol Sci. 1986;17:811–825. [Google Scholar]
- 7.Klose D, Siepmann F, Elkharraz K, Krenzlin S, Siepmann J. How porosity and size affect the drug release mechanisms from PLGA-based microparticles. Int J Pharm. 2006;314:198–206. doi: 10.1016/j.ijpharm.2005.07.031. [DOI] [PubMed] [Google Scholar]
- 8.Ravivarapu HB, Burton K, DeLuca PP. Polymer and microsphere blending to alter the release of a peptide from PLGA microspheres. Eur J Pharm Biopharm. 2000;50:263–270. doi: 10.1016/s0939-6411(00)00099-0. [DOI] [PubMed] [Google Scholar]
- 9.Ravivarapu HB, Lee H, DeLuca PP. Enhancing initial release of peptide from poly(d,l-lactide-co-glycolide) (PLGA) microspheres by addition of a porosigen and increasing drug load. Pharm Dev Technol. 2000;5:287–296. doi: 10.1081/pdt-100100543. [DOI] [PubMed] [Google Scholar]
- 10.Rosa GD, Iommelli R, La Rotonda MI, Miro A, Quaglia F. Influence of the co-encapsulation of different non-ionic surfactants on the properties of PLGA insulin-loaded microspheres. J Control Release. 2000;69:283–295. doi: 10.1016/s0168-3659(00)00315-1. [DOI] [PubMed] [Google Scholar]
- 11.Quaglia F, De Rosa G, Granata E, Ungaro F, Fattal E, La Rotonda M Immacolata. Feeding liquid, non-ionic surfactant and cyclodextrin affect the properties of insulin-loaded poly(lactide-co-glycolide) microspheres prepared by spray-drying. J Control Release. 2003;86:267–278. doi: 10.1016/s0168-3659(02)00414-5. [DOI] [PubMed] [Google Scholar]
- 12.Ungaro F, De Rosa G, Miro A, Quaglia F, La Rotonda MI. Cyclodextrins in the production of large porous particles: development of dry powders for the sustained release of insulin to the lungs. Eur J Pharm Sci. 2006;28:423–432. doi: 10.1016/j.ejps.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 13.Koushik K, Dhanda DS, Cheruvu NP, Kompella UB. Pulmonary delivery of deslorelin: large-porous PLGA particles and HPbetaCD complexes. Pharm Res. 2004;21:1119–1126. doi: 10.1023/b:pham.0000032997.96823.88. [DOI] [PubMed] [Google Scholar]
- 14.Wang J, Ben-Jebria A, Edwards DA. Inhalation of estradiol for sustained systemic delivery. J Aerosol Med. 1999;12:27–36. doi: 10.1089/jam.1999.12.27. [DOI] [PubMed] [Google Scholar]
- 15.Newhouse MT, Hirst PH, Duddu SP, Walter YH, Tarara TE, Clark AR, Weers JG. Inhalation of a dry powder tobramycin PulmoSphere formulation in healthy volunteers. Chest. 2003;124:360–366. doi: 10.1378/chest.124.1.360. [DOI] [PubMed] [Google Scholar]
- 16.Tsapis N, Bennett D, O'Driscoll K, Shea K, Lipp MM, Fu K, Clarke RW, Deaver D, Yamins D, Wright J, Peloquin CA, Weitz DA, Edwards DA. Direct lung delivery of para-aminosalicylic acid by aerosol particles. Tuberculosis (Edinb) 2003;83:379–385. doi: 10.1016/j.tube.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 17.Arnold MM, Gorman EM, Schieber LJ, Munson EJ, Berkland C. NanoCipro encapsulation in monodisperse large porous PLGA microparticles. J Control Release. 2007;121:100–109. doi: 10.1016/j.jconrel.2007.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim SK, Lee DY, Kim CY, Nam JH, Moon HT, Byun Y. A newly developed oral heparin derivative for deep vein thrombosis: non-human primate study. J Control Release. 2007;123:155–163. doi: 10.1016/j.jconrel.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 19.Yang T, Mustafa F, Bai S, Ahsan F. Pulmonary delivery of low molecular weight heparins. Pharm Res. 2004;21:2009–2016. doi: 10.1023/b:pham.0000048191.69098.d6. [DOI] [PubMed] [Google Scholar]
- 20.Bai S, Thomas C, Ahsan F. Dendrimers as a carrier for pulmonary delivery of enoxaparin, a low-molecular weight heparin. J Pharm Sci. 2007;96:2090–2106. doi: 10.1002/jps.20849. [DOI] [PubMed] [Google Scholar]
- 21.Hoffart V, Lamprecht A, Maincent P, Lecompte T, Vigneron C, Ubrich N. Oral bioavailability of a low molecular weight heparin using a polymeric delivery system. J Control Release. 2006;113:38–42. doi: 10.1016/j.jconrel.2006.03.020. [DOI] [PubMed] [Google Scholar]
- 22.Hoffart V, Ubrich N, Lamprecht A, Bachelier K, Vigneron C, Lecompte T, Hoffman M, Maincent P. Microencapsulation of low molecular weight heparin into polymeric particles designed with biodegradable and nonbiodegradable polycationic polymers. Drug Deliv. 2003;10:1–7. doi: 10.1080/713840325. [DOI] [PubMed] [Google Scholar]
- 23.Kreitz MR, Domm JA, Mathiowitz E. Controlled delivery of therapeutics from microporous membranes. II. In vitro degradation and release of heparin-loaded poly(D,L-lactide-co-glycolide) Biomaterials. 1997;18:1645–1651. doi: 10.1016/s0142-9612(97)00106-3. [DOI] [PubMed] [Google Scholar]
- 24.Jiao YY, Ubrich N, Marchand-Arvier M, Vigneron C, Hoffman M, Maincent P. Preparation and in vitro evaluation of heparin-loaded polymeric nanoparticles. Drug Deliv. 2001;8:135–141. doi: 10.1080/107175401316906892. [DOI] [PubMed] [Google Scholar]
- 25.Cui C, Schwendeman SP. One-step surface modification of poly(lactide-co-glycolide) microparticles with heparin. Pharm Res. 2007;24:2381–2393. doi: 10.1007/s11095-007-9378-1. [DOI] [PubMed] [Google Scholar]
- 26.Gonda I. Physico-chemical principles in aerosol delivery. In: Crommelin DJA, Midha KK, editors. Topics in Pharmaceutical Sciences. Medpharm GmbH Scientific Publisher; Stuttgart: 1991. pp. 95–115. [Google Scholar]
- 27.Cadene M, Boudier C, de Marcillac GD, Bieth JG. Influence of low molecular mass heparin on the kinetics of neutrophil elastase inhibition by mucus proteinase inhibitor. J Biol Chem. 1995;270:13204–13209. doi: 10.1074/jbc.270.22.13204. [DOI] [PubMed] [Google Scholar]
- 28.Scudiero DA, Shoemaker RH, Paull KD, Monks A, Tierney S, Nofziger TH, Currens MJ, Seniff D, Boyd MR. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer Res. 1988;48:4827–4833. [PubMed] [Google Scholar]
- 29.Crotts G, Park TG. Preparation of porous and nonporous biodegradable polymeric hollow microspheres. J Control Release. 1995;35:91–105. [Google Scholar]
- 30.Kusonwiriyawong C, Atuah K, Alpar OH, Merkle HP, Walter E. Cationic stearylamine-containing biodegradable microparticles for DNA delivery. J Microencapsul. 2004;21:25–36. doi: 10.1080/02652040410001653777. [DOI] [PubMed] [Google Scholar]
- 31.Ham HT, Choi YS, Chung IJ. An explanation of dispersion states of single-walled carbon nanotubes in solvents and aqueous surfactant solutions using solubility parameters. J Colloid Interface Sci. 2005;286:216–223. doi: 10.1016/j.jcis.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 32.De Rosa G, Quaglia F, La Rotonda MI, Appel M, Alphandary H, Fattal E. Poly(lactide-co-glycolide) microspheres for the controlled release of oligonucleotide/polyethylenimine complexes. J Pharm Sci. 2002;91:790–799. doi: 10.1002/jps.10063. [DOI] [PubMed] [Google Scholar]
- 33.Pistel KF, Kissel T. Effects of salt addition on the microencapsulation of proteins using W/O/W double emulsion technique. J Microencapsul. 2000;17:467–483. doi: 10.1080/026520400405723. [DOI] [PubMed] [Google Scholar]
- 34.Mitchell JP, Nagel NM. Cascade impactors for the size characterization of aerosols from medical inhalers: their uses and limitations. J Aerosol Med. 2004;16:341–377. doi: 10.1089/089426803772455622. [DOI] [PubMed] [Google Scholar]
- 35.Van Campen L, Venthoye G. Inhalation, Dry Powder. Marcel Dekker Inc.; New York: 2002. [Google Scholar]
- 36.Nihant N, Schugens C, Grandfils C, Jerome R, Teyssie P. Polylactide microparticles prepared by double emulsion/evaporation technique. I. Effect of primary emulsion stability. Pharm Res. 1994;11:1479–1484. doi: 10.1023/a:1018912426983. [DOI] [PubMed] [Google Scholar]
- 37.Yang T, Hussain A, Bai S, Khalil IA, Harashima H, Ahsan F. Positively charged polyethylenimines enhance nasal absorption of the negatively charged drug, low molecular weight heparin. J Control Release. 2006;115:289–297. doi: 10.1016/j.jconrel.2006.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Rosa G, Quaglia F, La Rotonda M, Besnard M, Fattal E. Biodegradable microparticles for the controlled delivery of oligonucleotides. Int J Pharm. 2002;242:225–228. doi: 10.1016/s0378-5173(02)00162-x. [DOI] [PubMed] [Google Scholar]
- 39.Park PIP, Jonnalagadda S. Predictors of Glass Transition in the Biodegradable Polylactide and Poly-lactide-co-glycolide Polymers. Appl Polym Sci. 2006;100:1983–1987. [Google Scholar]
- 40.Bianchini P, Bergonzini GL, Parma B, Osima B. Relationship between plasma antifactor Xa activity and the antithrombotic activity of heparins of different molecular mass. Haemostasis. 1995;25:288–298. doi: 10.1159/000217175. [DOI] [PubMed] [Google Scholar]
- 41.Isobe M, Yamazaki Y, Oida S, Ishihara K, Nakabayashi N, Amagasa T. Bone morphogenetic protein encapsulated with a biodegradable and biocompatible polymer. J Biomed Mater Res. 1996;32:433–438. doi: 10.1002/(SICI)1097-4636(199611)32:3<433::AID-JBM17>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 42.Tice TR, Tabibi SE. Parenteral drug delivery: Injectables. In: Kydonieus A, editor. Treatise on Controlled Drug Delivery. Marcel Dekker, Inc.; New York: 1992. pp. 315–339. [Google Scholar]
- 43.Maulding HV, Tice TR, Cowsar DR, Fong JW, Pearson JE, Nazareno JP. Biodegradable microcapsules: Acceleration of polymeric excipient hydrolytic rate by incorporation of a basic medicament. J Control Release. 1986;3:103–117. [Google Scholar]