

Abstract
ETHE1 (ethylmalonic encephalopathy protein 1) is a β-lactamase fold-containing protein that is essential for the survival of a range of organisms. In spite of the apparent importance of this enzyme, very little is known about its function or biochemical properties. In this study Arabidopsis ETHE1 was over-expressed and purified and shown to bind tightly to 1.2 ± 0.2 equivalents of iron. 1H NMR and EPR studies demonstrate that the predominant oxidation state of Fe in ETHE1 is Fe(II), and NMR studies confirm that two histidines are bound to Fe(II). EPR studies show that there is no antiferromagnetically-coupled Fe(III)Fe(II) center in ETHE1. Gel filtration studies reveal that ETHE1 is a dimer in solution, which is consistent with previous crystallographic studies. Although very similar in terms of amino acid sequence to glyoxalase II, ETHE1 exhibits no thioester hydrolase activity, and activity screening assays reveal that ETHE1 exhibits low level esterase activity. Taken together, ETHE1 is a novel, mononuclear Fe(II)-containing member of the β-lactamase fold superfamily.
Keywords: β-lactamase fold, Fe(II), ETHE1, NMR spectroscopy, EPR spectroscopy
Introduction
Mutations in the ethylmalonic encephalopathy protein 1 (ETHE1) are responsible for the complex human disease ethylmalonic encephalopathy (EE) [1, 2]. The symptoms of EE are well characterized, yet the underlying physiological cause of the disease remains unknown [1]. Symptoms associated with EE include chronic diarrhea, a delay in neural development, symmetric brain lesions, relapsing petechiae, lactic academia, and acrocyanosis of the hands and feet, all of which lead to death within the first few years of life [3]. Biochemical alterations associated with of EE include elevated levels of C4 and C5 plasma acylcarnitines and elevated levels of ethylmalonic acid (EMA). Based on these biochemical alterations, it has been suggested that ETHE1 may be involved in the β-oxidation of short-chain acyl-CoAs [1].
ETHE1-like enzymes are found in most organisms including archaebacteria, insects, plants, and animals, suggesting that ETHE1 serves a fundamental biochemical role in nature; however, the exact biochemical role of ETHE1 is currently unknown. ETHE1 is most similar to the glyoxalase II family of proteins that catalyze the second step of the glyoxalase pathway. The glyoxalase pathway, which consists of glyoxalase I (lactoylglutathione lyase) (GLX1) and glyoxalase II (hydroxacylglutathione hydrolase) (GLX2), is thought to be involved in the detoxification of 2-oxoaldehydes, toxic by-products of carbohydrate and lipid metabolism [4]. The primary physiological substrate appears to be methylglyoxal (2-oxopropanal), a reactive α-oxoaldehyde [4].
GLX2 enzymes belong to the metallo-β-lactamase fold family of proteins, which are typically dinuclear metallohydrolases. The metallo-β-lactamases typically bind two equivalents of Zn(II) and hydrolyze β-lactams [5, 6]. GLX2 enzymes, which hydrolyze SLG, contain dinuclear metal centers that can bind Zn, Fe, and Mn [7]. Additional protein families containing the β-lactamase fold include the rubredoxin:oxygen oxidoreductase (ROO) and ZiPD families, which have been shown to bind divalent Fe and Zn(II), respectively [8, 9]. Therefore, the β-lactamase fold can accommodate several different metals and is present in enzymes that can catalyze a wide range of reactions.
Arabidopsis ETHE1 is 54% identical to human ETHE1. Crystal structure comparisons between Arabidopsis ETHE1 [10] and Arabidopsis GLX2-5, a mitochondrial GLX2 enzyme [11], revealed that while the proteins show only 13% sequence identity, ETHE1 is structurally very similar to GLX2 [12]. ETHE1 and GLX2 enzymes contain a very similar metal binding domain and the β-lactamase fold consisting of two central mixed β-sheets surrounded on both sides by helices [12, 13]. Interestingly, even though ETHE1 and GLX2 enzymes exhibit extensive similarity in their metal binding regions, the ETHE1 crystal structure showed it only bound a single metal atom [12]. In fact, the crystal structure was best refined to a metal occupancy of 0.5 [12]. However, the metal content of purified ETHE1 was reported to be 2 equivalents of metal per equivalent of protein, raising the question of the actual metal content of this enzyme.
We have over-expressed, purified, and biochemically- and spectroscopically-characterized Arabidopsis ETHE1 to further investigate structural and biochemical properties of ETHE1 enzymes and to better understand the functional role of ETHE1. The results of these studies show that Arabidopsis ETHE1 is homodimeric in solution, exhibits low-level esterase activity, and specifically binds a single Fe(II) atom in the active site.
Materials and Methods
Over-expression and purification of A.t. ETHE1
The Arabidopsis ETHE1 cDNA was obtained from Arabidopsis bud RNA and cloned into pET24b as Nde1 and XhoI fragments following reverse transcription and PCR amplification using the primers GLX2-3 (TCTTCTCATATGAAGCTTCTCTTTCGTCAAC) and a 3′poly (A) anchor primer. During cloning the N-terminal leader peptide was removed for high-level expression of ETHE1 in E. coli, resulting in the amino terminal methionine, which corresponds to amino acid 50 of the predicted protein sequence. This is the same form of the protein that was used for crystal structure determination [12]. After verification by DNA sequencing, pET24b-ETHE1 was transformed into BL21-Codon Plus (DE3)-RIL cells and used for protein over-expression and purification as previously described [7]. Protein purity was determined by SDS-polyacrylamide gel electrophoresis, and protein concentrations were determined by using an extinction coefficient (ε280nm) of 7,240 M−1cm−1, which was determined using amino acid analyses.
Metal analyses
Metal analyses were performed on the purified enzyme using a Varian-Liberty 150 inductively coupled plasma spectrometer with atomic emission spectroscopy detection (ICP-AES) as described previously [13]. The purified protein was diluted to 10 μM in 50 mM TRIS, pH 7.2, and analyzed for the presence of zinc, manganese, iron, and copper. The data presented in this report represent the average of at least three preparations for each metal addition experiment.
Native molecular weight determination
The native molecular weight of ETHE1 was determined by utilizing a Sephacryl S200 column in 10 mM MOPS, pH 7.2, containing 0.15 M NaCl. ETHE1 (1 mg), purified as described above, was mixed with 1 mg of each of the protein standards: Blue Dextran, bovine serum albumin, ovalbumin, aldolase, and ribonuclease A. One milliliter fractions were collected with a flow rate of 0.5 ml/min, and samples containing protein were identified by monitoring A280 and by SDS-PAGE gel analysis.
Substrate analysis
Enzymatic assays were conducted at 25 ºC in 10 mM MOPS, pH 7.2, on a Cary IE UV-Vis spectrophotometer. A series of thioesters of glutathione were synthesized as previously described [14], and all other substrates were purchased commercially. The hydrolysis of S-D-lactoylglutathione (Sigma), S-D-acetylglutathione, S-D-acetoacetylglutathione, S-D-formylglutathione, S-D-glyocosylgutathione or S-D-pyruvylglutathione was monitored at 240 nm. S-D-mandeloylglutathione hydrolysis was monitored at 263 nm [14]. Hydrolysis of p-nitrophenyl phosphate (Sigma), p-nitrophenyl sulfate (Sigma), and p-nitrophenyl acetate (Sigma) was monitored at 400 nm [15–17]. The hydrolysis of L-alanine-p-nitroanilide (Sigma) was measured at 404 nm [16]. Ala-ala-ala-p-nitroanilide (Sigma) and y-L-Glu-p-nitroanalide (Sigma) hydrolysis was monitored at 410 nm. Hydrolysis of benzoylglycyl-L-phenylalanine was monitored at 254 nm [18]. Nitrocefin (Becton-Dickinson) hydrolysis was monitored at 485 nm [19]. Methylglyoxal was assayed colorimetrically by using the 2,3-dinitrophenylhydrazine-alkali reaction [20]. Assays were performed for 5 min using 80 μM substrate and varying concentrations of pure enzyme both as-isolated and after loading with excess iron.
Additional substrate screening was performed using the Micronaut-Taxa Profile E (Merlin GmbH) microtiter plate [8]. Each well was filled with 25 μl of 40 μM ETHE1, and reactions prepared according to manufacturer’s directions. A negative control was performed using MOPS, pH 7.2, to rule out non-specific reactions. The reactions were monitored visually for 24 hours at 37 ºC, and positive reactions were recorded. Each reaction was performed in duplicate using enzyme loaded with iron.
EPR Spectroscopy
EPR spectra were obtained at 9.63 GHz and 10 K using a Bruker EleXsys E580 spectrometer equipped with an ER4116DM cavity, and an Oxford ESR900 liquid helium flow cryostat and ITC503 temperature controller. Acquisition parameters included 12 G (1.2 mT) field modulation at 100 kHz. Samples contained 1.6 mM ETHE1 protein in 50 mM TRIS, pH 7.2.
1H NMR Spectroscopy
NMR spectra were collected on a Bruker Avance 500 spectrometer operating at 500.13 MHz, 298 K, and a magnetic field of 11.7 tesla, recycle delay (AQ), 41 ms, and sweep width, 400 ppm. Concentrated samples of ETHE1 (1.4 mM) contained 10% D2O for locking or 90% D2O for monitoring of solvent-exchangeable peaks. Protein chemical shifts were calibrated by assigning the H2O signal a value of 4.70 ppm, and a modified presaturation pulse sequence (zgpr) was used to suppress the proton signals that originated from the water molecules.
Results
Over-expression, purification, and characterization of Arabidopsis ETHE1
Based on publicly-available localization prediction programs (pSORT II, Mitoprot), recombinant ETHE1 was cloned into the pET24b expression vector after removing the predicted 50 amino acid N-terminal leader sequence, which generated an N-terminus of MKLLFRQ (Figure 1). This plasmid was transformed into E. coli BL21(RIL) Rosetta cells, and ETHE1 was over-expressed as described in Material and Methods. ETHE1 was purified using FPLC Q-Sepharose chromatography, eluting from the column at ~125 mM NaCl. Purified ETHE1 protein was obtained in high yield (~40–60 mg of protein/L) and was >95% pure (data not shown).
Figure 1. Sequence Alignment of ETHE1 and select metallo-β-lactamase fold proteins.

Protein sequences were aligned using CLUSTAL W (1.83). Identical residues are highlighted with black and similar residues with gray highlight. Conserved metal binding ligands are indicated by closed triangles. Residues labeled with an asterisk are mutated in patients with EE. Predicted N-terminal processing site is indicated by an arrow.
ETHE1 is a dimer in solution
In contrast to the GLX2 enzymes, which exist as monomers in solution [21, 22], the crystal structure of Arabidopsis ETHE1 suggested that it has a dimeric quaternary structure [12]. Gel filtration studies were performed on the recombinant ETHE1 enzyme to test this hypothesis. ETHE1 eluted from a Sephacryl S200 column between standard proteins adolase (158 kDa) and ovalbumin (44 kDa), resulting in a calculated molecular weight of 58.3 kDa, which is roughly twice the recombinant monomeric weight of 26.8 kDa (Figure 2). Therefore, Arabidopsis ETHE1 exists as a dimer in solution.
Figure 2. Gel filtration elution profile of internal protein standards and ETHE1.
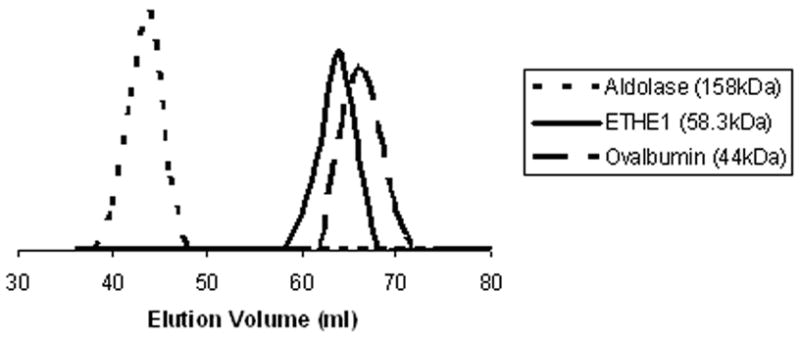
Approximately 1 mg of each of the following internal protein standards was separated on a Sephacryl S200 column: blue dextran, bovine serum albumin, ovalbumin, aldolase, and ribonuclease A. Protein containing fractions were monitored by A280 as well as by SDS-PAGE.
Metal Analyses
GLX2 enzymes have been shown to bind iron, zinc, and manganese; therefore, ETHE1 was analyzed for all three of these metals and also copper [11, 23]. The preparation of ETHE1 in rich medium containing no added metal ions resulted in an isolated enzyme that bound 0.2 equivalents of iron and no other metal ions at levels detectable with ICP-AES. When grown and over-expressed in rich medium containing 250 μM Fe(NH4)2(SO4)2 and Zn(SO4)2 or 250 μM MnCl2, ETHE1 was found to contain 0.33 ± 0.10 equivalents of iron and less than 0.003 equivalents of zinc, manganese, and copper. While low, this result suggested that ETHE1 may preferentially bind iron. The purified enzyme was incubated with a 4-molar excess of Fe(II) and dialyzed extensively (4X1L for four hours each) to remove loosely- or unbound Fe, and ETHE1 was found to bind 1.2 ± 0.2 equivalents of iron, and no detectable traces of zinc, manganese, or copper. This result is consistent with the crystal structure [12], which showed that when prepared this way, ETHE1 only binds 1 equivalent of iron. Unlike the GLX2 enzymes, the addition of excess zinc or manganese to the isolated enzyme resulted in no additional metal binding, suggesting that ETHE1 may be specific for iron.
ETHE1 Does Not Hydrolyze SLG
ETHE1 had previously been predicted to be a GLX2-like enzyme [10]. However, ETHE1 is lacking several highly-conserved amino acids known to participate in the hydrogen bonding of SLG in GLX2 [12, 13]. Furthermore, crystallographic analysis showed that the substrate binding pocket of ETHE1 is too small to accommodate SLG [12]. These results suggested that Arabidopsis ETHE1 does not utilize SLG as a substrate. ETHE1, both as-isolated and after incubation with excess iron, was assayed for thioesterase activity with SLG and various other thioester derivatives of glutathione to test this hypothesis. Consistent our prediction, purified ETHE1 did not hydrolyze any of the glutathione thioesters.
As-isolated ETHE1 and Fe-enriched ETHE1 were also assayed against 188 different substrates, including 95 substrates for peptidases, 17 substrates for diverse reactions, and 76 substrates for glycolytic enzymes, phosphatases, and esterases using a commercially-available substrate screening plate [8] to investigate potential substrates. Three potential substrates were identified through this screening process: Ala-Ala-Ala-p-nitroanilide (NA), Glu-pNA, and p-nitrophenyl acetate (NPA). Upon further characterization, Ala-Ala-Ala-pNA and Glu-pNA were found not to be substrates for ETHE1. However, ETHE1 did exhibit a low level of activity against p-NPA (2.02 ± 0.46 nmols/min/mg of enzyme). This low level of activity is similar to the esterase activity (25.8 nmols/min/mg) observed in recombinant rat carbonic anhydrase III when reacted with p-nitrophenyl acetate [24]. The activity of ETHE1 towards p-nitrophenyl acetate was inhibited by the presence of a metal chelator, 1,10-o-phenanthroline, indicating that p-nitrophenyl acetate hydrolysis by ETHE1 requires bound metal. Therefore, based on these results and those of the crystal structure, we predict that the ETHE1 substrate may be a relatively small ester.
Spectroscopic Studies on ETHE1
The metal binding site of iron-bound ETHE1 was investigated using 1H NMR spectroscopy (Figure 3). The spectrum revealed the presence of 3 paramagnetically-shifted, down field resonances between 200 and 20 ppm and no observable up field resonances > −30 ppm. In this spectrum peaks a and c integrate to 1 proton each, while peak b integrates to 2–3 protons (Figure 3). Spectra were obtained at 7 °C and 35 °C, and while all three peaks shifted up field with increasing temperature, peak b did not split into 2 or more peaks, suggesting that this peak is due to at least 2 chemically-equivalent protons. To further investigate the identity of these protons, an NMR spectrum of ETHE1 in 80–90% D2O was obtained (Figure 3). Peaks a and c broadened slightly but still integrate to 1 proton each. The intensity of peak b decreased by 85%. Based on the resonance positions and line widths, peak b can be assigned to two N-H protons on histidines bound to Fe(II) or possibly to an antiferromagnetically-coupled Fe(III)Fe(II) center [25, 26]. The ETHE1 crystal structure shows that both of the histidine ligands are bound through the ε nitrogen, and therefore, peaks a and c are likely due to the β-CH2 protons on the bound Asp ligand (Figure 3). We cannot rule out the possibility that these peaks are due to ortho protons on metal bound histidines, but these peaks are usually too broad to detect [27]. The NMR data were surprising, since we initially predicted that ETHE1 would have a metal binding site similar to those in the GLX2 family. NMR spectra from GLX2-5, which can contain a dinuclear iron center, shows at least eight paramagnetically-shifted resonances in between 110 and −30 ppm that correspond to protons on ligands bound to a Fe(III)Fe(II) antiferromagnetically-coupled center [11]. The recent crystal structure of ETHE1 demonstrated that His74 (His56 in GLX2-5) may not be in a position to coordinate a bound metal ion [12], which is consistent with NMR spectrum of ETHE1.
Figure 3. 1H NMR spectra of 1.4 mM iron-bound ETHE1 at pH 7.2 in water and 80–90% D2O.

The spectra were obtained on a Bruker Avance 500 spectrometer operating at 500.13 MHz, 298 K, and a magnetic field of 11.7 tesla, recycle delay (AQ), 41 ms; sweep width, 400 ppm.
EPR spectra on several different forms of ETHE1 were obtained to further investigate the ETHE1 metal center. The spectrum of as-isolated ETHE1 containing 0.33 equivalents of Fe (‘Feiso-ETHE1’) indicated the presence of rhombic Fe(III) (Figure 4). The signal was dominated by a broad derivative feature at geff = 4.27; no features that could be considered diagnostic for protein-bound Fe(III) were observed and the origin of the signal is, therefore, unclear. Although precise quantitation of high-spin systems for which more than one Kramers’ doublet is populated is not trivial, double integration and comparison with a similar EPR signal due to Fe(III) from a well-characterized (4-hydroxyphenyl) pyruvate dioxygenase [21] indicated an Fe(III) content of about 0.025 mM. Therefore about 95 % of the iron in Feiso-ETHE1 is EPR-silent.
Figure 4. EPR spectram of 1.6 mM ETHE1 under different conditions.
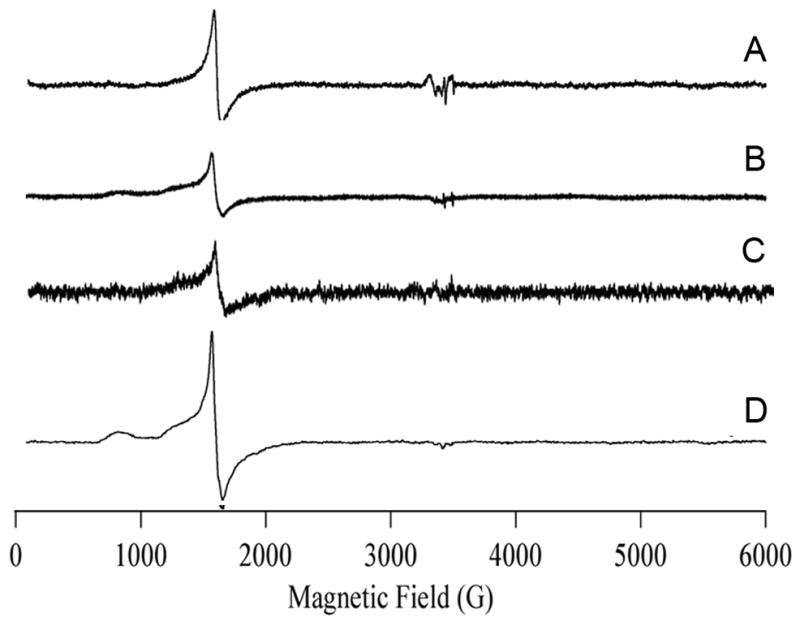
A, ETHE1 as-isolated containing 0.33 equivalents of Fe; B, iron-enriched ETHE1 containing 1.2 equivalents of Fe; C, iron-enriched ETHE1 after 2 cycles of freeze/thaw; D, iron-enriched ETHE1 after 4 days at 4º C. Spectra were collected on a Bruker ESP-300E spectrometer containing an ESR-900 helium flow cryostat operating at 4.7 K with 2 milliwatts of microwave power at 9.48 GHz and 10 G-field modulation at 100 kHz.
An Fe(III) signal with a resonance at g ~ 4.3 and additional absorption features at lower field were observed from the analysis of ETHE1 enzyme containing 1 equivalent of Fe (Fe1-ETHE1) (Figure 4B). In this spectrum, the resonance positions indicated a dominant zero-field splitting term (i.e., D,E ≫ gβBS; the lower field resonances centered at g ~ 5.5 and g ~ 9 terminate abruptly at g = 6 and g = 10, respectively); the lack of resolved structure and the broad absorption from g ~ 4.3 to g ~ 10 indicate a fairly broad distribution of the rhombic zero-field splitting parameter (i.e., strains in E/D) and thermal population of at least two Kramers’ doublets [22]. Again, the signal was not definitive for site-specific binding of Fe(III) by ETHE1, although the form of the signal, particularly the inflection on the g ~ 4.3 crossover due to incomplete rhombicity (i.e., E/D is slightly less than 1/3), is highly reminiscent of the spectrum due to Fe(III) bound to the active site of the metallo-β-lactamase GOB [26]. The intensity of the signal was somewhat lower than that of the as-isolated ETHE1 and accounted for < 1 % of the total iron. Interestingly, there was no evidence for a ‘g ~ 1.7, 1.8, 1.9’ signal due to an Fe(II)-Fe(III) center [28].
We attempted to disrupt the protein structure of Fe1-ETHE1 by applying freeze-thaw cycles, but the EPR signal remained relatively constant (Figure 4C; fewer scans were averaged). Efforts to produce more Fe(III) in the sample by addition of hydrogen peroxide resulted in rapid protein precipitation. The intensity of the Fe(III) signal was found to increase by a factor of 4 upon aerobic incubation for 100 h at 4 °C (Figure 4D). The form of this spectrum is clearly distinct from that of Feiso-ETHE1, but still accounts for < 5 % of the total Fe. Because of a weak background signal due to trace Mn(II) and Cu(II), the possibility of the presence of a signal due to an Fe(II)-Fe(III) center cannot be ruled out completely, but the population of such a center, if not zero, must be very low.
Discussion
Results presented in this work represent the first detailed characterization of an ETHE1 protein from any organism. ETHE1 is most similar to the GLX2-family of proteins, which are members of the metallo-β-lactamase superfamily. This superfamily consists of proteins that catalyze a wide range of reactions but share the common metal binding motif H-x-[EH]-x-D-[CRSH]-X50-70-[CSD]-X, which is part of the common β-lactamase fold [29]. This motif typically consists of two metal ions that are essential for the activity of the majority of the enzymes [29]. In most β-lactamase fold containing proteins, the coordination of the first metal (Zn1 site) is tetrahedral and consists of three histidines and a bridging hydroxide, while the second metal binding site (Zn2 site) is trigonal pyramidal. The site 2 metal binding ligands are more variable but always contain a histidine and aspartic acid (Figure 5A) [29]. Because of the similarity of ETHE1 to the GLX2 family of enzymes, it was predicted that ETHE1 would also bind two equivalents of metal. Metal analyses of ETHE1 done prior to this paper were based on a calculated extinction coefficient of 10,240 M−1cm−1 [12]. Metal analyses using this extinction coefficient indicated that iron-bound ETHE1 contained two equivalents of iron; however, the ETHE1 crystal structure reported only a 0.5 iron metal occupancy [12]. It was originally thought that this discrepancy resulted from the loss of metal during the crystallization processes [12]. However, results presented here suggest that the extinction coefficient used for these experiments is inaccurate. When using amino acid analyses, an extinction coefficient of 7,240 M−1cm−1 was obtained, which leads to a metal:protein stoichiometry of 1.2 ± 0.2. This number is consistent with the ETHE1 crystal structure [12].
Figure 5.
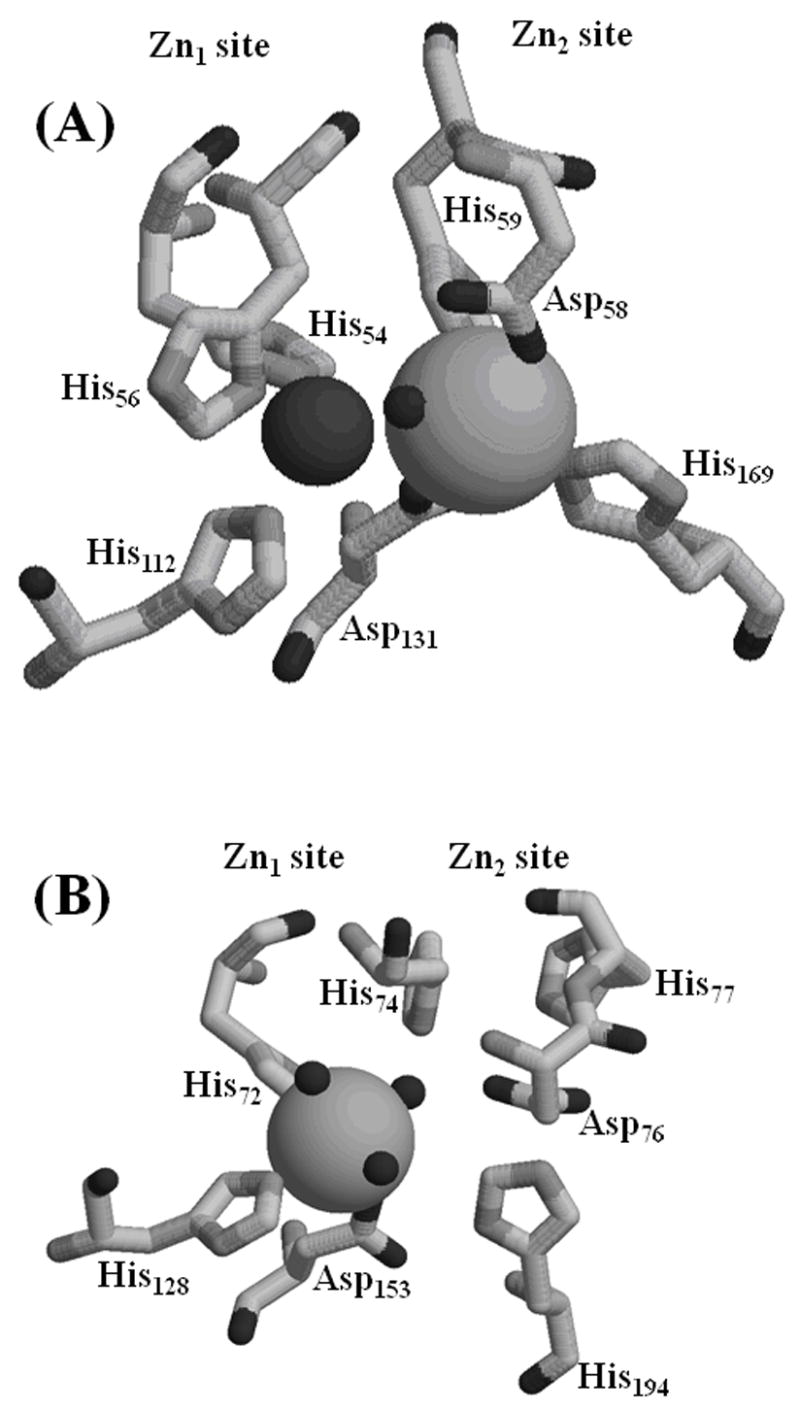
Metal binding sites of (A) A. thaliana GLX2-5 and (B) A. thaliana ETHE1. In GLX2-5, Zn(II) is coordinated by His54, His56, His112, Asp131, and a bridging hydroxide, and Fe(III) is coordinated by Asp58, His59, His169, Asp131, and the bridging hydroxide. In ETHE1, Fe(II) is coordinated by His72, His128, Asp153, and three solvent molecules.
The crystal used to solve the structure of ETHE1 took several months to grow, and we were concerned that the resulting structure was due to an analog of the protein that was not biologically-relevant. However, consistent with the crystal structure of ETHE1, our spectroscopic results suggest that ETHE1 contains a single Fe(II) that is coordinated by two histidines (Figure 3) [12]. GLX2 enzymes enriched in iron typically contain an antiferromagnically-coupled Fe(II)-Fe(III) center in their dinuclear metal binding site [11, 23, 30], due to the positive cooperative metal binding of these proteins. Results from our EPR studies argue against the presence of an Fe(II)-Fe(III) site in ETHE1. Two distinct EPR signals were observed for ETHE1, both due to magnetically-isolated Fe(III) and both accounting for only a very small proportion (1 – 5 %) of the total iron. Even extensive exposure to air was unsuccessful in increasing the Fe(III) signal. The very low intensities of the Fe(III) signals can either be due to most of the iron being in the Fe(II) state or most of the iron residing in an anti-ferromagnetically-coupled S′ = 0 dinuclear site. The lack of signals due to an Fe(III)-Fe(II) center and the crystallographic identification of a monometallated active site [12] both argue against a predominant Fe(III)-Fe(III) S′ = 0 species and support Fe(II) as the predominant oxidation state of the metal in ETHE1.
Even though ETHE1 contains all of the highly-conserved metal binding ligands of the metallo-β-lactamase family of proteins [19], ETHE1 does not bind two equivalents of metal. In agreement with this result, the crystal structure identified changes in the tertiary structure of the metal binding domain that do not allow for the coordination of a second metal atom (Figure 5B) [12]. Based on the amino acid sequence comparison, we anticipated that the Zn1 site in ETHE1 would be made up of His72, His74, His128, and Asp153, while the Zn2 site would have Asp76, His77, Asp153, and His194. In ETHE1, Fe binds to 3 of the 4 ligands in the Zn1 site (His72, His128, and Asp153). A single-turn helix in ETHE1 containing His74 is pulled away from the active site, displacing His74 4.4 Å away from the metal atom in ETHE1 relative to GLX2-5. This unwinding of the helix also displaces the side chains of Asp76 and His77, and the comparable amino acids in GLX2-5 (Asp58 and His59) bind metal in the Zn2 site (Figure 5) [12]. Therefore, subtle changes in protein conformation have displaced several of the metal binding ligands, ultimately limiting the ability of ETHE1 to bind two metal ions.
There are other examples of metallo-β-lactamases that only bind one metal. CphA from Aeromonas hydrophila and GOB-1 from Elizabethkinga meningosptica both only bind a single Zn(II) atom in the Zn2 site (Asp, Cys, His) [31, 32]. However, in these enzymes, the Zn1 site is altered either by the replacement of a histidine with an asparagine in CphA or a histidine with glutamine in GOB-1 [31–33]. To our knowledge ETHE1 is the first example of a β-lactamase family protein that contains all of the conserved metal-binding ligands, yet only binds one metal ion.
In addition, it has been shown that the presence of soft or hard ligands in the metal binding site can affect the specificity of the metal binding [29]. All of the metallo-β-lactamases exclusively bind Zn(II) [29]. The incorporation of an aspartic acid and an additional histidine in the Zn2 site likely allows for the variable binding of Fe, Zn, and Mn seen in the GLX2 enzymes (Figure 5A) [7, 11, 13, 30]. Likewise, the replacement of two soft ligands by an aspartic acid and a glutamate as observed in ROO allows the formation of a di-iron center [9, 29]. Interestingly, in the ETHE1 crystal structure, the Fe ion is bound in the Zn1 site that has been modified by the removal of a histidine and the shifting the GLX2 bridging aspartic acid to specifically coordinate the iron (Figure 5B) [12]. The Fe(II) ion is further coordinated by three water molecules resulting in an octahedrally-bound metal [12, 34], unlike the tetrahedral coordination of metal normally seen in the Zn1 site of metallo-β-lactamases.
Therefore, ETHE1 proteins appear to represent a new class in the metallo-β-lactamase fold family of proteins. Although structurally very similar to the GLX2 enzymes, ETHE1 appears to have evolved to bind a single iron atom in an octahedral configuration. Metal analyses and spectroscopic data suggest that unlike GLX2 enzymes ETHE1 tightly binds to a single Fe(II) atom in a modified Zn1 site of the metallo-β-lactamase metal binding motif. Finally we show that ETHE1 is homodimeric in solution and exhibits low levels of esterase activity, suggesting that ETHE1 might hydrolyze a short-chain ester. The metal binding site in ETHE1, with 2 His, 1 Asp, and 3 water molecules (Figure 5B), resemble the 2-His-1-carboxylate facial triad, which has been reported in a number of O2-utilizing reactions [35, 36]. However, ETHE1 does not share similar tertiary structural or amino acid sequence motifs with any of the enzymes known to have the 2-His-1-carboxylate facial triad. In addition, a 1H NMR spectrum on a sample of ETHE1 containing α-ketoglutarate showed no change in the resonance positions of the paramagnetically-shifted protons, suggesting that ETHE1 is not an α-ketoglutarate-dependent enzyme (data not shown). Further experiments are required to further classify the ETHE1 substrate and ultimately understand its biochemical role.
Acknowledgments
This work was supported by the National Institutes of Health (EB001980, AI056231 to B.B., GM40052 to M.W.C.), the National Science Foundation (MCB-9817083 to C.A.M.) and the Ohio Plant Biotechnology Consortium (to C.A.M.). The authors thank Sriram Devanathan and Zhenxin Hu for obtaining some of the 1H NMR spectra.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tiranti V, D’Adamo P, Briem E, Ferrari G, Mineri R, Lamantea E, Mandel H, Balestri P, Garcia-Silva MT, Vollmer B, Rinaldo P, Hahn SH, Leonard J, Rahman S, Dionisi-Vici C, Garavagilia B, Gasparini P, Zeviani M. Am J Hum Genet. 2004;74:239–252. doi: 10.1086/381653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tiranti V, Briem E, Lamantea E, Mineri R, Papaleo E, Gioia LD, Forlani F, Rinaldo P, Dickson P, Abu-Libdeh B, Cindro-Heberle L, Owaidha M, Jack RM, Christensen E, Burlina A, Zeviani M. J Med Genet. 2006;43:340–346. doi: 10.1136/jmg.2005.036210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burlina A, Zacchello F, Dionisi-Vici C, Bertini E, Sabetta G, Bennet MJ, Hale DE, Schmidt-Sommerfield E, Rinaldo P. Lancet. 1991;338:1522–1523. doi: 10.1016/0140-6736(91)92338-3. [DOI] [PubMed] [Google Scholar]
- 4.Thornalley P. Mol Aspects Medicine. 1993;14:287–371. doi: 10.1016/0098-2997(93)90002-u. [DOI] [PubMed] [Google Scholar]
- 5.Aravind L. In Silico Biol. 1999;1:69–91. [PubMed] [Google Scholar]
- 6.Crowder MW, Spencer J, Vila AJ. Acc Chem Res. 2006;39:721–728. doi: 10.1021/ar0400241. [DOI] [PubMed] [Google Scholar]
- 7.Zang TM, Hollman DA, Crawford PA, Crowder MW, Makaroff CA. J Biol Chem. 2001;276:4788–4795. doi: 10.1074/jbc.M005090200. [DOI] [PubMed] [Google Scholar]
- 8.Vogel A, Borchers T, Marcus K, Meyer HE, Krebs B, Spener F. Arch Biochem Biophys. 2002;401:164–172. doi: 10.1016/S0003-9861(02)00046-2. [DOI] [PubMed] [Google Scholar]
- 9.Frazao C, Silva G, Gomes CM, Matias P, Coelho R, Sieker L, Macedo S, Liu MY, Oliveira S, Teixeira M, Xavier AV, Rodrigues-Pousada C, Carrondo MA, Le Gall J. Nature Struct Biol. 2000;7:1041–1045. doi: 10.1038/80961. [DOI] [PubMed] [Google Scholar]
- 10.Maiti MK, Krishnasamy S, Owen HA, Makaroff CA. Plant Mol Biol. 1997;35:471–481. doi: 10.1023/a:1005891123344. [DOI] [PubMed] [Google Scholar]
- 11.Marasinghe GPK, Sander IM, Bennett B, Periyannan G, Yang KW, Makaroff CA, Crowder MW. J Biol Chem. 2005;280:40668–40675. doi: 10.1074/jbc.M509748200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCoy JG, Bingman CA, Bitto E, Holdorf MM, Makaroff CA, Phillips GNJ. Acta Crystallogr D Biol Crystallogr. 2006;62:964–970. doi: 10.1107/S0907444906020592. [DOI] [PubMed] [Google Scholar]
- 13.Cameron AD, Ridderstrom M, Olin B, Mannervik B. Structure. 1999;7:1067–1078. doi: 10.1016/s0969-2126(99)80174-9. [DOI] [PubMed] [Google Scholar]
- 14.Uotila L. Methods Enzymol. 1981;77:424–430. doi: 10.1016/s0076-6879(81)77044-7. [DOI] [PubMed] [Google Scholar]
- 15.Anderson RA, Bosron WF, Kennedy FS, Vallee BL. Proc Natl Acad Sci. 1975;72:2989–2993. doi: 10.1073/pnas.72.8.2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brandt JJ, Chatwood LL, Yang KW, Crowder MW. Anal Biochem. 1999;272:94–99. doi: 10.1006/abio.1999.4166. [DOI] [PubMed] [Google Scholar]
- 17.Armstrong JM, Myers DV, Verpoorte JA, Edsall JT. J Biol Chem. 1966;241:5137–5149. [PubMed] [Google Scholar]
- 18.Sabastian JF, Lo WY. Can J Biochem. 1978;56:329–333. doi: 10.1139/o78-051. [DOI] [PubMed] [Google Scholar]
- 19.Crowder MW, Wang Z, Franklin SL, Zovinka EP, Benkovic SJ. Biochemistry. 1996;35:12126–12132. doi: 10.1021/bi960976h. [DOI] [PubMed] [Google Scholar]
- 20.Cooper RA. In: Method Enzymol. Abelson JN, Simon MI, editors. Academic Press; New York: 1975. pp. 502–508. [Google Scholar]
- 21.Purpero VM, Moran GR. Biochemistry. 2006;45:6044–6055. doi: 10.1021/bi052409c. [DOI] [PubMed] [Google Scholar]
- 22.Copik AJ, Waterson S, Swierczek SI, Bennett B, Holz RC. Inorg Chem. 2005;44:1160–1162. doi: 10.1021/ic0487934. [DOI] [PubMed] [Google Scholar]
- 23.Schilling O, Wenzel N, Naylor M, Vogel A, Crowder M, Makaroff C, Meyer-Klaucke W. Biochemistry. 2003;42:11777–11786. doi: 10.1021/bi034672o. [DOI] [PubMed] [Google Scholar]
- 24.Kim G, Selengut J, Levine RL. Arch Biochem Biophys. 2000;377:334–340. doi: 10.1006/abbi.2000.1793. [DOI] [PubMed] [Google Scholar]
- 25.Bertini I, Turano P, Vila AJ. Chem Rev. 1993;93:2833–2932. [Google Scholar]
- 26.Moran-Barrio J, Gonzalez JM, Lisa MN, Costello AL, Peraro MD, Carloni P, Bennett B, Tierney DL, Limansky AS, Viale AM, Vila AJ. J Biol Chem. 2007;282:18286–18293. doi: 10.1074/jbc.M700467200. [DOI] [PubMed] [Google Scholar]
- 27.Wang Z, Ming MJ, Que L, Vincent JB, Crowder MW, Averill BA. Biochemistry. 1992;31:5263–5268. doi: 10.1021/bi00138a004. [DOI] [PubMed] [Google Scholar]
- 28.Crowder MW, Vincent JB, Averill BA. Biochemistry. 1992;31:9603–9608. doi: 10.1021/bi00155a012. [DOI] [PubMed] [Google Scholar]
- 29.Gomes CM, Frazao C, Xavier AV, Legall J, Teixeira M. Protein Sci. 2002;11:707–712. doi: 10.1110/ps.31202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wenzel NF, Carenbauer AL, Pfiester MP, Schilling O, Meyer-Klaucke W, Makaroff CA, Crowder MW. J Biol Inorg Chem. 2004;9:429–438. doi: 10.1007/s00775-004-0535-2. [DOI] [PubMed] [Google Scholar]
- 31.Garau G, Bebrone C, Anne C, Galleni M, Frere JM, Dideberg O. J Mol Biol. 2005;345:785–795. doi: 10.1016/j.jmb.2004.10.070. [DOI] [PubMed] [Google Scholar]
- 32.Bellais S, Poirel L, Leotard S, Naas T, Nordmann P. Antimicro Agents Chemo. 2000;44:3028–3034. doi: 10.1128/aac.44.11.3028-3034.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vanhove M, Zakhem M, Devreese B, Franceschini N, Anne C, Bebrone C, Amicosante G, Rossolini GM, Van Beeumen J, Frere JM, Galleni M. Cell Mol Life Sci. 2003;60:2501–2509. doi: 10.1007/s00018-003-3092-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gomes CM, Silva G, Oliveira S, LeGall J, Liu MY, Xavier AV, Rodrigues-Pousada C, Teixeira MJ. J Biol Chem. 1997;272:22502–22508. doi: 10.1074/jbc.272.36.22502. [DOI] [PubMed] [Google Scholar]
- 35.Koehntop KD, Emerson JP, Que L. J Biol Inorg Chem. 2005;10:87–93. doi: 10.1007/s00775-005-0624-x. [DOI] [PubMed] [Google Scholar]
- 36.Hegg EL, Que L. Eur J Biochem. 1997;250:625–629. doi: 10.1111/j.1432-1033.1997.t01-1-00625.x. [DOI] [PubMed] [Google Scholar]