

Abstract
Dendritic cells (DCs) shape T-cell response patterns and determine early, intermediate, and late outcomes of immune recognition events. They either facilitate immunostimulation or induce tolerance, possibly determined by initial DC activation signals, such as binding Toll-like receptor (TLR) ligands. Here we report that DC stimulation through the TLR3 ligand dsRNA [poly(I:C)] limits CD4 T-cell proliferation, curtailing adaptive immune responses. CD4+ T cells instructed by either LPS or poly(I:C)-conditioned DCs promptly upregulated the activation marker CD69. Whereas LPS-pretreated DCs subsequently sustained T-cell clonal expansion, proliferation of CD4+ T cells exposed to poly(I:C)-pretreated DCs was markedly suppressed. This proliferative defect required DC-T cell contact, was independent of IFN-α, and was overcome by exogenous IL-2, indicating T-cell anergy. Coinciding with the downregulation, CD4+ T cells expressed the inhibitory receptor PD-1. Antibodies blocking the PD-1 ligand PD-L1 restored proliferation. dsRNA-stimulated DCs preferentially induced PD-L1, whereas poly(I:C) and LPS both upregulated the costimulatory molecule CD86 to a comparable extent. Poly(dA-dT), a ligand targeting the cytoplasmic RNA helicase pattern-recognition pathway, failed to selectively induce PD-L1 upregulation, assigning this effect to the TLR3 pathway. Poly(I:C)-conditioned DCs promoted accumulation of phosphorylated SHP-2, the intracellular phosphatase mediating PD-1 inhibitory effects. The ability of dsRNA to bias DC differentiation toward providing inhibitory signals to interacting CD4+ T cells may be instrumental in viral immune evasion. Conversely, TLR3 ligands may have therapeutic value in silencing pathogenic immune responses.
Keywords: dendritic cell, T cell, Toll-like receptor 3, PD-L1
Introduction
Dendritic cells (DCs) are potent antigen-presenting cells (APCs) which bridge innate and adaptive immune responses and shape the balance between induction of immunity and tolerance [1]. As sentinels, DCs alert the immune system; capture, process and present antigens; and transport them to lymphoid tissues where they prime antigen-reactive T-cell clones. Driven by pathogens and inflammatory signals, DCs undergo a complex maturation process, which not only leads to enhanced expression of costimulatory molecules and increased formation of stable MHC-peptide complexes but also to cytokine secretion modulating T-cell activation and expansion, synthesis of chemokines and chemokine receptors, and regulation of T-cell and DC trafficking. Through these mechanisms, DCs guide lymphocytes along their differentiation process, thus ultimately determining the quantity and quality of the emerging immune response [2, 3].
Microbial signaling through Toll-like receptors (TLRs) is an effective way to induce DC maturation [1]. TLRs are a well-characterized family of pattern-recognition receptors able to detect pathogen-associated molecular motifs. Ten different TLRs have been described in humans; some of them, such as TLRs 1, 2, 4, 5, and 6, preferentially bind bacterial products [e.g. lipopolysaccharide (LPS), flagellin], whereas others such as TLRs 3, 7, 8, and 9 mainly detect nucleic acids (e.g. single- and double-stranded RNA) [4]. Intracellular signal transmission of most TLRs involves recruitment of the adapter molecule MyD88. TLR triggering of DCs results in activation, as determined by the upregulation of maturation markers (e.g. CD83) and costimulatory molecules (e.g. CD86, CD80, and CD40). Depending on which TLR is triggered, however, functional differences in DC maturation and in the subsequent DC-induced T-cell responses may occur. For instance, TLR2 preferentially induces DCs to produce IL-10, which in turn inhibits a number of pro-inflammatory cytokines. TLR4, on the other hand, is a strong inducer of IL-12p70 and IFN-α [5].
TLR3 mediates responses to double-stranded (ds)RNA and to its synthetic analog polyinosinic: polycytidylic acid [poly (I:C)]. dsRNA constitutes the genome of one class of viruses (e.g. Reoviridae) but is also a replication intermediate of most other viruses, assigning TLR3 a key role in anti-viral defense [6]. This notion is supported by in vitro and in vivo studies demonstrating the pivotal position of TLR3 in host responses against a number of viruses (e.g. West Nile, respiratory syncytial, and influenza) [6, 7]. Despite effective sensing mechanisms alerting the host to viral infections, many viral pathogens, including HIV, lymphocytic choriomeningitis virus (LCMV), and hepatitis C virus, are capable of evading effective immunity, causing chronic infection [8-12]. How DCs contribute to this immune failure is incompletely understood, but recent studies characterizing defective CD8+ T-cell responses in chronically infected hosts have shed light on virus-induced immune deviation [8]. Specifically, insufficient viral clearance has been linked to the induction and maintenance of “exhausted” CD8+ cytotoxic T cells (CTL), which may be tolerized by the inhibitory receptor programmed death-1 (PD-1). This type I immunoglobulin receptor belongs to the CD28/CTLA-4 family and transmits co-inhibitory signals through interaction with its ligands PD-L1 (B7-H1) and PD-L2 (B7-DC) [10-12]. Besides its role in impairing anti-viral immune responses, the PD-1/PD-L pathway is a major immunosuppressive mechanism [8, 9, 13]. PD-1/PD-L1 interactions have been implicated in the initiation and the reversal of T-cell anergy [14]. Also, PD-1 signaling emerges as a major tolerance mechanism in regulating autoimmune disease. In several murine models, aggravated disease has been reported in PD-1-deficient mice, including autoimmune encephalomyelitis, lupus-like syndrome with arthritis and nephritis [15], and intensified dilated cardiomyopathy [16-18]. Finally, the PD-1/PD-L1 pathway is involved in regulating early T-cell fate decisions [19].
CD4+ T cells play a critical role in adaptive immunity; however, relatively little is known about the impact of DC TLR triggering on CD4+ T-cell responses, especially during the course of viral challenges. CD4+ helper T cells contribute to anti-pathogen responses by promoting the generation of functionally mature DCs, a process described as DC licensing [20]. In the present study, we explored the consequences of TLR3 stimulation of monocyte-derived DCs on CD4+ T-cell function and compared it with the outcome resulting from TLR4-triggering using LPS. Our results indicate that TLR3-triggered DCs, as opposed to those stimulated via TLR4, downregulate CD4+ T-cell proliferation. Blockade of the co-inhibitory molecule PD-L1 on TLR3-stimulated DCs almost completely restored CD4+ T-cell expansion, thus implicating the PD-1/PD-L1 pathway in regulating CD4+ T-cell nonresponsiveness imposed by TLR3-conditioned DCs.
Materials and Methods
Cells
To generate immature monocyte-derived DCs (iDCs), CD14+ cells were isolated from peripheral blood mononuclear cells using positive selection with magnetic beads (Miltenyi, Auburn, CA) and cultured at a density of 1.5-2×106 cells/mL in RPMI (Mediatech, Cellgro, Herndon, VA) supplemented with 1000 U/mL IL-4 and 800 U/mL GM-CSF (both R&D Systems, Minneapolis, MN). Cells were harvested on day 6 and stimulated with poly(I:C) (Sigma Aldrich, St. Louis, MO) or LPS (Escherichia Coli, 0127:B8, Sigma Aldrich, St. Louis, MO) at the concentrations and durations described. All experiments were performed with naked poly(I:C) which is recognized by TLR3 but not by intracellular RIG-I-like receptors. Poly (dA-dT), a ligand for RIG-I-like receptors, was purchased from InvivoGen (San Diego, CA) and used at concentrations between 0.01 and 1 μg/mL. All patients provided informed consent, and biological specimens were handled according to Institutional Review Board-approved protocols.
T-cell proliferation assays
CD4+ T lymphocytes were purified from peripheral blood mononuclear cells with magnetic beads (Miltenyi). Cell purity, assessed by flow cytometry, was routinely >95%. iDCs were cultured in 96-well round-bottom microtiter plates (Falcon; BD Bioscience, San Jose, CA) (6.5 × 103 iDCs/well) in the presence of either 1 µg/mL LPS or 0.5, 5, 50, or 100 µg/mL poly(I:C). In selected cultures T-cell stimulation was facilitated by the bacterial superantigen toxic shock syndrome toxin-1 (TSST-1) (final concentration 0.04 ng/mL; Toxin Technology, Sarasota, FL). After 5 hours, CD4+ T cells were added (1 × 105 cells/well). Cultures were pulsed with 1 µCi/well [3H]thymidine on day 2, harvested onto glass fiber filters after 24 hours, and [3H]thymidine uptake was measured using a 1450 MicroBeta scintillation counter (PerkinElmer Wallac, Gaithersburg, MD). Where indicated, cultures were supplemented with 20 ng/mL recombinant human (rh) IL-2 (Chiron, Emeryville, CA). All experiments were performed in triplicates.
To determine the direct effects of poly(I:C) and IFN-α on T-cell proliferation, CD4+ T cells were stimulated (100,000 CD4+/well) in a 96-well flat-bottom plate coated with 1 µg/mL anti-CD3 (OKT-3, Ortho-Clinical Diagnostics, Raritan, NJ) and 0.5 µg/mL anti-CD28 monoclonal antibody (mAb) (BD Biosciences, San Jose, CA) and cultured with 100 U/mL IFN-α (BD Pharmingen, San Diego, CA), 100 µg/mL poly(I:C), or both. Additionally, supernatants harvested from T cell-DC cocultures containing either untreated DC, LPS-stimulated DC, or poly(I:C)- stimulated DC were transferred onto CD4+ T cells driven with anti-CD3/anti-CD28, and proliferation was assessed 3 days later.
Blockade of PD-L1 and PD-L2
Anti-human PD-L1 (clone MIH1) and PD-L2 (clone MIH18) mAbs were purchased from eBioscience (San Diego, CA). iDCs were stimulated with poly(I:C) or LPS for 5 hours and incubated with anti-PD-L1 mAb (1 µg/mL), anti-PD-L2 mAb (10 µg/mL), or isotype controls (mouse IgG1, BD Pharmingen, San Diego, CA) for 1 hour before allogeneic CD4+ T cells were added. Cultures were pulsed with 1 µCi/well of [3H]thymidine after 48 hours. In the kinetic studies, anti-PD-L1 mAb was added 12, 24, or 48 hours after initiating the DC-CD4+ T cell cocultures.
Quantitative PCR/qPCR
iDCs were stimulated with poly(I:C) in a 96-well plate for 48 hours. Total RNA was purified using TRIzol (Life Technologies, Gaithersburg, MD) and reverse-transcribed using AMV reverse transcriptase (Roche Molecular Biochemicals, Mannheim, Germany). qPCR were performed using the Mx3000 instrument (Stratagene, La Jolla, CA) in triplicates: 1 μl cDNA was mixed in a total volume of 50 μl SYBR Green Master Mix (5 μl 10 × PCR buffer, 2.5 mM MgCl2, 0.2 mM each dNTP, 0.025% BSA, 0.2 μl 5 U/μl Plt. Taq, 1:20,000 SYBR Green, and 0.1 μM of each primer).The following primers were used: β-actin, 5′-ATGGCCACGGCTGCTTCCAGC-3′ and 5′-CATGGTGGTGCCGCCAGACAG-3′; PD-L1, 5′-AACTACCTCTGGCACATCCTC-3′ and 5′-CACATCCATCATTCTCCCTTTT-3′; CD86, 5′-GATTCGGACAGTTGGAC-3′ and 5′-GTAACCGTGTATAGATGAGC-3′; IFN-α, 5′-ATGCGGACTCCATCTTG-3′ and 5′-CGTGACCTGGTGTATGAG-3′. Results were normalized to 2 × 105 copies of β actin.
Flow cytometry
mAbs used for flow cytometric analysis were phycoerythrin (PE)-conjugated anti-human PD-L1 (MIH1, eBioscience, San Diego, CA), PE-conjugated anti-human PD-L2 (MIH18, eBioscience, San Diego, CA), fluorescein isothiocyanate (FITC)-conjugated anti-human CD83 (HB15e, BD Pharmingen, San Diego, CA), allophycocyanin-conjugated anti-human CD86 (FUN-1, BD Pharmingen, San Diego, CA), FITC-conjugated anti-human CD69 (L78, BD Pharmingen, San Diego, CA), and PE-conjugated anti-human PD-1 (MIH4, BD Pharmingen, San Diego, CA). Cells were analyzed on an LSR II system (Becton Dickinson, Franklin Lake, NJ). Data analysis was performed with WinMDI software.
Western Blot
iDCs were stimulated with poly(I:C) (100 μg/mL) or LPS (1 μg/mL). Where indicated, control IgG1 or blocking anti-PD-L1 antibodies (1 μg/mL) were added after 5 hours of stimulation. Allogeneic CD4+ T cells were added 1 hour later, and the cocultures were incubated for an additional 12 or 24 hours. Cells were lysed in cell extraction buffer [10 mM Tris, pH 7.4, 100 mM NaCl, 1 mM EDTA, 1mM EGTA, 1mM NaF, 20 mM Na4P2O7, 2 mM Na3VO4, 1% Tritin X-100, 10% glycerol, 0.1% SDS, and 0.5% deoxycholate (BioSource International Inc., Camarillo, CA)] supplemented with 1 mM phenylmethanesulfonyl fluoride and a protease inhibitor cocktail (Sigma-Aldrich, St Louis, MO). Lysates were centrifuged at 14,000g for 10 minutes at 4°C. 30 μg of each supernatant sample was resolved by SDS-PAGE under reducing conditions, transferred to polyvinylidene fluoride membranes, and incubated with rabbit anti-phospho-SHP-2 (Tyr542) antibodies (Cell Signaling Technology, Danvers, MA) overnight at 4°C followed by incubation with horseradish peroxidase-conjugated goat anti-rabbit antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) at room temperature for 1 hour. Blots were visualized by Immobilon Western chemiluminescent detection system (Millipore Corporation, Billerica, MA, USA). The membranes were reprobed for total SHP-2 by using rabbit anti-SHP-2 antibody (Cell Signaling Technology, Danvers, MA). Densitometry was performed using the ImageJ software (available at http://rsb.info.nih.gov/; developed by Wayne Rasband, National Institutes of Health, Bethesda, MD).
Statistical Analysis
Two-sided Student's t test and Mann-Whitney U test were used, where appropriate. P less than 0.05 was considered to be statistically significant. Results are shown as either mean ± SD or box plots with medians and percentiles, when either parametric or nonparametric tests were used.
Results
DC stimulation with TLR 3 and 4 ligands induces divergent T-cell proliferative responses
To examine how conditioning of DCs through different TLR ligands affects CD4+ T cells, we compared poly(I:C)- and LPS-stimulated DCs for their ability to elicit CD4+ T-cell proliferation. In vitro-generated iDCs were exposed to optimal concentrations of LPS (1 μg/mL) or increasing concentrations of poly(I:C) for 5 hours before allogeneic CD4+ T cells were added; T-cell proliferation was assessed 3 days later. LPS-induced DC maturation increased their stimulatory capacity and enhanced T-cell proliferation rates. In contrast, TLR3-stimulated DCs were less effective at inducing T-cell expansion than unstimulated DCs. The inhibitory effect of poly(I:C)-triggered DCs was dose-dependent with maximal effects seen at 100 µg/mL (Fig. 1A).
Figure 1. TLR3 stimulation reduces the ability of DCs to induce T-cell proliferation.
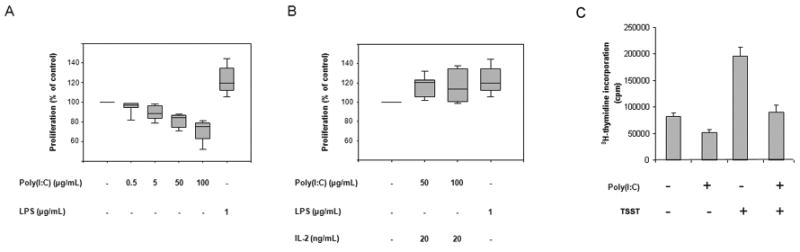
(A) 6.5 × 103 monocyte-derived DCs were left untreated (control) or stimulated with poly(I:C) (0.5, 5, 50 or 100 μg/mL) or LPS (1 μg/mL) for 5 hours. 1 × 105 allogeneic CD4+ T cells were added, and T-cell proliferative responses were measured by [3H] thymidine incorporation after 72 hours. Results from seven independent experiments are expressed relative to the T-cell proliferation induced by unstimulated DCs (on average 34,326 cpm). Box plots display medians, 25th and 75th percentiles as boxes, and 10th and 90th percentiles as whiskers. Untreated DC versus poly(I:C)-treated (50 μg/mL) DC, P = 0.04. Untreated DC versus poly(I:C)-treated (100 μg/mL) DC, P = 0.007. Untreated DC versus LPS-treated (1 μg/mL) DC, P = 0.003. (B) CD4+ T cells were cultured with DCs pretreated with poly(I:C) or LPS as described in (A). IL-2 (20 ng/mL) was added after 48 hours, and proliferation was assessed by [3H] thymidine incorporation 24 hours later. Box plots represent results from five independent experiments. Untreated DC versus poly(I:C)-treated (50 μg/mL) DC, P = 0.04. Untreated DC versus LPS-treated (1 μg/mL) DC, P = 0.01. (C) The effect of poly(I:C) on alloantigen- or superantigen-driven T-cell responses was compared by culturing CD4+ T cells with DC that were untreated, loaded with TSST-1, pretreated with poly (I:C) (100μg/mL), or pretreated with poly(I:C) plus TSST-1 (100μg/mL and 0.04 ng/mL, respectively). Cultures were initiated at a DC:T-cell ratio of 1:50, and proliferative responses were quantified by 3H-thymidine incorporation after 72 hours. Assays were performed in triplicates, and results are presented as mean ± SD. Untreated DC versus poly(I:C)-treated DC, P=0.02; TSST-1-loaded DC versus TSST-1 plus poly(I:C)-treated DC, P=0.01.
To distinguish whether diminished T-cell proliferation was the result of T-cell death or suppressed responsiveness, cultures were supplemented with exogenous IL-2. IL-2 was sufficient to restore T-cell proliferation and equalized proliferation levels of CD4+ T cells instructed by either TLR3- or TLR4-triggered DCs (Fig. 1B).
To assess whether the anti-proliferative effect of TLR3-conditioned DC was relevant in antigen-driven immune responses, we examined the impact of poly(I:C) stimulation on CD4+ T cells responding to the bacterial superantigen TSST-1. As TSST-1 binds to about 10% of all T-cell receptor molecules, T-cell proliferation was enhanced in the presence of this superantigen (Fig. 1C). Pre-conditioning of the DC with poly(I:C) inhibited T-cell proliferation under both conditions, alloreactivity and superantigen reactivity (Fig. 1C). These data strongly suggest that TLR3 triggering of DC modifies antigen-specific immune responses by suppressing T-cell expansion.
Poly(I:C) and LPS are equally sufficient at inducing DC maturation and cytokine production
Divergent proliferative responses of CD4+ T cells raised the question of whether the two classes of TLR ligands initiated distinct DC maturation programs, causing differential expression of costimulatory ligands and opposite effects on T-cell proliferation. We therefore investigated DC cell surface expression levels of CD83 and CD86. DCs were stimulated with 100 µg/mL poly(I:C) or 1 µg/mL LPS and at indicated times stained for surface expression of CD83 and CD86. As shown in Fig. 2, poly(I:C) and LPS rapidly upregulated expression of CD83 and CD86 on the cell surface with these costimulatory ligands appearing with similar kinetics and similar surface concentrations. Both TLR ligands effectively drove DCs into maturation as early as 6 hours after TLR crosslinking. By day 2, ample expression of CD83 and CD86 indicated full maturation of nearly the entire DC population. Thus, poly(I:C)-treated DCs were not inferior in providing T-cell costimulatory signals.
Figure 2. TLR3 and TLR4 ligands are equally competent at inducing DC maturation and cytokine production.
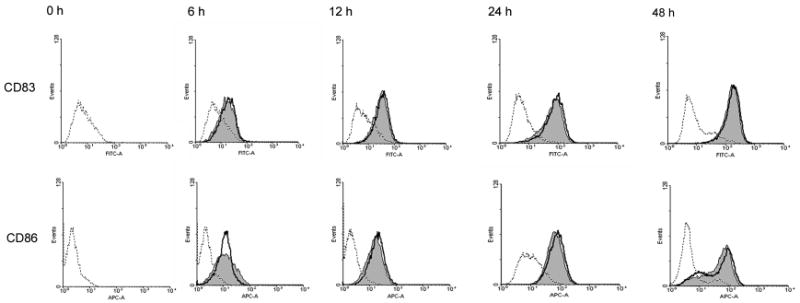
DCs were stimulated with poly(I:C) (100 μg/mL), LPS (1 μg/mL), or left untreated and at the indicated time-points stained using anti-CD83 FITC and anti-CD86-PE mAbs. Representative histograms of control cultures (dashed lines), cultures including poly(I:C) (shaded areas), or LPS (bold lines) are shown.
Inhibition of T-cell proliferation is not mediated by soluble factors
Given its role in recognizing viral infections, TLR3 typically elicits IFN-α/ß production when binding dsRNA. IFN-α has been reported to modulate the survival of activated and anergic T cells with sustained responsiveness to IL-2 [21-23]. We also considered that the diminished CD4+ T-cell proliferation could result from a direct effect of poly(I:C) on T cells [24]. Alternatively, a soluble product secreted by TLR3-triggred DCs could facilitate T-cell nonresponsiveness.
To assess whether IFN-α was exclusively produced by poly(I:C)-triggered DCs and had a direct role in inhibiting T-cell function, we quantified IFN-α transcripts. IFN-α transcription increased in a dose-dependent manner in poly(I:C)-activated DCs (Fig. 3A), confirming functional intactness of these DCs. Interestingly, LPS treatment of DCs also augmented expression of IFN-α transcripts to levels almost comparable to those of poly(I:C). This observation brought into question whether IFN-α had a direct role in mediating T-cell nonresponsiveness.
Figure 3. Induction of T-cell unresponsiveness by poly(I:C)-treated DCs is not dependent on soluble factors.
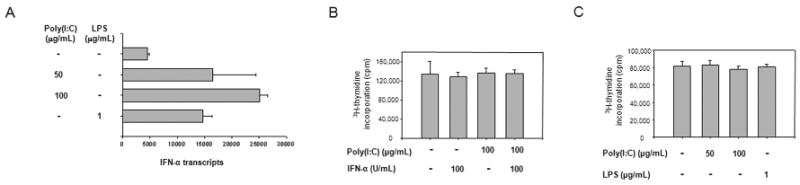
(A) DCs were cultured in medium (control), poly(I:C) (50 or 100 µg/mL), or LPS (1 µg/mL) for 48 hours. IFN-α transcripts were measured by quantitative PCR. Data represented are derived from a minimum of three individual experiments. Untreated DC versus poly(I:C)-treated (100 μg/mL) DC, P < 0.001; untreated DC versus LPS-treated (1 μg/mL) DC, P = 0.01; poly(I:C)-treated (100 μg/mL) DC versus LPS-treated (1 μg/mL) DC, P = 0.03. (B) T cells were stimulated with immobilized anti-CD3 and anti-CD28 Ab in the presence of IFN-α (100 U/mL) or poly(I:C) (100 μg/mL), or a combination of both. Proliferation was measured after 72 hours by [3H]thymidine incorporation. (C) CD4+ T cells were stimulated with untreated DCs or DCs activated with poly(I:C) or LPS. Supernatants from these cultures were added to fresh anti-CD3/CD28-stimulated CD4+ T cells. Cells were harvested and proliferation was determined by [3H]thymidine incorporation. Results are expressed as mean±SD of triplicate measurements and are representative of a minimum of three experiments.
To examine the functional impacts of IFN-α and poly(I:C), T cells were stimulated with immobilized anti-CD3/anti-CD28 Ab and IFN-α (100 U/mL) or poly(I:C) (100 μg/mL), or a combination of both. Neither IFN-α nor poly(I:C) nor their combination affected the proliferative capacity of CD4+ T cells (Fig. 3B).
To investigate whether TLR-3-activated DCs produced soluble factors that suppressed responding CD4+ T cells, supernatants were generated by coculturing poly(I:C)- or LPS-stimulated DCs and allogeneic CD4+ T cells and transferred to CD4+ T cells activated by TCR/CD28 stimulation. Proliferative responses remained unaffected (Fig. 3C).
TLR3 ligands preferentially induce expression of the co-inhibitory ligand PD-L1
Similarities in the expression of costimulatory ligands in TLR3- and TLR4-treated DCs raised the question of whether the differences in functional outcome of T-cell stimulation were related to differential expression of co-inhibitory ligands. One of the most important receptor-ligand pairs providing negative signals to T cells is the PD-1/PD-L1 pair. PD-1 is typically upregulated on T cells upon activation and could certainly be involved in directing CD4+ T cells towards nonresponsiveness [25]. We first assessed whether the TLR3 ligand-imposed suppression of T-cell activation was an early event, disrupting early steps of T-cell activation, or occurred during later stages. CD69 surface expression increased as early as after 3 hours. T cells interacting with poly(I:C)- and LPS-conditioned DCs showed a similar response in terms of CD69 induction (Fig. 4A). There was no difference between poly(I:C) and LPS stimulation until 24 hours, at which point LPS-triggered DCs outperformed poly(I:C)-triggered DCs in CD69 upregulation. CD69 surface expression steadily declined during the subsequent 24 hours. Thus, the downregulatory function of TLR3-treated DCs involved later steps of T-cell activation, affecting sustained T-cell stimulation. Similarly, PD-1 was barely detectable on resting T cells and remained markedly low for the first 12 hours of T-cell stimulation. After 24 hours, PD-1 appeared on the T-cell surface with similar kinetics in TLR3- and TLR4-activated cultures (Fig. 4A).
Figure 4. Poly(I:C) stimulation preferentially induces the co-inhibitory ligand PD-L1.
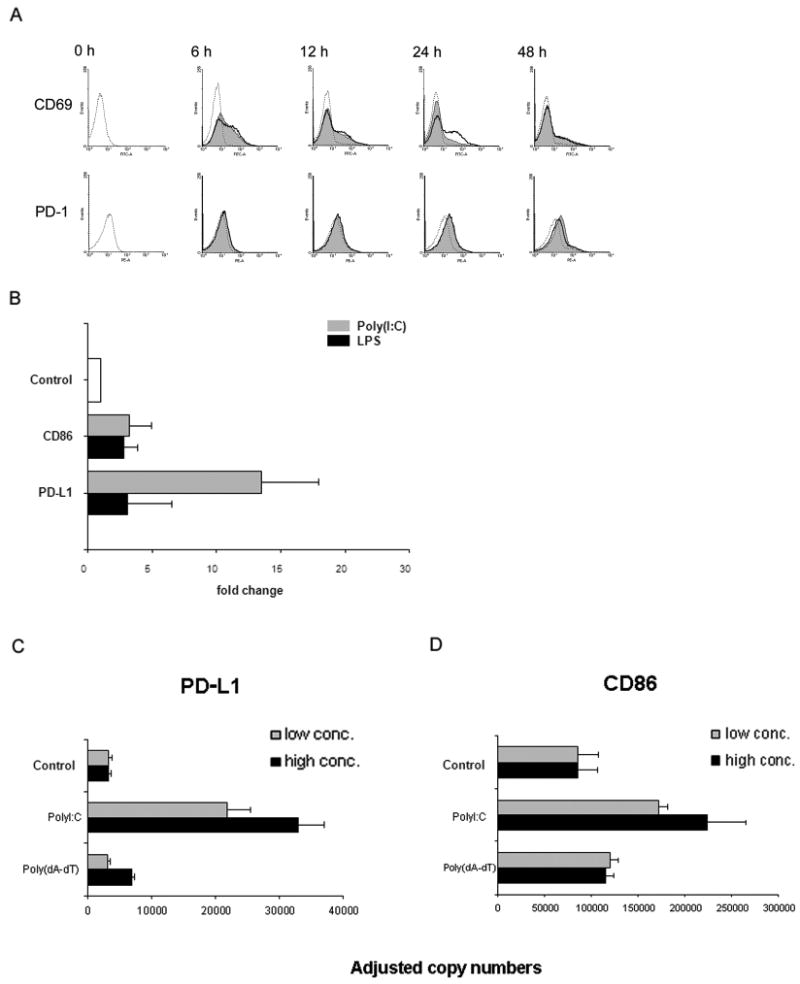
(A) Freshly isolated T cells were cocultured with TLR-stimulated DCs [poly(I:C) 100 μg/mL and LPS 1 μg/mL], and expression of the activation markers CD69 and PD-1 on CD4+ T cells was determined by flow cytometry at the indicated time points. Representative histograms of control cultures (dashed lines), cultures including poly(I:C) (shaded areas), or LPS (bold lines) are shown. Early activation events are indistinguishable but after 24 hours, LPS-triggered DCs mediate stronger CD69 expression than poly(I:C)-stimulated DCs. Induction of the co-inhibitory receptor PD-1 expression was similarly efficient by poly(I:C)- and LPS-stimulated DCs and peaked at 24 hours. (B) Dendritic cells were stimulated with TLR ligands poly(I:C) or LPS, and mRNA was harvested after 6 hours. Expression of PD-L1 and CD86 transcripts was assessed by quantitative PCR. Results are presented as mean±SD relative to transcription level of unstimulated DC and are representative of 3 independent experiments. CD86: Control versus LPS-stimulated (1 μg/mL) DC, P=0.04; Poly(I:C)-stimulated (100 μg/mL) DC versus LPS-stimulated (1 μg/mL) DC, P > 0.05. PD-L1: Poly(I:C)-stimulated (100 μg/mL) DC versus LPS-stimulated (1 μg/mL) DC, P = 0.01. (C and D) DC were stimulated with poly(I:C) or the RIG-I-like receptor ligand poly(dA-dT) for 4 hours, and transcript levels for PD-L1 (left) and CD86 (right) were quantified by qPCR. Two concentrations of each ligand were used: 25 and 100 μg/mL of poly(I:C) and 0.01 and 1 μg/mL of poly(dA-dT). Representative results from one of two independent experiments are shown as mean ± SD. PD-L1: Control versus poly(I:C)-treated DC (25 μg/mL), P=0.007; control versus poly(dA-dT)-stimulated DC (0.01 μg/mL), P=0.4. CD86: Control versus poly(I:C)-treated DC (25 μg/mL), P<0.01; control versus poly(dA-dT)-stimulated DC (0.01 μg/mL), P =0.07.
Differential signaling through the co-inhibitory receptor PD-1 by DC populations would require disparate availability of PD-L1 ligand. PD-L1 plays an important role in the induction of T-cell tolerance [25-29]. During the initial phase of DC stimulation, FACS was not sensitive enough to detect PD-L1 surface expression (data not shown). To detect the effect of TLR stimulation on PD-L1 and CD86 transcription, we therefore compared transcript levels in poly(I:C)- and LPS-stimulated DC (Fig. 4 B). TLR triggering induced a prompt response of DCs in terms of PD-L1 transcription. PD-L1-specific sequences were upregulated by poly(I:C) in a dose-dependent fashion with 9-fold to 13-fold increase compared with unstimulated DCs. Conversely, LPS stimulation resulted in only a minor increase of PD-L1-specific sequences. In contrast, both poly(I:C) and LPS stimulation proved equally efficient in upregulating CD86 expression (3-fold) compared with baseline. Thus, TLR3-mediated DC differentiation is associated with preferential induction of the co-inhibitory ligand PD-L1.
Because poly(I:C) not only interacts with TLR3 but can also be recognized by cytoplasmic RNA helicases, we wanted to know whether the preferential induction of PD-L1 can be achieved by targeting non-TLR pattern recognition receptors. RNA helicases, such as RIG-I/MDA5 induce the production of type I interferons via the adaptor IPS-1 [30]. All of our experiments were performed with naked poly(I:C) which targets TLR3. To stimulate the IPS-1 pathway, we utilized poly(dA-dT) and compared the effects of both polymers on the induction of co-stimulatory and co-inhibitory molecules. DC were stimulated with poly(I:C) (25 and 100 μg/mL) or poly(dA-dT) (0.01 and 1 μg/mL), and transcripts specific for PD-L1 and CD86 were quantified. As shown in Fig. 4C, poly(I:C) at both concentrations strongly amplified transcription of PD-L1. In contrast, the IPS-1 stimulator poly(dA-dT) had no or only a minimal effect on the activation-induced upregulation of PD-L1 sequences. The difference between both stimulators was maintained for CD86. Poly(I:C) functioned as a potent inducer of CD86 whereas poly(dA-dT) enhanced CD86 transcription by less than 2-fold (Fig. 4D).
These experiments confirmed that the differential induction of PD-L1 by poly(I:C) stimulation was mediated through TLR3. The selective enhancement of PD-L1 transcription by poly(I:C) raised the question of whether under conditions of combined stimulation of TLR3 and TLR4 ligands, PD-L1 induction was maintained, providing ideal conditions for suppressed T-cell expansion in hosts infected with both viral and bacterial microorganisms. To mimic these conditions, we stimulated DC with poly(I:C) in the absence and presence of LPS. Under all stimulation conditions, PD-L1 transcripts were induced to a similar level, indicating that LPS could not revert the selective induction of co-inhibitory molecules on the surface of the instructing DC (Supplemental Figure 1).
Blockade of PD-L1 restores the proliferative response of CD4+ T cells instructed by TLR3-conditioned DCs
To dissect the role of the PD-1/PD-L1 pathway in modulating the outcome of DC-T cell interactions, we used anti-PD-L1 and anti-PD-L2 mAbs to block access to PD-1. DCs triggered with either poly(I:C) or LPS for 5 hours were incubated with blocking mAb or isotype control antibodies before CD4+ T cells were added. Fig. 5A (left panel) shows that anti-PD-L1 effectively reversed the dose-dependent inhibition of T-cell responses by poly(I:C)-treated DC and led to a 2-fold increase in T-cell proliferation. Preventing PD-1 ligation resulted in T-cell proliferation levels comparable to those of untreated cultures. In contrast, anti-PD-L2 could not counteract the suppression of T-cell proliferation (Fig. 5A, right panel). Simultaneous addition of both blocking antibodies had no additive effect (data not shown). There was only a modest increase in T-cell proliferation if anti-PD-L1 was added to cocultures containing either LPS-preconditioned DCs or DCs that had not been exposed to TLR ligands.
Figure 5. Blockade of PD-L1 restores CD4+ T-cell proliferation.
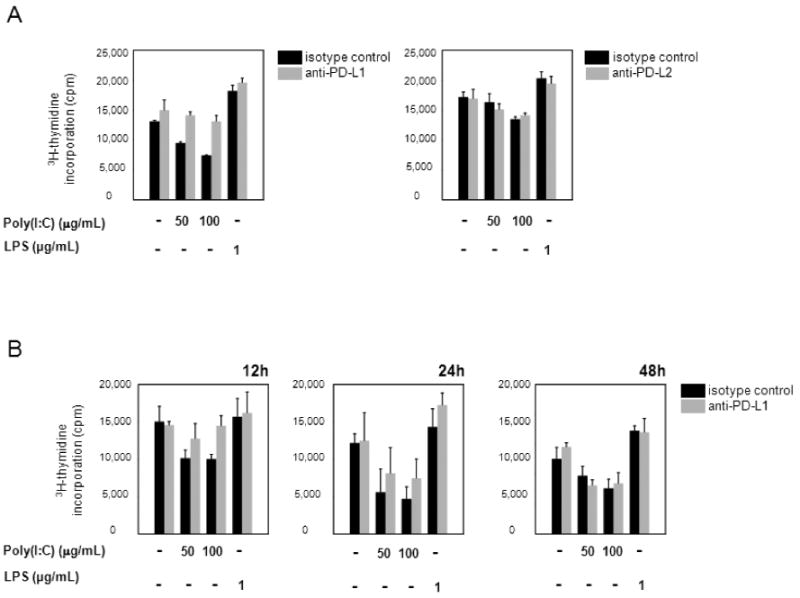
(A) DCs were stimulated with poly(I:C) or LPS for 5 hours and incubated with anti-PD-L1- (1 µg/mL) (left panel), or anti-PD-L2- (10 µg/mL) (right panel) blocking mAb, or the appropriate isotype control for 1 hour. 1×105 CD4+ T cells were added, and proliferation was assessed by [3H]thymidine incorporation 72 hours later. One representative experiment of three is shown. Left panel: Unstimulated DC versus poly(I:C)-stimulated (100 µg/mL) DC, P = 0.0003; unstimulated DC versus LPS-stimulated (1 µg/mL) DC, P = 0.008; poly(I:C)-stimulated (100 µg/mL) DC versus mAb-blocked poly(I:C)-stimulated (100 µg/mL) DC, P = 0.01. Right panel: Unstimulated DC versus poly(I:C)-stimulated (100 µg/mL) DC, P = 0.03; unstimulated DC versus LPS-stimulated (1 µg/mL) DC, P = 0.009. (B) Anti-PD-L1 blocking mAb was added at different time points after initiation of T cell-DC interaction, and T-cell proliferative responses were measured as described above. One representative experiment of three is shown. 12 hours: Unstimulated DC versus poly(I:C)-stimulated (100 µg/mL) DC, P = 0.04; poly(I:C)-stimulated (100 µg/mL) DC versus mAb-blocked poly(I:C)-stimulated (100 µg/mL) DC, P = 0.03. 24 hours: Unstimulated DC versus poly(I:C)-stimulated (100 µg/mL) DC, P = 0.01. 48 hours: Unstimulated DC versus poly(I:C)-stimulated (100 µg/mL) DC, P = 0.05.
To determine the kinetics of underlying inhibitory signaling mechanisms after PD-1 engagement on T cells, PD-L1-blocking mAb was added at different time points after initiation of the T cell-DC cocultures (Fig. 5B). Disruption of PD1-PD-L1 interaction even after 12 hours was sufficient to prevent negative signaling in T cells. Blocking access to PD-L1 after 24 hours resulted in partial restoration and after 48 hours had no effect, demonstrating that PD-1-mediated suppression of T-cell signaling occurred during a defined window of DC-T-cell interaction. These findings are consistent with the surface expression studies showing that PD-1 on T cells was not fully expressed until after 12 hours. The kinetics of the antibody-blocking studies confirmed that negative signals provided by TLR3-conditioned DCs were particularly important in dampening sustained TCR signaling but did not interfere with initial events.
dsRNA-conditioned DCs modulate CD4+ T-cell responses by inducing SHP-2 recruitment
If indeed PD-1 crosslinking is the critical step through which TLR3-conditioned DCs instruct T cells to enter a state of hyporesponsiveness, then T cells exposed to poly(I:C)-pretreated DCs should have evidence for PD-1 signaling. Engagement of both the TCR and PD-1 receptor is required for phosphorylation of an immunoreceptor tyrosine-based switch motif (ITSM) in the cytoplasmic domain of PD-1. Association of Src homology region 2 domain-containing phosphatase-2 (SHP-2) with the ITSM and phosphorylation-dependent stimulation of its tyrosine phosphatase activity are proposed to mediate inhibitory effects of PD-1 [11, 31, 32]. Cell lysates were prepared after 24 hours of DC-CD4+ T-cell coculture, and immunoblot analyses for total SHP-2 and phospho-SHP-2 were performed. Results are given in Fig. 6 and show that at the number of cells included, SHP-2 is nearly exclusively detected in T cells and only minimally in DCs. Exposure to poly(I:C)-treated DCs caused marked phosphorylation of SHP-2 (Fig. 6A, lane 4; D), whereas LPS-treated DCs did not transmit a signal to T cells enhancing SHP-2 phorphorylation (Fig. 6B, lane 2; D). No such effect was observed when DCs were left untreated. Generation of phospho-SHP-2 required 24 hours of coculture. Immunoblotting of cell extracts harvested after 12 hours did not show enhancement of SHP-2 phosphorylation under any of the culture conditions (data not shown). SHP-2 phosphorylation induced by TLR3-triggered DCs could be partially blocked by anti-PD-L1 blocking antibody (Fig. 6C, lanes 1 and 2; D). This observation was consistent with reinstitution of T-cell proliferation in the presence of this antibody. Taken together, these results indicated that TLR3-activated DCs modulate TCR-mediated signals by PD-1 engagement, and that crosslinking of PD-1 by its ligand PD-L1 has functional relevance in DC-T cell interactions.
Figure 6. TLR3-stimulated DCs enhance SHP-2 phosphorylation in CD4+ T cells downstream of PD-1/PD-L1 interaction.
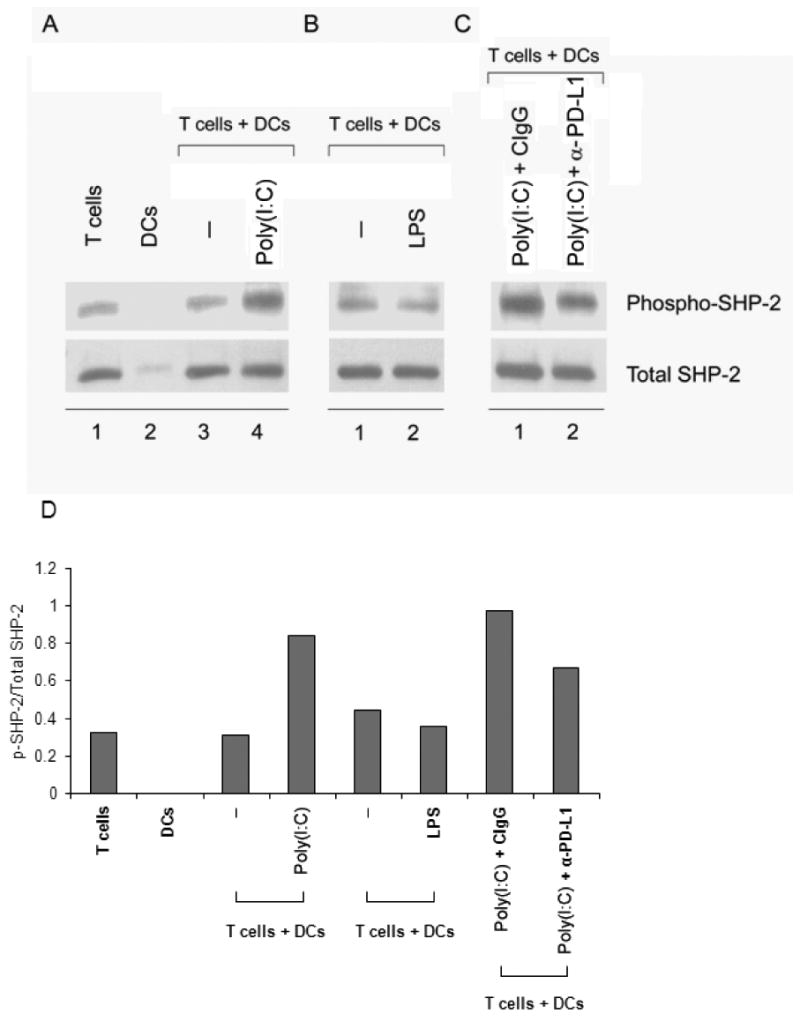
(A, B) CD4+ T cells were cocultured with unstimulated (A, lane 3 and B, lane 1), poly(I:C)-pretreated (A, lane 4), or LPS-pretreated (B, lane 2) DCs for 24 hours, lysed, and cell extracts were resolved on SDS-PAGE acrylamide gels. Proteins were transferred onto PVDF membranes and membranes probed for phospho-SHP-2 (upper row). Membranes were stripped and reprobed for total SHP-2 (lower row). Lanes 1 and 2 in (A) are T cells and DCs cultured alone. (C) CD4+ T cells were cocultured with DCs as in (A), lane 4, but with the addition of either control-IgG1 (lane 1) or anti-PD-L1 blocking mAb (lane 2) and probed for phospho- and total SHP-2. Results in (A), (B), and (C) are representative of three independent experiments. (D) Quantification of the phospho-SHP-2/SHP-2 ratios was performed using the ImageJ software (NIH).
Discussion
DCs hold a central position in alerting the host immune system to infection and in coordinating early and late immune responses. They are known as the most effective APCs in T-cell priming, but remain critical even during chronic immune reactions. The present study shows that the nature of the initial DC stimulus is crucial in preparing the DCs for the induction of T-cell responses. Indeed, ligands activating TLR3 induced DC maturation but also conditioned the DCs to inhibit CD4+ T-cell proliferation. This activity of the TLR3 ligand poly(I:C) contrasted with the robust effectiveness of the TLR4 ligand LPS in generating stimulatory DCs that drove CD4+ T cells into expansion. The inhibitory effect of TLR3-pretreated DCs involved intermediate steps in T-cell activation while early events were spared, guiding mechanistic studies towards receptor-ligand pairs that were by themselves T cell-activation dependent. Indeed, T-cell non-responsiveness was at least partially reversible by blocking access to PD-1, an inducible co-inhibitory receptor appearing on T cells after activation. CD4+ T cells primed with TLR3-conditioned DCs displayed a typical signature of phosphorylated SHP-2, a phosphatase inhibiting TCR-mediated signals.
The synthetic analog of double-stranded RNA poly(I:C) mimics a molecular pattern associated with viral infection. Poly(I:C) interacts with two types of pattern recognition receptors, TLRs and the RIG-I-like receptors (RLRs) [30]. Experiments probing the effects of the RLR stimulator poly(dA-dT) revealed that only poly(I:C) had the ability to selectively induce the co-inhibitory molecule PD-L1 (Fig. 4), assigning this mechanism to the TLR3 receptor. Thus, biologic consequences of accessing a particular pattern recognition system may be profound, and viral RNAs recognized through either endosomal TLR3 or cytoplasmic RNA helicases may modulate innate and subsequent adaptive immune responses in a very unique fashion.
TLR3 is expressed by a variety of different cell types (myeloid and monocyte-derived DCs, fibroblasts, epithelial cells, NK cells, and some T-cell subpopulations) [4, 33] and has been postulated to be a key player in anti-viral immunity [7]. Immune responses against viruses require an intricate orchestration of innate signals provided by APCs and adaptive virus-specific responses [34]. Evidence suggests that MHC class I-restricted recognition of virus-associated antigens by cytotoxic CD8+ T cells is a critical pathway in eliminating virus-infected cells and preventing further spread of the infection [34]. CD4+ T cells are thought to play an important auxiliary role in coordinating anti-viral adaptive immunity [35]. Our data indicate that the PD-1/PD-L1 pathway is employed by DCs for suppression of adjacent CD4+ T cells after licensing by TLR3 stimuli. Such DCs remain capable of initiating the early T-cell activation cascade and produce type I interferons, which may contribute to anti-viral immunity. This bypass of CD4+ helper T-cell inhibition might have evolved as a mechanism to prevent overwhelming immune stimulation. In the course of a viral infection, the host organism needs to detect and clear the pathogen, which involves not only broad APC stimulation, but also the expansion of self-reacting T cells. In this context, the concept has been put forward that autoimmune disease is precipitated by antiviral immunity. In view of the potentially detrimental outcome of immune activation, downregulation of CD4+ T-cell help by APCs after detection of the viral agents emerges as a powerful and targeted mechanism to prevent immunopathology. On the other hand, viruses may ultimately escape host immunity by exploiting such a pathway intended for maintenance of peripheral tolerance [8]. Data presented here suggest that anti-viral immune responses controlled by CD4+ T cells may be effectively compromised by triggering of intracellular TLR3 receptors with reprogramming of DCs away from T-cell priming functions towards suppression of CD4+ T cells. This hypothesis is consistent with recent reports demonstrating that wild-type mice are more susceptible to West Nile Virus-mediated brain inflammation than TLR3-deficient mice [36]. The role of TLR3-activated DCs in vivo and their modulatory function during the course of viral infection warrant validation from studies focusing particularly on the contribution of CD4+ T cells.
T cells exposed to TLR3-conditioned DCs did not undergo cell death but remained IL-2-responsive, a feature typically seen in anergic T cells. Also, blocking with anti-PD-L1 antibodies reversed their hyporesponsive state. Phenotyping of activated CD4+ T cells in cocultures confirmed that TLR3-conditioned DCs did not instruct T cells to die but to become hypoproliferative instead. T-cell cell cycle entry was comparable in cocultures driven by LPS and poly(I:C)-treated DCs, but the frequencies of CD69-expressing CD4+ T cells was eventually higher if DCs were conditioned with LPS. In essence, CD4+ T cells exposed to TLR3-triggered DCs entered the activation program but could not maintain it. Upregulation of PD-1 receptors was intact, making these CD4+ T cells sensitive to inhibitory signals. The cytoplasmic domain of PD-1 contains both an immunoreceptor tyrosine-based inhibitory motif (ITIM) and a switch motif (ITSM). The ITSM is considered to be important in mediating the inhibitory PD-1 function by the recruitment of SHP-2 [11, 31]. Subsequent phosphorylation of SHP-2 induces its phosphatase acitivity, thus mediating the negative signals of PD-1 by dephosphorylating and inactivating effector molecules downstream of TCR and CD28, such as ZAP-70 and phosphatidylinositol 3-kinase (PI3K) [27, 31, 37]. This eventually leads to impaired IL-2 synthesis, inhibition of glucose metabolism, and T-cell cycle arrest [32, 38].
Our results confirm that SHP-2 is the key adaptor in the inhibitory PD-1 signaling cascade which was activated by poly(I:C) stimulation. However, this also raises the question as to why LPS-pretreated DCs were not equally capable of initiating a similar process. First, we have found that poly(I:C) stimulation resulted in preferential upregulation of PD-L1 transcription, biasing the signaling potential of the differentiating DCs towards inhibition versus stimulation. TLR4 binding obviously enhanced CD86 and, to a lesser degree, PD-L1, and functionally skewed the balance towards amplification of TCR-mediated signals. Of note, it has been shown that the upregulation of costimulatory molecules by poly(I:C) is only partially dependent on the TLR3-TRIF-signaling axis, but also occurs in the absence of the adapter TRIF, whereas LPS always requires the presence of TRIF [39]. Second, in blocking LPS-treated cultures with anti-PD-L1 mAb, we did observe minor enhancement of T-cell proliferation. As the fate of T-cell stimulation ultimately depends on the integration of positive and negative signals, LPS-conditioned DCs favor providing amplificatory signals, which outweigh incoming inhibitory signals. The signaling studies shown in Fig. 6 support this notion. However, we cannot rule out the possibility that ligation of PD-1 may lead to a number of different scenarios. The ITSM motif on the cytoplasmic domain of PD-1 may interact with different adapter molecules, depending on the activating stimulus T cells receive; as a consequence, PD-L1 could transmit either positive or negative signals, or bind to a putative costimulatory receptor other than PD-1 [10, 40-42].
Data presented here have multiple implications which are also of clinical relevance. If TLR3 mediates anti-viral responses and the downregulation of CD4+ T-cell proliferation induced by TLR3-triggered DCs represents a viral escape mechanism, then blocking PD-1/PD-L1 interaction could be useful in enhancing anti-viral responses. In murine models, PD-1 and its ligands as critical contributors to the exhaustion of CD8+ T-cell memory responses have been implicated in the chronicity of a number of viral infections [8, 43]. In HIV-infected individuals, impaired virus-specific CD8+ T cells overexpressed PD-1, and the frequency of such T cells has been suggested as a marker of disease progression and response to HAART therapy [44, 45]. Blocking PD-1 in the LCMV model was sufficient to recover CD8+ T-cell proliferation and to improve their antiviral activity [44-47]. Similar findings were obtained in other human chronic viral infections such as hepatitis C virus infection [48]. PD-L1 was also found to be upregulated in patients with chronic HBV infection and induced defective HBV-specific T-cell responses [49].
Besides its role in anti-viral immunity, TLR3 has a number of critical functions in regulating adaptive immune responses and functional activities of nonimmune cells. TLR3 stimuli can augment antitumor immunity by directly triggering apoptosis of human breast cancer cells. Evidence has been provided that the PD-1/PD-L1 axis plays a role in cancer immunology. In patients with renal cell carcinoma, PD-L1 was found to be expressed by tumor cells and tumor-infiltrating lymphocytes, and the extent of PD-L1 expression correlated with tumor aggressiveness, thus suggesting that this could represent a mechanism through which tumors evade host immunity [13]. Notably, experimental studies in mice implanted with human ovarian cell carcinoma demonstrated that the transfer of T cells preconditioned with PD-L1-blocked DCs effectively reduced tumor growth [9]. Conversely, poly(I:C) has been explored as an adjuvant to enhance the specific anti-tumor immune response against peptide-based vaccines [50, 51]. Our findings suggest that poly(I:C) may mediate divergent effects, functioning as an immunostimulator under certain conditions but impairing immune responses, especially when deviating DC maturation toward PD-L1 high expressors.
It is expected that dsRNA-derived immune stimulation also has profound effects in inflammatory autoimmune diseases, such as experimental inflammatory arthritis [52]. The suppression of T-cell proliferative capacity in our studies is in line with a previous report in which poly(I:C) was used as an adjuvant component in experimental autoimmune encephalomyelitis and failed to induce T-cell proliferation [53]. In that disease model, poly(I:C) can be viewed as a potent immunoregulatory agent, significantly limiting disease severity and progression driven by CD4+ T cell-mediated damage. Mechanistic studies in animal models have suggested that the anti-inflammatory action of dsRNA in experimental autoimmune encephalomyelitis could be due to an upregulation of type I interferons leading to increased PD-L1 expression together with suppression of CD4+ T cells [54]. In these studies, blockade of PD-1/PD-L1 resulted in disease acceleration [16, 55]. Loss of peripheral tolerance is a sine qua non condition in the development of autoimmunity, but impairment of central tolerance mechanisms might also contribute. The PD-1/PD-L1 axis has a well-documented role in maintaining peripheral tolerance [56]. However, recent evidence points towards an involvement in central tolerance mechanisms; blockade may prevent T-cell anergy in lymphoid organs upon tolerogen recognition [14].
In conclusion, recognition of dsRNA by DCs results in a complex program of differentiation that can support both immunostimulatory and immunosuppressive pathways. DCs conditioned by TLR3 ligands support early activation events in CD4+ T cells but curtail their expansion by favoring the induction of co-inhibitory ligands. T cells exposed to such DCs display an anergic phenotype, and their inability to clonally expand is associated with accumulation of phosphorylated SHP-2. Thus, SHP-2 emerges as a critical mediator in T-cell anergy, allowing for fast reversal of T-cell nonresponsiveness. Two interventions, supplying exogenous IL-2 as well as preventing PD-1 ligation, restored proliferation in the present studies. Therapeutic targeting of the PD-1/PD-L1 pathway should be explored in attempts to enhance anti-viral immune responses but may also prove useful in modulating CD4 T-cell pathogenic immune responses in the setting of autoimmune disease.
Supplementary Material
Acknowledgments
This work was funded in part by grants from the National Institutes of Health (RO1 AR 41974, RO1 AR 42527, RO1 AI 57266, RO1 EY 11916, RO1 AG 15043, and UI9 AI 44142), a grant from the Dana Foundation (Dendritic Cell Function in Inflammatory Diseases of Blood Vessels), and a grant from the General Clinical Research Center (MO1 RR00039). The authors thank Tamela Yeargin for manuscript editing.
Funding: The authors declare no competing financial interests.
Footnotes
The original publication is available at www.springerlink.com.
References
- 1.Steinman RM, Nussenzweig MC. Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc Natl Acad Sci U S A. 2002;99:351–358. doi: 10.1073/pnas.231606698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sozzani S, Allavena P, Vecchi A, Mantovani A. The role of chemokines in the regulation of dendritic cell trafficking. J Leukoc Biol. 1999;66:1–9. doi: 10.1002/jlb.66.1.1. [DOI] [PubMed] [Google Scholar]
- 3.Turley SJ, Inaba K, Garrett WS, Ebersold M, Unternaehrer J, Steinman RM, Mellman I. Transport of peptide-MHC class II complexes in developing dendritic cells. Science (New York, NY) 2000;288:522–527. doi: 10.1126/science.288.5465.522. [DOI] [PubMed] [Google Scholar]
- 4.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nature immunology. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 5.Pulendran B. Variegation of the immune response with dendritic cells and pathogen recognition receptors. J Immunol. 2005;174:2457–2465. doi: 10.4049/jimmunol.174.5.2457. [DOI] [PubMed] [Google Scholar]
- 6.Kawai T, Akira S. Innate immune recognition of viral infection. Nature immunology. 2006;7:131–137. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- 7.Schroder M, Bowie AG. TLR3 in antiviral immunity: key player or bystander? Trends in immunology. 2005;26:462–468. doi: 10.1016/j.it.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 8.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 9.Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, Krzysiek R, Knutson KL, Daniel B, Zimmermann MC, David O, Burow M, Gordon A, Dhurandhar N, Myers L, Berggren R, Hemminki A, Alvarez RD, Emilie D, Curiel DT, Chen L, Zou W. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nature medicine. 2003;9:562–567. doi: 10.1038/nm863. [DOI] [PubMed] [Google Scholar]
- 10.Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nature medicine. 1999;5:1365–1369. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 11.Sharpe AH, Freeman GJ. The B7-CD28 superfamily. Nat Rev Immunol. 2002;2:116–126. doi: 10.1038/nri727. [DOI] [PubMed] [Google Scholar]
- 12.Wang S, Chen L. Co-signaling molecules of the B7-CD28 family in positive and negative regulation of T lymphocyte responses. Microbes and infection / Institut Pasteur. 2004;6:759–766. doi: 10.1016/j.micinf.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 13.Thompson RH, Gillett MD, Cheville JC, Lohse CM, Dong H, Webster WS, Krejci KG, Lobo JR, Sengupta S, Chen L, Zincke H, Blute ML, Strome SE, Leibovich BC, Kwon ED. Costimulatory B7-H1 in renal cell carcinoma patients: Indicator of tumor aggressiveness and potential therapeutic target. Proc Natl Acad Sci U S A. 2004;101:17174–17179. doi: 10.1073/pnas.0406351101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsushima F, Yao S, Shin T, Flies A, Flies S, Xu H, Tamada K, Pardoll DM, Chen L. Interaction between B7-H1 and PD-1 determines initiation and reversal of T-cell anergy. Blood. 2007;110:180–185. doi: 10.1182/blood-2006-11-060087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–151. doi: 10.1016/s1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 16.Carter LL, Leach MW, Azoitei ML, Cui J, Pelker JW, Jussif J, Benoit S, Ireland G, Luxenberg D, Askew GR, Milarski KL, Groves C, Brown T, Carito BA, Percival K, Carreno BM, Collins M, Marusic S. PD-1/PD-L1, but not PD-1/PD-L2, interactions regulate the severity of experimental autoimmune encephalomyelitis. Journal of neuroimmunology. 2007;182:124–134. doi: 10.1016/j.jneuroim.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 17.Keir ME, Francisco LM, Sharpe AH. PD-1 and its ligands in T-cell immunity. Current opinion in immunology. 2007;19:309–314. doi: 10.1016/j.coi.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 18.Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, Sasayama S, Mizoguchi A, Hiai H, Minato N, Honjo T. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science (New York, NY) 2001;291:319–322. doi: 10.1126/science.291.5502.319. [DOI] [PubMed] [Google Scholar]
- 19.Goldberg MV, Maris CH, Hipkiss EL, Flies AS, Zhen L, Tuder RM, Grosso JF, Harris TJ, Getnet D, Whartenby KA, Brockstedt DG, Dubensky TW, Jr, Chen L, Pardoll DM, Drake CG. Role of PD-1 and its ligand, B7-H1, in early fate decisions of CD8 T cells. Blood. 2007;110:186–192. doi: 10.1182/blood-2006-12-062422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hervas-Stubbs S, Olivier A, Boisgerault F, Thieblemont N, Leclerc C. TLR3 ligand stimulates fully functional memory CD8+ T cells in the absence of CD4+ T-cell help. Blood. 2007;109:5318–5326. doi: 10.1182/blood-2006-10-053256. [DOI] [PubMed] [Google Scholar]
- 21.Hasan UA, Caux C, Perrot I, Doffin AC, Menetrier-Caux C, Trinchieri G, Tommasino M, Vlach J. Cell proliferation and survival induced by Toll-like receptors is antagonized by type I IFNs. Proc Natl Acad Sci U S A. 2007;104:8047–8052. doi: 10.1073/pnas.0700664104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lombardi G, Dunne PJ, Scheel-Toellner D, Sanyal T, Pilling D, Taams LS, Life P, Lord JM, Salmon M, Akbar AN. Type 1 IFN maintains the survival of anergic CD4+ T cells. J Immunol. 2000;165:3782–3789. doi: 10.4049/jimmunol.165.7.3782. [DOI] [PubMed] [Google Scholar]
- 23.Marrack P, Kappler J, Mitchell T. Type I interferons keep activated T cells alive. The Journal of experimental medicine. 1999;189:521–530. doi: 10.1084/jem.189.3.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gelman AE, Zhang J, Choi Y, Turka LA. Toll-like receptor ligands directly promote activated CD4+ T cell survival. J Immunol. 2004;172:6065–6073. doi: 10.4049/jimmunol.172.10.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carter L, Fouser LA, Jussif J, Fitz L, Deng B, Wood CR, Collins M, Honjo T, Freeman GJ, Carreno BM. PD-1:PD-L inhibitory pathway affects both CD4(+) and CD8(+) T cells and is overcome by IL-2. Eur J Immunol. 2002;32:634–643. doi: 10.1002/1521-4141(200203)32:3<634::AID-IMMU634>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 26.Okazaki T, Iwai Y, Honjo T. New regulatory co-receptors: inducible co-stimulator and PD-1. Current opinion in immunology. 2002;14:779–782. doi: 10.1016/s0952-7915(02)00398-9. [DOI] [PubMed] [Google Scholar]
- 27.Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, Iwai Y, Long AJ, Brown JA, Nunes R, Greenfield EA, Bourque K, Boussiotis VA, Carter LL, Carreno BM, Malenkovich N, Nishimura H, Okazaki T, Honjo T, Sharpe AH, Freeman GJ. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nature immunology. 2001;2:261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 28.Rodig N, Ryan T, Allen JA, Pang H, Grabie N, Chernova T, Greenfield EA, Liang SC, Sharpe AH, Lichtman AH, Freeman GJ. Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur J Immunol. 2003;33:3117–3126. doi: 10.1002/eji.200324270. [DOI] [PubMed] [Google Scholar]
- 29.Mazanet MM, Hughes CC. B7-H1 is expressed by human endothelial cells and suppresses T cell cytokine synthesis. J Immunol. 2002;169:3581–3588. doi: 10.4049/jimmunol.169.7.3581. [DOI] [PubMed] [Google Scholar]
- 30.Lee MS, Kim YJ. Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annual review of biochemistry. 2007;76:447–480. doi: 10.1146/annurev.biochem.76.060605.122847. [DOI] [PubMed] [Google Scholar]
- 31.Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. 2004;173:945–954. doi: 10.4049/jimmunol.173.2.945. [DOI] [PubMed] [Google Scholar]
- 32.Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, Linsley PS, Thompson CB, Riley JL. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25:9543–9553. doi: 10.1128/MCB.25.21.9543-9553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wesch D, Beetz S, Oberg HH, Marget M, Krengel K, Kabelitz D. Direct costimulatory effect of TLR3 ligand poly(I:C) on human gamma delta T lymphocytes. J Immunol. 2006;176:1348–1354. doi: 10.4049/jimmunol.176.3.1348. [DOI] [PubMed] [Google Scholar]
- 34.Brown DM, Roman E, Swain SL. CD4 T cell responses to influenza infection. Semin Immunol. 2004;16:171–177. doi: 10.1016/j.smim.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 35.Bennett SR, Carbone FR, Karamalis F, Flavell RA, Miller JF, Heath WR. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 1998;393:478–480. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- 36.Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nature medicine. 2004;10:1366–1373. doi: 10.1038/nm1140. [DOI] [PubMed] [Google Scholar]
- 37.Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci U S A. 2001;98:13866–13871. doi: 10.1073/pnas.231486598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, Qiu Y, Jussif JM, Carter LL, Wood CR, Chaudhary D. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 2004;574:37–41. doi: 10.1016/j.febslet.2004.07.083. [DOI] [PubMed] [Google Scholar]
- 39.Hoebe K, Janssen EM, Kim SO, Alexopoulou L, Flavell RA, Han J, Beutler B. Upregulation of costimulatory molecules induced by lipopolysaccharide and double-stranded RNA occurs by Trif-dependent and Trif-independent pathways. Nature immunology. 2003;4:1223–1229. doi: 10.1038/ni1010. [DOI] [PubMed] [Google Scholar]
- 40.Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–548. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- 41.Wang S, Bajorath J, Flies DB, Dong H, Honjo T, Chen L. Molecular modeling and functional mapping of B7-H1 and B7-DC uncouple costimulatory function from PD-1 interaction. The Journal of experimental medicine. 2003;197:1083–1091. doi: 10.1084/jem.20021752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tseng SY, Otsuji M, Gorski K, Huang X, Slansky JE, Pai SI, Shalabi A, Shin T, Pardoll DM, Tsuchiya H. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. The Journal of experimental medicine. 2001;193:839–846. doi: 10.1084/jem.193.7.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maier H, Isogawa M, Freeman GJ, Chisari FV. PD-1:PD-L1 interactions contribute to the functional suppression of virus-specific CD8+ T lymphocytes in the liver. J Immunol. 2007;178:2714–2720. doi: 10.4049/jimmunol.178.5.2714. [DOI] [PubMed] [Google Scholar]
- 44.Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, Bessette B, Boulassel MR, Delwart E, Sepulveda H, Balderas RS, Routy JP, Haddad EK, Sekaly RP. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nature medicine. 2006;12:1198–1202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- 45.Zhang JY, Zhang Z, Wang X, Fu JL, Yao J, Jiao Y, Chen L, Zhang H, Wei J, Jin L, Shi M, Gao GF, Wu H, Wang FS. PD-1 up-regulation is correlated with HIV-specific memory CD8+ T-cell exhaustion in typical progressors but not in long-term nonprogressors. Blood. 2007;109:4671–4678. doi: 10.1182/blood-2006-09-044826. [DOI] [PubMed] [Google Scholar]
- 46.Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, Mncube Z, Duraiswamy J, Zhu B, Eichbaum Q, Altfeld M, Wherry EJ, Coovadia HM, Goulder PJ, Klenerman P, Ahmed R, Freeman GJ, Walker BD. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 47.Petrovas C, Casazza JP, Brenchley JM, Price DA, Gostick E, Adams WC, Precopio ML, Schacker T, Roederer M, Douek DC, Koup RA. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. The Journal of experimental medicine. 2006;203:2281–2292. doi: 10.1084/jem.20061496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Urbani S, Amadei B, Tola D, Massari M, Schivazappa S, Missale G, Ferrari C. PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. J Virol. 2006;80:11398–11403. doi: 10.1128/JVI.01177-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen L, Zhang Z, Chen W, Zhang Z, Li Y, Shi M, Zhang J, Chen L, Wang S, Wang FS. B7-h1 up-regulation on myeloid dendritic cells significantly suppresses T cell immune function in patients with chronic hepatitis B. J Immunol. 2007;178:6634–6641. doi: 10.4049/jimmunol.178.10.6634. [DOI] [PubMed] [Google Scholar]
- 50.Hokey DA, Larregina AT, Erdos G, Watkins SC, Falo LD., Jr Tumor cell loaded type-1 polarized dendritic cells induce Th1-mediated tumor immunity. Cancer Res. 2005;65:10059–10067. doi: 10.1158/0008-5472.CAN-05-1692. [DOI] [PubMed] [Google Scholar]
- 51.Cui Z, Qiu F. Synthetic double-stranded RNA poly(I:C) as a potent peptide vaccine adjuvant: therapeutic activity against human cervical cancer in a rodent model. Cancer Immunol Immunother. 2005:1–13. doi: 10.1007/s00262-005-0114-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yarilina A, DiCarlo E, Ivashkiv LB. Suppression of the effector phase of inflammatory arthritis by double-stranded RNA is mediated by type I IFNs. J Immunol. 2007;178:2204–2211. doi: 10.4049/jimmunol.178.4.2204. [DOI] [PubMed] [Google Scholar]
- 53.Hansen BS, Hussain RZ, Lovett-Racke AE, Thomas JA, Racke MK. Multiple toll-like receptor agonists act as potent adjuvants in the induction of autoimmunity. Journal of neuroimmunology. 2006;172:94–103. doi: 10.1016/j.jneuroim.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 54.Touil T, Fitzgerald D, Zhang GX, Rostami A, Gran B. Cutting Edge: TLR3 stimulation suppresses experimental autoimmune encephalomyelitis by inducing endogenous IFN-beta. J Immunol. 2006;177:7505–7509. doi: 10.4049/jimmunol.177.11.7505. [DOI] [PubMed] [Google Scholar]
- 55.Salama AD, Chitnis T, Imitola J, Ansari MJ, Akiba H, Tushima F, Azuma M, Yagita H, Sayegh MH, Khoury SJ. Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. The Journal of experimental medicine. 2003;198:71–78. doi: 10.1084/jem.20022119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, Koulmanda M, Freeman GJ, Sayegh MH, Sharpe AH. Tissue expression of PD-L1 mediates peripheral T cell tolerance. The Journal of experimental medicine. 2006;203:883–895. doi: 10.1084/jem.20051776. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.