

Abstract
We reported recently that the addition of ester-functionalized, ‘charge-shifting’ side chains to linear poly(ethyleneimine) (LPEI) can be used to design polyamines that promote both self-assembly and self-disassembly with DNA in aqueous environments. This investigation sought to characterize the influence of charge-shifting side chains on the ability of LPEI to mediate cell transfection and understand the extent to which increases (or decreases) in levels of transfection could be understood in terms of time-dependent changes in the net charges of these polymers. We report that the addition of ‘charge-shifting’ side chains to LPEI leads to significant increases in levels of LPEI-mediated transfection. In particular, polymer 1e, functionalized with 20 mol% ester-functionalized side chains, mediates levels of transgene expression in vitro up to eight-fold higher than LPEI. Experiments using an amide-functionalized analog of polymer 1e demonstrated that the esters in polymer 1e play an important role in promoting increased levels of transfection. These results, in combination with the results of additional gel electrophoresis experiments, provide support for the view that increases in transfection result from time-dependent changes in the net charge of polymer 1e and the disruption of ionic interactions in polyplexes. Additional support for this view is provided by the results of confocal microscopy experiments and measurements of fluorescence resonance energy transfer, which suggest that polymer 1e promotes the disruption of polyplexes in intracellular environments effectively. The approach reported here provides a means of addressing one important ‘late-stage’ obstacle to polyplex-mediated transfection (polyplex unpackaging). If integrated successfully with methods that have been developed to address other important barriers to transfection, this general approach could lead to the development of multifunctional polyplexes that mimic more effectively the range of functions of viruses as agents for the delivery of DNA.
Introduction
Cationic polymers can interact with DNA through electrostatic interactions to form nanometer-scale aggregates (called ‘polyplexes’) with sizes, charges, and other properties that render them useful for the delivery of DNA to cells.1-3 The formation of polyplexes is spontaneous and entropically driven and can, in general, be used to (i) condense and compact large DNA constructs, (ii) provide protection against the degradation of DNA in biological environments, and (iii) help promote the internalization of DNA by cells. The use of cationic polymers to formulate polyplexes also confers several additional practical advantages in the context of cell transfection, because cationic polymers provide macromolecular scaffolds from which to incorporate (and, thus, co-deliver to cells) chemical or biological functionality that can be used to address or overcome other important intracellular barriers to DNA delivery. The incorporation of new design elements into the structures of cationic polymers has, over the last decade, resulted in significant progress toward the design of functional polyplexes that mimic several of the functions of viruses in their abilities to facilitate the transport of DNA to cells and direct the intracellular trafficking of DNA in vitro and in vivo.1-3
Despite significant advances toward the design of functional cationic polymers, several critical challenges remain with respect to the development and optimization of cationic polymers for DNA delivery.1-4 The work reported here was motivated by our interest in developing cationic polymers that address one specific ‘late-stage’ obstacle to polyplex-mediated transfection – the need for a cationic polymer to ultimately release (or dissociate from) DNA once it has reached a safe and suitable location within a cell. At a certain point in the transfection process (e.g., after a polyplex has been transported into the cytoplasm or nucleus of a cell), the cationic polymer component of a polyplex may no longer be needed and, in fact, may serve to hinder the access to DNA that is required for cellular machinery to process it. The fact that significant levels of gene expression are often observed upon the internalization of polyplexes formed using conventional cationic polymers demonstrates that DNA is ultimately made accessible to the transcriptional machinery of cells to at least some extent. Experimental evidence suggests, however, that polyplex ‘unpackaging’ is a relatively inefficient process,5,6 and it seems apparent that, in general, the development of new materials that promote or provide control over the disassembly of polymer/DNA complexes could lead to materials that promote higher levels of cell transfection.
There is currently no general consensus in the literature regarding the timing or the intracellular locations that might be optimal for the release of DNA from a polyplex. There is, however, a growing consensus that the same charge-based interactions that are used to promote polyplex formation also introduce inherent and fundamental charge-based obstacles to the subsequent disassembly of polyplexes in intracellular environments. Over the last several years, there has been an increase in reports on new materials and molecular-level principles that can be used to promote the disruption or disassembly of polyplexes.5,7-21 Several groups have reported, for example, that hydrolytically8,10-12,15-17,21 or reductively degradable9,13,14,19,20 polyamines can be used to form polyplexes that release DNA upon incubation in aqueous media or exposure to reducing environments, and that polymer molecular weight plays an important role in governing polyplex stability and disassembly.5,7,18 Other researchers have reported that controlled and permanent reductions in the charge densities of polymers such as branched poly(ethyleneimine) (BPEI) can be used to modulate charge-based polymer/DNA interactions in ways that result in significant increases in cell transfection in vitro.22,23 Here, we report that controlled and dynamic reductions in the net charge of linear poly(ethyleneimine) (LPEI) – achieved by the addition of hydrolyzable, ‘charge-shifting’ side chains to the polymer backbone – can be used to promote polyplex disassembly and increase levels of LPEI-mediated cell transfection significantly.
We24,25 and others26-29 have demonstrated recently that cationic polymers designed to undergo controlled and dynamic reductions in net charge can be used fabricate polyplexes24,26-29 and other ionic assemblies25 that disassemble and release DNA in physiologically relevant environments. Hennink and coworkers have reported in a series of recent studies, for example, a family of poly(methacrylate) derivatives bearing amine-functionalized side chains attached to the backbone through hydrolyzable carbonate linkages.26-28 Hydrolysis of the side chains of these polymers was reported to result in a loss of amine functionality and, as a result, a gradual reduction of the net charge of the polymer (e.g., from ‘cationic’ to ‘less cationic’). Polyplexes formulated from these ‘charge-shifting’ polymers were observed to (i) release DNA upon incubation in aqueous media in ways that could be understood in terms of time-dependent changes in the nature of the electrostatic interactions in these assemblies and (ii) mediate increased levels of cell transfection in vitro.26-28 More recent reports from our group25 and Shim and Kwon29 have demonstrated similar approaches to the design of ‘charge-shifting’ cationic polymers and the application of these materials to surface-mediated25 and polyplex-mediated29 DNA delivery.
We also reported recently a different approach to the design of ‘charge-shifting’ cationic polymers based on the addition of hydrolyzable ester functionality to the cationic backbone of LPEI (see polymer 1, Scheme 1).24 In contrast to the approach described above (for which reductions in net charge result from the loss of cationic charge), dynamic reductions in net charge using this approach result from the gradual introduction of negative charge resulting from ester hydrolysis. We demonstrated in an initial report that this approach could be used to promote the disassembly of polyplexes in a defined physiological buffer, and that it was possible to control the rates and extents to which disassembly occurred by varying the molar amount of ester functionality added to the polymer.24
Scheme 1.

Schematic illustration of a ‘charge-shifting’ cationic polymer (polymer 1). Top: Gradual hydrolysis of ester-functionalized side-chains introduces carboxylate functionality and reduces the net charge of the polymer. Relative changes in net charge shown here are exemplary and are provided for illustrative purposes only (see text). Bottom: Polymer 1 can self-assemble with DNA to form nanometer-scale ‘polyplexes’. Time-dependent changes in the net charge of polymer 1 result in changes in the nature of the ionic interactions in polyplexes and promote polyplex disassembly and/or the release of DNA.
Here, we report on the ability of ‘charge-shifting’ derivatives of polymer 1 to promote the delivery of DNA to cells. We demonstrate that this modular approach to the functionalization of LPEI can yield significant improvements in levels of transgene expression (e.g., up to eight-fold higher compared to unmodified LPEI) when used to transport DNA into cells in vitro. We demonstrate further that improved transfection results from the hydrolyzable nature of the side chains, and we report evidence based upon confocal microscopy experiments and measurements of fluorescence resonance energy transfer (FRET) that provide support for the view that appropriately functionalized polymers can promote polyplex disassembly in intracellular environments more effectively than LPEI. The modular nature of the approach reported here should facilitate the introduction of ‘charge-shifting’ character to other conventional gene delivery polymers and, if integrated successfully with methods that have been developed to address other important barriers to transfection, could lead to the development of multifunctional polyplexes with the ability to mimic more completely the range of functions of viruses as agents for the delivery of DNA.
Experimental Section
Materials and General Considerations
Linear poly(ethyleneimine) was synthesized by hydrolysis of poly(2-ethyloxazoline) [MW = 50,000; Polysciences, Inc., Warrington, PA] and purified prior to use in analogy to procedures described previously.24,30 The synthesis of polymers 1a-e, 2, and 3 (Mw ≈ 33,000; PDI = 3.23) was performed as described previously.24 Plasmid DNA encoding firefly luciferase [pCMV-Luc, >95% supercoiled] was obtained from Elim Biopharmaceuticals, Inc. (San Francisco, CA). Deionized water (18 MΩ) was used to prepare all buffers and salt solutions. Bicinchoninic acid (BCA) protein assay kits were purchased from Pierce (Rockford, IL). Steady-Glo Luciferase Assay Systems were purchased from Promega Corporation (Madison, WI). 3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) was purchased from Acros (Geel, Belgium). Cy3 and Cy5 Label-IT nucleic acid labeling kits were purchased from Mirus (Madison, WI). Labeling density was determined using an Ultraspec2100 Pro spectrophotometer (Amersham Biosciences, Piscataway, NJ). Hoechst 34580 and Calcein AM fluorescent stains were purchased from Invitrogen (Carlsbad, CA). CellScrub Buffer was purchased from Gene Therapy Systems (San Diego, CA). All commercial materials were used as received without further purification unless otherwise noted. Flow cytometric analyses were performed using a BD FACSCalibur flow cytometer (BD Bioscience, San Jose, CA). Glass inset dishes used for laser scanning confocal microscopy (LSCM) were purchased from MatTek (Ashland, MA). Laser scanning confocal microscopy was performed using a Bio-Rad Radiance 2100 MP Rainbow laser scanning confocal microscope equipped with a multiphoton laser. Images were processed using the Bio-Rad LaserSharp 2000 processing kit and Adobe Photoshop 8.0.
Formation of Polymer/DNA Complexes
DNA/polymer complexes were formed by adding 50 μL of a plasmid DNA solution (2 μg/50 μL in water) to a gently vortexing solution of polyamine (50 μL in either 20 mM HEPES, pH = 7.2 or 20 mM acetate buffer, pH = 5.0). For these experiments, the concentration of polymer in this volume of buffer was adjusted to yield a desired DNA/polymer weight ratio (e.g., 1:1, 1:2, 1:3, etc.). The resulting samples were incubated at room temperature for 30 minutes prior to biophysical characterization or use in transfection experiments.
Characterization of Polyplexes by Agarose Gel Electrophoresis
For experiments designed to characterize complexes by agarose gel electrophoresis, a 30 μL aliquot of each sample (prepared as described above) was mixed with a loading buffer and analyzed on a 1% agarose gel (HEPES, 20 mM, pH = 7.2, 108 V, 50 min). DNA bands were visualized by ethidium bromide staining. For experiments in which poly(acrylic acid) (PAA) was used as a competitive agent to determine the relative strength of interactions between DNA and the different polyamines, a solution of PAA was added to the samples of polyplexes prior to characterization to yield a desired polycation/PAA weight ratio (e.g., 1:0.5, 1:0.75, 1:1, etc.). Samples used to evaluate temporal changes in DNA/polymer interactions (see text) were prepared by doubling the amounts and volumes in the above protocol. These samples were incubated at 37 °C and aliquots were removed at desired times (typically 0 hrs, 24 hrs, and 48 hrs) for characterization by agarose gel electrophoresis.
Characterization of Polyplexes by Dynamic Light Scattering
Dynamic light scattering was performed using a 100mW, 532nm laser (Coherent Compass 315M-100) illuminating a temperature-controlled glass cell at 37 °C filled with a refractive-index matching fluid (decahydronaphthalene). The scattering of light was measured at an angle of 90°. Autocorrelation functions for the intensity of scattered light were collected using a BI-9000AT digital autocorrelator (Brookhaven Instruments Corporation, Holtsville, NY) and analyzed using the CONTIN software package31,32 to yield a distribution of particle sizes by assuming that the relaxation processes in solution correspond to center-of-mass diffusion. In a typical experiment, polyplexes prepared as described above (100 μL) were added to HEPES buffer (20 mM, pH = 7.2) or HEPES buffer (20 mM, pH = 7.2, [NaCl] = 150 mM) (total volume = 1 mL). Measurements of autocorrelation functions were recorded at 37 °C directly on these samples.
General Protocol for Cell Transfection Assays
COS-7 cells used in transfection experiments were grown in opaque polystyrene 96-well culture plates at initial seeding densities of 15,000 cells/well in 200 μL of growth medium (90% Dulbecco's modified Eagle's medium, 10% fetal bovine serum, penicillin 100 U/mL, streptomycin 100 μg/mL). All cells were incubated at 37 °C for 24 hr, after which the growth medium was removed and replaced with 200 μL of a serum free medium (OptiMEM). Formulations of DNA/polymer complexes (30 μL) prepared at desired pCMV-Luc/polyamine ratios were added to assigned wells via pipette, and the cells were placed in an incubator at 37 °C. After 4 hrs, polyplex-containing culture media was aspirated from all wells and replaced with 200 μL of fresh serum-containing medium. Cells were then incubated for an additional 48 hrs. Luciferase protein expression was determined using a commercially available luminescence-based luciferease assay kit using the manufacturer's specified protocol. Samples, in replicates of three, were normalized against total cell protein in each respective well using a commercially available BCA assay kit. Experiments designed to characterize the influence of polyplexes on cell metabolism and growth were conducted using a protocol identical to that described above, with the following exceptions: at a predetermined time (described below), 25 μL of a 5 mg/mL solution of MTT in sterile PBS buffer was added to each well. The samples were incubated at 37 °C for two hours, and 100 μL of extraction buffer (20% w/v SDS in DMF/water (1:1), pH = 4.7) was added to each well. Samples were incubated at 37 °C for 24 hours. Optical absorbance in each well was measured at 560 nm with a microplate reader and expressed as a percent relative to control cells. For characterization of the short-term effects of polyplexes, MTT was added immediately after the replacement of polyplex-containing culture media with fresh media. For characterization of the longer-term effects of polyplexes, MTT was added 48 hours after the replacement of polyplex-containing media. The statistical significance of results reported in the text was determined using Student's t-test.
Preparation of Fluorescently-Labeled Plasmid DNA
Plasmid DNA was labeled with fluorescent dyes using commercially available Label-IT nucleic acid labeling kits (Mirus, Madison, WI). For DNA used to characterize the internalization of polyplexes, DNA was labeled with Cy5 according to the manufacturer's protocol (labeling density ≈ 50 labels per plasmid). For DNA used in experiments designed to characterize changes in fluorescence resonance energy transfer (FRET), DNA was double-labeled with both Cy3 and Cy5 (labeling densities ≈ 1 Cy3 label: 1 Cy5 label: 200 base pairs). Labeled DNA was purified by ethanol precipitation, and labeling densities were determined using a UV-Vis spectrophotometer, as described by the manufacturer.
Characterization of Polyplex Internalization Efficiency
Experiments designed to characterize the influence of changes in polyamine structure on extents of internalization of polyplexes by cells were conducted in the following general manner. COS-7 cells were seeded in 12-well plates at initial seeding densities of 7.5 × 104 cells/mL in 1.0 mL of growth medium [90% (v/v) Dulbecco's modified Eagle's medium, 10% (v/v) fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin]. Cells were allowed to grow overnight to approximately 90% confluence, and growth medium was replaced with 1.0 mL of a serum-free culture medium (OptiMEM). A solution of polyplexes (100 μL) prepared using Cy5-labled DNA at desired DNA/polymer ratios was added to assigned wells via pipette. Cells were incubated for 4 hrs at 37 °C, polyplex-containing culture media was aspirated from all wells, and cells were washed using CellScrub Buffer (Gene Therapy Systems, San Diego, CA) according to the manufacturer's specified protocol. Washed cells were harvested by treatment with trypsin, collected in microcentrifuge tubes, washed twice with phosphate-buffered saline (PBS), and finally resuspended in PBS containing 1% bovine serum albumin (BSA). Cell samples were characterized by flow cytometry. Data were collected for populations of at least 10,000 cells and analyzed using WinMDI software (Joe Trotter, Scripps Institute). Results are expressed as the percentage of cells positive for Cy5 fluorescence relative to all cells observed.
Characterization of Cells by Confocal Microscopy and Observation of Changes in FRET
Experiments designed to characterize cells using laser scanning confocal microscopy (LSCM) and characterize changes in FRET were conducted in the following general manner. COS-7 cells were seeded in glass inset confocal dishes at initial seeding densities of 7.5 × 104 cells/mL in 2.0 mL of growth medium [90% (v/v) Dulbecco's modified Eagle's medium, 10% (v/v) fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin]. Cells were allowed to grow overnight to approximately 90% confluence, and growth medium was replaced with 2.0 mL of a serum-free culture medium (OptiMEM). Polyplexes were prepared at desired DNA/polymer ratios using plasmid DNA double-labeled with both Cy5 and Cy3. Solutions of these polyplexes (100 μL) were added to assigned dishes, and cells were incubated at 37 °C for 1.5 hrs. After incubation, polyplex-containing culture media was aspirated from all wells and cells were washed with CellScrub Buffer using the manufacturer's specified protocol. Fresh growth medium (2 mL) was then added to each dish and cells were incubated at 37 °C for additional desired time periods (typically 1 hr, 8 hrs and 24 hrs) before characterization by LSCM. Immediately prior to imaging, cells were stained using Calcein AM live cell stain and Hochest 34580 nuclear stain according to each manufacturer's protocols. LSCM images (optical thicknesses = 1.33 μm) were acquired using a 60X/1.40 NA oil immersion objective. Calcein AM, Cy3, and Hoechst probes were excited sequentially using laser lines at 488, 543, and 800 nm (multiphoton laser), respectively. Fluorescence emission signals were collected for three individual channels using direct scanning mode (N = 1, scan speed = 50 lps). For experiments designed to characterize changes in FRET, Cy3 was excited and the emission of Cy3 (at 570 nm) and the emission of Cy5 (at 670 nm) were detected simultaneously. Changes in FRET were determined by monitoring and comparing changes in the emission of Cy5 over time.
Results and Discussion
We reported recently that the conjugate addition of the secondary amines of LPEI to methyl acrylate could be used to functionalize LPEI with side chains terminated with a hydrolyzable methyl ester group (e.g., polymer 1).24 This synthetic approach is modular and permits broad control over the mole percentage of ‘charge-shifting’ ester functionality incorporated into the polymer structure. In both our past study and this current study, we evaluated derivatives of polymer 1 having approximately 100, 80, 60, 40, and 20 mol% side chain substitution (referred to hereafter as polymers 1a-e, respectively). In the paragraph below, we summarize briefly several important results arising from our past investigation of polymers 1a-e in defined physiological media to provide a framework for interpreting the results of this current investigation.
Our past studies demonstrate that the ester groups in polymers 1a-e hydrolyze gradually when incubated in aqueous media at 37 °C and that potential amide-forming reactions between backbone secondary amines and ester side chains do not occur under these conditions.24 Characterization of polyplexes formed from plasmid DNA and polymers 1a-d by agarose gel electrophoresis demonstrated that DNA was released upon the incubation of polyplexes in HEPES buffer (pH = 7.2). The results of these experiments provided a basis from which to understand structure/property relationships for these charge-shifting materials. In general, polymers 1a and 1b (which are functionalized at higher mole percentages and, thus, able to undergo larger shifts in net charge upon ester hydrolysis) released DNA more completely and more rapidly than polymers such as 1c and 1d (which are functionalized at lower mole percentages). Polymer 1e, functionalized at 20 mol%, did not release DNA under the conditions used in this past study. In addition, polyplexes formulated using an analog of polymer 1 having non-hydrolyzable, amide-functionalized side chains did not release DNA. The results of these experiments, when combined, provide support for the view that the hydrolyzable ester groups in polymer 1 play important roles in regulating the stability of polyplexes incubated in aqueous media. Additional specific results arising from these past studies can be found in a past publication.24 Finally, we note here for clarity that the relative change in the net charge of the polymer shown in Scheme 1 is exemplary and provided for illustrative purposes only. In general, the overall net charges of polymer 1 would depend specifically upon mol% substitution and the extent of ester hydrolysis, as well as additional environmental factors such as pH and ionic strength. We return to each of the above considerations again in the discussion below.
Our past studies demonstrate that the addition of charge-shifting side chains to LPEI can be used to promote both self-assembly and self-disassembly with DNA in defined physiological buffers. In this current investigation, we sought to characterize (i) the influence of the addition of charge-shifting side chains on the ability of LPEI to mediate cell transfection, (ii) the extent to which any observed increases (or decreases) in levels of transgene expression could be understood in terms of time-dependent changes in the net charges of these polymers, and (iii) the extent to which these charge-shifting polymers are able to promote the self-disassembly of polyplexes in intracellular environments.
Characterization of Transgene Expression Using Polyplexes Formulated from Polymers 1a-e
We performed a series of initial screening experiments using the COS-7 cell line and polyplexes formed from polymers 1a-e and a plasmid DNA construct (pCMV-Luc) encoding firefly luciferase to quantify relative differences in the abilities of these materials to transfect cells in vitro. These initial experiments were conducted using polyplexes of each polymer formed at ten different DNA/polymer ratios (w/w) ranging from 1:1 to 1:10. Polyplexes were incubated in the presence of cells in a serum-free growth medium for four hours, after which growth medium was replaced with fresh serum-containing growth medium. Fifty-two hours after cells were first exposed to polyplexes, levels of luciferase expression were characterized quantitatively using a luminescence-based protocol. Figure 1 shows levels of luciferase expression (expressed as relative light units normalized to total cell protein) for polyplexes formulated using polymers 1a-e and LPEI at the optimal DNA/polymer ratio determined to mediate the highest levels of transfection for each polymer in this initial screen.
Figure 1.
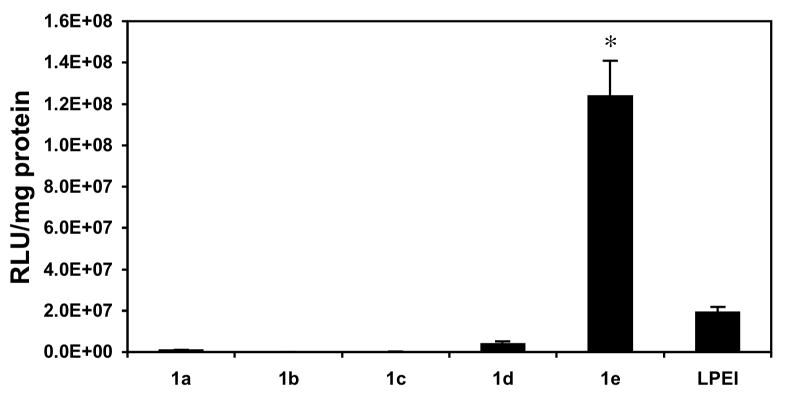
Normalized luciferase expression (expressed as relative light units per mg of total cell protein) in COS-7 cells treated with polyplexes formed using ester-functionalized polymers 1a-e or LPEI. Cells were treated with polyplexes in serum-free media for four hours, followed by incubation in serum-containing media for 48 hours. These experiments were conducted in replicates of three using complexes formed at ten different DNA/polymer ratios (w/w) ranging from 1:1 to 1:10. Values of normalized luciferase expression shown correspond to the DNA/polymer ratio determined to mediate the highest levels of transfection for each polymer and correspond to ratios of 1:1 (for polymers 1a, 1c, 1e, and LPEI), 1:9 (for polymer 1b), and 1:7 (for polymer 1d) and are reported as averages with standard deviation. Figure S1 of the Supporting Information shows a plot of these data using a log scale on the y-axis. Results are presented as an average of three experiments ± standard deviation. Results marked with an asterisk (*) indicate results determined to be statistically different from all others (p<0.05) using Student's t-test.
Inspection of Figure 1 reveals large differences in the levels of transgene expression mediated by polymers 1a-e. These data demonstrate that polymers 1a-d, corresponding to polymers substituted with ester-functionalized side chains at percent substitutions of 100%, 80%, 60%, and 40%, do not mediate high levels of gene expression relative to LPEI. Further inspection of these data, however, reveals that polymer 1e, having 20 mol% substitution, mediates levels of luciferase expression that are ∼six-fold higher than LPEI. These differences in levels of transfection were observed consistently in multiple different repetitions of this experiment. In general, we observed polymer 1e to mediate levels of transfection ranging from five-fold to eight-fold higher than LPEI under these conditions.
We considered several potential explanations for the large differences in the behavior of polymers 1a-e in the experiments above. Characterization of polyplex sizes by dynamic light scattering (at the specific DNA/polymer ratios used to generate the data shown in Figure 1) in HEPES buffer revealed average particle sizes ranging from ∼75 nm to ∼150 nm (see Figure S2, Supporting Information). The sizes of these polyplexes fall generally within the range of sizes considered to be suitable for the internalization of polyplexes by endocytosis.2-4,33 However, characterization of polyplexes immediately after addition to HEPES buffer containing 150 mM NaCl (used here to approximate the ionic strength of serum-free cell culture media) revealed changes in polyplex size and/or structure. Autocorrelation functions collected on solutions of polyplexes formed from polymer 1a were not sufficient to calculate particle sizes, suggesting that these polyplexes were disrupted significantly under these higher ionic strength conditions. Particle sizes measured for polyplexes formed using less-substituted polymers 1b-e were larger than those measured at lower ionic strength, but generally within the range of ∼100 nm to ∼250 nm in size (Figure S2, Supporting Information). These results suggest that differences in transfection observed using polymers 1b-e do not arise principally from differences in the initial sizes of the polyplexes or from the formation of larger-scale aggregates. Characterization of the influence of polyplexes on cell metabolism and growth using an MTT assay demonstrated that the elevated levels of transfection mediated by polymer 1e did not result from differences in the cytotoxicities of polymer 1e and LPEI (see Figure S3, Supporting Information).
We also considered the possibility that polyplex unpackaging and the release of DNA from polyplexes formed using charge-shifting polymers 1c-e could occur sufficiently rapidly upon incubation that it could influence the rates and/or extents to which polyplexes are internalized by cells (or, possibly, prevent polyplex internalization altogether). The results of our past studies demonstrate that polyplexes formulated from polymers 1c-e did not release observable amounts of DNA over the first four hours of incubation in defined HEPES buffer.24 It remains possible, however, that ester hydrolysis and polyplex disassembly could occur more rapidly in the presence of cells, or that changes in polyplex size or surface charge (zeta potential) upon incubation in cell culture media could influence the rates and extents to which these polyplexes are internalized over the four-hour exposure period used in this current study. Characterization of polyplex internalization efficiencies directly using flow cytometry and polyplexes formulated using fluorescently labeled DNA demonstrated large differences in the extents of internalization of polyplexes that correlated with the large differences in transfection shown in Figure 1.
Table 1 shows the results of flow cytometry experiments using polyplexes formulated using Cy5-labeled plasmid at the specific DNA/polymer ratios used in Figure 1 (percent internalization of polyplexes is expressed as the percent of Cy5-positive cells relative to all cells interrogated, and geometric mean fluorescence intensities of Cy5-positive populations are shown for comparison). These data demonstrate that polyplexes formed from polymers 1d, 1e, and LPEI are internalized much more efficiently (and to much greater extents) than polyplexes formed from polymers 1a-c. The very low percentages of internalization of polyplexes formed from polymers 1a-c are consistent with the notion that polyplex unpackaging could occur prematurely for these more highly substituted materials (e.g., as induced by changes in ionic strength or through hydrolysis-mediated disassembly, as discussed above). We caution, however, that these experiments do not rule out other possibilities (such as more subtle changes in polyplex size or charge upon incubation in the presence of cells) and that additional experiments will be required to evaluate the physical behavior of polyplexes formed from polymers 1a-c in cell culture media more completely. In the context of this current study, we do conclude that the addition of a low mole percentage of charge-shifting side chains (e.g., 20 mol%, polymer 1e) yields significant increases in polyplex-mediated gene expression relative to unmodified LPEI. In the following sections, we focus our attention specifically on the evaluation and characterization of the behavior of polyplexes formed from polymer 1e.
Table 1. Characterization of polyplex internalization.a.
| Polymer Usedb | Percent Internalization of Polyplexesc | Geometric Mean Fluorescenced |
|---|---|---|
| 1a | 1.89% (±0.06%) | 46.5 (±0.9) |
| 1b | 1.46% (±0.06%) | 45.0 (±1.9) |
| 1c | 21.53% (±0.89%) | 49.8 (±0.4) |
| 1d | 96.13% (±1.16%) | 452.7 (±5.3) |
| 1e | 92.88% (±0.43%) | 219.7 (±1.0) |
| LPEI | 90.42% (±1.30%) | 225.7 (±2.3) |
Based on flow cytometry experiments using COS-7 cells and polyplexes formed using Cy-5 labeled DNA (see main text and Materials and Methods for additional experimental details).
Polyplexes were prepared at polymer/DNA ratios (w/w) identical to those shown in Figure 1, and correspond to those ratios determined to mediate the highest levels of transfection for each polymer (1:1 for polymers 1a, 1c, 1e, and LPEI; 1:9 for polymer 1b; 1:7 for polymer 1d.)
Values are expressed as the percent of Cy5-positive cells relative to all cells interrogated.
Geometric mean fluorescence intensity of Cy5-positive cells gated against negative controls.
Investigation of the Importance of Ester-Functionalized Side Chains and ‘Charge-Shifting’ Character on Levels of Cell Transfection
The results above demonstrate that the addition of low mole percentages of ester-functionalized side chains to LPEI can improve cell transfection efficiency significantly. These results are consistent with the notion that changes in the charge state of polymer 1e, resulting from ester hydrolysis, could contribute to the destabilization of internalized polyplexes and to increased levels of transgene expression. To provide further support for this proposition and investigate the role of the esters in polymer 1e more directly, we performed an additional series of transfection experiments using polyplexes formed from polymers 2 and 3. Polymer 2 is an analog of polymer 1e functionalized at 20 mol% with amide-functionalized side chains that do not hydrolyze readily in physiologically relevant media. Polymer 3 is a derivative of polymer 1e that was completely hydrolyzed to present carboxylic acid-functionalized side chains. Figure 2 shows a comparison of levels of luciferase expression for polyplexes formulated using either LPEI or polymers 1e, 2, or 3 (formulated at the DNA/polymer ratio found to be optimal for each polymer, as discussed above).
Figure 2.
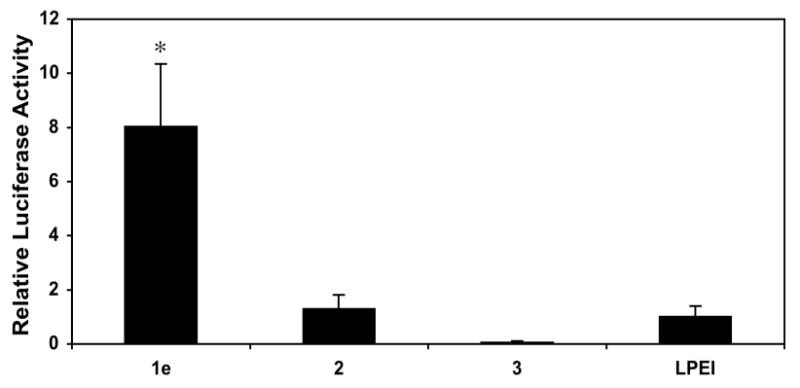
Plot of relative levels of normalized luciferase expression in COS-7 cells mediated by polyplexes formulated using polymers 1e, 2, 3, and LPEI. Relative levels of expression are normalized to the transfection efficiency of LPEI. Transfection experiments were conducted in replicates of three the manner described for data collected in Figure 1. Values of normalized luciferase expression used correspond to those obtained using the DNA/polymer ratio (w/w) determined to mediate the highest levels of transfection for each polymer (1:1 for polymers 1e, 3, and LPEI; 1:3 for polymer 2) and are reported as averages with standard deviation. Results are presented as an average of three experiments ± standard deviation. Results marked with an asterisk (*) indicate results determined to be statistically different from all others (p<0.05) using Student's t-test.

Characterization of the efficiency of internalization of polyplexes by flow cytometry demonstrated that polyplexes formulated using amide-functionalized polymer 2 were internalized at levels [95.55% (±0.92%)] that were similar to those observed using polymer 1e and LPEI (described above). However, inspection of Figure 2 reveals large differences in the levels of luciferase expression mediated by these three materials. In particular, we note that polyplexes formed using amide-functionalized polymer 2 mediated levels of normalized luciferase expression that were approximately eight-fold lower than those mediated by polymer 1e, and that were, in general, similar to levels of expression mediated by unmodified LPEI. These results, when combined, demonstrate that (i) the enhanced gene delivery activity of polymer 1e does not arise simply from the conjugate addition of short side chains to LPEI, and (ii) the ester groups in polymer 1e play an important role in promoting transgene expression. Because the amide-functionalized groups present in polymer 2 should not hydrolyze readily in an intracellular environment, these results also provide support for the view that the elevated levels of transgene expression mediated by polymer 1e occur through a hydrolysis-based ‘charge-shifting’ process that results in time-dependent changes in the nature of the ionic interactions in these polyplexes.
Further inspection of Figure 2 reveals that polyplexes formulated using carboxylic acid-substituted polymer 3 mediate very low levels of transgene expression. Characterization of the efficiency of internalization of polyplexes formed using polymer 3 by flow cytometry demonstrated that polyplexes were internalized by cells at very low levels [2.76% (±0.06%)]. These results provide one clear explanation for the low levels of transfection observed using this polymer. These results also hint, however, at the importance of designing a ‘charge-shifting’ polymer such as polymer 1e that is capable of undergoing a gradual shift (rather than an abrupt shift) in net charge – that is, it is likely not simply the final charge state of the polymer that is important, but also the rates and extents to which charge-shifting and polyplex disassembly are made to occur. Polyplexes formed from polymers with side chains that hydrolyze or undergo a change in charge state too rapidly could disassemble in extracellular environments or reduce transfection efficiency by unpackaging in undesirable (or less desirable) intracellular locations.
Characterization of Release of DNA and Evidence for the Disruption of Polyplexes in Intracellular Environments
The results above are consistent with a process in which the time-dependent hydrolysis of the side chains of polymer 1e leads to changes in polyplex stability and, as a result, either the intracellular release of DNA or DNA that is made more accessible to the transcriptional machinery of the cell. We performed several additional experiments to determine the extent to which polyplexes formed using polymer 1e become destabilized upon side chain hydrolysis and, subsequently, whether it was possible to characterize the disruption of polyplexes or the release of DNA in intracellular environments.
In our past investigation of polyplexes of polymer 1 and plasmid DNA, we used an agarose gel electrophoresis gel shift assay to characterize extents of time-dependent polyplex disruption upon incubation in HEPES buffer (pH = 7.2, 37 °C).24 We note here that the results of this past study demonstrated that polyplexes formed using polymer 1e did not release observable levels of DNA after incubation under these conditions for up to 48 hours (as determined by visual inspection of ethidium bromide-stained gels). We note further, however, that conclusions based on these past measurements are limited strictly to those regarding changes in the nature of the interactions between DNA and polymer 1e in a defined buffer system, and that these results may not reflect the extent to which polyplex disruption could occur in more complex media or in an intracellular environment. Several past studies have suggested, for example, that the intracellular disassembly of polyplexes formed using conventional (non-charge-shifting) cationic polymers could be aided by competitive interactions with negatively charged macromolecular components of the intracellular milieu.34-37
To characterize more completely any time-dependent differences in the relative strength of binding interactions between DNA and polymer 1e or unmodified LPEI, we conducted a series of gel electrophoresis experiments in which polyplexes formed from these polymers were incubated in the presence of varying amounts of poly(acrylic acid) (PAA). PAA and other anionic polymers have been used in several past studies to characterize relative binding strengths between conventional cationic polymers and DNA.23,36,38 Figure 3 shows the results of gel electrophoresis assays conducted using polyplexes formed from LPEI or polymer 1e (prepared at DNA/polymer ratios (w/w) of 1:1, as used in the transfection experiments above) and incubated in HEPES buffer (pH = 7.2) at 37 °C in (i) the absence of PAA or (ii) the presence of increasing concentrations of PAA for 0, 24, or 48 hours.
Figure 3.
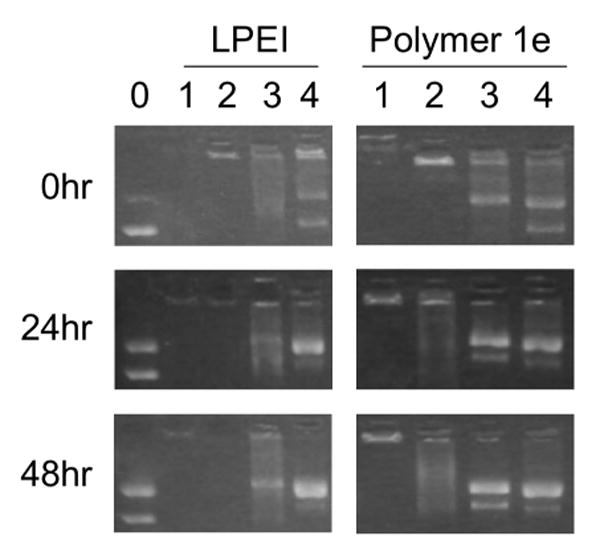
Agarose gel electrophoresis experiments showing time courses for the release of DNA from polyplexes formed using LPEI and polymer 1e at 0, 24, or 48 hours. All polyplexes were formed at DNA/polymer ratios (w/w) of 1:1, as used in transfection experiments and described in the text. The leftmost lane (lane 0) corresponds to a plasmid DNA control (no polymer added). Lanes labeled 1-4 in each column correspond to polyplexes incubated in the presence of increasing amounts of PAA. DNA/polymer/PAA ratios (w/w) for each lane are 1:1:0 (for lanes marked 1), 1:1:0.5 (for lanes marked 2), 1:1:0.75 (for lanes marked 3), and 1:1:1 (for lanes marked 4). The DNA control in lane 0 (shown for experiments using LPEI) also serves as the internal DNA control for experiments using polymer 1e. These experiments were conducted using the same gel but have been separated in the figure to enhance clarity.
The data in the left column of Figure 3 correspond to experiments using polyplexes formed from LPEI. Inspection of these data reveals that polyplexes formed using LPEI do not release DNA (that is, DNA is retarded and unable to migrate into the gel) for up to 48 hours when incubated in the absence of PAA (lane 1) or the presence of low amounts of PAA [lane 2, DNA/LPEI/PAA = 1:1:0.5 (w/w/w)]. The data in lanes 3 and 4 of this column, however, demonstrate that DNA is released from polyplexes upon incubation in the presence of higher LPEI/PAA ratios (lanes 3 and 4 correspond to DNA/LPEI/PAA ratios of 1:1:0.75 and 1:1:1, respectively). These data contrast to the data shown in the right column of Figure 3, which correspond to experiments using polyplexes formed using polymer 1e. These data demonstrate that DNA is not released when polyplexes are incubated for up to 48 hours in the absence of PAA (lane 1), but that DNA is released – and that it is released in a manner that is time-dependent – when polyplexes are incubated at DNA/1e/PAA ratios as low as 1:1:0.5 (e.g., lane 2, middle column). When combined, these data demonstrate that there are significant differences in the relative strengths of the ionic interactions in polyplexes formed from DNA and either LPEI or polymer 1e. In particular, these data demonstrate that the interactions between LPEI and DNA are stronger than those between polymer 1e and DNA, and that the nature of the interactions between polymer 1e and DNA changes upon incubation in a manner that is time-dependent.
We note that the results shown in Figure 3 reveal significant time-dependent reductions in the supercoiled content of both the control DNA (lane 0) and the DNA released from polyplexes (lanes 1-4) and an increase in the relative amounts of open-circular, or nicked, plasmid. These changes in plasmid structure likely result, at least in part, from hydrolysis of the phosphodiester bonds in DNA that occurs upon the prolonged incubation of DNA in aqueous media. We note further, however, that changes in plasmid structure appear to be more significant for DNA released from polyplexes formed from either LPEI or polymer 1e (lanes 1-4) as compared to naked DNA (lane 0; incubated in the absence of polymer). These observations are consistent with observations reported previously and described in full in our past study on the incubation and release of DNA from polyplexes formed using polymer 1.24 Additional experiments will be required to understand the origins of this behavior and the extent to which it could also occur under conditions used during the transfer of DNA to cells. Because the conversion of supercoiled plasmid DNA to an open-circular topology could influence levels of transgene expression, the potential for this form of plasmid damage to occur should be borne in mind with respect to the continued development of these new materials as agents for the delivery of DNA.24
The results shown in Figure 3 are consistent with our ‘charge-shifting’ hypothesis and suggest that differences in the levels of transfection mediated by polymer 1e and LPEI (e.g., in Figures 1 and 2) could result from the ability of polyplexes formed from polymer 1e to unpack more efficiently in complex media where competitive interactions are present or possible. We next performed a series of experiments using confocal microscopy and polyplexes formed using fluorescently labeled DNA to determine whether it was possible to observe evidence of the unpackaging of polyplexes formed from polymer 1e in intracellular environments.
Several groups have reported recently that it is possible to characterize changes in the strength of interactions between cationic polymers and DNA in intracellular environments by observing changes in fluorescence resonance energy transfer (FRET) in appropriately-labeled polyplexes.23,39-42 These past studies have made use of fluorescently-labeled polyplexes prepared using three basic approaches: (i) using polyplexes formed from DNA and polymer each labeled individually with either the donor or the acceptor of a known FRET pair,41,42 (ii) using polyplexes formed using mixtures of polymer samples labeled individually with donors and acceptors,23 or by (iii) using polyplexes formed from unlabeled polymer and plasmid DNA that is double-labeled with both the donor and the acceptor of a known FRET pair.39,40 Itaka et al. recently demonstrated that this third approach, in combination with laser scanning confocal microscopy (LSCM), could be used to observe changes in FRET upon the internalization of polyplexes formed using LPEI,40 and that these changes in FRET could be interpreted in terms of changes in the strength of interactions between LPEI and DNA (that is, polyplex formation brings DNA-associated donors and acceptors in closer proximity and increases FRET, whereas weaker interactions result in an increase in the distance between donors and acceptors and, as a result, a decrease in FRET).39,40 In this current study, we used polyplexes formed using DNA double-labeled with both Cy3 (donor) and Cy5 (acceptor) fluorescent labels. We selected this method on the basis of the results of Itaka et al. described above and because this method does not require chemical or structural modifications that could influence the physicochemical behavior of the cationic polymer component of our polyplexes. We note here that Cy3 and Cy5 are used frequently as a donor/acceptor pair to characterize FRET in physiological media.43
Figure 4 shows representative LSCM results for experiments conducted using polyplexes formed using LPEI or polymer 1e at DNA/polymer ratios of 1:1 (as used in the transfection experiments described above). For these experiments, COS-7 cells were incubated in the presence of polyplexes for 1.5 hours, after which growth medium was replaced with serum-containing growth medium. Figure 4A shows the results of cells characterized by LSCM one hour after the removal of polyplexes. The images in Column I of Figure 4A (green channel) correspond to cells labeled with calcein AM, a whole-cell stain added prior to imaging to identify the locations of cells in these experiments. The images in Columns II and III correspond to cells excited at a wavelength of 543 nm (the excitation wavelength of Cy3) and observed simultaneously at wavelengths of 570 nm (the emission wavelength of Cy3; Column II, false-colored red) and 670 nm (the emission wavelength of Cy5; Column III, false-colored yellow). The presence of numerous red punctate fluorescent spots in nearly all cells in Column II (i) demonstrates that DNA is internalized efficiently by cells treated with polyplexes formed from both LPEI and polymer 1e, and (ii) suggests that these polyplexes are likely sequestered in endosomes or lysosomes. These results are, in general, consistent with the results of our flow cytometry-based experiments, described above. The images in Column III show the presence of punctate yellow spots in each cell arising from FRET from Cy3 to Cy5 and establish a baseline from which to characterize time-dependent changes in FRET at longer incubation times.
Figure 4.
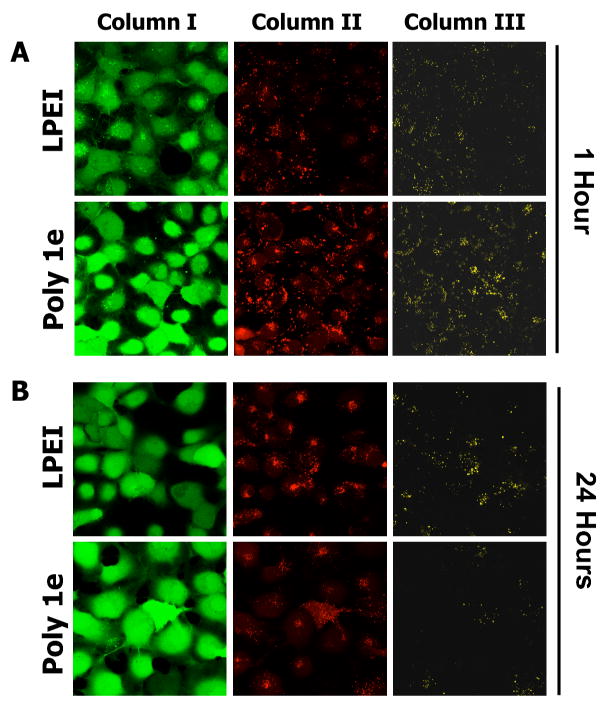
Laser scanning confocal microscopy images showing time-dependent changes in FRET in COS-7 cells treated with polyplexes formed using LPEI or polymer 1e. A) Cells imaged one hour after internalization of polyplexes. B) Cells imaged 24 hours after internalization of polyplexes. Polyplexes were formed using DNA double-labeled using Cy3 and Cy5 dyes at DNA/polymer ratios (w/w) of 1:1, as described in the text. Cells were stained with calcein AM prior to imaging (Column I, green channel) to aid in locating cells. Columns II and III are false-colored images showing relative levels of Cy3 and Cy5 emission, respectively, when cells were excited at a wavelength of 543 nm (the excitation wavelength of Cy3). Observation of yellow fluorescence in Column III results from FRET from Cy3 to Cy5 (see text).
Figure 4B shows representative LSCM images of cells described above after incubation for an additional 24 hours. Inspection of the images in Column III reveals significant differences in the levels of Cy5 fluorescence associated with cells treated with polyplexes formed using LPEI and polymer 1e. Whereas a large number of yellow punctate structures (arising from FRET from Cy3 to Cy5) were still observed in cells treated with polyplexes formed using LPEI, significantly less FRET was observed in cells treated with polyplexes formed using polymer 1e. Because the Cy3 and Cy5 labels are both conjugated to the DNA in these experiments, reductions in FRET can be interpreted to arise from changes in polyplex structure that result in the release of DNA or in DNA that is more loosely bound by polymer (that is, DNA that becomes less compacted or less condensed will result in an increase in the average distance between Cy3 and Cy5 labels and, as a result, a reduction in FRET).39,40 We note that the time-dependent reduction in FRET observed using polyplexes formed from polymer 1e could also arise from the rapid degradation of DNA in these experiments. The rapid degradation of DNA is not consistent, however, with the dramatic increases in transfection using polymer 1e observed in Figures 1 and 2 above, and, as a result, is a less likely explanation for the reductions in FRET observed in these experiments.
The time-dependent reductions in FRET shown in Figure 4B for polyplexes formed using polymer 1e were observed in multiple different repetitions of this experiment, and suggest that this polymer can promote the disruption of polyplexes in intracellular environments effectively. Although we cannot rule out other potential contributions to the enhanced gene delivery activity of polymer 1e, the results of these confocal microscopy experiments, in combination with the results of the other experiments described above, provide additional support for the view that the addition of hydrolyzable, ester-functionalized side chains to LPEI increases levels of transfection through a ‘charge-shifting’ mechanism that promotes more the effective intracellular dissociation of polyplexes and greater access to DNA.
Summary and Conclusions
The extent to which charge-based interactions between cationic polymers and DNA can (or cannot) be controlled underlies the success (or failure) of many polymer-based methods for gene delivery. We recently reported that the addition of ester-functionalized, ‘charge-shifting’ side chains to LPEI can be used to design polyamines that promote both self-assembly and time-dependent self-disassembly with DNA in defined physiological environments.24 In this current investigation, we demonstrated that the addition of ‘charge-shifting’ side chains to LPEI can be used to increase levels of LPEI-mediated cell transfection significantly. In particular, polymer 1e, functionalized with 20 mol% ester-functionalized side chains, mediates levels of transgene expression in vitro up to eight-fold higher relative to polyplexes formed using LPEI. Transfection experiments using amide-functionalized polymer 2 demonstrated that the esters in polymer 1e play an important role in promoting increased levels of transfection. These results, in combination with the results of additional gel electrophoresis experiments, provide support for the view that observed increases in levels of transgene expression can be understood in terms of time-dependent changes in the net charge of polymer 1e, the subsequent disruption of polyplexes, and greater access to DNA. Additional support for this view is provided by the results of confocal microscopy experiments and measurements of fluorescence resonance energy transfer, which suggest that polymer 1e is able to promote the disruption or disassembly of polyplexes in intracellular environments effectively.
Our results demonstrate proof-of-concept and illustrate the potential of this new approach. However, several additional important points deserve comment and provide additional directions for future investigations. First, our results demonstrate clearly that the amount of charge-shifting functionality incorporated is critical to promoting high levels of cell transfection. As discussed briefly in the introduction to this paper, there is, as yet, no general consensus regarding the timing or the intracellular locations that might be considered optimal for the release of DNA from a polyplex. It is clear, however, that polymers that undergo reductions in charge state too rapidly could disassemble prematurely in extracellular environments or, alternatively, lead to low levels of transfection by unpackaging in undesirable intracellular locations. In this context, the observation of increases in transfection reported using polymer 1e in this current study are somewhat serendipitous – that is, polymer 1e was not designed to target a specific time course or deposit DNA in any specific intracellular location. We note, however, that new methods such as those reported here could provide a platform from which to begin to address these important issues. For example, although we used hydrolyzable methyl esters in this current study, the use of other, more bulky or chemically diverse ester functionality could be used to tune or provide temporal control over charge-shifting and polyplex disassembly and, as a result, further increase levels of cell transfection.
Finally, the work reported here was motivated by our interest in developing new principles and new polymers that can be used to address one single ‘late-stage’ obstacle to polyplex-mediated transfection (polyplex unpackaging). We note, however, that polyplex-mediated transfection is, in fact, a multistep process and that, ultimately, polyplexes must be endowed with functionality capable of addressing numerous different extracellular and intracellular obstacles to gene delivery.1-4 In our current investigation, we used LPEI as a scaffold from which to incorporate new ‘charge-shifting’ functionality because LPEI is a well-studied and effective gene delivery vector that is already capable of addressing several other barriers to transfection.44 The modular nature of the synthetic approach reported here should, however, facilitate the introduction of ‘charge-shifting’ character to a variety of other polyamines used for gene delivery. If integrated successfully with methods that have been developed to address other important barriers to transfection, this general approach could lead to the development of multifunctional polyplexes that mimic more effectively the range of functions of viruses as agents for the delivery of DNA.
Supplementary Material
Supporting Information Available. Plots of additional data, including results of light scattering and cytotoxicity experiments. This information is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
Financial support was provided by the Arnold and Mabel Beckman Foundation, the National Institutes of Health (1 R21 EB002746 and 1 R01 EB006820), and the University of Wisconsin. We are grateful to the NSF (CHE-9208463) and the NIH (NIH 1 S10 RR0 8389-01) for support of the UW NMR spectroscopy facilities. We thank Ryan Flessner and Chris Jewell for many helpful discussions and for technical assistance with confocal microscopy experiments. D. M. L. is an Alfred P. Sloan Research Fellow.
References
- 1.Luo D, Saltzman WM. Nat Biotechnol. 2000;18:33–37. doi: 10.1038/71889. [DOI] [PubMed] [Google Scholar]
- 2.Pack DW, Hoffman AS, Pun S, Stayton PS. Nat Rev Drug Discov. 2005;4:581–593. doi: 10.1038/nrd1775. [DOI] [PubMed] [Google Scholar]
- 3.Putnam D. Nat Mater. 2006;5:439–451. doi: 10.1038/nmat1645. [DOI] [PubMed] [Google Scholar]
- 4.Wolff JA. Nat Biotechnol. 2002;20:768–769. doi: 10.1038/nbt0802-768. [DOI] [PubMed] [Google Scholar]
- 5.Schaffer DV, Fidelman NA, Dan N, Lauffenburger DA. Biotechnol Bioeng. 2000;67:598–606. doi: 10.1002/(sici)1097-0290(20000305)67:5<598::aid-bit10>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 6.Suh J, Wirtz D, Hanes J. P Natl Acad Sci USA. 2003;100:3878–3882. doi: 10.1073/pnas.0636277100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.MacLaughlin FC, Mumper RJ, Wang JJ, Tagliaferri JM, Gill I, Hinchcliffe M, Rolland AP. J Control Release. 1998;56:259–272. doi: 10.1016/s0168-3659(98)00097-2. [DOI] [PubMed] [Google Scholar]
- 8.Lynn DM, Langer R. J Am Chem Soc. 2000;122:10761–10768. [Google Scholar]
- 9.Gosselin MA, Guo W, Lee RJ. Bioconjugate Chem. 2001;12:989–94. doi: 10.1021/bc0100455. [DOI] [PubMed] [Google Scholar]
- 10.Lim YB, Kim SM, Lee Y, Lee WK, Yang TG, Lee MJ, Suh H, Park JS. J Am Chem Soc. 2001;123:2460–2461. doi: 10.1021/ja005715g. [DOI] [PubMed] [Google Scholar]
- 11.Lynn DM, Anderson DG, Putnam D, Langer R. J Am Chem Soc. 2001;123:8155–8156. doi: 10.1021/ja016288p. [DOI] [PubMed] [Google Scholar]
- 12.Lim YB, Kim SM, Suh H, Park JS. Bioconjugate Chem. 2002;13:952–7. doi: 10.1021/bc025541n. [DOI] [PubMed] [Google Scholar]
- 13.Pichon C, LeCam E, Guerin B, Coulaud D, Delain E, Midoux P. Bioconjugate Chem. 2002;13:76–82. doi: 10.1021/bc015503o. [DOI] [PubMed] [Google Scholar]
- 14.Oupicky D, Parker AL, Seymour LW. J Am Chem Soc. 2002;124:8–9. doi: 10.1021/ja016440n. [DOI] [PubMed] [Google Scholar]
- 15.Anderson DG, Lynn DM, Langer R. Angew Chem Int Edit. 2003;42:3153–3158. doi: 10.1002/anie.200351244. [DOI] [PubMed] [Google Scholar]
- 16.Akinc A, Lynn DM, Anderson DG, Langer R. J Am Chem Soc. 2003;125:5316–5323. doi: 10.1021/ja034429c. [DOI] [PubMed] [Google Scholar]
- 17.Forrest ML, Koerber JT, Pack DW. Bioconjugate Chem. 2003;14:934–940. doi: 10.1021/bc034014g. [DOI] [PubMed] [Google Scholar]
- 18.Koping-Hoggard M, Varum KM, Issa M, Danielsen S, Christensen BE, Stokke BT, Artursson P. Gene Ther. 2004;11:1441–1452. doi: 10.1038/sj.gt.3302312. [DOI] [PubMed] [Google Scholar]
- 19.Christensen LV, Chang CW, Kim WJ, Kim SW, Zhong ZY, Lin C, Engbersen JFJ, Feijen J. Bioconjugate Chem. 2006;17:1233–1240. doi: 10.1021/bc0602026. [DOI] [PubMed] [Google Scholar]
- 20.Lin C, Zhong ZY, Lok MC, Jiang XL, Hennink WE, Feijen J, Engbersen JFJ. Bioconjugate Chem. 2007;18:138–145. doi: 10.1021/bc060200l. [DOI] [PubMed] [Google Scholar]
- 21.Jain R, Standley SM, Frechet JMJ. Macromolecules. 2007;40:452–457. [Google Scholar]
- 22.Forrest ML, Meister GE, Koerber JT, Pack DW. Pharm Res. 2004;21:365–371. doi: 10.1023/b:pham.0000016251.42392.1e. [DOI] [PubMed] [Google Scholar]
- 23.Gabrielson NP, Pack DW. Biomacromolecules. 2006;7:2427–2435. doi: 10.1021/bm060300u. [DOI] [PubMed] [Google Scholar]
- 24.Liu XH, Yang JW, Miller AD, Nack EA, Lynn DM. Macromolecules. 2005;38:7907–7914. [Google Scholar]
- 25.Zhang JT, Lynn DM. Adv Mater. 2007;19:4218–4223. [Google Scholar]
- 26.Funhoff AM, van Nostrum CF, Janssen APCA, Fens MHAM, Crommelin DJA, Hennink WE. Pharm Res. 2004;21:170–176. doi: 10.1023/b:pham.0000012165.68765.e6. [DOI] [PubMed] [Google Scholar]
- 27.Luten J, Akeroyd N, Funhoff A, Lok MC, Talsma H, Hennink WE. Bioconjugate Chem. 2006;17:1077–1084. doi: 10.1021/bc060068p. [DOI] [PubMed] [Google Scholar]
- 28.Jiang X, Lok MC, Hennink WE. Bioconjugate Chem. 2007;18:2077–2084. doi: 10.1021/bc0701186. [DOI] [PubMed] [Google Scholar]
- 29.Shim MS, Kwon YJ. Biomacromolecules. 2008 doi: 10.1021/bm7007313. in press. [DOI] [PubMed] [Google Scholar]
- 30.Brissault B, Kichler A, Guis C, Leborgne C, Danos O, Cheradame H. Bioconjugate Chem. 2003;14:581–587. doi: 10.1021/bc0200529. [DOI] [PubMed] [Google Scholar]
- 31.Provencher SW. Comput Phys Commun. 1982;27:213–227. [Google Scholar]
- 32.Provencher SW. Comput Phys Commun. 1982;27:229–242. [Google Scholar]
- 33.Zauner W, Ogris M, Wagner E. Adv Drug Deliver Rev. 1998;30:97–113. doi: 10.1016/s0169-409x(97)00110-5. [DOI] [PubMed] [Google Scholar]
- 34.Okuda T, Niidome T, Aoyagi H. J Control Release. 2004;98:325–332. doi: 10.1016/j.jconrel.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 35.Huth S, Hoffmann F, von Gersdorff K, Laner A, Reinhardt D, Rosenecker J, Rudolph C. J Gene Med. 2006;8:1416–1424. doi: 10.1002/jgm.975. [DOI] [PubMed] [Google Scholar]
- 36.Bertschinger M, Backliwal G, Schertenleib A, Jordan M, Hacker DL, Wurm FM. J Control Release. 2006;116:96–104. doi: 10.1016/j.jconrel.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 37.Iida T, Mori T, Katayama Y, Niidome T. J Control Release. 2007;118:364–369. doi: 10.1016/j.jconrel.2006.12.027. [DOI] [PubMed] [Google Scholar]
- 38.Trubetskoy VS, Wong SC, Subbotin V, Budker VG, Loomis A, Hagstrom JE, Wolff JA. Gene Ther. 2003;10:261–271. doi: 10.1038/sj.gt.3301888. [DOI] [PubMed] [Google Scholar]
- 39.Itaka K, Harada A, Nakamura K, Kawaguchi H, Kataoka K. Biomacromolecules. 2002;3:841–845. doi: 10.1021/bm025527d. [DOI] [PubMed] [Google Scholar]
- 40.Itaka K, Harada A, Yamasaki Y, Nakamura K, Kawaguchi H, Kataoka K. J Gene Med. 2004;6:76–84. doi: 10.1002/jgm.470. [DOI] [PubMed] [Google Scholar]
- 41.Breunig M, Lungwitz U, Liebl R, Goepferich A. Eur J Pharm Biopharm. 2006;63:156–165. doi: 10.1016/j.ejpb.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 42.Ho YP, Chen HH, Leong KW, Wang TH. J Control Release. 2006;116:83–89. doi: 10.1016/j.jconrel.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sekar RB, Periasamy A. J Cell Biol. 2003;160:629–633. doi: 10.1083/jcb.200210140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kircheis R, Wightman L, Wagner E. Adv Drug Deliver Rev. 2001;53:341–358. doi: 10.1016/s0169-409x(01)00202-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available. Plots of additional data, including results of light scattering and cytotoxicity experiments. This information is available free of charge via the Internet at http://pubs.acs.org.