

Abstract
Paramyxoviruses are negative strand non-segmented RNA viruses. Several members of this family constitute major human pathogens that, collectively, are responsible for major morbidity and mortality worldwide. In an effort to ultimately develop novel therapeutics against measles virus (MV), a prominent member of the paramyxovirus family, we report a high-throughput screening protocol that allows hit identification using non-recombinant primary MV strains as targets. Implementation of the assay has yielded 60 hit candidates from a 137,500-entry library. Counterscreening and generation of dose-response curves narrows this pool to 35 compounds with active concentrations ≤15.3 μM against the MV-Alaska strain and specificity indices ranging from 36 to >500. Library mining for structural analogs of several confirmed hits combined with re-testing of identified candidates reveals a low false-negative rate and, thus, a high accuracy of primary hit identification. Eleven of the confirmed hits were found to interfere with the viral entry machinery, while the remaining 24 compounds target post-entry steps of the viral life cycle. Activity testing against selected members of the paramyxovirus family reveals three patterns of activity: 1) exclusively MV-specific blockers; 2) inhibitors of MV and related viruses of the same genus; 3) broader-range inhibitors with activity against a different paramyxovirinae genus. Representatives of the last class may open avenues for the development of broad-range paramyxovirus inhibitors through hit-to-lead chemistry.
Keywords: high-throughput screening, drug discovery, paramyxovirus, anti-infective drugs
Introduction
Members of the paramyxovirus family, enveloped, non-segmented negative strand RNA viruses, constitute major human and animal pathogens. Most paramyxoviruses are highly contagious airborne pathogens that spread via the respiratory route. Measles virus, a representative of the paramyxovirus family, in particular is one of the most infectious viruses identified (Griffin 2001; Hethcote 2000; van den Hof et al. 2002). Despite enhanced efforts for global implementation of a live-attenuated vaccine, MV is a principle cause of morbidity, and infection results in approximately 300,000 to 400,000 deaths annually worldwide, (CDC 2005; Wolfson et al. 2007), rendering the virus the leading cause of childhood death from a vaccine-preventable disease globally (CDC 2005).
Due to the exceptionally high infectivity of MV, a herd immunity of >95% is required to suppress endemic transmission (Griffin et al. 2008). Low vaccination coverage in parts of the developing world and insufficient or declining herd immunity in several developed countries, largely driven by parental concerns about vaccination safety, contribute to continued MV activity. In addition to a substantial immunosuppression that lasts several months, complications associated with MV infection include acute encephalitis and subacute sclerosing panencephalitis (SSPE), a late lethal sequela that manifests itself years after the primary infection (Hilleman 2001).
Currently, Ribavirin is the only drug approved for the treatment of some paramyxovirus infections (Chakrabarti et al. 2001; Shigeta et al. 1992). It has been used experimentally against MV but with limited efficacy (Barnard 2004). Thus, no therapeutic strategy is available to rapidly control local outbreaks or improve case management of severe measles.
In an effort to augment the existing vaccination program through small molecule antivirals that block MV, we have in previous work developed a protocol for automated screening for large compound libraries (White et al. 2007). Implementation of the protocol yielded compound AS-136A, a first-in-class non-nucleoside inhibitor of MV RNA-dependent RNA polymerase (RdRp) complex activity (Sun et al. 2007; White et al. 2007), providing overall proof-of-concept for our approach. However, limitations of this pilot protocol include dependence on a laboratory-adapted MV recombinant as target that stably expresses enhanced green fluorescent protein (eGFP), since blocked expression of virus-encoded eGFP served as the primary readout. This excludes direct screening of non-attenuated wild type viral isolates as target and confines identifiable hits to inhibitors of early steps of the viral life cycle, i.e. entry into target cells or RdRp-driven expression of viral proteins. In contrast, inhibitors of later stages of the viral life cycle such as particle assembly and egress are unlikely to be identified.
The precedence set by highly active antiretroviral therapy demonstrates that combined administration of antivirals with different target sites may boost efficacy and reduce the rate of viral escape from inhibition (Bartlett et al. 2001; Bartlett et al. 2006; Murphy et al. 2001). Moreover, the high potential of inhibitors of particle assembly for antiviral therapy has been demonstrated for a variety of different viral targets (Baba 2004; Garcia et al. 2003; Stray and Zlotnick 2006; Yang et al. 2005; Zhou et al. 2006). The identification of MV inhibitors distinct from the RdRp blocker AS-136A is thus desirable.
We report here an HTS protocol that permits screening against non-attenuated wild type MV strains and is equally suitable for the identification of compounds that interfere with different phases of the viral life cycle ranging from entry to particle assembly and release. Scale-up of the protocol has identified a structurally diverse panel of hit candidates. Following confirmation through independent secondary assays, confirmed hits were tested for interference with the entry versus post-entry phase of the viral life cycle. In parallel, overlapping cross-resistance with compound AS-136A and target specificity was assessed.
Materials and Methods
Cell culture, transfection and production of virus stocks
All cell lines were maintained at 37°C and 5% CO2 in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. Vero (African green monkey kidney epithelial) cells (ATCC CCL-81) stably expressing human SLAM (Vero-SLAM cells (Ono et al. 2001)) or canine SLAM (Vero-dogSLAM cells (Seki et al. 2003)) were incubated at every third passage in the additional presence of G-418 (Geneticin) at a concentration of 100 μg/ml. Lipofectamine 2000 (Invitrogen) was used for transient transfection experiments. All experimentation with life virus was conducted under BSL2 biosafety conditions. To prepare stocks of wild type MV strain Alaska (MVi/Alaska.USA/16.00 (Rota et al. 2002)) or CDV strain 752 (kind gift of S. Niewiesk), Vero or Vero-dogSLAM cells, respectively, were infected at a multiplicity of infection (MOI) of 0.001 plaque-forming units (pfu)/cell and incubated at 37°C. Cells were scraped in OPTI-MEM (Invitrogen), virus released by two freeze-thaw cycles, and titers determined by 50% tissue culture infective dose (TCID50) titration according to the Spearman-Karber method (Spearman 1908) as previously described (White et al. 2007). To prepare stocks of modified vaccinia virus Ankara expressing T7 polymerase (MVA-T7, (Sutter et al. 1995), DF-1 cells (ATCC CRL-12203) were infected at an MOI of 1.0 pfu/cell and cell-associated viral particles harvested 40 hours post-infection.
High throughput screening
For screening, Vero-SLAM cells were seeded in 384-well clear bottom black wall microtiter plates at a density of 1,500 cells per well in 30 μl growth medium. After a 12-hour incubation period at 37°C and 5% CO2, compound was added in 0.1 μl/well doses (3.3 μM final concentration) with a Beckman NX automated liquid handler, followed by infection with MV-Alaska at an MOI of 0.2 pfu/cell in 20 μl serum-free medium. Final solvent (DMSO) concentrations were 0.2% at which no adverse effect on cell viability or virus growth could be detected in control samples. Following a 72-hour incubation period at 37°C, media was removed from all wells and cells stained with 0.1% crystal violet in 20% ethanol for two hours at room temperature. Plates were washed, air-dried and absorbance at 560 nm quantified using an Envision Multilabel microplate reader (Perkin Elmer). Absorbance values were then transformed into % of control by normalizing them to the absorbance of control wells on each plate. To evaluate individual compounds, primary HTS scores were calculated based on the formula: primary HTS score = (% control compound - Mean % control)/SD % control. As control compound and to validate the assay, the MV RdRp inhibitor AS-136A and an inactive analog of AS-136A were included on each plate in eight replicates each. For validation, z’ values were calculated according to the formula z’ = 1-(3SD(C)+3SD(B))/(Mean(C)-Mean(B)), with C: control and B: background (Zhang et al. 1999). The compound library used is a diversity set obtained from the NIH/MLSCN (Molecular Library Screening Center Network).
Generation of dose-response inhibition curves for counterscreening
Two different assays, based on suppression of viral CPE and reduction of virus yields, were employed to assess the sensitivity of MV to candidate compounds. For dose-response curves based on suppression of CPE, cells (Vero-SLAM for MV-Alaska; Vero-dogSLAM for CDV 752; Vero for HPIV3) were infected in two replicates per compound concentration in a 96-well plate format at an MOI of 0.2 pfu/cell in the presence of compound in two-fold dilutions with starting concentrations of 37.5 μM (concentration range examined 0.15 μM to 37.5 μM). At 96 hours post-infection, virus-induced CPE was quantified at 560 nm through staining of cells with crystal violet. For each compound concentration, viral CPE was then calculated according to the formula [% CPE = 100-(experimental-background)/(maximum-background)*100], in which “background” refers to wells containing growth media only and “maximum” refers to solvent-only treated mock-infected wells. Fifty-percent effective concentrations (EC50) at which viral CPE was reduced by 50% were then determined from the dose-response curves using Excel software (MS Office package).
Quantification of compound cytotoxicity
To determine cytotoxicity, all confirmed hit compounds were subjected to a quantitative cytotoxicity assay (CytoTox 96 Non-Radioactive Cytotoxicity Assay, Promega). In a 96-well plate format, 12,000 cells per well were incubated at 37°C for 24 hours (to ensure active growth of mock-treated cells in control wells) in four replicates per concentration tested in the presence of a range of compound concentrations in 2-fold dilutions (150 μM highest). A tetrazolium salt (INT) substrate (20 μl/well) was then added and conversion into a colored formazan product by cellular lactate dehydrogenase quantified at 490 nm using a BioRad plate reader. Values were calculated according to the formula [% viability = 100-((experimental-background)/(maximum-background)*100)], in which “background” refers to wells containing only growth media and “maximum” constitutes solvent-only treated cells.
Chemical synthesis of selected hit candidates
Selected hit candidates were re-synthesized as depicted in figures 2A and 2B.
Figure 2.

Chemical synthesis pathways of two promising confirmed hit compounds, 14722514 (A) and 22407448 (B).
Quantitative cell-to-cell fusion assay
To quantify fusion activity, an effector Vero cell population (1.1 × 105 cells/well; 24-well plate format) was co-transfected with 2 μg each of MV H and F glycoprotein expression plasmid (Cathomen et al. 1998), and target Vero cells (6 × 105 cells/well; 6-well plate format) were transfected with 4 μg of the reporter plasmid encoding firefly luciferase under the control of the T7 promoter. Single-transfections of H-encoding plasmid served as controls. Two hours post-transfection, the effector cells were infected with MVA-T7 at an MOI of 1.0 pfu/cell and 200 μM (final concentration) fusion inhibitory peptide (FIP) was added. Following incubation for 12-16 hours at 30°C, target cells were detached, overlaid on PBS-washed effector cells at a (1:1)-ratio and incubated at 37°C in the presence or absence of candidate compounds as indicated. Five hours post-overlay, cells were lysed using Bright Glo Lysis Buffer (Promega) and the luciferase activity determined using a luminescence counter (PerkinElmer) and the Britelite Reporter Gene Assay System (PerkinElmer). The instrument’s arbitrary values were analyzed by subtracting relative background provided by values of the controls, and these values were normalized against solvent-only (DMSO) treated reference samples.
Results
To screen for inhibitors of non-attenuated wild type MV isolates, a cell-based HTS strategy was developed on the basis of a previously reported protocol that relies on an attenuated, recombinant viral variant expressing eGFP (Duprex et al. 1999; Ehrengruber et al. 2001).
Assay evaluation confirms suitability for automated screening
To establish a screen against non-attenuated viral isolates, a new protocol was developed that directly quantifies suppression of the cytopathic effect (CPE) associated with MV infection. An active blocker preserves intact cell monolayers through inhibition of viral CPE, which is readily visualized through staining of fixed cells with crystal violet 72 hours post-infection. Inactive compounds or substances with intrinsic cytotoxicity result in reduced staining intensity due to the breakdown of the cell monolayer. This protocol was scaled-up to a 384-well plate format and then evaluated for its suitability for automated screening using compound AS-136A, the previously identified inhibitor of MV polymerase activity, as positive control (figure 1). This exercise returned z’ values of 0.71 and a signal to background (S/B) ratio of 10.2, indicating that the protocol is appropriate for automated hit identification.
Figure 1.

HTS-assay set-up and evaluation. Cells seeded in a 384-well plate format were infected with wild-type MV strain Alaska at an MOI of 0.2 when reaching a density of 3,000/well. Prior to infection, wells were treated with the MV RdRp inhibitor AS-136A (final concentration 5 μM) or an equal volume of solvent (DMSO) as indicated. Mock-infected wells received equal volumes of media and DMSO, and media control wells (w/o) lacked target cells. Plates were fixed and stained with crystal violet 72 hours post-infection and absorbance at 560 nm determined. Two representative wells each are shown, numbers below the wells reflect averages ± SD of 16 replicates.
Primary HTS and counterscreening reveals 35 confirmed hit candidates
In an implementation of the protocol, 137,500 compounds of the NIH/MLSCN collection were screened at a final concentration of 3.3 μM. On all plates, mock-infected wells and wells treated with AS-136A or an inactive analog of AS-136A were included as internal standards. Automated absorbance reading returned a short-list of 2070 compounds with a standard score of ≥1.9. Overall z’ values were 0.72 and a signal to background (S/B) ratio of 15.3 corresponding closely to the value obtained at assay evaluation. Since crystal violet staining is permanent, these wells were re-examined visually to verify the results of the automated reading and ensure presence of an undisturbed, homogenous cell monolayer. The combined exercise resulted in the identification of 60 distinct hit candidates that passed both examinations, equaling a primary hit identification rate of ∼0.04% (table 1).
Table 1.
Hit identification through primary HTS and counterscreening
| PubChem ID (SID) | aStructure | Primary HTS Score | bEC50 [μM] (MV-Alaska) | cCC50 [μM] | SI (CC50/EC50) |
|---|---|---|---|---|---|
| 14735307 |

|
6.62 | d3.3 ± 0.9 | >150 | >56 |
| 4242806 |

|
7.48 | 1.4 ± 0.2 | 147 | 105 |
| 22407448 |
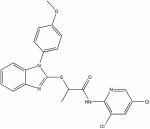
|
1.93 | d0.2 ± 0.1 | >150 | >500 |
| 22404943 |
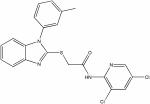
|
3.9 | 0.7 ± 0.1 | >150 | >214 |
| 22407466 |

|
6.55 | 2.1 ± 0.1 | >150 | >71 |
| 22415419 |

|
10.02 | 4.5 ± 0.9 | >150 | >33 |
| 22415706 |

|
9.45 | e6.7 ± 0.3 | ND | ND |
| 4265637 |
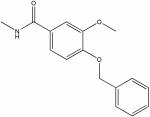
|
10.08 | 3 ± 0.2 | >150 | >50 |
| 14743644 |

|
5.9 | 2.7 ± 0.5 | ∼150 | 56 |
| 3711163 |
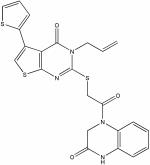
|
5.76 | f4.6 ± 0.7 | ND | ND |
| 17505573 |
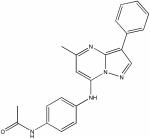
|
8.63 | 3.8 ± 0.5 | 54 | 14 |
| 4245209 |
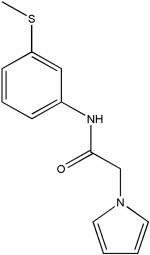
|
8.61 | e5.9 ± 1.7 | ND | ND |
| 14723513 |

|
13.68 | 3.7 ± 0.1 | >150 | >41 |
| 22400459 |
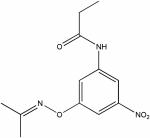
|
8.24 | e5.4 ± 1.2 | ND | ND |
| 22402685 |
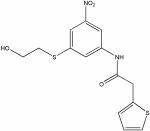
|
8.86 | 1.9 ± 0.2 | >150 | >79 |
| 17416393 |
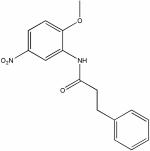
|
9.72 | 1.5 ± 0.3 | >150 | >100 |
| 846635 |
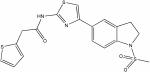
|
9.45 | 2.7 ± 0.2 | ∼150 | 56 |
| 852230 |

|
6.97 | 3.3 ± 0.1 | ∼150 | 46 |
| 22410899 |
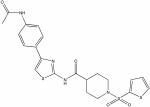
|
6.58 | 3.9 ± 0.1 | >150 | >39 |
| 22408576 |
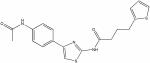
|
8.51 | 2.3 ± 0.4 | >150 | >65 |
| 4256670 |

|
8.6 | 2.2 ± 0.3 | ∼150 | 68 |
| 17433375 |
|
9.73 | 3.7 ± 0.2 | 70 | 19 |
| 7973070 |

|
6.31 | 0.4 ± 0.1 | ∼150 | 375 |
| 4261934 |

|
9.49 | 2.4 ± 0.5 | >150 | >63 |
| 17431538 |

|
5.78 | e6.7 ± 2 | ND | ND |
| 14722514 |

|
9.76 | 0.7 ± 0.1 | ∼150 | 214 |
| 3714418 |

|
9.03 | f3.4 ± 0.6 | ND | ND |
| 3717588 |

|
7.75 | e15.3 ± 0.1 | ND | ND |
| 4259333 |

|
10.19 | 3.3 ± 0.4 | 118 | 36 |
| 7976354 |

|
5.8 | 4.6 ± 0.1 | >150 | >33 |
| 14740150 |
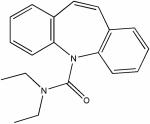
|
3.7 | 4.4 ± 0.1 | >150 | >34 |
| 14741996 |

|
7.69 | 3.2 ± 0.4 | >150 | >47 |
| 17409378 |

|
7.09 | 0.7 ± 0.1 | 26 | 37 |
| 17507322 |
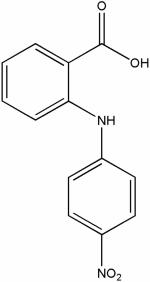
|
11.73 | e15.3 ± 1.4 | ND | ND |
| 22406106 |

|
4.63 | 1.2 ± 0.1 | 139 | 116 |
| 22406746 |

|
7.96 | 4.1 ± 0.2 | ∼150 | 37 |
| 22415156 |
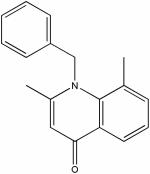
|
5.35 | 3.6 ± 0.1 | >150 | >42 |
| 842929 | not shown | 5.05 | no activity detected | ND | ND |
| 851385 | not shown | 4.33 | no activity detected | ND | ND |
| 851657 | not shown | 3.93 | no activity detected | ND | ND |
| 856055 | not shown | 10.8 | no activity detected | ND | ND |
| 861621 | not shown | 8.83 | no activity detected | ND | ND |
| 862353 | not shown | 6.33 | no activity detected | ND | ND |
| 3713991 | not shown | 8.71 | no activity detected | ND | ND |
| 3714527 | not shown | 6.55 | no activity detected | ND | ND |
| 3714591 | not shown | 8.28 | no activity detected | ND | ND |
| 4264593 | not shown | 6.82 | no activity detected | ND | ND |
| 7969913 | not shown | 4.65 | no activity detected | ND | ND |
| 7970555 | not shown | 9.47 | no activity detected | ND | ND |
| 7972435 | not shown | 7.59 | no activity detected | ND | ND |
| 17386222 | not shown | 8.75 | no activity detected | ND | ND |
| 17408757 | not shown | 4.31 | no activity detected | ND | ND |
| 17515899 | not shown | 9.75 | no activity detected | ND | ND |
| 22405307 | not shown | 3.66 | no activity detected | ND | ND |
| 22406518 | not shown | 3.44 | no activity detected | ND | ND |
| 22408307 | not shown | 5.58 | no activity detected | ND | ND |
| 22409661 | not shown | 9.21 | no activity detected | ND | ND |
| 22410208 | not shown | 5.24 | no activity detected | ND | ND |
| 22410903 | not shown | 5.59 | no activity detected | ND | ND |
| 24708194 | COMPOUND NOT AVAILABLE FOR COUNTERSCREENING | ||||
structures of compounds lacking activity in counter-screening are not shown
values represent averages of two experiments ± range; highest concentration assessed 37.5 μM
values represent averages of two experiments ± range; highest concentration assessed 150 μM; ND: not determined
values represent averages of four experiments ± SD; highest concentration assessed 37.5 μM
CC50 not determined (ND) when EC50 >5 μM
CC50 not determined (ND) due to insufficient amount of compound available
Fifty-nine of the selected 60 compounds could be obtained for counterscreening assays. For hit confirmation, dose-response curves were generated for each of these hit candidates, examining their ability to inhibit live MV in a 96-well plate format. In parallel, MTT assays were employed to determine compound-induced cytotoxicity in the absence of virus infection. Of the 59 hit candidates, 22 (37%) were found inactive in the counterscreening assay. Another two were excluded from further consideration since they showed considerable cytotoxicity, resulting in specificity indexes (SI = CC50/EC50) <20. Primary HTS and counterscreening thus returned 35 confirmed hits with EC50 concentrations ≤15.3 μM and a specificity index (CC50/EC50) > 20 from the NIH/MLSCN library. This equals a true-hit identification rate of ∼0.03%.
Chemical re-synthesis of selected hits
Degradation of compounds upon storage is known to potentially compromise the quality of compound libraries and the results of screening exercises. We therefore assessed the structural integrity of two of the most promising candidate compounds by chemical re-synthesis (figures 2A and B) and biotesting. Both re-synthesized compounds, 14722514 and 22407448, returned activities essentially identical to the original substances stored in the library (data not shown), confirming their structural accuracy.
In-silico data mining indicates a high accuracy of primary hit identification
Structurally, the identified compounds represent 10 distinct chemical classes (see structures in table 1). Several classes were represented repeatedly through independently identified analogs present in the randomized library. This provides confidence in the accuracy of hit identification by the primary screen. To further address this question, the occurrence of false-negative data points was assessed. We based this approach on the consideration that, although chemical analogs can vastly differ in their biological activities, of all library entries found inactive by primary HTS, analogs of confirmed hits have the highest likelihood to represent false-negative screening data.
Starting with confirmed hit compounds representing eight distinct structural classes, we identified all library entries with ≥70% structural similarity through in-silico screening (25 compounds in total, summarized in table 2). Four of these compounds had independently passed both primary evaluations and were confirmed in counterscreening. The remaining 21 analogs were obtained and dose-response curves against MV-Alaska generated. Of the 25 compounds total identified in-silico, this exercise returned a single compound, 22406048, as weakly active in counterscreening (EC50 = 14.4 μM ± 2.6 μM) but with a primary HTS score below the cut-off of 1.9 (table 2). Ten additional compounds had a primary HTS score >1.9, but were ruled out upon visual re-examination. Importantly, of these only two compounds, 17403091 and 22404620, revealed active concentrations <3.3 μM and thus the screening concentration in this exercise. This corresponds to a false-negative rate of 12% in this heavily biased subgroup, generating confidence for the overall accuracy of the strategies implemented for primary hit identification.
Table 2.
Assessment of accuracy of primary hit identification
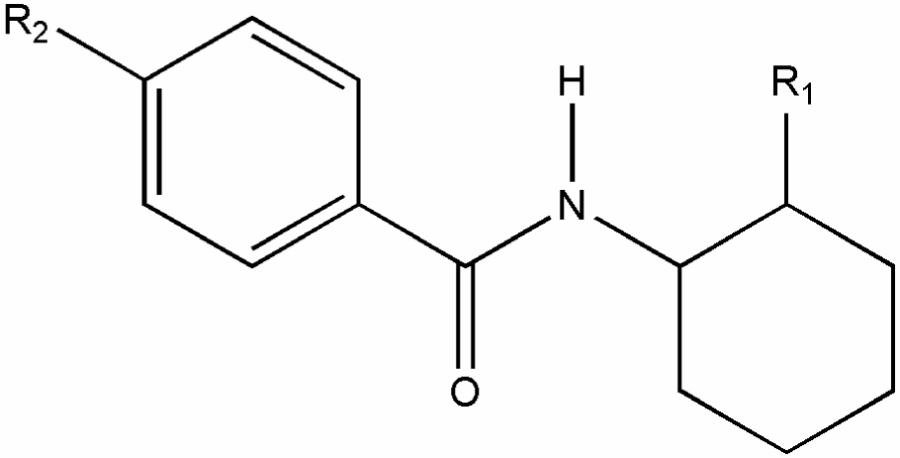 | |||||
|---|---|---|---|---|---|
| R1 | R2 | aPubChem ID (SID) | b% Similarity | Primary HTS Score | cEC50 [μM] (MV-Alaska) |
| -H |

|
14722514 | 100 | 9.76 | 0.7 ± 0.1 |
| -OH |

|
(&)7976541 | 78 | 1.31 | no activity detected |
| -OH |
|
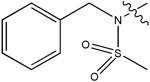
(&)7974606 | 78 | -0.72 | no activity detected |
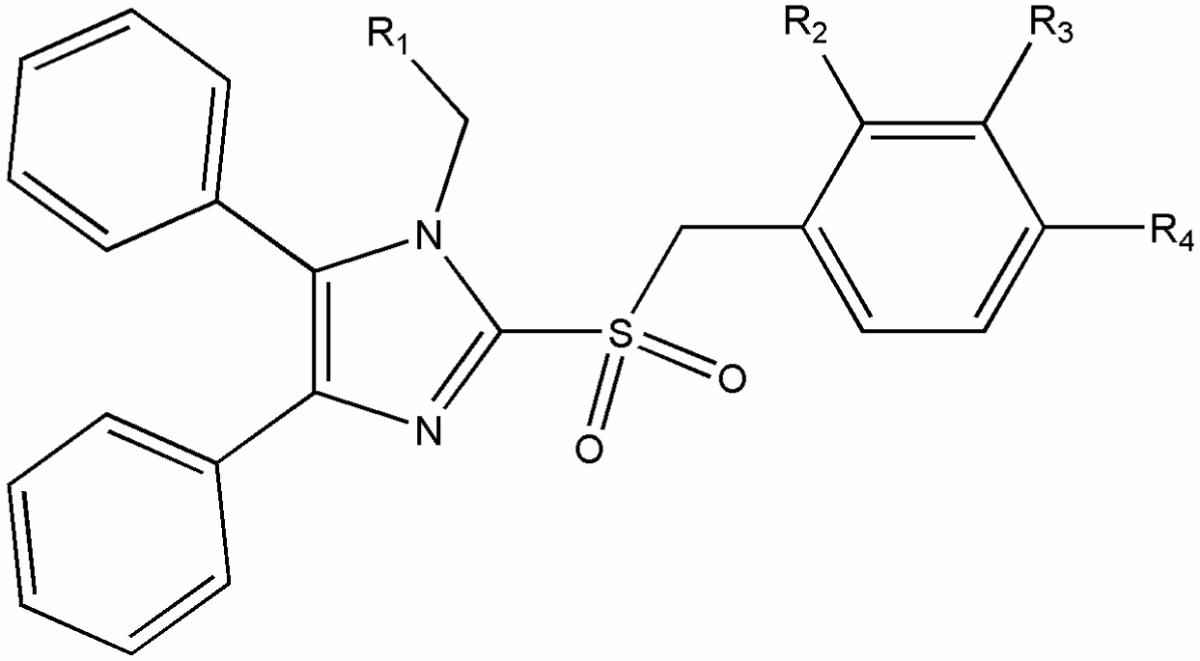
 | |||||||
|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 | aPubChem ID (SID) | b% Similarity | Primary HTS Score | cEC50 [μM] (MV-Alaska) |
| -CH3 | -OCH3 | -OCH3 | -H | 4242806 | 100 | 7.48 | 1.4 ± 0.2 |
| -CH3 | -OC2H5 | -H | -H | (#)14735476 | 82 | 5.19 | 6.8 ± 1 |
| -CH3 | -H | -H | -H | (*)14735307 | 80 | 6.62 | d3.3 ± 0.9 |
| -CH3 | -H | -H | -OC2H5 | (#)14731162 | 78 | 6.16 | 8.8 ± 0.4 |
| -C2H5 | -OC2H5 | -H | -H | (#)14733051 | 72 | 4.29 | 9.7 ± 0.4 |
 | |||||
|---|---|---|---|---|---|
| R1 | R2 | aPubChem ID (SID) | b% Similarity | Primary HTS Score | cEC50 [μM] (MV-Alaska) |
| -COOC2H5 | -F | 7973070 | 100 | 6.31 | 0.4 ± 0.1 |
| -CH2CN | -Cl | (&)847134 | 74 | -0.41 | no activity detected |
| -CH3 | -H | (&)14734775 | 70 | -0.27 | no activity detected |
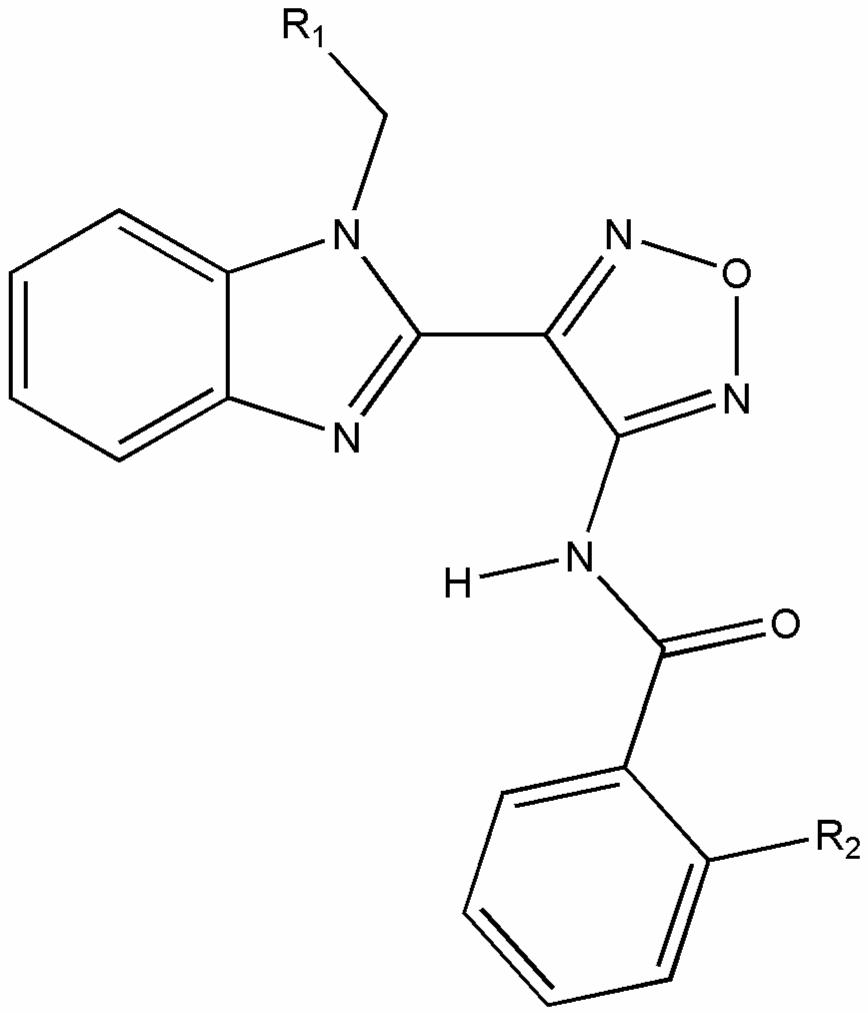
 | |||||||
|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 | aPubChem ID (SID) | b% Similarity | Primary HTS Score | cEC50 [μM] (MV-Alaska) |
| -CCH | -H | -H | -F | 17409378 | 100 | 7.09 | 0.7 ± 0.1 |
| -CCH | -Cl | -H | -H | (&)17410432 | 89 | 0.3 | no activity detected |
| -CCH | -H | -H | -CH3 | (#)17403091 | 81 | 2.22 | 1.9 ± 0.1 |
| -COOC2H5 | -H | -H | -Cl | (&)17411209 | 74 | -0.47 | no activity detected |
| -COOC2H5 | -H | -H | -F | (#)17409348 | 74 | 3.6 | no activity detected |
| -CCH | -H | -H | -OCH3 | (&)17409555 | 73 | -0.1 | no activity detected |
| -COOC2H5 | -H | -F | -H | (#)17409398 | 73 | 3.1 | no activity detected |
 | ||||||
|---|---|---|---|---|---|---|
| R1 | R2 | R3 | aPubChem ID (SID) | b% Similarity | Primary HTS Score | cEC50 [μM] (MV-Alaska) |
| -H | -H | -H | 22407466 | 100 | 6.55 | 2.1 ± 0.1 |
| -CH3 | -H | -H | (#)22404620 | 91 | 4.14 | 0.7 ± 0.1 |
| -H | -CH3 | -H | (*)22404943 | 89 | 3.9 | 0.7 ± 0.1 |
| -OCH3 | -H | -CH3 | (*)22407448 | 72 | 1.93 | d0.2 ± 0.1 |
 | |||||
|---|---|---|---|---|---|
| R1 | R2 | aPubChem ID (SID) | b% Similarity | Primary HTS Score | cEC50 [μM] (MV-Alaska) |
| -H | -CHCH2 | 22406106 | 100 | 4.63 | 1.2 ± 0.1 |
| -F | -CHCH2 | (&)22406048 | 88 | 1.28 | 14.4 ± 2.6 |
| -H | -CH3 | (*)22406746 | 84 | 7.96 | 4.1 ± 0.2 |
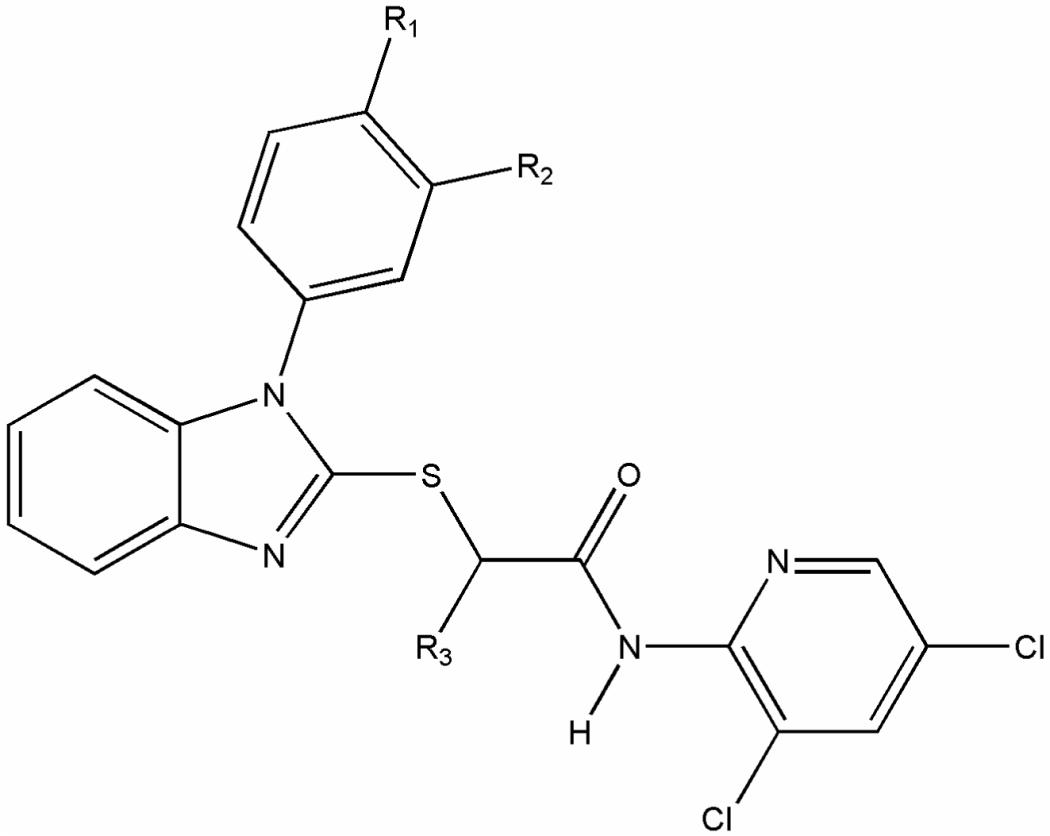
 | ||||||
|---|---|---|---|---|---|---|
| R1 | R2 | R3 | aPubChem ID (SID) | b% Similarity | Primary HTS Score | cEC50 [μM] (MV-Alaska) |

|
-H |

|
22408576 | 100 | 8.51 | 2.3 ± 0.4 |
| -CH3 | -H |
|
(&)22409879 | 76 | -0.49 | no activity detected |
| -F | -H |
|
(#)22404024 | 74 | 2.21 | no activity detected |

|
-H |

|
(&)14725916 | 73 | 0.1 | no activity detected |
| -OCH3 | -H |

|
(#)22408901 | 73 | 2.26 | no activity detected |
| -H |

|
|
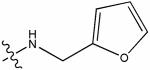
(#)845362 | 70 | 3.56 | no activity detected |
 | ||||
|---|---|---|---|---|
| R1 | aPubChem ID (SID) | b% Similarity | Primary HTS Score | cEC50 [μM] (MV-Alaska) |

|
22402685 | 100 | 8.86 | 1.9 ± 0.2 |
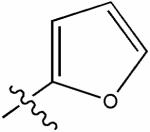
|
(&)22403476 | 82 | 0.79 | no activity detected |
unmarked compounds represent the group-defining confirmed hit; (*) bona-fide hits (primary HTS score >1.9, passed visual inspection); (#) excluded (primary HTS score >1.9, excluded at visual inspection); (&) negatives (primary HTS score < 1.9); shading highlights compounds found to be “false negatives”
based on chemical structures
values represent averages of two experiments ± range; highest concentration assessed 37.5 μM
values represent averages of four experiments ± SD; highest concentration assessed 37.5 μM
Confirmed hits comprise inhibitors of entry and post-entry steps
Transient expression of plasmid-encoded MV glycoproteins was employed for basic classification of confirmed hits into compounds that interfere with viral entry or block post-entry steps of the viral life cycle. Entry inhibitors are characterized by inhibition of virus glycoprotein-mediated cell-to-cell fusion under these conditions as reported previously (Plemper et al. 2004), while compounds blocking RdRp activity or particle assembly, for example, would lack activity. As first-pass screen, cells transiently transfected with expression plasmids encoding the MV envelope glycoproteins under the control of the constitutive CMV promoter were incubated in the presence of different concentrations of confirmed hit compounds. The extent of cell-to-cell fusion was assessed microscopically 36 hours post-transfection. Of 35 compounds tested, 11 suppressed syncytium formation to various degrees in a dose-dependent fashion (data not shown).
These compounds were short listed for a firefly luciferase reporter-based cell-to-cell fusion assay to quantify their inhibitory potential. For comparison, cells were treated with an inhibitory tripeptide that is known to block MV glycoprotein-mediated membrane fusion at higher concentrations (Richardson and Choppin 1983). Ten of the 11 compounds caused a dose-dependent reduction in luciferase expression in this assay (figure 3), with active concentrations ranging from 1.5 to 14.5 μM. These are thus very likely blockers of the viral entry machinery. One compound (PubChem ID # 4256670) did not suppress reporter expression at all at 5 μM and returned an overall EC50 of 22 μM in this reporter assay, prohibiting classification with confidence (figure 3). Against live virus, this compound returned an active concentration of 2.2 μM, suggesting that its antiviral effect may be based predominantly on interference with post-entry steps of the viral life cycle.
Figure 3.

Quantitative assessment of entry inhibitor candidates using a firefly luciferase reporter-based cell-to-cell fusion assay. Effector cells expressing T7 polymerase and plasmid-encoded MV H and F envelope glycoproteins were overlaid in the presence of different candidate compound concentrations with target cells harboring the luciferase reporter construct under the control of the T7 promoter. Luciferase activities in cell lysates were determined as an indicator for the extend of cell-to-cell fusion five hours post-overlay. Graphs represent averages of four independent experiments, EC50 concentrations are shown.
Little cross-resistance with MV inhibitor AS-136A
To determine whether targets of the newly identified hits overlap with a previously identified MV entry inhibitor (Plemper et al. 2004) or the RdRp inhibitor (Sun et al. 2007; White et al. 2007) class, cross-resistance profiles were determined. Of the 35 confirmed hits, the more potent compounds (EC50 < 5 μM) and all 11 fusion inhibitor candidates (30 compounds total) were selected for this assay. All compounds were tested against an MV-Alaska variant, MV-AII-5, that was adapted to grow in the presence of the MV RdRp inhibitor AS-136A (will be described in detail elsewhere). In a parallel screen, the 11 fusion inhibitor candidates (figure 3) were additionally tested against MV-Ibd, a strain that we previously found to be resistant to a small-molecule MV entry inhibitor class (Doyle et al. 2006).
Resistance to the entry inhibitor AS-48 resulted in all cases in reduced sensitivity or complete resistance to the fusion inhibitor candidates, indicating strong cross-resistance (table 3). In contrast, dose-response curves reveal inhibition of the adapted MV-AII-5 variant by all but one (14740150) confirmed HTS hits with active concentrations mostly resembling the values obtained against the non-adapted parent virus (table 3). Thus, these data demonstrate a nearly complete lack of cross-resistance between the newly identified HTS hits and the AS-136A polymerase inhibitor class.
Table 3.
Assessment of cross-resistance with known MV inhibitors
| aPubchem ID (SID) | bEC50 [μM] (MV-AII-5) | cEC50 [μM] (MV-Ibd) | dEC50 [μM] (MV-Alaska) |
|---|---|---|---|
| 846635 | 3.7 ± 0.1 | no activity detected | 2.7 ± 0.2 |
| 852230 | 4.5 ± 0.2 | no activity detected | 3.3 ± 0.1 |
| 4242806 | 2.1 ± 0.1 | ND | 1.4 ± 0.2 |
| 4245209 | 14.3 ± 0.8 | no activity detected | 5.9 ± 1.7 |
| 4256670 | 4.0 ± 0.1 | 26.6 ± 9.3 | 2.2 ± 0.3 |
| 4259333 | 4.3 ± 0.3 | ND | 3.3 ± 0.4 |
| 4261934 | 3.9 ± 0.1 | ND | 2.4 ± 0.5 |
| 4265637 | 4.7 ± 0.3 | no activity detected | 3 ± 0.2 |
| 7973070 | 2.0 ± 0.2 | ND | 0.4 ± 0.1 |
| 7976354 | 1.0 ± 0.1 | ND | 4.6 ± 0.1 |
| 14722514 | 3.6 ± 1.6 | ND | 0.7 ± 0.1 |
| 14723513 | 3.6 ± 0.2 | no activity detected | 3.7 ± 0.1 |
| 14735307 | 5.4 ± 1.6 | ND | 3.3 ± 0.9 |
| 14740150 | no activity detected | ND | 4.4 ± 0.1 |
| 14741996 | 7.7 ± 0.1 | ND | 3.2 ± 0.4 |
| 14743644 | 4.6 ± 0.1 | ND | 2.7 ± 0.5 |
| 17409378 | 1.4 ± 0.3 | ND | 0.7 ± 0.1 |
| 17416393 | 2.9 ± 0.5 | no activity detected | 1.5 ± 0.3 |
| 17433375 | 3.2 ± 0.1 | ND | 3.7 ± 0.2 |
| 17505573 | 1.6 ± 0.1 | no activity detected | 3.8 ± 0.5 |
| 22402685 | 1.6 ± 0.2 | no activity detected | 1.9 ± 0.2 |
| 22404943 | 0.5 ± 0.1 | ND | 0.7 ± 0.1 |
| 22406746 | 2 ± 0.1 | ND | 4.1 ± 0.2 |
| 22407448 | 0.1 | ND | 0.2 ± 0.1 |
| 22407466 | 1.0 | ND | 2.1 ± 0.1 |
| 22408576 | 2.0 ± 0.1 | ND | 2.3 ± 0.4 |
| 22410899 | 3.8 ± 0.3 | ND | 3.9 ± 0.1 |
| 22415156 | 2.4 ± 0.2 | ND | 3.6 ± 0.1 |
| 22415419 | 2.2 ± 0.3 | no activity detected | 4.5 ± 0.9 |
| 22415706 | 4.1 | no activity detected | 6.7 ± 0.3 |
shading highlights compound identified in pre-screening as fusion inhibitor candidates
MV-Alaska variant adapted to grow in the presence of MV RdRp inhibitor AS-136A (Sun et al. 2007; White et al. 2007); values represent averages of two experiments ± range; highest concentration assessed 37.5 μM
MV-Ibd, naturally resistant to MV fusion inhibitor AS-48 (Plemper et al. 2004) values represent averages of two experiments ± range; highest concentration assessed 37.5 μM; only compounds identified as fusion inhibitor candidates were tested against MV-Ibd (ND: not determined)
for comparison, EC50 values against MV-Alaska (table 1) are shown
Three distinct patterns of target specificity
To classify the target range of this pool of 30 confirmed compounds, their ability to inhibit two other members of the paramyxovirinae, canine distemper virus (CDV) and human parainfluenzavirus type 3 (HPIV3) was assessed. CDV is closely related to MV, belongs like MV to the genus morbillivirus and both viruses share approximately 61% overall identity on the protein level. HPIV3 is a representative of the more distantly related genus respirovirus and shows only approximately 27% overall protein identity to MV.
Generation of dose-response curves for these viruses revealed three distinct patterns of hit activity (table 4). A subgroup of 11 compounds was highly MV-specific with active concentrations ranging from 0.7 to 3.9 μM. Members of this subgroup were completely inactive even against closely related CDV. A second class of eight compounds showed a slightly expanded target range, since these hits blocked both morbilliviruses tested, MV and CDV, but not HPIV3. Inhibitory activity of compounds of the 11-entry third class extended to all three viruses, MV, CDV and HPIV3 with active concentrations ranging from 0.2 to 4.6 μM.
Table 4.
Three distinct patterns of target specificity
| Pubchem ID (SID) | aEC50 [μM] (MV-Alaska) | aEC50 [μM] (CDV-752) | aEC50 [μM] (HPIV3) |
|---|---|---|---|
| MV specific | |||
| 14722514 | 0.7 ± 0.1 | no activity detected | no activity detected |
| 22408576 | 2.3 ± 0.4 | no activity detected | no activity detected |
| 4261934 | 2.4 ± 0.5 | no activity detected | no activity detected |
| 14743644 | 2.7 ± 0.5 | no activity detected | no activity detected |
| 846635 | 2.7 ± 0.2 | no activity detected | no activity detected |
| 14735307 | b3.3 ± 0.9 | no activity detected | no activity detected |
| 852230 | 3.3 ± 0.1 | no activity detected | no activity detected |
| 4259333 | 3.3 ± 0.4 | no activity detected | no activity detected |
| 17433375 | 3.7 ± 0.2 | no activity detected | no activity detected |
| 17505573 | 3.8 ± 0.5 | no activity detected | no activity detected |
| 22410899 | 3.9 ± 0.1 | no activity detected | no activity detected |
| MV and CDV (genus morbillivirus) specific | |||
| 17416393 | 1.5 ± 0.3 | 12.9 ± 0.7 | no activity detected |
| 22402685 | 1.9 ± 0.2 | 2.1 ± 0.3 | no activity detected |
| 4265637 | 3.0 ± 0.2 | 6.7 ± 0.1 | no activity detected |
| 22415156 | 3.6 ± 0.1 | 11.0 ± 0.1 | no activity detected |
| 14723513 | 3.7 ± 0.1 | 26.8 ± 8.7 | no activity detected |
| 22415419 | 4.5 ± 0.9 | 5.0 ± 0.2 | no activity detected |
| 4245209 | 5.9 ± 1.7 | 31.1 | no activity detected |
| 22415706 | 6.7 ± 0.3 | 12.9 | no activity detected |
| active against different paramyxovirus genera | |||
| 22407448 | b0.2 ± 0.1 | 0.2 ± 0.1 | 0.2 ± 0.1 |
| 7973070 | 0.4 ± 0.1 | 1.0 ± 0.1 | 1.9 ± 0.1 |
| 17409378 | 0.7 ± 0.1 | no activity detected | 1.7 ± 0.2 |
| 22404943 | 0.7 ± 0.1 | 0.4 ± 0.1 | 0.6 ± 0.1 |
| 4242806 | 1.4 ± 0.2 | 2.1 ± 0.1 | 2.0 ± 0.1 |
| 22407466 | 2.1 ± 0.1 | 0.9 ± 0.1 | 1.0 ± 0.1 |
| 4256670 | 2.2 ± 0.3 | 3.9 ± 0.2 | 5.8 ± 0.6 |
| 14741996 | 3.2 ± 0.4 | 4.2 ± 0.1 | 3.9 ± 0.1 |
| 22406746 | 4.1 ± 0.2 | 2.3 ± 0.2 | 2 ± 0.1 |
| 14740150 | 4.4 ± 0.1 | 1.9 ± 0.1 | 3.0 ± 0.1 |
| 7976354 | 4.6 ± 0.1 | 0.9 ± 0.1 | 3.5 ± 0.1 |
values represent averages of two experiments ± range; highest concentration assessed 37.5 μM
values represent averages of four experiments ± SD; highest concentration assessed 37.5 μM
Thus, these data indicate different degrees of specificity for the confirmed hits, suggesting that different components of the pathogen or, in case of hits with broader activity, possibly of the host cell are targeted by the compounds.
Discussion
In this study, we describe a robust automated assay for the identification of MV inhibitors. The protocol implemented here does not necessitate modification of the pathogen itself and is suitable for the screen of non-attenuated wild type viral isolates. This is a major advantage over a previously reported screening strategy (White et al. 2007), which, although suitable for hit identification, relied on an attenuated MV recombinant expressing eGFP as an additional transcription unit. The current protocol thus maximizes the likelihood that identified inhibitors show activity against primary virus strains currently endemic in the field. While optimized here for the identification of MV blockers, the protocol should be readily adaptable to other viral targets, provided infection coincides with a strong cytopathic effect and triggers breakdown of the host cell monolayer.
Fixation and permanent stain of assay plates with crystal violet is cost-effective and eliminates tight time constraints for obtaining automated readouts. It furthermore allows re-assessment of plates visually and microscopically to ensure complete absence of infectious centers in individual candidate wells. This double assessment has greatly reduced the amount of primary hit candidates (from 2070 to 60), while minimizing labor-intensive manual counterscreening efforts. At the same time, its accuracy is reflected in the high 59% hit confirmation rate obtained in secondary assays.
Importantly, in-silico data base mining followed by biotesting confirmed that our hit identification regimen is not over-stringent, which can lead to extensive discarding of potentially promising compounds. Our assessment of the frequency of compounds falsely tested inactive in the primary screen was based on the assumption that structural analogs of confirmed hits should have a higher likelihood for antiviral activity than randomly selected library entries. The highest density of false-negatives should thus be found among those analogs, biasing the assessment heavily towards a higher false-negative rate. However, dose-response curves generated for eight independent sets of analogs revealed that most analogs designated inactive by the primary assay indeed had no active in counterscreening exercises. In this biased subgroup, the false-negative rate was 12%, generating confidence that a majority of active structural compound classes present in the library may have been discovered.
Highly effective antiretroviral therapy has confirmed that combined administration of antivirals with distinct targets is desirable. Both overall boosting of inhibitory activity and a reduced rate of viral escape through the development of resistance have been observed (Bartlett et al. 2001; Bartlett et al. 2006; Murphy et al. 2001). By analogy, similar beneficial effects will most likely apply to inhibitors of other viral pathogens. Our protocol has therefore been designed with a maximum potential for the identification of a mechanistically diverse array of paramyxovirus blockers in a single screen. Since virus-induced cytopathicity is monitored after completion of multiple infectious cycles, inhibitors of viral entry (through blockage of receptor binding or membrane fusion), the viral RdRp complex, or of particle assembly are likely to be identified.
Basic classification of confirmed hits in blockers of the viral entry machinery and inhibitors acting post-entry suggests that the screen has indeed returned a mechanistically diverse group of compounds that interfere with different steps of the viral life cycle. At present, we cannot completely exclude that general suppression of host cell protein biosynthesis by entry blocker candidates biases the plasmid-based cell-to-cell fusion assay. Naturally, this would coincide with high cytotoxicity, however. Thus, it appears unlikely based on our cytotoxicity assessment. Importantly, none of the hits shows cross-resistance with the MV RdRp activity inhibitor AS-136A. This may reflect that either none of the new hits blocks RdRp activity or that those hits blocking RdRp share no overlapping target sites with AS-136A. In either case, these findings open future avenues for enhancing antiviral activity through combination of functionally distinct hit classes with each other and/or with AS-136A.
The potential for the identification of assembly inhibitors in particular stands in contrast to the previously reported rMV-eGFP based screening protocol, which made the identification of compounds acting downstream of RdRp activity and thus eGFP expression unlikely. Interestingly, activity of confirmed hits discovered in the previous screen is restricted to the morbillivirus genus (MV and to a lesser degree the closely related CDV) and does not extend to more distantly related members of the paramyxovirinae (White et al. 2007). In this context it is intriguing to speculate that newly identified hits with a broader target range may interfere with late stages in the viral life cycle, possibly by targeting cellular components that are uniformly exploited by different members of the family for particle assembly.
Such specific targeting of host cell components stands in contrast to the mechanism of activity of promiscuous compounds that are frequently found in HTS exercises (McGovern et al. 2002; McGovern and Shoichet 2003). The latter are thought to function through non-specific recruitment of effector molecules examined in the assay to larger compound aggregates (McGovern et al. 2002; McGovern and Shoichet 2003). Reflecting activity through aggregate formation, promiscuous compounds are characterized by active concentrations in the micromolar range and essentially flat structure-activity profiles. In the case of the hit compounds identified in our screen, future in-depth mechanistic characterization will be required to uncover the molecular nature of the targeted pathogen or host components. Nonetheless, high target specificity of the MV- and morbillivirus-specific compounds with active concentrations in the nanomolar range of the most potent representatives of the group with broader target range (i.e. compound 22407448) supports inhibition through specific docking and argues against a promiscuous, aggregation-based mechanism of action.
In conclusion, we have developed a robust, cost-effective protocol for automated screening of MV inhibitors that should be readily transferable to other members of the paramyxovirus family. Implementation of the assay has confirmed high accuracy of primary hit identification and has yielded a diverse set of confirmed hits that target distinct steps of the viral life cycle. Efficacy testing and future hit-to-lead chemistry will explore the developmental potential of selected compound classes.
Acknowledgements
We thank S. Niewiesk for CDV strain (CDV 752), Ray Dingledine and Haian Fu from the Emory Chemical Biology Drug Discovery Center for support, Iestyn Lewis for HTS data analysis, and A. L. Hammond for critical reading of the manuscript. This work was supported by public health service grant HG003918 (to JPS, Ray Dingledine and Haian Fu), and grants AI071002 and MH080836-01 from NIH/NIAID (to RKP).
References
- CDC PIB 2005 http://www.cdc.gov/programs/global06.htm.
- Baba M. Recent progress in anti-HIM-1 research. Uirusu. 2004;54:59–66. doi: 10.2222/jsv.54.59. [DOI] [PubMed] [Google Scholar]
- Barnard DL. Inhibitors of measles virus. Antivir Chem Chemother. 2004;15:111–119. doi: 10.1177/095632020401500301. [DOI] [PubMed] [Google Scholar]
- Bartlett JA, DeMasi R, Quinn J, Moxham C, Rousseau F. Overview of the effectiveness of triple combination therapy in antiretroviral-naive HIV-1 infected adults. Aids. 2001;15:1369–1377. doi: 10.1097/00002030-200107270-00006. [DOI] [PubMed] [Google Scholar]
- Bartlett JA, Fath MJ, Demasi R, Hermes A, Quinn J, Mondou E, Rousseau F. An updated systematic overview of triple combination therapy in antiretroviral-naive HIV-infected adults. Aids. 2006;20:2051–2064. doi: 10.1097/01.aids.0000247578.08449.ff. [DOI] [PubMed] [Google Scholar]
- Cathomen T, Naim HY, Cattaneo R. Measles viruses with altered envelope protein cytoplasmic tails gain cell fusion competence. Journal of virology. 1998;72:1224–1234. doi: 10.1128/jvi.72.2.1224-1234.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC Progress in Reducing Measles Mortality --- Worldwide, 1999-2003. MMWR. 2005;54:200–203. [PubMed] [Google Scholar]
- Chakrabarti S, Collingham KE, Holder K, Fegan CD, Osman H, Milligan DW. Pre-emptive oral ribavirin therapy of paramyxovirus infections after haematopoietic stem cell transplantation: a pilot study. Bone Marrow Transplant. 2001;28:759–763. doi: 10.1038/sj.bmt.1703216. [DOI] [PubMed] [Google Scholar]
- Doyle J, Prussia A, White LK, Sun A, Liotta DC, Snyder JP, Compans RW, Plemper RK. Two domains that control prefusion stability and transport competence of the measles virus fusion protein. Journal of virology. 2006;80:1524–1536. doi: 10.1128/JVI.80.3.1524-1536.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duprex WP, McQuaid S, Hangartner L, Billeter MA, Rima BK. Observation of measles virus cell-to-cell spread in astrocytoma cells by using a green fluorescent protein-expressing recombinant virus. Journal of virology. 1999;73:9568–9575. doi: 10.1128/jvi.73.11.9568-9575.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrengruber MU, Hennou S, Bueler H, Naim HY, Deglon N, Lundstrom K. Gene transfer into neurons from hippocampal slices: comparison of recombinant Semliki Forest Virus, adenovirus, adeno-associated virus, lentivirus, and measles virus. Molecular and cellular neurosciences. 2001;17:855–871. doi: 10.1006/mcne.2001.0982. [DOI] [PubMed] [Google Scholar]
- Garcia CC, Candurra NA, Damonte EB. Differential inhibitory action of two azoic compounds against arenaviruses. International journal of antimicrobial agents. 2003;21:319–324. doi: 10.1016/s0924-8579(02)00390-4. [DOI] [PubMed] [Google Scholar]
- Griffin DE. Measles Virus. Lippincott; Philadelphia, PA: 2001. [Google Scholar]
- Griffin DE, Pan CH, Moss WJ. Measles vaccines. Front Biosci. 2008;13:1352–1370. doi: 10.2741/2767. [DOI] [PubMed] [Google Scholar]
- Hethcote HW. The mathematics of infectious disease. SIAM Review. 2000;42:599–653. [Google Scholar]
- Hilleman MR. Current overview of the pathogenesis and prophylaxis of measles with focus on practical implications. Vaccine. 2001;20:651–665. doi: 10.1016/s0264-410x(01)00384-x. [DOI] [PubMed] [Google Scholar]
- McGovern SL, Caselli E, Grigorieff N, Shoichet BK. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J Med Chem. 2002;45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- McGovern SL, Shoichet BK. Kinase inhibitors: not just for kinases anymore. J Med Chem. 2003;46:1478–1483. doi: 10.1021/jm020427b. [DOI] [PubMed] [Google Scholar]
- Murphy EL, Collier AC, Kalish LA, Assmann SF, Para MF, Flanigan TP, Kumar PN, Mintz L, Wallach FR, Nemo GJ. Highly active antiretroviral therapy decreases mortality and morbidity in patients with advanced HIV disease. Ann Intern Med. 2001;135:17–26. doi: 10.7326/0003-4819-135-1-200107030-00005. [DOI] [PubMed] [Google Scholar]
- Ono N, Tatsuo H, Hidaka Y, Aoki T, Minagawa H, Yanagi Y. Measles viruses on throat swabs from measles patients use signaling lymphocytic activation molecule (CDw150) but not CD46 as a cellular receptor. Journal of virology. 2001;75:4399–4401. doi: 10.1128/JVI.75.9.4399-4401.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plemper RK, Erlandson KJ, Lakdawala AS, Sun A, Prussia A, Boonsombat J, Aki-Sener E, Yalcin I, Yildiz I, Temiz-Arpaci O, Tekiner B, Liotta DC, Snyder JP, Compans RW. A target site for template-based design of measles virus entry inhibitors. Proc Natl Acad Sci U S A. 2004;101:5628–5633. doi: 10.1073/pnas.0308520101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson CD, Choppin PW. Oligopeptides that specifically inhibit membrane fusion by paramyxoviruses: studies on the site of action. Virology. 1983;131:518–532. doi: 10.1016/0042-6822(83)90517-2. [DOI] [PubMed] [Google Scholar]
- Rota P, Liffick S, Rota J, Katz R, Redd S, Papania M, Bellini W. Molecular epidemiology of measles viruses in the United States, 1997-2001. Emerg Infect Dis. 2002;8:902–908. doi: 10.3201/eid0809.020206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki F, Ono N, Yamaguchi R, Yanagi Y. Efficient isolation of wild strains of canine distemper virus in Vero cells expressing canine SLAM (CD150) and their adaptability to marmoset B95a cells. Journal of virology. 2003;77:9943–9950. doi: 10.1128/JVI.77.18.9943-9950.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigeta S, Mori S, Baba M, Ito M, Honzumi K, Nakamura K, Oshitani H, Numazaki Y, Matsuda A, Obara T, et al. Antiviral activities of ribavirin, 5-ethynyl-1-beta-D-ribofuranosylimidazole-4-carboxamide, and 6′-(R)-6′-C-methylneplanocin A against several ortho- and paramyxoviruses. Antimicrobial agents and chemotherapy. 1992;36:435–439. doi: 10.1128/aac.36.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spearman C. The method of right and wrong cases (constant stimuli) without Gauss’s formula. Br J Phsychol. 1908;2:227–242. [Google Scholar]
- Stray SJ, Zlotnick A. BAY 41-4109 has multiple effects on Hepatitis B virus capsid assembly. J Mol Recognit. 2006;19:542–548. doi: 10.1002/jmr.801. [DOI] [PubMed] [Google Scholar]
- Sun A, Chandrakumar N, Yoon JJ, Plemper RK, Snyder JP. Non-nucleoside inhibitors of the measles virus RNA-dependent RNA polymerase complex activity: Synthesis and in vitro evaluation. Bioorganic & medicinal chemistry letters. 2007;17:5199–5203. doi: 10.1016/j.bmcl.2007.06.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutter G, Ohlmann M, Erfle V. Non-replicating vaccinia vector efficiently expresses bacteriophage T7 RNA polymerase. FEBS Lett. 1995;371:9–12. doi: 10.1016/0014-5793(95)00843-x. [DOI] [PubMed] [Google Scholar]
- van den Hof S, Conyn-van Spaendonck MA, van Steenbergen JE. Measles epidemic in the Netherlands, 1999-2000. J Infect Dis. 2002;186:1483–1486. doi: 10.1086/344894. [DOI] [PubMed] [Google Scholar]
- White LK, Yoon JJ, Lee JK, Sun A, Du Y, Fu H, Snyder JP, Plemper RK. Nonnucleoside inhibitor of measles virus RNA-dependent RNA polymerase complex activity. Antimicrobial agents and chemotherapy. 2007;51:2293–2303. doi: 10.1128/AAC.00289-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfson LJ, Stebel PM, Gacic-Dobo M, Hoekstra EJ, McFarland JW, Hersh BS. Has the 2005 measles mortality reduction goal been achieved? A natural history modelling study. Lancet. 2007;369:191–200. doi: 10.1016/S0140-6736(07)60107-X. [DOI] [PubMed] [Google Scholar]
- Yang G, Pevear DC, Davies MH, Collett MS, Bailey T, Rippen S, Barone L, Burns C, Rhodes G, Tohan S, Huggins JW, Baker RO, Buller RL, Touchette E, Waller K, Schriewer J, Neyts J, DeClercq E, Jones K, Hruby D, Jordan R. An orally bioavailable antipoxvirus compound (ST-246) inhibits extracellular virus formation and protects mice from lethal orthopoxvirus Challenge. Journal of virology. 2005;79:13139–13149. doi: 10.1128/JVI.79.20.13139-13149.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JH, Chung TDY, Oldenburg KR. A simple statistical parameter for used in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Zhou J, Chen CH, Aiken C. Human immunodeficiency virus type 1 resistance to the small molecule maturation inhibitor 3-O-(3′,3′-dimethylsuccinyl)-betulinic acid is conferred by a variety of single amino acid substitutions at the CA-SP1 cleavage site in Gag. Journal of virology. 2006;80:12095–12101. doi: 10.1128/JVI.01626-06. [DOI] [PMC free article] [PubMed] [Google Scholar]