

Abstract
We first demonstrate the transcription, expression, and DNA binding properties of the PPARγ subtype of the peroxisome proliferator-activated nuclear receptor family to the spinal cord with real time PCR, western blot, and electrophoretic mobility shift assay. To test the hypothesis that activation of spinal PPARγ decreases nerve injury-induced allodynia, we intrathecally administered PPARγ agonists and/or antagonists in rats following transection of the tibial and common peroneal branches of the sciatic nerve. Single injection of either a natural (15-deoxy-prostaglandin J2, 15d-PGJ2) or synthetic (rosiglitazone) PPARγ agonist dose-dependently decreased mechanical and cold hypersensitivity. These effects were maximal at a dose of 100μg and peaked at ~60 min after injection, a rapid time course suggestive of transcription-independent mechanisms of action. Concurrent administration of a PPARγ antagonist (bisphenol A diglycidyl ether, BADGE) reversed the effects of 15d-PGJ2 and rosiglitazone, further indicating a receptor-mediated effect. In animals without nerve injury, rosiglitazone did not alter motor coordination, von Frey threshold, or withdrawal response to a cool stimulus. Intraperitoneal and intracerebroventricular administration of PPARγ agonists (100μg) did not decrease mechanical and cold hypersensitivity, arguing against effects subsequent to diffusion from the intrathecal space. We conclude that ligand-induced activation of spinal PPARγ rapidly reverses nerve injury-induced mechanical allodynia. New or currently-available drugs targeted at spinal PPARγ may yield important therapeutic effects for the management of neuropathic pain.
PERSPECTIVE
PPARγ receptor agonists such as rosiglitazone and pioglitazone remain FDA approved as insulin sensitizers. We demonstrate PPARγ expression in the spinal cord, and report that activation of these receptors inhibits allodynia. BBB-permeant PPARγ agonists may yield important therapeutic effects for the management of neuropathic pain.
INTRODUCTION
Peroxisome proliferator-activated receptors (PPARs) are transcription factors belonging to the nuclear receptor superfamily 15. PPARs are activated by fatty acids, eicosanoids, and synthetic ligands. Activated PPARs form functional heterodimers with retinoid X receptors (RXR). This complex interacts with various co-activators and a specific peroxisome proliferator response element (PPRE) on the promoter region of target genes to alter transcription 34.
Three PPARreceptors have been identified – α, β/δ, and γ 2, 22. The PPARγ isotype mediates numerous physiological functions, and of particular clinical significance is its role as a lipid sensor. PPARγ activation leads to adipocyte differentiation and drives the gene expression of enzymes that facilitate lipid uptake and synthesis 16. Dysregulation of PPARγ function is associated with numerous diseases such as type 2 diabetes. Indeed, synthetic PPARγ agonists of the thiazolidinedione (TZD) class act as insulin sensitizers. Although our understanding of the glucose lowering properties of PPARγ ligands remains an intense area of investigation 31, TZDs such as rosiglitazone and pioglitazone represent an important pharmacotherapy for the treatment of glucose intolerance 21.
It is becoming increasingly clear that PPARγ ligands represent a promising therapeutic strategy for other diseases as well, including atherosclerosis, cancer, cardiovascular complications, inflammation, spinal cord injury, and neurodegenerative disorders such as Alzeimer’s disease 1. Our previous data suggested that PPARα ligands exert additional CNS actions on nociceptive signalling pathways 37, possibly by acting at PPARreceptors in the brain 36. In specific, we reported that PPARα ligands reduced behavioral signs of inflammatory pain in rats 37, findings that were recently confirmed with PPARα deletion-mutant mice 18. Whether PPARγ ligands can similarly exert analgesic actions is unclear. An important clue comes from very recent studies in a rat model of spinal cord injury (SCI). Vemuganti et al found that pre-treatment with the TZD pioglitazone prevented numerous consequences of SCI, including neuronal damage, motor dysfunction, myelin loss, inflammation, and most notably, thermal hyperalgesia. Co-administration of the PPARγ antagonist GW9662 reversed these actions 26. Whether pioglitazone acted directly at spinal cord sites to reduce nociceptive signalling cannot be deduced from these studies, however, because of its multiple effects on SCI pathology. To directly address this question, the present studies were designed to test the hypothesis that spinally-directed delivery of PPARγ ligands decreases allodynia and hyperalgesia in an animal model of peripheral neuropathic pain. Furthermore, we extend a single immunohistochemical finding suggesting PPARγ immunoreactivity in a key site of pain transmission, lamina II of the dorsal horn 25, with additional real time PCR, western, and EMSA studies.
MATERIALS AND METHODS
Animals
Male Sprague-Dawley rats (Charles Rivers Laboratories, Inc) were 280-320g at the time of nerve injury and intrathecal catheter implantation, and 340-380g during pharmacological testing. Animals were housed in individual cages on a 12-hour light/dark cycle starting at 6 a.m., and were given food and water ad libitum. All animal use protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of Tulane University.
Spared nerve injury surgery
Rats were anesthetized with isoflurane (5% induction, then 1.5% maintenance in oxygen). As previously described 7, an incision was made in the skin at the level of the trifurcation of the left sciatic nerve. The overlying muscles were retracted, exposing the common peroneal, tibial, and sural nerve. The common peroneal and tibial nerves were ligated with 6-0 silk (Ethicon, Somerville, NJ) and then the knot and adjacent nerve (2mm) were transected. Care was taken to avoid touching the sural nerve branch. The muscle was next sutured with 4-0 absorbable sutures (Ethicon) and the wound was closed with metal clips.
Intrathecal catheter implantation
At the time of nerve injury, animals were re-anesthetized with isoflurane (Baxter, Deerfield, IL), and then placed in a stereotaxic apparatus (Stoelting, Wood Dale, IL). As previously described 19, rats were implanted with polyethylene-10 (Clay Adams, Sparks, MD) intrathecal catheters. Briefly, the animal head was flexed forward in the stereotaxic apparatus. An incision was made in the skin at the back of the rat’s head and neck. Next, the cisternal membrane was exposed by dissection. The membrane was gently punctured with the tip of a 15 blade. A 7.5 cm polyethylene-10 catheter was passed through the opening in the cisternal membrane and passed into the intrathecal space. The catheter was loosely sutured to subcutaneous tissue and the skin then approximated using 4-0 absorbable sutures (Ethicon).
Intracerebroventricular cannulae implantation
Guide cannulae (Plastics one, Roanoke, VA) for intracerebroventricular (i.c.v.) injection were placed one wk before experimentation, as previously described 35. Surgical anesthesia was achieved with isoflurane (5% for induction; 1.5-2% for maintenance). Rats were placed in a stereotaxic apparatus fitted with blunt ear bars (Stoelting, Kiel, WI). After an incision to expose the cranium, the dorsal surface of the skull was leveled by zeroing the dorso-ventral coordinate at lambda and bregma. A stainless steel guide cannula (Plastics One, Roanoke, VA) was lowered to the right lateral brain ventricle using the following stereotaxic coordinates: 0.7 posterior to bregma, 1.5 mm lateral from midline and 3.3-4.0 below the skull surface 27. Initial placement of the cannula was verified by slow downward movement of saline when the tubing was opened and raised. The cannula was fixed to the skull with 2-3 small screws and dental cement. After hardening of the cement and suturing of the incision, a 30G stylet (Plastics One, Roanoke, VA) was secured within the guide.
Drug administration and materials
Following surgery, rats were given 7 days to recover prior to drug administration and experimentation. Drugs were administered via remote injection to minimize effects of animal handling. PE-10 tubing, filled with saline or drug, was used to connect a Hamilton microsyringe to a 30G microinjector. 15-20 μL was delivered to the lumbar region of the spinal cord via the intrathecal catheter. Progress of the injection was visually confirmed by observation of movement of a small air bubble within the PE-10 tubing. Injectors were left in place an additional min to minimize backflow within the catheter. The animal was returned to its testing box.
All drugs solutions were prepared fresh daily. 15d-PGJ2 was reconstituted in isotonic saline after methyl acetate solvent was evaporated using a gentle nitrogen stream, and injected at doses of 25 μg (8 μmol), 50 μg (16 μmol), 100 μg (32 μmol), and 200 μg (63 μmol) per injection. Rosiglitazone was diluted in a 30% DMSO and 70% isotonic saline solution and injected at doses of 25 μg (7 μmol), 50 μg (14 μmol), and 100 μg (28 μmol) per injection. BADGE was diluted in 100% DMSO. 15d-PGJ2, rosiglitazone, BADGE, and DMSO were obtained from Cayman Chemicals (Ann Arbor, MI).
Behavioral tests of mechanical and cold allodynia
Animals were acclimated to a stainless steel grid within individual Plexiglas boxes for 30-60 min, and then tested for mechanical allodynia followed by cold allodynia.
Mechanical allodynia
In all animals, we first assessed mechanical allodynia using von Frey filaments (Stoelting, Inc). The plantar region of each hind paw was stimulated with an incremental series of 8 monofilaments of logarithmic stiffness. In SNI rats, we further localized the stimulus region to the sural innervation territory of the lateral aspect of the plantar hind paw. The 50% withdrawal threshold was determined using the up-down method of Dixon, modified by Chaplan et al. 5. First, an intermediate von Frey monofilament (number 4.31, exerts 2.0g of force) was applied perpendicular to the plantar skin, causing a slight bending. In case of a positive response (rapid withdrawal of the paw within 6 sec), a smaller filament was tested. In case of a negative response, a larger filament was tested. Less than 5% animals did not develop mechanical allodynia on the day of pharmacological testing after nerve injury (VF threshold > 5.0 g on the nerve-injured side). In such cases, von Frey testing was either terminated or its data was not included in the final analysis. Data were plotted as actual mean values and as change in threshold between baseline and post-drug values (Δ Threshold). Δ Threshold was used for the statistical analysis.
Cold allodynia
Using a syringe connected to PE-90 tubing, flared at the tip to a diameter of 3.5 mm, we applied a drop of acetone to the plantar paw. Surface tension maintained the volume of the drop to 10-12 μl. The length of time the animal lifted or shook its paw was recorded. The duration of paw withdrawal was recorded for 30 sec. Three observations were averaged. Less than 5% of SNI animals did not develop cold allodynia on the day of pharmacological testing after nerve injury (response duration < 2 sec). In such cases, acetone testing was either terminated or its data was not included in the final analysis. Data is plotted as actual mean values and as change in response between baseline and post-drug values (Δ Response).
Paw withdrawal latency to heat
We evaluated the latency to paw withdrawal from an infrared heat stimulus (Ugo Basile) with the rat resting on a glass platform, as described previously, and present the average of 4 measurements 33 37.
Rotarod test
In order to evaluate the effect of PPARγ agonists on ataxia, rats were placed on an accelerating rotarod (Stoelting, Wood Dale, Ill). The rotating bar was subdivided into four compartments for the simultaneous assessment of four animals. Initially rotating at a rate of 4 rpm, the system was adjusted to accelerate to a maximum speed of 40 rpm over a period of 10 min. When a rat fell off the bar, the time was recorded. Animals were acclimated to the rotarod one day before testing for 5-10 trials to yield average latencies of approximately 3 min. On the day of testing, rotarod duration was measured three times prior to drug or vehicle administration and averaged. After drug administration, rotarod duration was measured in triplicate at 60 and 120 min and averaged for each time point.
Real time PCR
PCR conditions were optimized to ensure the linearity of the serial dilutions and the efficient amplification of a single PCR product. Rats were terminally anesthetized with ketamine / xylazine [1 ml/kg of a ketamine/xylazine mixture [ketamine (88.9 mg/ml)/xylazine (11.1 mg/ml)] (Butler, Columbus, OH)]. Following decapitation, the liver, brain, spleen and lumbar spinal cord were removed. On ice, the lumbar enlargements (L4–L6) were dissected and divided into hemispheres along the horizontal midline, just above the central canal. Samples were placed in RNAlater tissue storage reagent (Ambion, Austin, TX), and stored at 4°C overnight. Isolation of total RNA was performed using the RiboPure Kit (Ambion, Austin, TX) according to the manufacturer’s instructions. Purity and concentration of resulting samples was determined spectrophotometrically. Next, cDNA was prepared from 2 μg of total RNA by reverse transcription using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA) according to the manufacturer’s instructions. cDNA samples were diluted 1:10 in DNase- and RNase-free water prior to further analysis. Quantitative real-time PCR was performed using the iCycler iQ Real Time Detection System (Bio-Rad, Hercules, CA). Gene specific primer sequences were as follows: PPARγ, forward primer 5′-TGAAGGCTCATATCTGTCTCCG-3′; PPARγ reverse primer 5′-CATCGAGGAC ATCCAAGACAAC-3′; β-Actin forward primer 5′-GAG GCTCTCTTCCAGCCTTCCT TCCT-3′; and β-Actin reverse primer 5′-CCTGCTTGCTGAT CCACATCTGCTGG-3′. PCR reactions were carried out using 5 μL of cDNA, 10 μM of each primer, and 2× SYBR Green Supermix (Bio-Rad, Hercules, CA) in 25 μL reactions. Thermal cycling conditions were 95°C for 3 min, followed by 40 cycles of 95°C for 20 sec and 61.5°C for 1 min. A final melting curve verified single product formation. Gene starting quantity was based on the cycle threshold (Ct) method. A control cDNA dilution series of known concentration was created for each gene to establish a standard curve, plotting the logarithm of the standard concentration against the Ct values. Unknown samples were quantified from measured Ct values by interpolation, using the regression equation. Each value was normalized to β-Actin, a housekeeping gene, to control for the amount of input cDNA.
Western blotting
Lumbar L4-L5 spinal cord was removed and dissected as above. Nuclear extracts were obtained using a NE-PER Nuclear and Cytoplasmic Extraction Reagents kit (Pierce), and the resulting supernatant was stored at -80° C. They were diluted 1:1 in 2X Sample Buffer (Rockland, Gilbertsville, PA) to a final concentration of 12 μg total protein in 15 μl solution. Samples were boiled for 5 min, and then resolved on 10 % Tris-HCl Readygels in a mini-electrophoresis chamber (Bio-Rad) for 90 min at 90 V, which provided maximum resolution at 67 kD, the apparent molecular weight of PPARγ. Separated proteins were transferred to polyvinylidine fluoride (PVDF) membranes at 20 V for 1 hr. After washing, the membrane was blocked in buffer containing 5% non-fat dry milk and probed with a mouse monoclonal antibody directed at the C-terminus of PPARγ (Primary antibody: H-100, Santa Cruz Biotechnology; Secondary ab: HRP-conjugated goat-anti-mouse secondary antibody) or phospho(ser117)-PPARγ (Primary: sc-28001-R, 1:500; Secondary: goat anti-rabbit, 1:1000). All antibodies were from Santa Cruz Biotechnology. Immunodetection was performed using Supersignal West-pico chemiluminescence reagents (Pierce). Films were scanned and band intensities quantified using a Bio-Rad Gel-Doc system
Electrophoretic Mobility Shift Assay
Spinal cord was removed and dissected as above. Nuclear extracts from rat L4-L5 spinal cord were obtained using a NE-PER Nuclear and Cytoplasmic Extraction Reagents kit (Pierce) and then stored at -80° C. Samples were prepared using a LightShift Chemiluminescent EMSA kit (Pierce), by adding in the following order: ultra-pure water (volume-adjusted to yield 20 μL total sample volume), 2 μL 10X binding buffer, 1 μL polydI-dC, 1 μL 50% glycerol, 1 μL 1% NP-40, 1 μL of 1 M KCl, 1 μL 100 mM MgCl2, 1 μL 200 mM EDTA, pH 8.0, 4 pmol unlabeled probe, 2 μL nuclear extract, 20 fmol biotin-labeled probe, and 1 μg PPARγ antibody (sc-6284x or sc-7273, Santa Cruz). A consensus PPARγ response element 5′-CTGACACAGGCTAAAGGTCATCTGAAGAAG-3′ (PPRE) bearing a 3′ biotin tag was annealed to its biotinylated antisense partner for 20 min at room temperature, forming the biotin-labeled probe. Samples were then incubated at room temperature for 20 min, after which 5 μL of loading buffer is added. Before sample loading, 4-20% Ready Gel® TBE (Tris-buffered EDTA) gels (Bio-Rad) were electrophoresed for 1 hr at 100 V. 15 μL of each sample was loaded into the pre-electrophoresed gel and resolved for ~90 min (or to optimal specific oligonucleotide resolution). DNA was then transferred to a Zetaprobe membrane (Bio-Rad), UV cross-linked (Fisher Scientific), probed with streptavidin-horseradish peroxidase (HRP) conjugate, and incubated with LightShift chemiluminescent substrate (Pierce, Rockford, IL).
Statistical analysis
Data are presented as mean ± SEM. Differences between means were analyzed by two-way repeated-measures analysis of variance (ANOVA), with drug treatment as the between-subjects variable and time as the repeated measure. If significant (P<0.05), the analyses were followed by post hoc t-tests. Using GraphPad Prism, sigmoidal dose-response curves were generated, and used to calculate ED50 (dose of drug producing 50% of maximal effect).
RESULTS
PPARγ expression and DNA binding in the rat lumbar spinal cord
To demonstrate the expression of PPARγ in the dorsal horn, we measured its levels of mRNA and protein, as well as PPARγ – DNA interactions, in spinal cord tissue. As illustrated in Fig 1A, we found detectable levels of PPARγ mRNA in the spinal cord that was 10-12 fold lower than spleen or liver (p < 0.05), but essentially equivalent to brain. Fig 1B confirms the presence of PPARγ protein (~67kD) in dorsal L4-L5 spinal cord tissues; as expected, spinal levels were relatively low compared to liver. The EMSA of Figs 1C-D reveals PPARγ-DNA binding capability in the dorsal horn.
Figure 1. PPARγ expression in the rat dorsal horn.
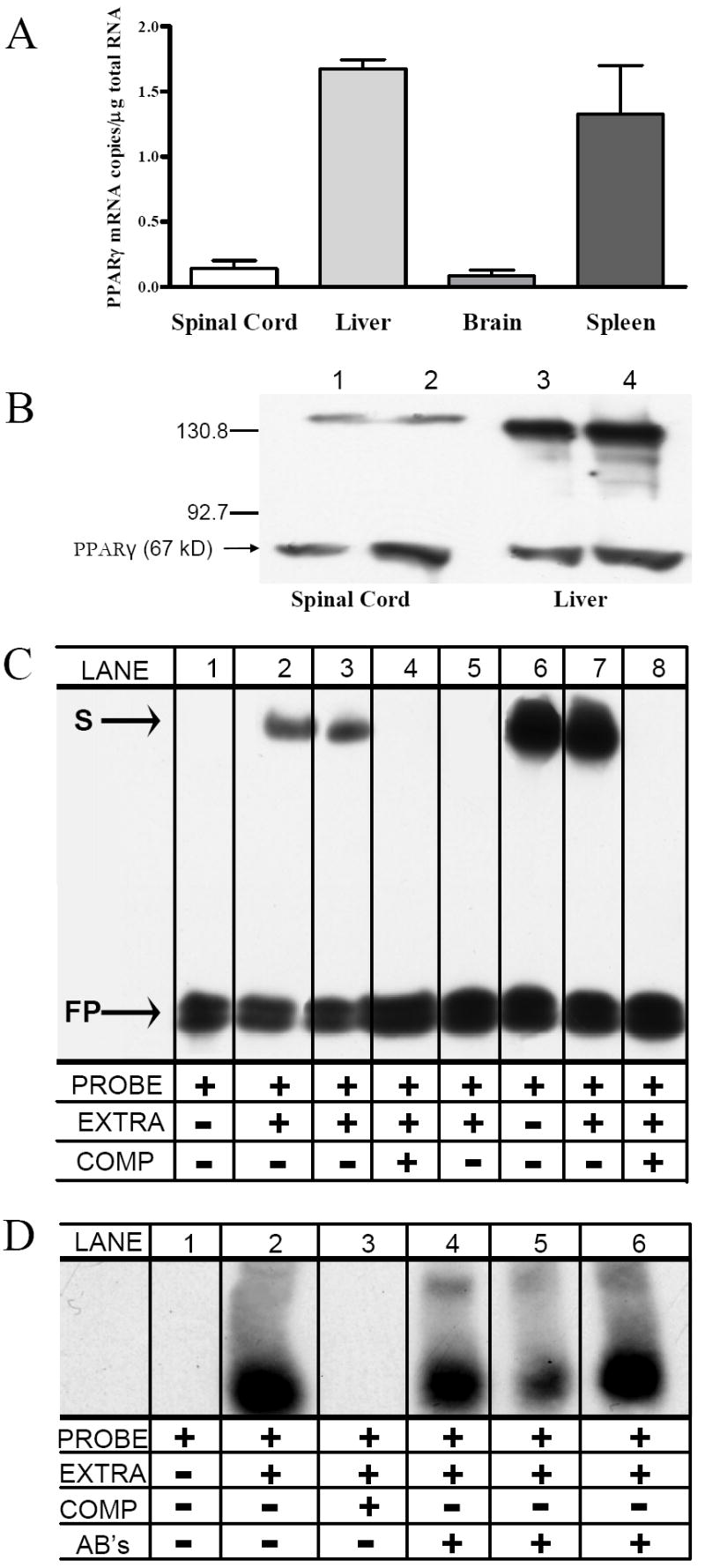
(A) Expression of PPARγmRNA in spinal cord. Positive controls include liver, brain, and spleen. (B) Representative Western blots of PPARγ immunoreactivity. This gel illustrates the results of two rats from an enriched nuclear fraction of lumbar dorsal spinal cord (lanes 1-2) or liver (lanes 3-4). MW markers (kD) are indicated on the left. A band of expected size (~67 kD) for PPARγ was present. The higher MW band (=~135 kD) was no longer apparent with the addition of β-mercaptoethanol (not shown). These experiments were repeated several times with similar results. (C) Specificity of protein-binding to DNA in spinal cord (lanes 1-4) and liver (lanes 5-8). Lanes 1-5 show the location of the biotinylated probe alone (free probe, “FP”). Lanes 2-3 and 6-7 illustrate examples of shift bands (S), indicating that protein is bound to the probe, thus reducing its mobility. Lanes 4-8 show that addition of a competitor untagged oligonucleotide (COMP), in high molar excess relative to the probe, leads to disappearance of S. This indicates that all transcriptional complexes are bound to unlabeled probe rather than labeled probe, and demonstrate specificity of the nuclear protein complex (likely a PPARγ/RXR heterodimer) to the oligonucleotide probe. (D) Specificity of DNA-binding to PPARγ in spinal cord. To further demonstrate specificity of the consensus PPARresponse element (probe) to PPARγ heterodimer complexes, a supershift assay was performed. Lane 3 repeats our finding that excess unlabeled probe competes with labeled probe, thus eliminating the S band. Lanes 4-6 show that 2 μg of PPARγ-specific antibodies (sc-6284x and sc-7273x) further decrease electrophoretic mobility, leading to the appearance of supershift (SS) bands. The formation of this antibody/protein/DNA complex indicates that a PPARγ-heterodimer binds to the DNA probe.
Intrathecal 15d-PGJ2 acts at PPARγ receptors to inhibit neuropathic pain
We first tested the effects of 15-deoxy-Δ12,14-PGJ2 (15d-PGJ2, a cyclopentanone prostaglandin), a naturally-occurring PPARγ agonist 30, on behavioral signs of neuropathic pain. As illustrated in Fig 2, SNI reduced mechanical VF thresholds from baseline (~15 g) to 1-2 g. Intrathecal injection of 15d-PGJ2 increased mechanical threshold in animals with spared nerve injury (F(4,154) = 18.8, P<0.001, overall effect of Dose in Fig 2A). Effects began within 30 min, peaked at approximately 60 min, and were of moderate duration, resolving within 4 hr. Fig 2C illustrates that the effect was dose-dependent, with an ED50 = 73.97 μg. The highest doses of 15d-PGJ2 did not return threshold to baseline levels (only to ~10 g), hence the anti-allodynic effect was incomplete. Adverse effects were not discernable by the observer, except for ataxia in a single animal following the 200 μg dose.
Figure 2. The naturally-occurring PPARγ agonist, 15d-PGJ2, dose-dependently increases mechanical threshold.

Panel A illustrates the time course of mechanical threshold following intrathecal injection of saline or 15d-PGJ2 in rats, 7d after spared nerve injury (SNI). Panel B illustrates the change in mechanical threshold averaged from 30 to 90 min after injection (Mean Δ in Threshold). Panel C illustrates the log dose-response curve at 60 min after injection. The 100 μg and 200 μg doses significantly increased mechanical threshold at the 60 and 90 min time points. Values represent mean ± SEM, n=5-7. *P < 0.05 vs saline control.
To test whether the anti-allodynic effects of 15d-PGJ2 were PPARγ receptor mediated, we co-administered a PPARγ antagonist, bisphenol A diglycidyl ether (BADGE) 41, with an efficacious dose of 15d-PGJ2 (100 μg). As illustrated in Fig 3, BADGE dose-dependently reduced the anti-allodynic effect of 15d-PGJ2 (F(6,33) = 16.1, P<0.001, overall effect of Dose in Fig 3A). Subsequent one-way analyses, as illustrated in Fig 2B, revealed a significant effect of the 5, 25, and 100 μg BADGE doses as compared to 15d-PGJ2 alone. Fig 3C illustrates that the effect was dose-dependent, with an ED50 = 11.44 μg. Adverse effects of BADGE or 15d-PGJ2/BADGE combinations were not discernable by the observer.
Figure 3. The PPARγ antagonist, bisphenol A diglycidyl ether (BADGE), dose-dependently reverses the antiallodynic effect of 15d-PGJ2.

Panel A illustrates the time course of mechanical threshold following intrathecal co-injection of 15d-PGJ2 (100 μg) and BADGE, 7d after spared nerve injury (SNI). Panel B illustrates the change in mechanical threshold averaged from 30 to 90 min after injection (Mean Δ in Threshold), and indicates that thresholds in the 5, 25, and 100 μg BADGE+15d-PGJ2 groups were different from 15d-PGJ2 controls. Panel C illustrates the log dose-response curve at 60 min after injection. Vehicle for BADGE was DMSO. Values represent mean ± SEM, n=4-8. *P < 0.05.
Intrathecal rosiglitazone acts at PPARγ receptors to inhibit neuropathic pain
Next, we tested the effects of the synthetic PPARγ agonist, rosiglitazone (ROSI), on nerve injury-induced allodynia. As illustrated in Fig 4A, intrathecal injection of rosiglitazone dose-dependently reversed mechanical hypersensitivity (F(3,140) = 12.5, P<0.0001, overall effect of Dose). As illustrated in Fig 4B, rosiglitazone dose-dependently reversed cold hypersensitivity (F(3,140) = 19.2, P<0.0001, overall effect of Dose). These effects peaked at approximately 90 min and resolved within 4 hr. As with 15d-PGJ2, the anti-allodynic effect was robust, but incomplete. Gross adverse effects were not observed, except for ataxia in a single animal following the 100 μg dose.
Figure 4. The PPARγ agonist, rosiglitazone, dose-dependently inhibits behavioral hypersensitivity.

Panel A illustrates the time course of mechanical threshold following intrathecal injection of saline or drug in rats, 7d after spared nerve injury (SNI). Panel B illustrates the time course of cold response following intrathecal injection of saline or drug in rats, 7d after spared nerve injury (SNI). The 100 μg dose significantly increased mechanical threshold at the 60 and 90 min time points, and significantly decreased cold response at the 60, 90, and 120 min time points. The 50 μg dose significantly decreased cold response at the 60 and 90 min time points. Vehicle represents 5μL 30% DMSO +10μL saline flush Values represent mean ± SEM, n=6. *P < 0.05 vs saline control.
As illustrated in Fig 5, co-administration of 25 μg BADGE reversed the inhibitory effect of 100 μg rosiglitazone on mechanical (F(2,119) = 22, P<0.0001, overall effect of Dose) and cold (F(2,119) = 46, P<0.0001, overall effect of Dose) hypersensitivity.
Figure 5. BADGE dose-dependently reversed the antiallodynic effect of rosiglitazone.

Panel A illustrates the time course of mechanical threshold following intrathecal co-injection of drugs, 7d after spared nerve injury (SNI). Panel B illustrates the change in mechanical threshold at 90 min after injection. Panel C illustrates the time course of cold response following intrathecal co-injection of drugs, 7d after spared nerve injury (SNI). Panel D illustrates the change in cold response at 90 min after injection. A 100 μg dose of rosiglitazone (Rosi) was used. The vehicle (DMSO/saline) and Rosi + 25 μg BADGE groups were significantly different from Rosi at the 90 min time point for mechanical allodynia. The vehicle and Rosi + 100 μg BADGE groups were significantly different from Rosi at the 90 min time point for cold response. Vehicle represents 5μL 30% DMSO + 5μL 100% DMSO + 10μL saline flush Values represent mean ± SEM, n=4-8. *P < 0.05.
Intrathecal 15d-PGJ2 and rosiglitazone do not alter motor coordination, von Frey threshold, or heat or cold withdrawal response
As mentioned above, neither 15d-PGJ2 nor rosiglitazone produced gross behavioral changes or other obvious deleterious effects. To assess more subtle behavioral effects, we evaluated motor coordination and sensory thresholds in uninjured rats. As illustrated in Fig 6, anti-allodynic doses of rosiglitazone (100-300 μg) did not change the response to mechanical or thermal stimuli, and did not reduce motor coordination (P>0.05). Only a supra-maximal dose of 1000 μg reduced motor coordination: this effect was small, not statistically significant, and not associated with gross behavioral changes or other obvious deleterious effects.
Figure 6. Rosiglitazone does not affect mechanical threshold, cold response, IR latency, or motor coordination in animals without nerve injury.

Panels A, B, and C illustrates the time course of mechanical threshold, cold response, and IR latency, respectively, following intrathecal injection of drug, 7d after sham SNI surgery. The intrathecal administration of rosiglitazone did not change mechanical threshold, cold response, or IR latency in sham animals. Panel D illustrates the effects of saline/DMSO or 100 μg, 300 μg, or 1000 μg rosiglitazone, administered intrathecally, on motor coordination. Motor coordination is measured by duration spent walking on rotarod. The administration of intrathecal rosiglitazone at anti-allodynic doses does not change motor coordination, when compared to baseline measures (P > 0.05). Vehicle represents 5μL 30% DMSO + 10μL saline flush.
Supraspinal or peripheral sites do not mediate the anti-allodynic actions of intrathecally-administered PPARγ agonists
As with many other compounds injected into the spinal CSF, PPARγ agonists could conceivably diffuse into the CNS 28 and periphery. To test this, identical doses of these drugs were administered by the intracerebroventricular and systemic routes. As illustrated in Fig 7, i.c.v. administration of neither rosiglitazone nor 15d-PGJ2 changed the paw withdrawal response to von Frey hairs or topical acetone. As a positive control, we show that i.c.v administration of morphine essentially reversed mechanical and cold allodynia. Similarly, intraperitoneal injection of neither drug changed mechanical or cold responses.
Figure 7. Supraspinal or peripheral administration of PPARγ agonists (at the intrathecal doses used above) did not reduce mechanical or cold hypersensitivity.

An intracerebroventricular (i.c.v.) or intraperitoneal (i.p.) injection of saline, 15d-PGJ2, rosiglitazone, and/or morphine was administered to rats, 7d after spared nerve injury (SNI). Panels A and C illustrate the time course of mechanical threshold. Panels B and D illustrate the time course of cold response. In contrast to morphine, a positive control that reduced mechanical and cold allodynia (Panels A-B, P<0.05), neither 15d-PGJ2, rosiglitazone, nor vehicle significantly changed mechanical threshold or cold response. Values represent mean ± SEM, n=4.
DISCUSSION
Evidence for PPARγ expression in the rat dorsal horn
Only a single study using a single technique (immunohistochemistry) has reported the presence of PPARγ protein in the dorsal horn 25. Our results confirm this finding using western blot. Furthermore, we demonstrate for the first time expression of PPARγ mRNA, and also we show that a PPARγ heterodimer binds to a specific consensus PPARγ response element. Our findings provide the precedence for further studies to determine the relationship between allodynia and spinal PPARγ expression in models of neuropathic pain. One clue comes from Mey and colleagues 38, who reported that peripheral nerve injury involving sciatic nerve crush increases transcript and protein levels of RXR, which forms functional heterodimers with activated PPARs 34, 43. Whether this result extends to PPARγ expression in the spinal cord, however, remains to be determined.
Intrathecal administration of 15d-PGJ2 and rosiglitazone act at PPARγ in the spinal cord to decrease neuropathic pain
The present studies are the first to report that intrathecal administration of PPARγ agonists reduces nerve injury-induced cold and/or mechanical allodynia in a dose- and receptor-dependent manner. Although 15d-PGJ2 and rosiglitazone only partially reduced mechanical allodynia, the highest dose of rosiglitazone essentially reversed cold allodynia. And although the PPARγ receptor-selectively of BADGE is controversial 10, and BADGE exerts agonist properties in an ECV304 cell line 4, we believe that its ability to reverse the anti-allodynic actions of agonists strongly supports a PPARγ -dependent mechanism. In summary, we conclude that intrathecal administration of rosiglitazone and 15d-PGJ2 act at PPARγ in the spinal cord to inhibit behavioral signs of established peripheral neuropathic pain.
For three reasons, we do not believe that non-specific side effects contribute to PPAR ligand-induced reduction of behavioral hypersensitivity. First, the anti-allodynic effects of 15d-PGJ2 and rosiglitazone returned to pre-injection baseline within 4 hr; such a reversible nature argues against drug-induced injury to the nervous system. Second, rotarod testing demonstrated that rosiglitazone (at anti-allodynic doses) did not produce ataxia. And third, behavioral somatosensory responses to mechanical or thermal stimuli remained intact in uninjured animals.
Other actions of PPARγ agonists in the spinal cord
We do not understand the molecular mechanisms responsible for the novel analgesic effects of PPARγ agonists in peripheral neuropathic pain. An intriguing line of thought comes from a review of PPARγ-mediated actions in the CNS. Such studies are intriguing because the central sensitization mechanisms of central neuropathic and peripheral neuropathic pain are similar in many ways 42. Indeed, both arise in part from microglia or astrocyte activation and neuro-immune activation in the spinal cord 9, 20, 23, 40. A rapidly growing data stream indicates that, in the setting of brain injury, pioglitazone and other PPARγ agonists reduce the local transcription and synthesis of pro-inflammatory systems such nitric oxide, prostaglandins, cytokines, and chemokines 1, 3, 8, 13, 29. The PPARγ -sensitive events underlying spinal cord injury are less well-described 38, but do include local microglial activation, protein expression of several inflammatory mediators including interleukin-1β, interleukin-6, and the chemokine MCP-1, and, most notably, heat hyperalgesia 26. We speculate that similar actions in the spinal cord mediate the anti-allodynic actions of PPARγ agonists. Future investigations in our laboratory are designed to determine the spinal mediators affected by pioglitazone treatment after peripheral nerve injury.
Supraspinal actions of PPARγ agonists
The CNS actions of PPARγ agonists are not restricted to the spinal cord. Indeed, the in vivo administration of pioglitazone exerts neuroprotective effects in the brain in animal models of Alzheimer’s disease, Parkinson’s disease, stroke and multiple sclerosis 3, 32. Furthermore, immunohistochemical studies indicate a broad distribution of PPARγ-like immunoreactivity and/or mRNA transcripts throughout the CNS 6, 25. As with peripheral nerve injury, brain ischemia increases PPARγ expression in neurons, supporting the idea that injury alters neuronal PPARγ signalling 39. We do not believe that diffusion from the intrathecal space to supraspinal (or peripheral) sites contributes to the antiallodynic actions of 15d-PGJ2 (200 μg) or rosiglitazone (100 μg), because neither i.c.v. (nor i.p.) injection of these doses changed allodynia. Although this data does not preclude analgesic actions of higher supraspinal doses, they do strongly support a spinal site of action following intrathecal administration.
Transcription-independent mechanism of steroid receptor action
Steroid receptor ligands are well-known to exert numerous actions via long-term, transcription-dependent, genomic mechanisms. However, there is a growing recognition for the importance of transcription-independent mechanisms. As one of the first examples, progesterone triggers an acrosomal reaction in sperm cells within min of its administration 17. Furthermore, estrogens or glucocorticoids increase ion flux and the activation of protein kinases, leading to phosphorylation of cAMP response element binding protein (CREB) and activation of extracellular signal-regulated kinase / mitogen activated-protein kinase (ERK/MAPK) 17, indicating that signal transduction cascades contribute to nongenomic actions of steroid receptors. More recent data argue for the contribution of nongenomic mechanisms to the actions of PPARligands 11; PPARγ acts in a nongenomic manner to suppress NF-κB, STAT-1, and AP-1 signalling pathways 24. Importantly, LoVerme et al. demonstrated in mice that systemic administration of PPARα agonists rapidly (within 30 min) reduced behavioral signs of nociception after the intraplantar injection of dilute formalin 18. Similarly, the present studies show that both 15d-PGJ2 and rosiglitazone reduce neuropathic pain within 60 min of administration. Because this relatively short time course may be insufficient for altered transcription and translation of pain-related proteins, we suggest that nongenomic mechanisms in the spinal cord contribute to the anti-allodynic actions of PPARγ ligands. To test this hypothesis, future studies are needed to investigate the antiallodynic actions of PPARγ agonists at earlier time points after administration.
Clinical Significance
Neuropathic pain is not effectively controlled with currently available analgesic drugs. For example, first-line medical therapies such as gabapentin and opioids only reduce neuropathic pain by 26 to 38 percent 12. Other than very recent findings in models of spinal cord injury, the current findings comprise the first published report to describe inhibitory actions of PPARγ agonists on spinal pain transmission. We suggest that PPARγ agonists act as anti-allodynic agents and will yield important therapeutic effects. A particularly exciting candidate for PPARγ analgesia is pioglitazone; unlike rosiglitazone, it can cross the blood brain barrier to exert CNS actions 14. And because pioglitazone and rosiglitazone remain FDA approved, perhaps this and future basic science studies can be quickly translated to clinical trials, not only to treat hyperglycemia but also the neuropathic pain associated with diabetes.
Acknowledgments
The study was supported by NIH grants DA10356, NS43383, and DA19656 (BKT).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abdelrahman M, Sivarajah A, Thiemermann C. Beneficial effects of PPAR-gamma ligands in ischemia-reperfusion injury, inflammation and shock. Cardiovasc Res. 2005;65:772–81. doi: 10.1016/j.cardiores.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 2.Berger J, Akiyama T, Meinke P. PPARs: therapeutic targets for metabolic disease. Trends Pharmacol Sci. 2005;26:244–51. doi: 10.1016/j.tips.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 3.Bernardo A, M L. PPAR-gamma agonists as regulators of microglial activation and brain inflammation. Curr Pharm Des. 2006;12:93–109. doi: 10.2174/138161206780574579. [DOI] [PubMed] [Google Scholar]
- 4.Bishop-Bailey D, Hla T, Warner TD. Bisphenol A diglycidyl ether (BADGE) is a PPARgamma agonist in an ECV304 cell line. British journal of pharmacology. 2000;131:651–4. doi: 10.1038/sj.bjp.0703628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. Journal of neuroscience methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 6.Cullingford TE, Bhakoo K, Peuchen S, Dolphin CT, Patel R, Clark JB. Distribution of mRNAs encoding the peroxisome proliferator-activated receptor alpha, beta, and gamma and the retinoid X receptor alpha, beta, and gamma in rat central nervous system. J Neurochem. 1998;70:1366–75. doi: 10.1046/j.1471-4159.1998.70041366.x. [DOI] [PubMed] [Google Scholar]
- 7.Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. 2000;87:149–58. doi: 10.1016/S0304-3959(00)00276-1. [DOI] [PubMed] [Google Scholar]
- 8.Dehmer T, Heneka MT, Sastre M, Dichgans J, Schulz JB. Protection by pioglitazone in the MPTP model of Parkinson’s disease correlates with I kappa B alpha induction and block of NF kappa B and iNOS activation. J Neurochem. 2004;88:494–501. doi: 10.1046/j.1471-4159.2003.02210.x. [DOI] [PubMed] [Google Scholar]
- 9.DeLeo JA, Tanga FY, Tawfik VL. Neuroimmune activation and neuroinflammation in chronic pain and opioid tolerance/hyperalgesia. Neuroscientist. 2004;10:40–52. doi: 10.1177/1073858403259950. [DOI] [PubMed] [Google Scholar]
- 10.Fehlberg S, Trautwein S, Goke A, Goke R. Bisphenol A diglycidyl ether induces apoptosis in tumour cells independently of peroxisome proliferator-activated receptor-gamma, in caspase-dependent and -independent manners. The Biochemical journal. 2002;362:573–8. doi: 10.1042/0264-6021:3620573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gardner OS, Dewar BJ, Graves LM. Activation of mitogen-activated protein kinases by peroxisome proliferator-activated receptor ligands: an example of nongenomic signaling. Molecular pharmacology. 2005;68:933–41. doi: 10.1124/mol.105.012260. [DOI] [PubMed] [Google Scholar]
- 12.Gilron I, Bailey JM, Tu D, Holden RR, Weaver DF, Houlden RL. Morphine, gabapentin, or their combination for neuropathic pain. N Engl J Med. 2005;352:1324–34. doi: 10.1056/NEJMoa042580. [DOI] [PubMed] [Google Scholar]
- 13.Heneka MT, Landreth GE, Feinstein DL. Role for peroxisome proliferator-activated receptor-gamma in Alzheimer’s disease. Annals of neurology. 2001;49:276. doi: 10.1002/1531-8249(20010201)49:2<276::aid-ana53>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 14.Heneka MT, Sastre M, Dumitrescu-Ozimek L, Hanke A, Dewachter I, Kuiperi C, O’Banion K, Klockgether T, Van Leuven F, Landreth GE. Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1-42 levels in APPV717I transgenic mice. Brain. 2005;128:1442–53. doi: 10.1093/brain/awh452. [DOI] [PubMed] [Google Scholar]
- 15.Kota BP, H T, Roufogalis BD. An overview on biological mechanisms of PPARs. Pharmacological Research. 2005;51:85–94. doi: 10.1016/j.phrs.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 16.Lehrke M, L M. The many faces of PPARgamma. Cell. 2005;123:993–99. doi: 10.1016/j.cell.2005.11.026. [DOI] [PubMed] [Google Scholar]
- 17.Losel RM, Falkenstein E, Feuring M, Schultz A, Tillmann HC, Rossol-Haseroth K, Wehling M. Nongenomic steroid action: controversies, questions, and answers. Physiological reviews. 2003;83:965–1016. doi: 10.1152/physrev.00003.2003. [DOI] [PubMed] [Google Scholar]
- 18.LoVerme J, R R, La Rana G, Fu J, Farthing J, Mattace-Raso G, Meli R, Hohmann A, Calignano A, Piomelli D. Rapid broad-spectrum analgesia through activation of peroxisome proliferator-activated receptor-alpha. J Pharmacol Exp Ther. 2006;319:1051–61. doi: 10.1124/jpet.106.111385. [DOI] [PubMed] [Google Scholar]
- 19.Malkmus SA, Yaksh TL. Intrathecal catheterization and drug delivery in the rat. Methods Mol Med. 2004;99:109–21. doi: 10.1385/1-59259-770-X:011. [DOI] [PubMed] [Google Scholar]
- 20.Marchand F, Perretti M, McMahon SB. Role of the immune system in chronic pain. Nat Rev Neurosci. 2005;6:521–32. doi: 10.1038/nrn1700. [DOI] [PubMed] [Google Scholar]
- 21.Martens FM, Visseren FL, Lemay J, de Koning EJ, Rabelink TJ. Metabolic and additional vascular effects of thiazolidinediones. Drugs. 2002;62:1463–80. doi: 10.2165/00003495-200262100-00004. [DOI] [PubMed] [Google Scholar]
- 22.Michalik L, Wahli W. Involvement of PPARnuclear receptors in tissue injury and wound repair. The Journal of clinical investigation. 2006;116:598–606. doi: 10.1172/JCI27958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moalem G, Tracey DJ. Immune and inflammatory mechanisms in neuropathic pain. Brain Res Rev. 2006;51:240–64. doi: 10.1016/j.brainresrev.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 24.Moraes LA, Piqueras L, Bishop-Bailey D. Peroxisome proliferator-activated receptors and inflammation. Pharmacology & therapeutics. 2006;110:371–85. doi: 10.1016/j.pharmthera.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 25.Moreno S, Farioli-Vecchioli S, Ceru MP. Immunolocalization of peroxisome proliferator-activated receptors and retinoid X receptors in the adult rat CNS. Neuroscience. 2004;123:131–45. doi: 10.1016/j.neuroscience.2003.08.064. [DOI] [PubMed] [Google Scholar]
- 26.Park SW, Y J, Miranpuri G, Satriotomo I, Bowen K, Resnick DK, Vemuganti R. Thiazolidinedione Class of Peroxisome Proliferator-Activated Receptor {gamma} Agonists Prevents Neuronal Damage, Motor Dysfunction, Myelin Loss, Neuropathic Pain, and Inflammation after Spinal Cord Injury in Adult Rats. J Pharmacol Exp Ther. 2007;320:1002–12. doi: 10.1124/jpet.106.113472. [DOI] [PubMed] [Google Scholar]
- 27.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Orlando, FL: Academic Press, Inc.; 1997. [Google Scholar]
- 28.Proescholdt MG, Hutto B, Brady LS, Herkenham M. Studies of cerebrospinal fluid flow and penetration into brain following lateral ventricle and cisterna magna injections of the tracer [14C]inulin in rat. Neuroscience. 2000;95:577–92. doi: 10.1016/s0306-4522(99)00417-0. [DOI] [PubMed] [Google Scholar]
- 29.Schutz B, Reimann J, Dumitrescu-Ozimek L, Kappes-Horn K, Landreth GE, Schurmann B, Zimmer A, Heneka MT. The oral antidiabetic pioglitazone protects from neurodegeneration and amyotrophic lateral sclerosis-like symptoms in superoxide dismutase-G93A transgenic mice. J Neurosci. 2005;25:7805–12. doi: 10.1523/JNEUROSCI.2038-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soares AF, Nosjean O, Cozzone D, D’Orazio D, Becchi M, Guichardant M, Ferry G, Boutin JA, Lagarde M, Geloen A. Covalent binding of 15-deoxy-delta12,14-prostaglandin J2 to PPARgamma. Biochem Biophys Res Commun. 2005;337:521–5. doi: 10.1016/j.bbrc.2005.09.085. [DOI] [PubMed] [Google Scholar]
- 31.Staels B, Fruchart JC. Therapeutic roles of peroxisome proliferator-activated receptor agonists. Diabetes. 2005;54:2460–70. doi: 10.2337/diabetes.54.8.2460. [DOI] [PubMed] [Google Scholar]
- 32.Sundararajan S, Jiang Q, Heneka M, Landreth G. PPARgamma as a therapeutic target in central nervous system diseases. Neurochem Int. 2006;49:136–44. doi: 10.1016/j.neuint.2006.03.020. [DOI] [PubMed] [Google Scholar]
- 33.Taiwo OB, Taylor BK. Antihyperalgesic effects of intrathecal neuropeptide Y during inflammation are mediated by Y1 receptors. Pain. 2002;96:353–63. doi: 10.1016/S0304-3959(01)00481-X. [DOI] [PubMed] [Google Scholar]
- 34.Tan NS, Michalik L, Desvergne B, Wahli W. Multiple expression control mechanisms of peroxisome proliferator-activated receptors and their target genes. The Journal of steroid biochemistry and molecular biology. 2005;93:99–105. doi: 10.1016/j.jsbmb.2004.12.025. [DOI] [PubMed] [Google Scholar]
- 35.Taylor BK, Holloway D, Printz MP. A unique central cholinergic deficit in the spontaneously hypertensive rat: physostigmine reveals a bradycardia associated with sensory stimulation. The Journal of pharmacology and experimental therapeutics. 1994;268:1081–90. [PubMed] [Google Scholar]
- 36.Taylor BK, Kriedt C, Nagalingam S, Dadia N, Badr M. Central administration of perfluorooctanoic acid inhibits cutaneous inflammation. Inflamm Res. 2005;54:235–42. doi: 10.1007/s00011-005-1350-0. [DOI] [PubMed] [Google Scholar]
- 37.Taylor BK, D N, Yang CB, Krishnan S, Badr M. Peroxisome proliferator-activated receptor agonists inhibit inflammatory edema and hyperalgesia. Inflammation. 2002;26:121–27. doi: 10.1023/a:1015500531113. [DOI] [PubMed] [Google Scholar]
- 38.van Neerven S, Mey J. RAR/RXR and PPAR/RXR Signaling in Spinal Cord Injury. PPARresearch. 2007;2007:29275. doi: 10.1155/2007/29275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Victor NA, Wanderi EW, Gamboa J, Zhao X, Aronowski J, Deininger K, Lust WD, Landreth GE, Sundararajan S. Altered PPARgamma expression and activation after transient focal ischemia in rats. The European journal of neuroscience. 2006;24:1653–63. doi: 10.1111/j.1460-9568.2006.05037.x. [DOI] [PubMed] [Google Scholar]
- 40.Wieseler-Frank J, Maier SF, Watkins LR. Central proinflammatory cytokines and pain enhancement. Neuro-Signals. 2005;14:166–74. doi: 10.1159/000087655. [DOI] [PubMed] [Google Scholar]
- 41.Wright HM, C C, Mikami T, Hauser S, Yanagi K, Hiramatsu R, Serhan CN, Spiegelman BM. A synthetic antagonist for the peroxisome proliferator-activated receptor gamma inhibits adipocyte differentiation. J Biol Chem. 2000;275:1873–77. doi: 10.1074/jbc.275.3.1873. [DOI] [PubMed] [Google Scholar]
- 42.Yezierski RP. Pain following spinal cord injury: pathophysiology and central mechanisms. Prog Brain Res. 2000;129:429–49. doi: 10.1016/S0079-6123(00)29033-X. [DOI] [PubMed] [Google Scholar]
- 43.Zhelyaznik N, Mey J. Regulation of retinoic acid receptors alpha, beta and retinoid X receptor alpha after sciatic nerve injury. Neuroscience. 2006;141:1761–74. doi: 10.1016/j.neuroscience.2006.05.021. [DOI] [PubMed] [Google Scholar]