

Abstract
The lipid A and inner core regions of Rhizobium leguminosarum lipopolysaccharide contain four galacturonic acid (GalA) residues. Two are attached to the outer unit of the 3-deoxy-D-manno-octulosonic acid (Kdo) disaccharide, one to the mannose residue, and one to the 4′-position of lipid A. The enzymes RgtA and RgtB, described in the accompanying article, catalyze GalA transfer to the Kdo residue, whereas RgtC is responsible for modification of the core mannose unit. Heterologous expression of RgtA in Sinorhizhobium meliloti 1021, a strain that normally lacks GalA modifications on its Kdo disaccharide, resulted in detectable GalA transferase activity in isolated membrane preparations, suggesting that the appropriate GalA donor substrate is available in S. meliloti membranes. In contrast, heterologous expression of RgtA in Escherichia coli yielded inactive membranes. However, RgtA activity was detectable in the E. coli system when total lipids from R. leguminosarum 3841 or S. meliloti 1021 were added. We have now purified and characterized dodecaprenyl (C60) phosphate-GalA as a minor novel lipid of R. leguminosarum 3841 and S. meliloti. This substance is stable to mild base hydrolysis and was purified by DEAE-cellulose column chromatography. Its structure was established by a combination of electrospray ionization mass spectrometry and gas-liquid chromatography. Purified dodecaprenyl phosphate-GalA supports the efficient transfer of GalA to Kdo2-1-dephospho-lipid IVA by membranes of E. coli cells expressing RgtA, RgtB, and RgtC. The identification of a polyisoprene phosphate-GalA donor substrate suggests that the active site of RgtA faces the periplasmic side of the inner membrane. This work represents the first definitive characterization of a lipid-linked GalA derivative with the proposed structure dodecaprenyl phosphate-β-D-GalA.
Polyisoprene phosphate-linked sugars are donor substrates for a wide variety of glycosyltransferases found in prokaryotic and eukaryotic cells (1–4). These compounds usually function in compartments where sugar nucleotides are limited or not available. In Escherichia coli, undecaprenyl diphosphate-sugars are substrates for the polymerization of peptidoglycan or O-antigen in the periplasm (1–3, 5). Undecaprenyl monophosphate-sugars are donors in the biosynthesis of mycobacterial lipoglycans (6–8), glycosylation of teichoic acids (9), or bacteriophage-mediated modification of O-antigens (10, 11). The structurally related dolichyl monophosphate- and diphosphate-sugars of eukaryotic cells are required for protein glycosylation (4, 12–14) and for the enzymatic assembly of phosphatidylinositol glycans (15, 16).
As shown in the preceding article (17), the membrane-bound enzymes RgtA, RgtB, and RgtC are responsible for the transfer of three GalA moieties to the Rhizobium leguminosarum LPS2 core (Fig. 1). A fourth rgt gene, designated rgtD, is proposed to encode the enzyme that adds Gal to the lipid A 4′-position in this organism (Fig. 1). RgtA, RgtB, and RgtC were identified by an expression-cloning strategy involving an R. leguminosarum 3841 genomic DNA library harbored in Sinorhizobium meliloti 1021 (17, 18). These transferases appear to use a membrane-associated Gal donor, possibly a polyisoprene phosphate derivative (17, 19). The Rgt enzymes show distant sequence similarity to ArnT, an Escherichia coli glycosyltransferase that uses undecaprenyl phosphate-4-amino-4-deoxy-L-arabinose to modify lipid A in polymyxin-resistant mutants (20, 21).
FIGURE 1. Presence of GalA units in the lipid A and inner core oligosaccharide of R. leguminosarum.

The inner core oligosaccharide (panel A) is modified with three GalA residues (52–54). The lipid A of R. leguminosarum is a mixture of species designated B, C, D-1, and E (panel B). B and D-1 differ in their proximal sugar unit. C and E are the 3-O-deacylated versions of B and D-1, respectively. All lipid A species contain a GalA residue at the 4′-position (29, 32, 55).
The membrane-associated GalA donor is present not only in R. leguminosarum but also in S. meliloti 1021 membranes (17). It is absent in E. coli. Although S. meliloti apparently does not modify its LPS inner core with GalA residues, it does synthesize several surface polysaccharides, some of which may contain GalA units (22). This fact may explain why RgtA, RgtB, and RgtC activity can be detected in membranes of S. meliloti 1021 expressing the R. leguminosarum GalA transferase genes (17). We now report the purification and characterization of a novel, minor lipid from R. leguminosarum 3841 that supports the RgtA-catalyzed conversion of Kdo2-[1-dephospho]lipid IVA to GalA-Kdo2-[1-dephospho]lipid IVA in vitro. Electrospray ionization mass spectrometric (ESI/MS) and gas-liquid chromatographic (GC/MS) analyses clearly demonstrate that the proposed donor is dodecaprenyl (C60) phosphate-GalA. Based on the sequence similarity of RgtA to better characterized enzymes that utilize dolichyl phosphate-mannose as donor substrates (12, 15), we suggest that the GalA unit of our dodecaprenyl phosphate-GalA has the β-anomeric configuration. The same donor lipid is present in S. meliloti 1021, along with a less abundant C65 analogue. The presence of a polyisoprene phosphate-linked GalA donor lipid suggests that GalA addition to the LPS core occurs on the periplasmic leaflet of the inner membrane. A putative lipid-linked GalA derivative, generated in R. leguminosarum bv. trifolii extracts from endogenous lipids and UDP-[14C]glucuronic acid, was reported previously (23), but its covalent structure and biological functions were not characterized.
EXPERIMENTAL PROCEDURES
Chemicals and Materials
[γ-32P]ATP was obtained from PerkinElmer Life Sciences. Silica Gel 60 (0.25 mm) TLC plates were from Merck. Chloroform, ammonium acetate, and sodium acetate were from EM Science. Triton X-100, Nonidet P-40, and the BCA protein determination kit were purchased from Pierce. Yeast extract and tryptone were from Difco. All other chemicals were reagent-grade and were obtained from either Sigma or Mallinckrodt.
Bacterial Strains and Growth Conditions
All bacterial strains used in this study are described in Table 1. All Rhizobium and Sinorhizobium strains were grown at 30 °C in TY medium (5 g/liter tryptone, 3 g/liter yeast extract, and 10 mM CaCl2) supplemented with the antibiotics nalidixic acid (20 μg/ml) and streptomycin (200 μg/ml). The subclone pRK-RgtA in S. meliloti 1021 (17) was grown under additional selection with tetracycline (12.5 μg/ml). The E. coli strain NovaBlue (DE3) (Novagen), harboring either the empty vector pET23a or the plasmid pRgtA, pRgtB, or pRgtC (containing the rgtA, rgtB, or rgtC gene), was grown at 37 °C from a single colony in 200 ml of LB broth (10 g/liter tryptone, 5 g/liter yeast extract, and 10 g/liter NaCl), supplemented with ampicillin (100 μg/ml), until the A600 reached ~0.6. The culture was split into two equal portions, one of which was induced with 1 mM isopropyl 1-thio-β-D-galactopyranoside. Both cultures were further incubated with shaking at 225 rpm for an additional 4 h at 25 °C.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain/plasmid | Genotype or description | Source or Ref. |
|---|---|---|
| Strains | ||
| R. leguminosarum 3841 | Wild type strain 300 bv. viciae Smr | 51 |
| S. meliloti 1021 | SU47 Smr | S. Long |
| E. coli | ||
| NovaBlue (DE3) | E. coli host strain used for expression | Novagen |
|
| ||
| Plasmids | ||
| pET-23a | E. coli expression vector, T7lac promoter, Ampr | Novagen |
| pRgtA | pET-23a derivative harboring rgtA behind the T7lac promoter | 17 |
| pRgtB | pET-23a derivative harboring rgtB behind the T7lac promoter | 17 |
| pRgtC | pET-23a derivative harboring rgtC behind the T7lac promoter | 17 |
| pRK-RgtA | rgtA in pRK-404a | 17 |
Galacturonic Acid Transferase Assay
Standard assay conditions for the galacturonic acid transferase activity were as follows. The reaction mixture (10–20 μl) contained 50 mM MES, pH 6.5, 0.05% Nonidet P-40, 2 mM MgCl2, and carrier-free Kdo-1-dephospho-[4′-32P]lipid IVA (50,000 cpm/nmol), unless otherwise indicated. Radiolabeled substrates were prepared as described in the accompanying article (17). Washed membranes (0.25 mg/ml final concentration) were used as the enzyme source. Lipids at different stages of purity were dissolved in chloroform/methanol (4:1, v/v), and a volume corresponding to 0.25 mg of lipid was added to each assay tube. The lipids were dried down in a SpeedVac appliance (Savant). The rest of the assay mixture was then added, and the lipids were resuspended by vigorous mixing and bath sonication. Reactions were incubated at 30 °C for varying times and terminated by spotting 4 μl onto a Silica Gel 60 TLC plate, which was developed in the solvent chloroform, pyridine, 88% formic acid, water (30:70:16:10, v/v). After drying and overnight exposure of the plate to a PhosphorImager screen, product formation was detected and quantified with a PhosphorImager (Storm 840, Amersham Biosciences), equipped with Image-Quant software.
Extraction and Purification of Total Lipids from S. meliloti 1021 and R. leguminosarum 3841
One liter of mid-logarithmic phase cells was harvested by centrifugation at 4,000 × g for 20 min and washed once with phosphate-buffered saline. Lipids were extracted from the cell pellets, as described previously (24).
DEAE-cellulose Column Chromatography
Dried total R. leguminosarum lipids were redissolved in 50 ml of chloroform, methanol, water (2:3:1, v/v) and loaded onto a 5-ml DEAE-cellulose column equilibrated in the acetate form (25, 26). After application of the sample, the column was washed with 5 column volumes of chloroform, methanol, water (2:3:1, v/v). The product was then eluted with 8 column volumes of the same solvent system but with the aqueous portion consisting of 30, 60, 90, 120, 240, or 480 mM ammonium acetate, in ascending order. Each elution step was collected as one fraction of 40 ml. Each fraction was then converted to a two-phase acidic Bligh-Dyer mixture (27, 28) by the addition of appropriate amounts of chloroform, water, and 1 M HCl. The lower phase from each fraction was recovered, and the upper phase was washed once with fresh lower phase. The pooled lower phases from each fraction were neutralized with pyridine (1 drop per 2 ml of pooled lower phase), dried under a stream of nitrogen, and stored at −20 °C.
Mild Alkaline Hydrolysis of Lipids
Lipids that eluted in the 60 mM fraction of the DEAE-cellulose column were extracted and subjected to mild alkaline hydrolysis. An alkaline methanol solution was prepared by adding 0.4 ml of 5 M NaOH to 5 ml of methanol (final NaOH concentration of 0.37 M). To this we added 5 ml of chloroform. DEAE-cellulose column purified lipids (~3 mg) were redissolved in 1.5 ml of this mixture and incubated at room temperature for 1 h. The NaOH was neutralized with 3–4 drops of concentrated HCl, as judged by pH paper, and the system was converted to a two-phase Bligh-Dyer mixture (27). The extracted lipids were weighed, and a volume corresponding to 0.25 mg of lipid was used to assay for the ability to support GalA transferase activity (as described above).
ESI/MS Analysis
All mass spectra were acquired on a QSTAR XL quadrupole time-of-flight tandem mass spectrometer (ABI/MDS-Sciex, Toronto, Canada), equipped with an electrospray ionization source.
Spectra were acquired in the negative ion mode and were typically the accumulation of 60 scans, collected from 200 to 2000 atomic mass units. For MS analysis, the DEAE-cellulose-purified lipids were dissolved in 200 μl of chloroform/methanol (4:1, v/v). Typically, 20 μl of this solution was diluted into 200 μl of chloroform/methanol (1:1, v/v), and infused into the ion source at 5–10 μl/min. The negative ion analysis was carried out at − 4200 V. In the MS/MS mode, collision-induced dissociation tandem mass spectra were obtained, using a collision energy of −80 V (laboratory frame of energy). Nitrogen was used as the collision gas. Data acquisition and analysis were performed using Analyst QS software.
GC/MS Analysis
The column-purified lipid-linked hexuronic acid (HexA) donor was hydrolyzed in acidic methanol, treated with acetic anhydride, and converted to trimethylsilyl esters, according to a published procedure (29). Sugar standards (GalA and GlcA) were purchased from Sigma and prepared for analysis under identical conditions.
GC/MS was performed using a Finnigan Trace MS, coupled with a Trace GC 2000 gas chromatograph. The column used was a 30-m RTX-5MS (0.25-mm internal diameter; 0.25-μm phase thickness) from Restek (Bellefonte, PA). Helium was used as the carrier gas with a constant flow rate of 1 ml/min. The gas chromatography program started with the column oven temperature being held at 100 °C for 3 min, followed by an increase to 325 °C at a rate of 20 °C/min. The column was then held at 325 °C for 3 min. The injector was operated in the split mode (1:20 split), and the temperature of the injection port was kept at 200 °C. The instrument was operated in the electron impact mode with the electron energy set at 70 eV.
In Vitro Reconstitution of GalA Transferase Activity
The purified lipid donor, isolated from 4 liters of R. leguminosarum culture, was weighed (~0.2 mg) and redissolved in an appropriate volume of the GalA assay buffer to obtain a 500 μM stock solution, with the exception that the final Triton X-100 concentration in the buffer was increased from 0.05 to 0.2% to solubilize the purified lipid. Membranes (0.25 mg/ml) from E. coli NovaBlue (DE3), harboring either pRgtA or the vector pET23a, were assayed with 2.5 μM Kdo-1-dephospho-[4′-32P]lipid IVA (50,000 cpm/nmol). The purified donor substrate was added from the stock solution in increasing concentrations ranging from 0.1 to 100 μM. For complete reconstitution of Rgt activities, membranes (0.25 mg/ml) from E. coli NovaBlue (DE3), harboring either the vector pET23a or a combination of pRgtA, pRgtB, and pRgtC, were assayed with mannosyl-Kdo-1-dephospho-[4′-32P]lipid IVA (50,000 cpm/nmol) and 200 μM purified donor. Because we determined the lipid donor quantity by weighing a small amount (0.2 mg), the actual stock concentration may vary by as much as 2-fold from the estimated value. Reactions were incubated at 30 °C for 20 min and terminated by spotting 4 μl onto a Silica Gel 60 TLC plate that was developed and quantified as described.
RESULTS
RgtA-catalyzed Glycosylation Requires a Rhizobiaceae Membrane Lipid
As shown in the preceding article (17), RgtA expressed in E. coli NovaBlue (DE3) requires a Rhizobiaceae membrane component for activity. Based on the characteristics of the Rgt proteins and their sequence similarity to the E. coli ArnT (20, 21, 30), we hypothesized that a polyisoprene phosphate-linked GalA donor might be involved. We therefore extracted total lipids from S. meliloti 1021 or R. leguminosarum 3841, and we added them back to an assay system containing membranes of E. coli/pRgtA. Formation of the GalA-modified product was indeed dependent upon the presence of an added lipid component from either S. meliloti or R. leguminosarum (Fig. 2). The presence of a lipid-linked GalA donor in S. meliloti could reflect the use of a lipid-linked GalA donor for the addition of GalA moieties to some S. meliloti surface polysaccharides (22). It is well established that S. meliloti contains an epimerase (LpsL) that converts UDP-GlcA to UDP-GalA (22).
FIGURE 2. A Rhizobiaceae membrane-lipid component is required for RgtA activity in E. coli.
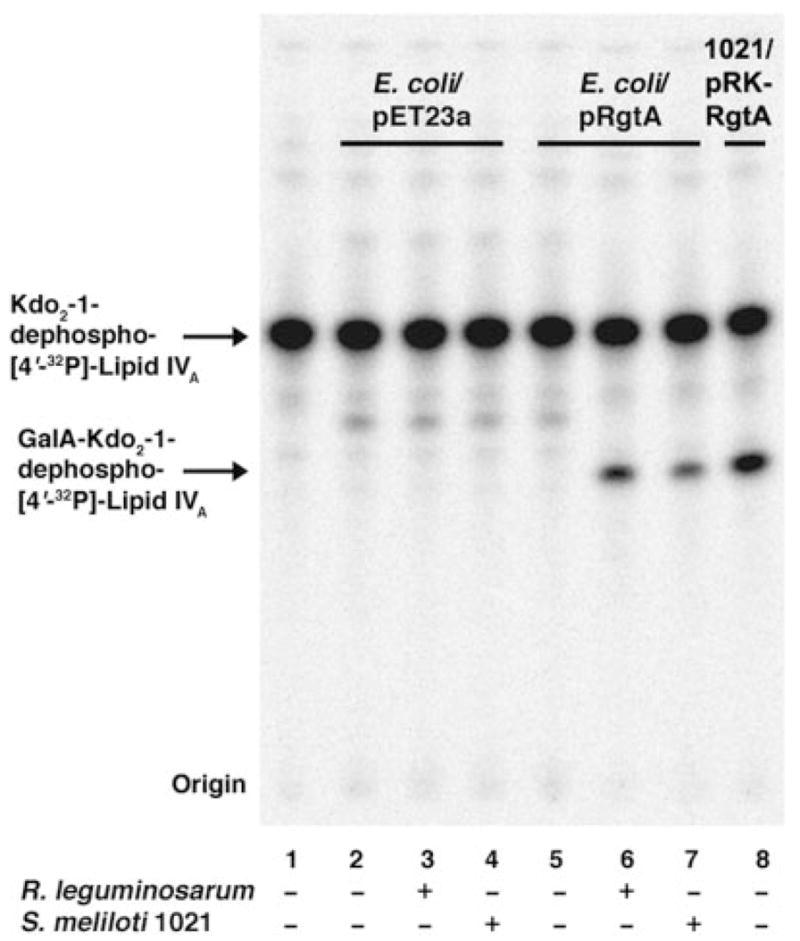
Membranes of E. coli harboring pRgtA or the vector control pET23a were assayed under standard conditions at 0.25 mg/ml with or without the addition of 0.25 mg of total lipids extracted from S. meliloti 1021 or R. leguminosarum 3841, as indicated. GalA addition was judged by the shift of the Kdo2-1-dephospho-[4′-32P]lipid IVA band. Membranes of S. meliloti 1021/pRK-RgtA without added lipids were used as the positive control (lane 8).
Purification and ESI/MS Analysis of the Lipid-linked GalA Donor
The reconstitution assay (Fig. 2) was used to monitor the presence of the GalA donor during the purification. The first step was DEAE-cellulose column chromatography, which separates lipids on the basis of their net negative charge (25). Lipids were loaded in chloroform, methanol, water (2:3:1, v/v) and eluted by replacing the water component with increasing concentrations of aqueous ammonium acetate (30–480 mM). Assays of the lipids contained in each fraction demonstrated that the GalA donor bound to the column and eluted with 60 mM ammonium acetate (Fig. 3). ESI/MS analysis of the 60 mM fraction revealed large amounts of cardiolipin and some phosphatidylglycerol (Fig. 4, panel A). In addition, two peaks were present that did not correspond to the mass of any known lipid. These two peaks were interpreted as arising from different ionization states of the same compound, a singly charged negative ion at m/z 1089.770 and a doubly charged negative ion at m/z 544.389 (Fig. 4, panel A, labeled as Donor Lipid).
FIGURE 3. Ability of DEAE-cellulose fractionated R. leguminosarum 3841 lipids to support RgtA activity.

Total lipids were extracted from R. leguminosarum 3841 and fractionated by DEAE-cellulose column chromatography. Lipids were eluted from the column with the solvent chloroform/methanol/aqueous ammonium acetate (2:3:1, v/v) by increasing the ammonium acetate concentration from 0 to 480 mM in the aqueous component. Lipids (0.25 mg) from each fraction were used in the reconstitution assay. NE, no enzyme; FT, flow-through.
FIGURE 4. Negative ion ESI/MS spectrum of the DEAE-cellulose fraction containing the donor substrate for RgtA.

The full spectrum of the R. leguminosarum lipids eluting in the 60 mM fraction from the DEAE-cellulose column is shown in panel A. The full spectrum of the base-stable lipids, eluting in the 60 mM fraction from a second DEAE-cellulose column, is shown in panel B. amu, atomic mass units.
The identity of these unknown lipid peaks was investigated by collision-induced dissociation tandem mass spectrometry (MS/MS) (Fig. 5). Among the fragments derived from the peak at m/z 544.3 are ions interpreted as (m/z 78.956), hexuronic acid (HexA) (m/z 193.031), HexA-phosphate (m/z 135.994, doubly charged), and dodecaprenyl phosphate (m/z 913.738) (Fig. 5). Additional peaks corresponding to a dehydrated HexA unit and a ladder of isoprene phosphate units were also observed (not labeled). Similar results were obtained from the MS/MS analysis of the ion at m/z 1089.7 (Fig. 4B) (data not shown), verifying that m/z 544.389 and m/z 1089.770 (Fig. 4) are indeed derived from the same parent lipid. The data are consistent with a novel dodecaprenyl phosphate-linked HexA, presumed to be the GalA donor (Fig. 5).
FIGURE 5. ESI/MS/MS analysis of the putative GalA donor substrate for RgtA.
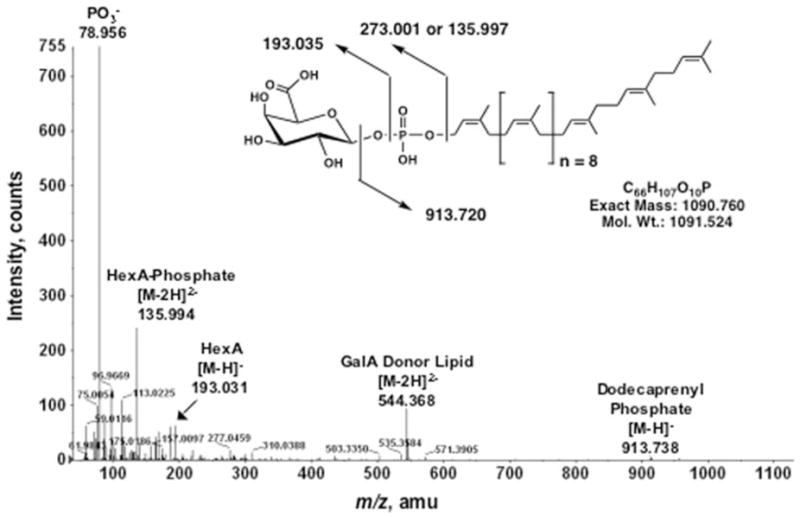
The MS/MS analysis of the predominant [M – 2H]2− ion at m/z 544.3 in Fig. 4, panel A, is shown together with the proposed fragment ion assignments and the covalent structure of the intact lipid. The proposed stereochemistry of the GalA linkage has not been established by NMR but is proposed based on the sequence similarity of the Rgt proteins to ArnT. amu, atomic mass units.
Because phospholipids are labile to mild alkaline treatment (31), we tested a range of alkaline hydrolysis conditions (0.1 to 0.54 N NaOH in chloroform/methanol, 1:1, v/v) to eliminate all phospholipid contaminants. The GalA donor is stable up to 0.4 N NaOH (data not shown). The alkali-treated lipids were recovered by two-phase Bligh-Dyer extraction. At post-purification, the donor lipid was the predominant species in the 60 mM fraction, as judged by ESI/MS (Fig. 4, panel B). The repurified material retained the ability to reconstitute GalA transferase activity when added to membranes of E. coli/pRgtA (see below).
GC/MS Analysis of the Dodecaprenyl Phosphate-linked HexA
The identity of the HexA moiety was determined by GC/MS (Fig. 6). The repurified donor lipid was derivatized as described previously (29). Assignments were based on peak retention times and electron impact mass spectra of the sugars released from the donor lipid compared with those of the standards (GalA and GlcA), prepared and run in parallel. The profile of the total ion current from the purified donor lipid exhibits major peaks with retention times exactly matching those of the GalA standard (Fig. 6).
FIGURE 6. Identification of GalA in the dodecaprenyl phosphate-linked HexA isolated from R. leguminosarum.

Trimethylsilyl ester derivatives of methyl glycosides, obtained by methanolic HCl hydrolysis of the purified dodecaprenyl phosphate-linked HexA, were separated by gas-liquid chromatography. Peaks were identified by comparison of retention times with standards prepared in parallel. The chromatogram for the lipid donor was compared with those of GalA (see asterisks) and GlcA standards. No GlcA peaks are present in the natural product.
Reconstitution of GalA Transferase Activity in Vitro with Purified Ddecaprenyl Phosphate-GalA
Transfer of the GalA moiety to Kdo2-1-dephospho-[4′-32P]lipid IVA proceeds rapidly when RgtA is overexpressed in S. meliloti 1021 but not in E. coli, reflecting the lack of the appropriate GalA donor in E. coli (17). The addition of the purified dodecaprenyl phosphate-GalA, prepared as described above, to membranes of E. coli NovaBlue (DE3)/pRgtA reconstituted robust GalA transferase activity in a concentration-dependent manner, whereas donor lipid addition to membranes of the vector control did not (Fig. 7). Upon addition of higher concentrations of the purified lipid donor (50–100 μM), RgtA exhibited a weak RgtB-like activity (the slower migrating band in Fig. 7, last 2 lanes, indicated by the asterisk). Given the low concentration of the lipid donor in vivo, we do not envisage that RgtA performs a second galacturonosylation in R. leguminosarum cells with significant efficiency. Isolation of mutants deleted in the rgtA or rgtB genes, in conjunction with the analysis of the modified Kdo species isolated from cells, should resolve this issue.
FIGURE 7. In vitro reconstitution of RgtA activity with purified dodecaprenyl phosphate-GalA.

RgtA was assayed using 2.5 μM of Kdo2-1-dephospho-[4′-32P]lipid IVA as the acceptor substrate under standard conditions for 5 min with 0.25 mg/ml membrane protein. The dodecaprenyl phosphate-GalA was dissolved in the same assay buffer, except that the Triton X-100 concentration was increased to 0.2% to dissolve the purified lipid. Membranes from E. coli NovaBlue (DE3) harboring pRgtA or the vector pET23a were used as indicated. The purified donor substrate was added in increasing concentrations, ranging from 0.1 to 100 μM. The reconstituted E. coli system was run alongside S. meliloti 1021/pRgtA control membranes.
The GalA transferase activities of all three Rgt proteins expressed in E. coli could be reconstituted upon addition of the purified donor to a combination of the respective membranes, when using mannosyl-Kdo2-1-dephospho-[4′-32P]lipid IVA as substrate (Fig. 8).
FIGURE 8. In vitro reconstitution of RgtA, RgtB, and RgtC activities with purified dodecaprenyl phosphate-GalA.
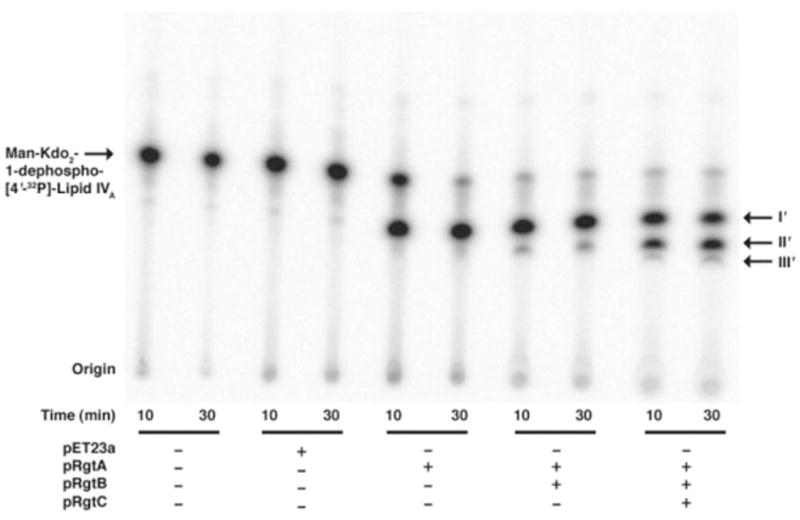
RgtA, RgtB, and RgtC were assayed using mannosyl-Kdo2-1-dephospho-[4′-32P]lipid IVA as the acceptor substrate (no carrier) under standard assay conditions for 30 min with 0.25 mg/ml of each membrane protein. Membranes from E. coli NovaBlue (DE3) harboring pRgtA, pRgtB, pRgtC, or the vector pET23a were used as indicated. The purified donor substrate was added at 200 μM. The three metabolites (products I′, II′, and III′) seen in the original clone (pMKGE) can be generated by the sequential action of RgtA, RgtB, and RgtC expressed in E. coli, when supplied with the purified GalA donor.
In summary, our findings demonstrate conclusively that the addition of the GalA moiety to the distal unit of R. leguminosarum Kdo disaccharide by RgtA requires the novel lipid, dodecaprenyl phosphate-GalA. Furthermore, this analysis verifies that RgtA, RgtB, and RgtC all use the same donor for GalA modification of the R. leguminosarum core.
DISCUSSION
Some Gram-negative bacteria synthesize LPS molecules in which selected phosphate groups of the lipid A and/or core domains are replaced with GalA residues (29, 32–37). By modifying their lipid A and core domains with GalA, bacteria lacking phosphorylated LPS provide an alternative source of negative charge, possibly enhancing divalent cation binding, LPS cross-bridging, and outer membrane stability (37, 38). GalA-deficient mutants of Klebsiella pneumoniae exhibit heightened sensitivity to vancomycin, SDS, and polymyxin. Furthermore, they display altered outer membrane protein profiles and release periplasmic β-lactamase (37, 39). Because many bacteria containing GalA-modified LPS are environmental isolates, this substitution may give these organisms an ecological advantage in low phosphate habitats. The presence of GalA units in place of phosphate residues may also protect endosymbionts like R. leguminosarum from cationic anti-microbial peptides (40).
Compelling evidence for the involvement of a lipid-linked GalA donor in the assembly of the R. leguminosarum LPS core (Fig. 1) was obtained by heterologous expression of the rgtA gene in E. coli versus S. meliloti 1021 (17). Although membranes of S. meliloti 1021 that express RgtA catalyze GalA transfer to Kdo2-1-dephospho-[4′-32P]lipid IVA from an endogenous source, membranes of E. coli expressing RgtA do not (Fig. 2). However, RgtA activity could be reconstituted by the addition of Rhizobiaceae membranes or total lipids (Fig. 3) to the E. coli system (17), suggesting the absence of the appropriate GalA donor substrate in E. coli.
We have now purified and structurally characterized dodecaprenyl phosphate-GalA, a novel minor lipid present in R. leguminosarum and S. meliloti. DEAE-cellulose column chromatography was the first purification step. Activity assays indicated the presence of the sugar donor in the 60 mM ammonium acetate fraction (Fig. 3). The ESI/MS spectrum of this fraction revealed the presence of a novel lipid with a calculated exact mass of 1090.78 (based on the spectrum in Fig. 4A), consistent with the predicted exact mass of 1090.76 (Fig. 5). MS/MS analysis of this minor lipid verified that it was indeed a C60-polyisoprene phosphate-linked HexA (Fig. 5).
To remove phospholipid contaminants, we used a mild alkaline hydrolysis procedure, which cleaves the fatty acid esters of phospholipids (31). By optimizing the base concentration, the phospholipids were deacylated, whereas the GalA donor lipid remained intact. After repurification over a second DEAE-cellulose column, dodecaprenyl phosphate-HexA was the major component remaining in the 60 mM ammonium acetate fraction (Fig. 4B). GC/MS analysis of the purified donor demonstrated that its HexA unit consisted solely of GalA (Fig. 6). ESI/MS of the total lipids from S. meliloti 1021 revealed the presence of the same dodecaprenyl phosphate-HexA species, as well as a less abundant C65 analogue (data not shown). Neither S. meliloti 1021 nor R. leguminosarum contain significant amounts of undecaprenyl phosphate, which is the predominant polyisoprene phosphate present in E. coli (3).
As shown in Fig. 7, purified dodecaprenyl phosphate-GalA supports the RgtA catalyzed conversion of Kdo2-1-dephospho-lipid IVA to GalA-Kdo2-1-dephospho-lipid IVA in a dose-dependent manner. Based on the sequence similarity of RgtA to other glycosyltransferases that utilize lipid-linked sugar donors, such as the yeast protein mannosyltransferases (41, 42) or Salmonella ArnT (20, 21), we propose that the GalA unit of dodecaprenyl phosphate-GalA has the β anomeric (equatorial) configuration (Fig. 5). NMR studies with larger amounts of material will be required to confirm this proposal.
The pathway for the formation of dodecaprenyl phosphate-β-D-GalA is unknown. One possibility is suggested by the presence of the orf3 gene on the same 7-kb R. leguminosarum 3841 DNA fragment containing the three rgt genes (17). The sequence of Orf3 displays significant similarity to eucaryotic dolichyl-phosphate mannose synthase (12, 15) and to E. coli ArnC (30). The latter generates undecaprenyl phosphate-4-deoxy-4-form-amido-L-arabinose from undecaprenyl phosphate and UDP-4-deoxy-4-formamido-L-arabinose (30). This similarity suggests that Orf3 might transfer the GalA moiety of UDP-GalA to a polyisoprene phosphate. Attempts to demonstrate this reaction in vitro with UDP-GalA and commercial undecaprenyl phosphate as the acceptor substrate have not been successful.3 It may be necessary to include the proper R. leguminosarum C60 polyisoprene phosphate (seen at m/z 913.74 in Fig. 4A) to synthesize lipid-linked GalA in an Orf3-dependent manner.
Based on all the available data, we propose the biosynthetic pathway and topography for GalA addition to R. leguminosarum LPS shown in Fig. 9. R. leguminosarum lipid A and most of the LPS core sugars are assembled on the cytoplasmic side of the inner membrane, where acyl-ACP and sugar nucleotides are available (43–46). The resulting intermediate, which contains phosphate groups at the lipid A 1 and 4′-positions, is then flipped across the inner membrane by the ABC transporter MsbA (47–49). On the periplasmic face of the inner membrane, the lipid A is dephosphorylated at the 1- and 4′-positions by LpxE and LpxF, respectively (40, 50). LpxE generates the preferred substrate for RgtA, which is followed in sequence by RgtB and RgtC, thereby generating the characteristic R. leguminosarum LPS core with its three GalA moieties. In parallel, RgtD, for which we do not yet have an in vitro assay, is proposed to transfer a fourth GalA unit to the 4′-position of 4′-dephosphorylated lipid A. RgtD has significant orthologues in Aquifex and Mesorhizobium, both of which have GalA moieties on their lipid A. Based on our reconstitution experiments (Figs. 7 and 8), all four Rgt proteins are presumed to utilize dodecaprenyl phosphate-β-D-GalA as their sugar donor. The flippase responsible for transferring dodecaprenyl phosphate-β-D-GalA across the inner membrane is unknown.
FIGURE 9. Proposed pathway and topography for attachment of GalA residues to the lipid A and core domains of R. leguminosarum LPS.

The simplest scenario for the biosynthesis of dodecaprenyl-β-D-GalA is as follows. 1 and 2, UDP-Glc is oxidized by the dehydrogenase Exo5 to UDP-GlcA, which is then converted by the C4-epimerase LpsL to UDP-GalA (22). 3, in analogy to PmrF (ArnC) (30) in E. coli, Orf3 transfers GalA from UDP-GalA to dodecaprenyl phosphate, generating dodecaprenyl phosphate-β-D-GalA, which is flipped to the periplasmic leaflet by an unknown mechanism. As yet, we have not been able to assay Orf3 in vitro. 4, lipid A and core LPS sugars are synthesized on the cytoplasmic leaflet of the inner membrane (43–46, 56, 57) and flipped to the periplasmic leaflet by MsbA. 5 and 6, on the periplasmic side of the inner membrane the 1- and 4′-phosphatases, LpxE and LpxF (40, 50), dephosphorylate lipid A, creating the substrate for GalA addition. 7–10, GalA is transferred from dodecaprenyl-β-D-GalA to the outer Kdo by RgtA and RgtB, and to mannose by RgtC. The same GalA donor is presumably used in the modification of lipid A by RgtD, but an in vitro assay is not yet available.
Our strategy for obtaining pure dodecaprenyl phosphate-β-D-GalA from R. leguminosarum should be applicable to other polyisoprene phosphate-linked sugars. Furthermore, the total lipids of bacteria containing GalA-modified LPS, such as Mesorhizobium haukuii, K. pneumoniae, and Aquifex pyrophilus (34, 36, 37), could be screened directly by ESI/MS for the presence of dodecaprenyl phosphate-β-D-GalA and related substances, using the protocols shown in Figs. 4 and 5. We anticipate the existence of many glycosyltransferases that utilize novel polyisoprene phosphate-linked sugars as donor substrates for modifying LPS and other complex glycoconjugates in bacterial membranes.
Acknowledgments
The mass spectrometry facility in the Department of Biochemistry of the Duke University Medical Center was supported by the LIPID MAPS Large Scale Collaborative Grant GM069338 from the National Institutes of Health. We thank Dr. T. A. Garrett and Dr. Z. Guan for their invaluable help with the ESI/MS and GC/MS analyses. We also thank Dr. C. M. Reynolds and Dr. D. A. Six for their help with the preparation of this manuscript.
Footnotes
This work was supported by National Institutes of Health Grant GM-51796 (to C. R. H. R.).
The abbreviations used are: LPS, lipopolysaccharide; ESI/MS, electrospray ionization/mass spectrometry; GalA, galacturonic acid; GC/MS, gas-liquid chromatography/mass spectrometry; GlcA, glucuronic acid; HexA, hexuronic acid; Kdo, 3-deoxy-D-manno-octulosonic acid; MES, 4-morpholineethanesulfonic acid; MS/MS, tandem mass spectrometry.
References
- 1.Bugg TD, Brandish PE. FEMS Microbiol Lett. 1994;119:255–262. doi: 10.1111/j.1574-6968.1994.tb06898.x. [DOI] [PubMed] [Google Scholar]
- 2.Lazar K, Walker S. Curr Opin Chem Biol. 2002;6:786–793. doi: 10.1016/s1367-5931(02)00355-1. [DOI] [PubMed] [Google Scholar]
- 3.Guan Z, Breazeale SD, Raetz CRH. Anal Biochem. 2005;345:336–339. doi: 10.1016/j.ab.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 4.Kelleher DJ, Karaoglu D, Gilmore R. Glycobiology. 2001;11:321–333. doi: 10.1093/glycob/11.4.321. [DOI] [PubMed] [Google Scholar]
- 5.Raetz CRH, Whitfield C. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolucka BA, McNeil MR, de Hoffmann E, Chojnacki T, Brennan PJ. J Biol Chem. 1994;269:23328–23335. [PubMed] [Google Scholar]
- 7.Wolucka BA, de Hoffmann E. J Biol Chem. 1995;270:20151–20155. doi: 10.1074/jbc.270.34.20151. [DOI] [PubMed] [Google Scholar]
- 8.Besra GS, Morehouse CB, Rittner CM, Waechter CJ, Brennan PJ. J Biol Chem. 1997;272:18460–18466. doi: 10.1074/jbc.272.29.18460. [DOI] [PubMed] [Google Scholar]
- 9.Yokoyama K, Araki Y, Ito E. Eur J Biochem. 1988;173:453–458. doi: 10.1111/j.1432-1033.1988.tb14020.x. [DOI] [PubMed] [Google Scholar]
- 10.Mavris M, Manning PA, Morona R. Mol Microbiol. 1997;26:939–950. doi: 10.1046/j.1365-2958.1997.6301997.x. [DOI] [PubMed] [Google Scholar]
- 11.Allison GE, Verma NK. Trends Microbiol. 2000;8:17–23. doi: 10.1016/s0966-842x(99)01646-7. [DOI] [PubMed] [Google Scholar]
- 12.Orlean P, Albright C, Robbins PW. J Biol Chem. 1988;263:17499–17507. [PubMed] [Google Scholar]
- 13.Imbach T, Schenk B, Schollen E, Burda P, Stutz A, Grunewald S, Bailie NM, King MD, Jaeken J, Matthijs G, Berger EG, Aebi M, Hennet T. J Clin Investig. 2000;105:233–239. doi: 10.1172/JCI8691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Helenius A, Aebi M. Science. 2001;291:2364–2369. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]
- 15.Orlean P. Biochem Cell Biol. 1992;70:438–447. doi: 10.1139/o92-067. [DOI] [PubMed] [Google Scholar]
- 16.Ferguson MA. J Cell Sci. 1999;112:2799–2809. doi: 10.1242/jcs.112.17.2799. [DOI] [PubMed] [Google Scholar]
- 17.Kanjilal-Kolar S, Basu SS, Kanipes MI, Guan Z, Garrett TA, Raetz CRH. J Biol Chem. 2006;281:12865–12878. doi: 10.1074/jbc.M513864200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Basu SS, Karbarz MJ, Raetz CRH. J Biol Chem. 2002;277:28959–28971. doi: 10.1074/jbc.M204525200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanjilal SJ, Basu SS, Kanipes MI, Raetz CRH. Glycobiology. 2005;15:1194. [Google Scholar]
- 20.Trent MS, Ribeiro AA, Lin S, Cotter RJ, Raetz CRH. J Biol Chem. 2001;276:43122–43131. doi: 10.1074/jbc.M106961200. [DOI] [PubMed] [Google Scholar]
- 21.Trent MS, Ribeiro AA, Doerrler WT, Lin S, Cotter RJ, Raetz CRH. J Biol Chem. 2001;276:43132–43144. doi: 10.1074/jbc.M106962200. [DOI] [PubMed] [Google Scholar]
- 22.Keating DH, Willits MG, Long SR. J Bacteriol. 2002;184:6681–6689. doi: 10.1128/JB.184.23.6681-6689.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Russomando G, Dankert MA. An Asoe Qufm Argent. 1992;80:175–186. [Google Scholar]
- 24.Kanipes MI, Lin S, Cotter RJ, Raetz CRH. J Biol Chem. 2001;276:1156–1163. doi: 10.1074/jbc.M009019200. [DOI] [PubMed] [Google Scholar]
- 25.Raetz CRH, Kennedy EP. J Biol Chem. 1973;248:1098–1105. [PubMed] [Google Scholar]
- 26.Raetz CRH, Purcell S, Meyer MV, Qureshi N, Takayama K. J Biol Chem. 1985;260:16080–16088. [PubMed] [Google Scholar]
- 27.Bligh EG, Dyer JJ. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 28.Reynolds CM, Kalb SR, Cotter RJ, Raetz CRH. J Biol Chem. 2005;280:21202–21211. doi: 10.1074/jbc.M500964200. [DOI] [PubMed] [Google Scholar]
- 29.Que NLS, Lin S, Cotter RJ, Raetz CRH. J Biol Chem. 2000;275:28006–28016. doi: 10.1074/jbc.M004008200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Breazeale SD, Ribeiro AA, McClerren AL, Raetz CRH. J Biol Chem. 2005;280:14154–14167. doi: 10.1074/jbc.M414265200. [DOI] [PubMed] [Google Scholar]
- 31.Do UH, Ramachandran S. J Lipid Res. 1980;21:888–894. [PubMed] [Google Scholar]
- 32.Bhat UR, Forsberg LS, Carlson RW. J Biol Chem. 1994;269:14402–14410. [PubMed] [Google Scholar]
- 33.Carlson RW, Reuhs B, Chen TB, Bhat UR, Noel KD. J Biol Chem. 1995;270:11783–11788. doi: 10.1074/jbc.270.20.11783. [DOI] [PubMed] [Google Scholar]
- 34.Plötz BM, Lindner B, Stetter KO, Holst O. J Biol Chem. 2000;275:11222–11228. doi: 10.1074/jbc.275.15.11222. [DOI] [PubMed] [Google Scholar]
- 35.Niedziela T, Lukasiewicz J, Jachymek W, Dzieciatkowska M, Lugowski C, Kenne L. J Biol Chem. 2002;277:11653–11663. doi: 10.1074/jbc.M111885200. [DOI] [PubMed] [Google Scholar]
- 36.Choma A, Sowinski P. Eur J Biochem. 2004;271:1310–1322. doi: 10.1111/j.1432-1033.2004.04038.x. [DOI] [PubMed] [Google Scholar]
- 37.Frirdich E, Bouwman C, Vinogradov E, Whitfield C. J Biol Chem. 2005;280:27604–27612. doi: 10.1074/jbc.M504987200. [DOI] [PubMed] [Google Scholar]
- 38.Nikaido H. Microbiol Mol Biol Rev. 2003;67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Regue M, Hita B, Pique N, Izquierdo L, Merino S, Fresno S, Benedi VJ, Tomas JM. Infect Immun. 2004;72:54–61. doi: 10.1128/IAI.72.1.54-61.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karbarz MJ, Kalb SR, Cotter RJ, Raetz CRH. J Biol Chem. 2003;278:39269–39279. doi: 10.1074/jbc.M305830200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dotson SB, Rush JS, Ricketts AD, Waechter CJ. Arch Biochem Biophys. 1995;316:773–779. doi: 10.1006/abbi.1995.1103. [DOI] [PubMed] [Google Scholar]
- 42.Strahl-Bolsinger S, Gentzsch M, Tanner W. Biochim Biophys Acta. 1999;1426:297–307. doi: 10.1016/s0304-4165(98)00131-7. [DOI] [PubMed] [Google Scholar]
- 43.Brozek KA, Carlson RW, Raetz CRH. J Biol Chem. 1996;271:32126–32136. doi: 10.1074/jbc.271.50.32126. [DOI] [PubMed] [Google Scholar]
- 44.Brozek KA, Kadrmas JL, Raetz CRH. J Biol Chem. 1996;271:32112–32118. [PubMed] [Google Scholar]
- 45.Kadrmas JL, Brozek KA, Raetz CRH. J Biol Chem. 1996;271:32119–32125. [PubMed] [Google Scholar]
- 46.Kadrmas JL, Allaway D, Studholme RE, Sullivan JT, Ronson CW, Poole PS, Raetz CRH. J Biol Chem. 1998;273:26432–26440. doi: 10.1074/jbc.273.41.26432. [DOI] [PubMed] [Google Scholar]
- 47.Doerrler WT, Reedy MC, Raetz CRH. J Biol Chem. 2001;276:11461–11464. doi: 10.1074/jbc.C100091200. [DOI] [PubMed] [Google Scholar]
- 48.Doerrler WT, Raetz CRH. J Biol Chem. 2002;277:36697–36705. doi: 10.1074/jbc.M205857200. [DOI] [PubMed] [Google Scholar]
- 49.Doerrler WT, Gibbons HS, Raetz CRH. J Biol Chem. 2004;279:45102–45109. doi: 10.1074/jbc.M408106200. [DOI] [PubMed] [Google Scholar]
- 50.Wang X, McGrath SC, Cotter RJ, Raetz CRH. J Biol Chem. 2006;281:9321–9330. doi: 10.1074/jbc.M600435200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Johnston AW, Beringer JE. J Gen Microbiol. 1975;87:343–350. doi: 10.1099/00221287-87-2-343. [DOI] [PubMed] [Google Scholar]
- 52.Bhat UR, Krishnaiah BS, Carlson RW. Carbohydr Res. 1991;220:219–227. doi: 10.1016/0008-6215(91)80020-n. [DOI] [PubMed] [Google Scholar]
- 53.Carlson RW, Bhat UR, Reuhs B. In: Plant Biotechnology and Development. Gresshoff PM, editor. CRC Press; Boca Raton, FL: 1992. pp. 33–44. [Google Scholar]
- 54.Carlson RW, Forsberg LS, Price NP, Bhat UR, Kelly TM, Raetz CRH. Prog Clin Biol Res. 1995;392:25–31. [PubMed] [Google Scholar]
- 55.Que NLS, Ribeiro AA, Raetz CRH. J Biol Chem. 2000;275:28017–28027. doi: 10.1074/jbc.M004009200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Price NPJ, Kelly TM, Raetz CRH, Carlson RW. J Bacteriol. 1994;176:4646–4655. doi: 10.1128/jb.176.15.4646-4655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Price NJP, Jeyaretnam B, Carlson RW, Kadrmas JL, Raetz CRH, Brozek KA. Proc Natl Acad Sci U S A. 1995;92:7352–7356. doi: 10.1073/pnas.92.16.7352. [DOI] [PMC free article] [PubMed] [Google Scholar]