

1. Introduction
A firm link between female reproductive history and increased risk of developing cancer in the breast and endometrium has been established from epidemiological studies (1-4). The longer women are exposed to estrogens, either through early menarche and late menopause and/or through estrogen replacement therapy, the higher is the risk of developing certain hormone-dependent cancers. It used to be thought that the purported benefits of estrogen replacement therapy, which included the relief of menopausal symptoms, decrease in coronary heart disease, osteoporosis, stroke, and Alzheimer’s disease, justified the use of long-term estrogen replacement therapy. However, the release of the initial results from the Women’s Health Initiative Study in July 2002 cast serious doubt on this paradigm for the treatment of post-menopausal women (5). The estrogen plus progestin arm was halted three years early due to significant increases in breast cancer, coronary heart disease, stroke, and pulmonary embolism, with more recent data suggesting an increase in vascular dementia in women over 65 on estrogen replacement therapy (6). In 2004, the estrogen arm was halted because of increased incidence of stroke (7). A recent analysis of data from the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) registries showed that age-adjusted incidence rate of breast cancer fell sharply (6.7%) in 2003 compared to 2002, which seemed to be related to the drop in the use of HRT (8). Finally, a reanalysis of nine prospective studies has shown that exposure to estrogens is associated with an increase in breast cancer risk with evidence of a dose-response relationship (9). These troubling findings highlight the urgent need for a full understanding of all the deleterious effects of estrogens including their potential to initiate and/or promote the carcinogenic process.
The mechanisms of estrogen carcinogenesis are not well understood. The central hypothesis of this review is that the formation of electrophilic/redox active quinones is an important mechanism of carcinogenesis for estrogens (Scheme 1, using the equine estrogen equilenin as an example). o-Quinones and quinone methides are known metabolites of estrogens and they have been shown to cause alkylation and/or oxidative damage to cellular proteins and DNA (2, 10-13). In addition, our data strongly suggests that estrogen receptors (ERs) appear to play a major role in catechol estrogen-induced DNA damage (14, 15). Binding and/or alkylation of ERs by the o-quinones generates a highly redox active “Trojan horse” which selectively targets estrogen sensitive genes. The long-range goal is to develop a better understanding of these reactive intermediates in vivo which will allow the rational development of estrogen replacement therapies that maintain the beneficial properties of estrogens without generating genotoxic species.
Scheme 1. Bioactivation of equilenin (EN) to redox active/electrophilic quinoids.

Interaction with estrogen receptors (ERs) and ER associated nuclear proteins, DNA, and cytosolic proteins which represent potential cellular targets of estrogen quinoids which could contribute to estrogen carcinogenesis.
2. The risk/benefits of traditional estrogen replacement therapy
Recently, the National Toxicology Program of NIEHS declared that steroidal estrogens, both of endogenous nature and as components of hormone replacement therapy (HRT) formulations, are “known to be human carcinogens”, causing breast and endometrial cancers (16). In July 2002, the Data and Safety Monitoring Board prematurely terminated a major clinical Women’s Health Initiative trial on the long-term risks and benefits of estrogen plus progestin therapy, a form of HRT for postmenopausal women who have an intact uterus (5). This decision was based, in part, on the significantly increased risk (24%) of invasive breast cancer, as well as a higher incidence of heart disease and stroke in women undergoing estrogen replacement therapy as compared to those receiving placebo. Even women without tumors were often found to have more abnormal mammograms than those on placebo (5, 17). Indeed, it has been known for some time that estrogens and estrogens plus progestin (18) can contribute significantly to the development of cancers (19, 20), especially of the breast (21-25) and other hormone-sensitive tissues (24) such as the ovary and uterus. These are some of the major types of cancers that afflict women in the United States (25).
Nevertheless, there are also significant benefits since estrogen replacement therapy relieves symptoms of menopause such as sleeplessness, hot flashes, and mood swings, provides protection against early menopausal bone loss, and lowers the risk of colon cancer (5, 17). For these reasons, women continue to use hormone replacement formulations (26) in spite of the well recognized risks (27). Although, the sales of standard dose Premarin prescriptions (0.625 mg/day) have decreased by 33% since July 2002 when NHLBI terminated the clinical trial on the long term risks and benefits of estrogen plus progestin therapy, more recently the sales of low-dose Premarin preparations (0.45 mg/day) have been rising (26).
3. Mechanisms of estrogen carcinogenesis
The molecular mechanisms of steroidal estrogen carcinogenesis are highly complex and ambiguous (2, 12, 25, 28, 29). Malignant phenotypes arise as a result of a series of mutations, most likely in genes associated with tumor suppressor, oncogene, DNA repair, or endocrine functions (30). One major pathway considered to be important is the extensively studied hormonal pathway, by which estrogen stimulates cell proliferation through nuclear ER-mediated signaling pathways, thus resulting in an increased risk of genomic mutations during DNA replication (4, 30-32). A similar “non-genomic pathway”, potentially involving newly discovered membrane associated ERs, also appears to regulate extranuclear estrogen signaling pathways (33, 34). The focus of this review will be the third pathway involving estrogen metabolism, mediated by cytochrome P450, that generates reactive electrophilic estrogen o-quinones and reactive oxygen species (ROS) through redox cycling of these o-quinones (Scheme 1, 2).
Scheme 2. Metabolism of estradiol to quinoids.

Circled quinoids represent the major isomers that react with DNA. Estrone is also metabolized to the corresponding catechols/quinoids.
Studies have shown that constitutive and TCDD-inducible P450 isozymes, P4501A1/1A2 and P4501B1 selectively catalyze hydroxylation at the 2- and 4-positions of estrone and 17β-estradiol, respectively (35-40) suggesting that excessive exposure to environmental pollutants could lead to enhanced production of these metabolites. This is particularly significant since 4-hydroxyestrone/estradiol was found to be carcinogenic in the male Syrian golden hamster kidney tumor model, whereas, 2-hydroxylated metabolites were without activity (41, 42). Similarly, Newbold and Liehr have shown that 4-hydroxyestradiol induced uterine tumors in 66% of CD-1 mice; whereas, mice treated with 2-hydroxyestradiol or 17β-estradiol had much lower uterine tumor incidence (43). Animal studies with catechol estrogens have produced contradictory results particularly with the ACI rat model for mammary carcinogenesis. For example, a lack of tumorigenicity for the catechol estrogens and the quinone from 4-hydroxyestrone was reported in female ACI rats (44, 45). In contrast, DNA adducts of catechol estrogen quinones have been detected in the mammary glands of ACI rats treated with 4-hydroxyestradiol or it’s quinone (46). In women, significantly higher amounts of GSH conjugates resulting from reaction of GSH with the 4-OHE/E2-o-quinones were detected in the non-tumor tissue from women with breast cancer compared to women without the disease (47). In addition, estrogen 4-hydroxylase levels (P4501B1 and 1A1) had higher expression in breast tissue of women with breast cancer, whereas expression of protective enzymes was lower (48). Finally, epidemiology studies have suggested a link between genetic polymorphism in estrogen 4-hydroxylases and a risk for developing breast cancer (49-51). These data suggest that estrogen metabolites are likely contributors to the development of cancer.
Most of the epidemiology studies on hormone replacement therapy and cancer risk have investigated the association with use of Premarin® (conjugated equine estrogens, Wyeth-Ayerest) or Prempro® (conjugated equine estrogens plus progestin). Both Premarin® and Prempro® are currently the most popular forms of HRT, widely prescribed in North America (52). Since Premarin® was approved by the Food and Drug Administration in the 1940s, very little is known about the metabolism and potential toxic metabolites that could be produced from the various equine estrogens, which make up approximately 50% of the estrogens in Premarin® (53-57) (Scheme 3). It is known that treating hamsters for 9 months with either estrone, equilin + equilenin, or sulfatase-treated Premarin®, resulted in 100% kidney tumor incidences and abundant tumor foci (54). Furthermore, in a small clinical trial of 596 postmenopausal women, a significant increase in endometrial hyperplasia was found in those women receiving a daily dose of 0.625 mg of Premarin® (58). Finally, we have shown that a major phase I metabolite of both equilenin and equilin (4-OHEN) can act as a complete carcinogen and tumor promoter in vitro, whereas the endogenous catechol estrogen metabolite, 4-hydroxyestrone was much less effective (59). As a result, it is quite possible that the B-ring unsaturated equine estrogens have very different biological properties in vivo compared to the endogenous catechol estrogens (10, 60).
Scheme 3. Primary phase I metabolic pathway for equine estrogens.

Interestingly, increasing unsaturation in the B ring leads to a change in metabolism from predominately 2-hydroxylation for estrone to mainly 4-hydroxylation for equilin and exclusively 4-hydroxylation for equilenin (55, 56, 61). Similar to what has been reported with endogenous estrogens the 4-hydroxylation pathway is predominately catalyzed by P4501B1/1A1 (61, 62). This could be problematic since 2-hydroxylation of endogenous estrogens is regarded as a benign metabolic pathway whereas 4-hydroxylation could lead to carcinogenic metabolites (41-43). It can be reasonably hypothesized that metabolism of equilenin and equilin (and their 17β-hydroxylated metabolites) to 4-OHEN and 4,17β-OHEN represents the major carcinogenic pathway for equine estrogens.
4. Oxidation of catechol estrogens to o-quinones
Once formed, the endogenous catechol estrogens can be oxidized by virtually any oxidative enzyme or metal ion giving o-quinones (2, 12). The o-quinone formed from 2-hydroxyestrone has a half-life of 47 s, whereas the 4-hydroxyestrone-o-quinone is considerably longer lived (t1/2 = 12 min) (63). In the absence of nucleophilic trapping agents, both o-quinones isomerize to quinone methides (Scheme 2) although the relative importance and biological targets of these potentially highly electrophilic intermediates has not been explored in detail (63-65). As far as the equine catechol estrogens are concerned, both 4-OHEN and 4-OHEQ autoxidize to o-quinones without the need for enzymatic or metal ion catalysis (Scheme 3) (56, 66). The o-quinone formed from 4-OHEN is much more stable (t1/2 = 2.3 h) than that from the endogenous catechol estrogens (66). It appears that the adjacent aromatic ring stabilizes 4-OHEN-o-quinone through extended π-conjugation. In support of this it has been shown that the catechol metabolite of benzo[a]pyrene rapidly undergoes air oxidation to yield a very stable o-quinone, benzo[a]pyrene-7,8-dione (67-70).
4-OHEQ-o-quinone readily isomerizes to 4-OHEN-o-quinone (Scheme 3). As a result, most of the biological effects caused by catechol metabolites of equilin are likely due to 4-OHEN-o-quinone formation (56). Finally, although 2-hydroxylation does occur with equilin producing 2-hydroxyequilin that will isomerize to 2-hydroxyequilenin, the latter catechol does not autoxidize to an o-quinone at any appreciable rate (71). This suggests that similar to what has been observed with endogenous catechol estrogens, 2-hydroxylation is likely a benign metabolic pathway for equilin.
5. DNA damage induced by catechol estrogen o-quinones
5.1 Oxidative damage
o-Quinones are also potent redox active compounds (72). They can undergo redox cycling with the semiquinone radical generating superoxide radicals mediated through cytochrome P450/P450 reductase (as shown in Scheme 1 for 4-OHEN). The reaction of superoxide anion radicals with hydrogen peroxide, formed by the enzymatic or spontaneous dismutation of superoxide anion radical, in the presence of trace amounts of iron or other transition metals gives hydroxyl radicals. The hydroxyl radicals are powerful oxidizing agents that may be responsible for damage to essential macromolecules. In support of this mechanism, various free radical toxicities have been reported in hamsters treated with 17β-estradiol including DNA single strand breaks (73, 74), 8-oxo-dG formation (13, 75, 76), and chromosomal abnormalities (29, 77, 78). Recently, it has also been shown that 4-hydroxyestradiol also induces oxidative stress and apoptosis in human mammary epithelial cells (MCF-10A), although the concentrations used in this study (25 μM) have questionable physiological relevance (79). We have shown that 4-OHEN is also capable of causing DNA single strand breaks and oxidative damage to DNA bases both in vitro and in vivo (14, 15, 80). Using the single cell gel electrophoresis assay (comet assay) to measure DNA damage, we found that 4-OHEN causes concentration-dependent DNA single strand cleavage in breast cancer cell lines and this effect could be enhanced by NADH or diethyl maleate (14, 15). We have also shown that injection of 4-OHEN into the mammary fat pads of Sprague-Dawley rats resulted in a dose-dependent increase in single strand breaks and oxidized bases as analyzed by the comet assay (80). In addition, extraction of mammary tissue DNA, hydrolysis to deoxynucleosides, and analysis by LC-MS-MS showed the formation of 8-oxo-dG as well as 8-oxo-dA. Finally, a recent study evaluated the potential of HRT to induce DNA damage in peripheral blood leukocytes of postmenopausal women using the comet assay (81). Significant increases in DNA damage were observed between women receiving 0.625 mg/day conjugated equine estrogens or conjugated equine estrogens plus medroxyprogesteron acetate as compared to the control group that had never received HRT. Finally, the excessive production of reactive oxygen species in breast cancer tissue has been linked to metastasis of tumors in women with breast cancer (82-84). These and other data are evidence for a mechanism of estrogen-induced tumor initiation/promotion by redox cycling of estrogen metabolites generating reactive oxygen species, which damage DNA.
5.2 Formation of estrogen DNA adducts
Estrogen quinoids can directly damage cellular DNA leading to genotoxic effects (11, 12, 46, 60, 85-87). Cavalieri’s group has shown that the major DNA adducts produced from 4-hydroxyestradiol-o-quinone are depurinating N7-guanine and N3-adenine adducts resulting from 1,4-Michael addition both in vitro and in vivo (12, 13, 46, 65, 88, 89). Interestingly, they have recently concluded that only the N3-adenine adduct is likely to induce mutations since this adduct depurinates instantaneously whereas the N7-guanine adduct takes hours to hydrolyze (89, 90). In contrast, the considerably more rapid isomerization of the 2-hydroxyestradiol-o-quinone to the corresponding quinone methides results in 1,6-Michael addition products with the exocyclic amino groups of adenine and guanine (65, 91). In contrast to the N3 and N7 purine DNA adducts, these adducts are stable which may alter their rate of repair and relative mutagenicity in vivo. A depurinating N3-adenine adduct of 2-hydroxyestradiol quinone methide has recently been reported in reactions with adenine and DNA (89). The levels of this adduct were considerably lower than corresponding depurinating adducts observed with similar experiments with 4-hydroxyestradiol-o-quinone which may explain why 2-hydroxylation is considered a benign metabolic pathway whereas 4-hydroxylation results in carcinogenesis. Finally, this same study (89) suggested that depurinating DNA adducts of estrogen quinoids were formed in much greater abundance compared to stable bulky adducts implying a causal role for these adducts in estrogen carcinogenesis; however, the depurinating adducts were analyzed by different methods (HPLC with electrochemical detection) as compared to the stable adducts (32P-postlabeling/TLC) making direct quantitative comparisons problematic. The mutagenic properties of 2-hydroxyestrogen quinone methide derived stable DNA adducts have been evaluated using oligonucleotides containing site specific adducts transfected into simian kidney (COS-7) cells where G -> T and A -> T mutations were observed (92). It is important to mention that stable DNA adducts have been detected by 32P-postlabeling in Syrian hamster embryo cells treated with estradiol and it’s catechol metabolites (93). The rank order of DNA adduct formation which correlated with cellular transformation was 4-OHE2 > 2-OHE2 > E2. Finally, stable bulky adducts of 4-OHE1 and 4-OHE2 corresponding to alkylation of guanine have been detected in human breast tumor tissue (94). These data suggest that the relative importance of depurinating adducts versus stable DNA adducts in catechol estrogen carcinogenesis remains unclear.
For the major equine estrogens (equilin and equilenin and 17β-ol derivatives) the data strongly suggests that the majority of DNA damage results from reactions of 4-OHEN-o-quinone through a combination of oxidative damage (i.e., single strand cleavage and oxidation of DNA bases) and through generation of apurinic sites as well as stable bulky cyclic adducts (Schemes 1, 4) (95). For example, a depurinating guanine adduct was detected in in vivo experiments with rats treated with 4-OHEN, following LC-MS-MS analysis of extracted mammary tissue (80). However, isolation of mammary tissue DNA, hydrolysis to deoxynucleosides, and analysis by LC-MS-MS also showed the formation of stable cyclic deoxyguanosine and deoxyadenosine adducts (Figure 1) as well as the above mentioned oxidized bases and single strand breaks. Interestingly, the ratio of the diasteriomeric adducts detected in vivo differs from in vitro experiments suggesting that there are differences in the response of these stereoisomeric lesions to DNA replication and repair enzymes (96-99). Finally, in a recent report, highly sensitive nano LC/MS-MS techniques were used to analyze the DNA in five human breast tumor and five adjacent tissue samples, including samples from donors with a known history of Premarin-based HRT (94). While the sample size is small, and the history of the patients is not fully known, cyclic 4-OHEN-dC, -dG, and -dA stable adducts were detected for the first time in four out of the ten samples. These results suggest that 4-OHEN has the potential to be carcinogenic through the formation of a variety of DNA lesions in vivo.
Figure 1. DNA adducts formed from estrogen quinoids.

6. Protein targets of catechol estrogen o-quinones
6.1 Redox sensitive enzymes
Other potential targets for the estrogen metabolites include redox sensitive enzymes, including GST P1-1 (100, 101), quinone reductase (QR, NAD(P)H dependent quinone oxidoreductase, NQO1) (102), and thioredoxin reductase (103). Protein-modifying stresses elicit characteristic changes in gene expression that reflect responses to stress or injury (104, 105). A number of studies have characterized gene expression changes in response to oxidizing and/or alkylating agents (106, 107); however, the mechanisms by which these protein damaging agents cause modulation of gene expression are largely unknown. Several studies have suggested that some proteins have two probably inter-related functions; as sensors for alkylating and oxidizing agents as well as regulators of specific kinases or transcription factors involved in stress signaling (106-108). GST P1-1 and QR are both involved in protein sensor-trigger systems and they have been implicated in genetic polymorphisms associated with increased breast cancer risk (109). In addition, estradiol and catechol estrogens can induce elevation of the levels of QR and GST, both in cell culture, as well as in vivo (110, 111). It has also been shown that thioredoxin-1, a target for estrogen quinones, regulates the constitutive and inducible expression of P4501B1 and 1A1, the enzymes responsible for formation of catechol estrogens, in breast cancer cells (112). Finally, we have determined that 4-OHEN at higher concentrations is an irreversible inactivator of these redox sensitive enzymes through mechanisms involving both alkylation and oxidation (100, 113). The effects on signaling mechanisms controlled by these enzymes could have major consequences on physiological levels as well as potential targets of estrogen o-quinones in vivo.
6.2 Estrogen receptors
It is quite likely that targets for catechol estrogen o-quinones could be crucial cellular proteins, particularly ERs and proteins associated with ERs (ER coregulators). The potential for 4-OHEN to covalently alkylate the ligand binding domain of ERα and ERβ was investigated using ESI-MS analysis (Figure 2, unpublished results). ERα showed more sensitivity to 4-OHEN-o-quinone-mediated modification compared to ERβ as determined by the number of covalently modified protein species. We also demonstrated that 4-OHEN caused oxidative damage to both ERs as bands related to disulfide bonded proteins appeared on non-reducing SDS-PAGE (unpublished results). These data show that 4-OHEN can oxidize and alkylate ERs although sites of alkylation are unknown at present.
Figure 2. ESI mass spectra of the His6-hERα and β ligand binding domains alkylated by 4-OHEN.

ERs (20 μM) were incubated with 4-OHEN (75 μM) for 15 min (pH 7.4, 37°C) and analyzed by ESI-MS (unpublished data).
Given that the ERs contain zinc finger structures rich in cysteine residues in the DNA binding domain, several laboratories have recently been investigating the reactivity of these cysteines to various electrophiles (114-116). It has been proposed that chemical modification of ER zinc fingers may inhibit the growth of some ER expressing cancers by preventing ER transcription of estrogen sensitive genes (115); however, if modified ER is translocated to the nucleus this could lead to local generation of ROS through redox cycling of catechol estrogen modified ER cysteine sulfhydryl groups near estrogen sensitive genes resulting in genotoxicity (see Section 7). Recently, Baldwin’s group has shown, through several elegant proteomics experiments, that the quinone, menadione, preferentially alkylates cysteine 240, which is located in the second zinc finger of the DNA binding domain of ERα (117). Although these experiments were carried out using the purified protein, their studies do show that quinones alkylate ERs. Due to the high affinity of catechol metabolites for ERs, we predict that estrogen catechols/o-quinones will selectivity target these proteins making them much more likely targets for estrogen o-quinones in vivo compared to non-estrogenic quinones like menadione.
7. Estrogen receptor as a “Trojan horse”
It is well known that ERs play a major role in the mechanism of estrogen-induced carcinogenesis (30, 118). Binding of estrogens to the ERs leads to an increase in cell proliferation in hormone-sensitive target tissues such as the breast and endometrium. In these rapidly dividing cells, the chances for mutations to occur increases dramatically leading to initiation/promotion of the carcinogenic process. We have hypothesized that ERs could also play a role in catechol estrogen/o-quinone-induced carcinogenesis through the “Trojan horse” mechanism (Scheme 1). It is quite possible that the catechol estrogen and/or o-quinone bind to the estrogen receptor, which carries it directly to estrogen sensitive genes, where DNA damage occurs resulting in mutations.
We have preliminary data that this mechanism may play a role in catechol estrogen-induced DNA damage (14, 15). We have examined the effect of ER status on the relative ability of 4-OHEN and 4,17β-OHEN to induce DNA damage in ER negative cells (MDA-MB-231), ERα positive cells (S30), and ERβ positive cells (β41). The data showed that both 4-OHEN and 4,17β-OHEN induced concentration dependent DNA single strand cleavage in all three cell lines. However, cells containing ERs had significantly higher DNA damage. Finally, the endogenous catechol estrogen metabolite 4-hydroxyestrone was considerably less effective at inducing DNA damage in breast cancer cell lines as compared to 4-OHEN (14). Our data suggest that the genotoxic effects of 4-OHEN could be related to its ability to induce DNA damage in hormone sensitive cells in vivo, and these effects may be potentiated by the ER.
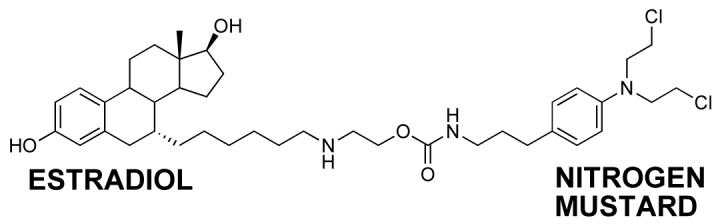
Essigmann’s group has reported on estradiol linked to a nitrogen mustard as a very effective chemotherapeutic agent with selective toxicity for ERα positive cells (Figure 3) (119, 120). It is proposed that the estradiol portion of the molecule binds to the ER, which then travels to the nucleus, binds to ERE, where the nitrogen mustard forms DNA adducts. Consistent with the ER playing a role in increasing toxicity and DNA damage in ERα positive cells, was the observation that the ER-independent nitrogen mustard, chlorambucil, was equally toxic in both ER positive and negative cells. These data are remarkably similar to observations of 4-OHEN-induced DNA damage in ER+ versus ER- cells and are consistent with the “Trojan Horse” hypothesis.
Figure 3. Essigmann’s ER “Trojan horse”.
8. Neoplastic transformation of non-tumorigenic breast epithelial cells
Treatment of MCF-10F cells, which are ER-negative immortalized human breast epithelial cells, with E2, 4-OHE2, or 2-OHE2 induced their neoplastic transformation in vitro (121, 122). The transformed cells exhibited specific mutations in several genes. Poorly differentiated adenocarcinomas develop when aggressively transformed MCF-10F cells were selected and injected into severe combined immune depressed (SCID) mice. Similar studies in our laboratory using ER negative MCF-10A cells treated with 4-OHEN showed a concentration dependent increase in colony formation using an anchorage-independent growth assay (123). Analysis of differentially expressed genes in these transformed cells showed modulation of several genes involved in cell transformation and oxidative stress, strengthening the hypothesis that this mechanism plays a considerable role in 4-OHEN-induced carcinogenesis. Whether the presence of ERs can enhance cellular transformation compared to ER negative cells is as yet unknown.
9. Summary
Recent data suggest that metabolism of estrogens to their catechols/o-quinones is a major mechanism of estrogen carcinogenesis. Oxidative enzymes, metal ions, and in some cases molecular oxygen can catalyze o-quinone formation, so that these electrophilic/redox active quinones can cause damage within cells by alkylation of cellular nucleophiles (proteins, DNA) to a significant extent in many tissues. In addition, the formation of reactive oxygen species especially through redox cycling between the quinones and semiquinone radicals, can cause oxidation of DNA and protein damage. DNA damage is significantly increased in cells containing ERs leading us to hypothesize a mechanism involving ER binding/alkylation of catechol/quinone resulting in a “Trojan horse”. The “Trojan horse” carries the highly redox active catechol to estrogen sensitive genes, where high amounts of ROS are generated causing selective DNA damage. Our data further suggests that other key protein targets for estrogen o-quinones could be redox sensitive enzymes including GST P1-1, QR, and thioredoxin reductase. These proteins are involved in stress response cascades that are known to contribute to regulation of cell proliferation and apoptosis. Finally, we and others have shown that catechol estrogens can transform breast epithelial cells into a tumorigenic phenotype and that these transformed cells had differential gene expression of several genes involved in oxidative stress. Given the direct link between excessive exposure to estrogens, metabolism of estrogens, and increased risk of breast cancer, it is crucial that factors that affect the formation, reactivity, and cellular targets of estrogen quinoids be thoroughly explored.
Acknowledgements
This work is supported by NIH Grants CA102590, CA79870 and CA73638.
Abbreviations
- E2
17β-estradiol
- ER
estrogen receptor
- ERE
estrogen response element
- ESI
electrospray ionization
- GST
glutathione-S-transferase
- hGST PI-1
human glutathione-S-transferase
- LBD
ligand binding domain
- ROS
reactive oxygen species
- 2-OHE
2-hydroxyestrone, 2,3-dihydroxy-1,3,5(10)-estratrien-17-one
- 4-OHE
4-hydroxyestrone, 3,4-dihydroxy-1,3,5(10)-estratrien-17-one
- estrone
3-hydroxy-1,2,5-(10)-estratrien-17-one
- 4-OHEN
4-hydroxyequilenin, 3,4-dihydroxy-1,3,5(10),6,8-estrapentaen-17-one
- equilenin
1,3,5(10),6,8-estrapentaen-3-ol-17-one
- equilin
1,3,5(10), 7-estratetraen-3-ol-17-one
- 9(11)-dehydro-4-OHE
9(11)-dehydro-4-hydroxyestrone, 3,4-dihydroxy-1,3,5(10),9(11)-estratetraen-17-one)
- 9(11)-dehydro-2-OHE
9(11)-dehydro-2-hydroxyestrone, 2,3-dihydroxy-1,3,5(10),9(11)-estratetraen-17-one)
- 2-OHE-QM1
3-hydroxy-1(10),3(4),5(6)-estratrien-2,17-dione
- 2-OHE-QM2
2-hydroxy-1(2),4(5),9(10)-estratrien-3,17-dione
- 4-OHE-QM2
4-hydroxy-1(2),4(5),9(10)-estratrien-3,17-dione
- 4-OHE-QM1
3-hydroxy-1(10),2(3),5(6)-estratrien-4,17-dione
- P450
cytochrome P450
- QM
quinone methide, 4-alkyl-2,5-cyclohexadien-1-one
- o-quinone
3,5-cyclohexadien-1,2-dione
- LC-MS
liquid chromatography mass spectrometry
- electrospray-MS
electrospray mass spectrometry
- dN
2′-deoxynucleoside
- dG
2′-deoxyguanosine
- dA
2′-deoxyadenosine
- dC
2′-deoxycytosine
- 8-OHdG
8-hydroxydeoxyguanosine
- 8-OHdA
8-hydroxydeoxyadenosine
- 2-OHdA
2-hydroxydeoxyadenosine
- 8-OHG
8-hydroxyguanine
References
- (1).Liehr JG. Genotoxic effects of estrogens. Mutation Res. 1990;238:269–276. doi: 10.1016/0165-1110(90)90018-7. [DOI] [PubMed] [Google Scholar]
- (2).Yager JD, Davidson NE. Estrogen carcinogenesis in breast cancer. N. Engl. J. Med. 2006;354:270–282. doi: 10.1056/NEJMra050776. [DOI] [PubMed] [Google Scholar]
- (3).Colditz GA. Relationship between estrogen levels, use of hormone replacement therapy, and breast cancer. J. Natl. Cancer Inst. 1998;90:814–823. doi: 10.1093/jnci/90.11.814. [DOI] [PubMed] [Google Scholar]
- (4).Feigelson HS, Henderson BE. Estrogens and breast cancer. Carcinogenesis. 1996;17:2279–2284. doi: 10.1093/carcin/17.11.2279. [DOI] [PubMed] [Google Scholar]
- (5).Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. Jama. 2002;288:321–333. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- (6).Shumaker SA, Legault C, Rapp SR, Thal L, Wallace RB, Ockene JK, Hendrix SL, Jones BN, Assaf AR, Jackson RD, Kotchen JM, Wassertheil-Smoller S, Wactawski-Wende J. Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women. The Women’s Health Initiative Memory Study: A Randomized controlled trial. JAMA. 2003;289:2651–2662. doi: 10.1001/jama.289.20.2651. [DOI] [PubMed] [Google Scholar]
- (7).Brass LM. Hormone replacement therapy and stroke: clinical trials review. Stroke. 2004;35:2644–2647. doi: 10.1161/01.STR.0000143218.20061.ac. [DOI] [PubMed] [Google Scholar]
- (8).Ravdin PM, Cronin KA, Howlader N, Berg CD, Chlebowski RT, Feuer EJ, Edwards BK, Berry DA. The decrease in breast-cancer incidence in 2003 in the United States. N. Engl. J. Med. 2007;356:1670–1674. doi: 10.1056/NEJMsr070105. [DOI] [PubMed] [Google Scholar]
- (9).Key T, Appleby P, Barnes I, Reeves G. Endogenous sex hormones and breast cancer in postmenopausal women: reanalysis of nine prospective studies. J. Natl. Cancer Inst. 2002;94:606–616. doi: 10.1093/jnci/94.8.606. [DOI] [PubMed] [Google Scholar]
- (10).Bolton JL, Pisha E, Zhang F, Qiu S. Role of quinoids in estrogen carcinogenesis. Chem. Res. Toxicol. 1998;11:1113–1127. doi: 10.1021/tx9801007. [DOI] [PubMed] [Google Scholar]
- (11).Bolton JL, Yu L, Thatcher GR. Quinoids formed from estrogens and antiestrogens. Methods Enzymol. 2004;378:110–123. doi: 10.1016/S0076-6879(04)78006-4. [DOI] [PubMed] [Google Scholar]
- (12).Cavalieri E, Chakravarti D, Guttenplan J, Hart E, Ingle J, Jankowiak R, Muti P, Rogan E, Russo J, Santen R, Sutter T. Catechol estrogen quinones as initiators of breast and other human cancers: implications for biomarkers of susceptibility and cancer prevention. Biochim. Biophys. Acta. 2006;1766:63–78. doi: 10.1016/j.bbcan.2006.03.001. [DOI] [PubMed] [Google Scholar]
- (13).Cavalieri E, Frenkel K, Liehr JG, Rogan E, Roy D. Estrogens as endogenous genotoxic agents--DNA adducts and mutations. J. Natl. Cancer Inst. Monogr. 2000;27:75–93. doi: 10.1093/oxfordjournals.jncimonographs.a024247. [DOI] [PubMed] [Google Scholar]
- (14).Chen Y, Liu X, Pisha E, Constantinou AI, Hua Y, Shen L, van Breemen RB, Elguindi EC, Blond SY, Zhang F, Bolton JL. A metabolite of equine estrogens, 4-hydroxyequilenin, induces DNA damage and apoptosis in breast cancer cell lines. Chem. Res. Toxicol. 2000;13:342–350. doi: 10.1021/tx990186j. [DOI] [PubMed] [Google Scholar]
- (15).Liu X, Yao J, Pisha E, Yang Y, Hua Y, van Breemen RB, Bolton JL. Oxidative DNA Damage Induced by Equine Estrogen Metabolites: Role of Estrogen Receptor alpha. Chem. Res. Toxicol. 2002;15:512–519. doi: 10.1021/tx0101649. [DOI] [PubMed] [Google Scholar]
- (16).U.S. Department of Health and Human Services, P. H. S. National toxicology report on carcinogens. 11 ed. 2005. [Google Scholar]
- (17).Hays J, Ockene JK, Brunner RL, Kotchen JM, Manson JE, Patterson RE, Aragaki AK, Shumaker SA, Brzyski RG, LaCroix AZ, Granek IA, Valanis BG. Effects of estrogen plus progestin on health-related quality of life. N. Engl. J. Med. 2003;348:1839–1854. doi: 10.1056/NEJMoa030311. [DOI] [PubMed] [Google Scholar]
- (18).Fletcher SW, Colditz GA. Failure of estrogen plus progestin therapy for prevention. J.A.M.A. 2002;288:366–368. doi: 10.1001/jama.288.3.366. [DOI] [PubMed] [Google Scholar]
- (19).Lupulescu A. Estrogen Use and Cancer Incidence: A Review. Cancer Invest. 1995;13:287–295. doi: 10.3109/07357909509094464. [DOI] [PubMed] [Google Scholar]
- (20).Service RF. New role for estrogen in cancer? Science. 1998;279:1631–1633. doi: 10.1126/science.279.5357.1631. [DOI] [PubMed] [Google Scholar]
- (21).Bergkvist L, Adami HO, Persson I, Hoover R, Schairer C. The risk of breast cancer after estrogen and estrogen-progestin replacement. N Engl J Med. 1989;321:293–297. doi: 10.1056/NEJM198908033210505. [DOI] [PubMed] [Google Scholar]
- (22).Ross RK, Paganini-Hill A, Wan PC, Pike MC. Effect of hormone replacement therapy on breast cancer risk: estrogen versus estrogen plus progestin. J Natl Cancer Inst. 2000;92:328–332. doi: 10.1093/jnci/92.4.328. [DOI] [PubMed] [Google Scholar]
- (23).Colditz GA. Relationship between estrogen levels, use of hormone replacement therapy, and breast cancer. J. Natl. Cancer Inst. 1998;90:814–823. doi: 10.1093/jnci/90.11.814. [DOI] [PubMed] [Google Scholar]
- (24).Colditz GA, Hankinson SE, Hunter DJ, Willett WC, Manson JE, Stampfer MJ, Hennekens C, Rosner B, Speizer FE. The use of estrogens and progestins and the risk of breast cancer in postmenopausal women. New Eng. J. Med. 1995;332:1589–1593. doi: 10.1056/NEJM199506153322401. [DOI] [PubMed] [Google Scholar]
- (25).Russo J, Hu YF, Yang X, Russo IH. Developmental, cellular, and molecular basis of human breast cancer. J. Natl. Cancer Inst. Monogr. 2000;27:17–37. doi: 10.1093/oxfordjournals.jncimonographs.a024241. [DOI] [PubMed] [Google Scholar]
- (26).Hersh AL, Stefanick ML, Stafford RS. National use of postmenopausal hormone therapy: annual trends and response to recent evidence. J.A.M.A. 2004;291:47–53. doi: 10.1001/jama.291.1.47. [DOI] [PubMed] [Google Scholar]
- (27).Zumoff B. Does postmenopausal estrogen administration increase the risk of breast cancer? Contributions of animal, biochemical, and clinical investigative studies to a resolution of the controversy. Proc. Soc. Exp. Biol. Med. 1998;217:30–37. doi: 10.3181/00379727-217-44202. [DOI] [PubMed] [Google Scholar]
- (28).Jefcoate CR, Liehr JG, Santen RJ, Sutter TR, Yager JD, Yue W, Santner SJ, Tekmal R, Demers L, Pauley R, Naftolin F, Mor G, Berstein L. Tissue-specific synthesis and oxidative metabolism of estrogens. J. Natl. Cancer Inst. Monogr. 2000;27:95–112. doi: 10.1093/oxfordjournals.jncimonographs.a024248. [DOI] [PubMed] [Google Scholar]
- (29).Russo J, Russo IH. The role of estrogen in the initiation of breast cancer. J. Steroid Biochem. Mol. Biol. 2006;102:89–96. doi: 10.1016/j.jsbmb.2006.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Henderson BE, Feigelson HS. Hormonal carcinogenesis. Carcinogenesis. 2000;21:427–433. doi: 10.1093/carcin/21.3.427. [DOI] [PubMed] [Google Scholar]
- (31).Nandi S, Guzman RC, Yang J. Hormones and mammary carcinogenesis in mice, rats, and humans: a unifying hypothesis. Proc. Natl. Acad. Sci. U S A. 1995;92:3650–3657. doi: 10.1073/pnas.92.9.3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Flototto T, Djahansouzi S, Glaser M, Hanstein B, Niederacher D, Brumm C, Beckmann MW. Hormones and hormone antagonists: mechanisms of action in carcinogenesis of endometrial and breast cancer. Horm. Metab. Res. 2001;33:451–457. doi: 10.1055/s-2001-16936. [DOI] [PubMed] [Google Scholar]
- (33).Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- (34).Song RX, Fan P, Yue W, Chen Y, Santen RJ. Role of receptor complexes in the extranuclear actions of estrogen receptor {alpha} in breast cancer. Endocr. Relat. Cancer. 2006;1(13 Suppl):S3–S13. doi: 10.1677/erc.1.01322. [DOI] [PubMed] [Google Scholar]
- (35).Spink DC, Spink BC, Cao JQ, Gierthy JF, Hayes CL, Li Y, Sutter TR. Induction of cytochrome P450 1B1 and catechol estrogen metabolism in ACHN human renal adenocarcinoma cells. J. Steroid Biochem. Molec. Biol. 1997;62:223–232. doi: 10.1016/s0960-0760(97)00024-1. [DOI] [PubMed] [Google Scholar]
- (36).Spink DC, Spink BC, Cao JQ, Depasquale JA, Pentecost BT, Fasco MJ, Li Y, Sutter TR. Differential expression of CYP1A1 and CYP1B1 in human breast epithelial cells and breast tumor cells. Carcinogenesis. 1998;19:291–298. doi: 10.1093/carcin/19.2.291. [DOI] [PubMed] [Google Scholar]
- (37).Shimada T, Watanabe J, Kawajiri K, Sutter TR, Guengerich FP, Gillam EM, Inoue K. Catalytic properties of polymorphic human cytochrome P450 1B1 variants. Carcinogenesis. 1999;20:1607–1614. doi: 10.1093/carcin/20.8.1607. [DOI] [PubMed] [Google Scholar]
- (38).Badawi AF, Cavalieri EL, Rogan EG. Effect of chlorinated hydrocarbons on expression of cytochrome P450 1A1, 1A2 and 1B1 and 2- and 4-hydroxylation of 17beta-estradiol in female sprague-dawley rats. Carcinogenesis. 2000;21:1593–1599. [PubMed] [Google Scholar]
- (39).Badawi AF, Cavalieri EL, Rogan EG. Role of human cytochrome P450 1A1, 1A2, 1B1, and 3A4 in the 2-, 4-, and 16alpha-hydroxylation of 17beta-estradiol. Metabolism. 2001;50:1001–1003. doi: 10.1053/meta.2001.25592. [DOI] [PubMed] [Google Scholar]
- (40).Cribb AE, Knight MJ, Dryer D, Guernsey J, Hender K, Tesch M, Saleh TM. Role of polymorphic human cytochrome P450 enzymes in estrone oxidation. Cancer Epidemiol. Biomarkers Prev. 2006;15:551–558. doi: 10.1158/1055-9965.EPI-05-0801. [DOI] [PubMed] [Google Scholar]
- (41).Liehr JG, Fang WR, Sirbasku DA, Ari-Ulubelen A. Carcinogenicity of catechol estrogens in Syrian hamsters. J. Steroid Biochem. 1986;24:353–356. doi: 10.1016/0022-4731(86)90080-4. [DOI] [PubMed] [Google Scholar]
- (42).Li JJ, Li SA. Estrogen carcinogenesis in Syrian hamster tissues: role of metabolism. Fed. Proc. 1987;46:1858–1863. [PubMed] [Google Scholar]
- (43).Newbold RR, Liehr JG. Induction of uterine adenocarcinoma in CD-1 mice by catechol estrogens. Cancer Res. 2000;60:235–237. [PubMed] [Google Scholar]
- (44).Turan VK, Sanchez RI, Li JJ, Li SA, Reuhl KR, Thomas PE, Conney AH, Gallo MA, Kauffman FC, Mesia-Vela S. The effects of steroidal estrogens in ACI rat mammary carcinogenesis: 17beta-estradiol, 2-hydroxyestradiol, 4-hydroxyestradiol, 16alpha-hydroxyestradiol, and 4-hydroxyestrone. J. Endocrinol. 2004;183:91–99. doi: 10.1677/joe.1.05802. [DOI] [PubMed] [Google Scholar]
- (45).El-Bayoumy K, Ji BY, Upadhyaya P, Chae YH, Kurtzke C, Rivenson A, Reddy BS, Amin S, Hecht SS. Lack of tumorigenicity of cholesterol epoxides and estrone-3,4-quinone in the rat mammary gland. Cancer Res. 1996;56:1970–1973. [PubMed] [Google Scholar]
- (46).Li KM, Todorovic R, Devanesan P, Higginbotham S, Kofeler H, Ramanathan R, Gross ML, Rogan EG, Cavalieri EL. Metabolism and DNA binding studies of 4-hydroxyestradiol and estradiol-3,4-quinone in vitro and in female ACI rat mammary gland in vivo. Carcinogenesis. 2004;25:289–297. doi: 10.1093/carcin/bgg191. [DOI] [PubMed] [Google Scholar]
- (47).Rogan EG, Badawi AF, Devanesan PD, Meza JL, Edney JA, West WW, Higginbotham SM, Cavalieri EL. Relative imbalances in estrogen metabolism and conjugation in breast tissue of women with carcinoma: potential biomarkers of susceptibility to cancer. Carcinogenesis. 2003;24:697–702. doi: 10.1093/carcin/bgg004. [DOI] [PubMed] [Google Scholar]
- (48).Singh S, Chakravarti D, Edney JA, Hollins RR, Johnson PJ, West WW, Higginbotham SM, Cavalieri EL, Rogan EG. Relative imbalances in the expression of estrogen-metabolizing enzymes in the breast tissue of women with breast carcinoma. Oncol. Rep. 2005;14:1091–1096. [PubMed] [Google Scholar]
- (49).Zheng W, Xie DW, Jin F, Cheng JR, Dai Q, Wen WQ, Shu XO, Gao YT. Genetic polymorphism of cytochrome P450-1B1 and risk of breast cancer. Cancer Epidemiol. Biomarkers Prev. 2000;9:147–150. [PubMed] [Google Scholar]
- (50).Kisselev P, Schunck WH, Roots I, Schwarz D. Association of CYP1A1 polymorphisms with differential metabolic activation of 17beta-estradiol and estrone. Cancer Res. 2005;65:2972–2978. doi: 10.1158/0008-5472.CAN-04-3543. [DOI] [PubMed] [Google Scholar]
- (51).Wen W, Ren Z, Shu XO, Cai Q, Ye C, Gao YT, Zheng W. Expression of cytochrome P450 1B1 and catechol-O-methyltransferase in breast tissue and their associations with breast cancer risk. Cancer Epidemiol. Biomarkers Prev. 2007;16:917–920. doi: 10.1158/1055-9965.EPI-06-1032. [DOI] [PubMed] [Google Scholar]
- (52).Wysowski DK, Governale LA. Use of menopausal hormones in the United States, 1992 through June, 2003. Pharmacoepidemiol. Drug Saf. 2005;14:171–176. doi: 10.1002/pds.985. [DOI] [PubMed] [Google Scholar]
- (53).Purdy RH, Moore PH, Williams MC, Goldzheher HW, Paul SM. Relative rates of 2- and 4-hydroxyestrogen synthesis are dependent on both substrate and tissue. FEBS Lett. 1982;138:40–44. doi: 10.1016/0014-5793(82)80390-6. [DOI] [PubMed] [Google Scholar]
- (54).Li JJ, Li SA, Oberley TD, Parsons JA. Carcinogenic activities of various steroidal and nonsteroidal estrogens in the hamster kidney: relation to hormonal activity and cell proliferation. Cancer Res. 1995;55:4347–4351. [PubMed] [Google Scholar]
- (55).Sarabia SF, Zhu BT, Kurosawa T, Tohma M, Liehr JG. Mechanism of cytochrome P450-catalyzed aromatic hydroxylation of estrogens. Chem. Res. Toxicol. 1997;10:767–771. doi: 10.1021/tx970021f. [DOI] [PubMed] [Google Scholar]
- (56).Zhang F, Chen Y, Pisha E, Shen L, Xiong Y, van Breemen RB, Bolton JL. The Major Metabolite of Equilin, 4-Hydroxyequilin, Autoxidizes to an o-Quinone Which Isomerizes to the Potent Cytotoxin 4-Hydroxyequilenin-o- quinone. Chem Res Toxicol. 1999;12:204–213. doi: 10.1021/tx980217v. [DOI] [PubMed] [Google Scholar]
- (57).Bhavnani BR. Pharmacokinetics and pharmacodynamics of conjugated equine estrogens: Chemistry and metabolism. Proc. Soc. Exp. Biol. Med. 1998;217:6–16. doi: 10.3181/00379727-217-44199. [DOI] [PubMed] [Google Scholar]
- (58).Judd HL, Mebane-Sims I, Legault C, Wasilauskas C, Johnson S, Merino M, Barrett-Connor B, Trabal J. Effects of hormone replacement therapy on endometrial histology in postmenopausal women. JAMA. 1996;275:370–375. doi: 10.1001/jama.1996.03530290040035. [DOI] [PubMed] [Google Scholar]
- (59).Pisha E, Liu X, Constantinou AI, Bolton JL. Evidence that a metabolite of equine estrogens, 4-hydroxyequilenin induces cellular transformation in vitro. Chem. Res. Toxicol. 2001;14:82–90. doi: 10.1021/tx000168y. [DOI] [PubMed] [Google Scholar]
- (60).Prokai-Tatrai K, Prokai L. Impact of metabolism on the safety of estrogen therapy. Ann. N Y Acad. Sci. 2005;1052:243–257. doi: 10.1196/annals.1347.018. [DOI] [PubMed] [Google Scholar]
- (61).Spink DC, Zhang F, Hussain MM, Katz BH, Liu X, Hilker DR, Bolton JL. Metabolism of equilenin in MCF-7 and MDA-MB-231 human breast cancer cells. Chem. Res. Toxicol. 2001;14:572–581. doi: 10.1021/tx000219r. [DOI] [PubMed] [Google Scholar]
- (62).Sissung TM, Price DK, Sparreboom A, Figg WD. Pharmacogenetics and regulation of human cytochrome P450 1B1: implications in hormone-mediated tumor metabolism and a novel target for therapeutic intervention. Mol. Cancer Res. 2006;4:135–150. doi: 10.1158/1541-7786.MCR-05-0101. [DOI] [PubMed] [Google Scholar]
- (63).Iverson SL, Shen L, Anlar N, Bolton JL. Bioactivation of estrone and its catechol metabolites to quinoid-glutathione conjugates in rat liver microsomes. Chem Res Toxicol. 1996;9:492–499. doi: 10.1021/tx950178c. [DOI] [PubMed] [Google Scholar]
- (64).Bolton JL, Shen L. p-Quinone methides are the major decomposition products of catechol estrogen o-quinones. Carcinogenesis. 1996;17:925–929. doi: 10.1093/carcin/17.5.925. [DOI] [PubMed] [Google Scholar]
- (65).Stack DE, Byun J, Gross ML, Rogan EG, Cavalieri EL. Molecular characteristics of catechol estrogen quinones in reactions with deoxyribonucleosides. Chem. Res. Toxicol. 1996;9:851–859. doi: 10.1021/tx960002q. [DOI] [PubMed] [Google Scholar]
- (66).Shen L, Pisha E, Huang Z, Pezzuto JM, Krol E, Alam Z, van Breemen RB, Bolton JL. Bioreductive activation of catechol estrogen-ortho-quinones: Aromatization of the B ring in 4-hydroxyequilenin markedly alters quinoid formation and reactivity. Carcinogenesis. 1997;18:1093–1101. doi: 10.1093/carcin/18.5.1093. [DOI] [PubMed] [Google Scholar]
- (67).Smithgall TE, Harvey RG, Penning TM. Spectroscopic identification of ortho-quinones as the products of polycyclic aromatic trans-dihydrodiol oxidation catalyzed by dihydrodiol dehydrogenase. J. Biol. Chem. 1988;263:1814–1820. [PubMed] [Google Scholar]
- (68).Penning TM. Dihydrodiol dehydrogenase and its role in polycyclic aromatic hydrocarbon metabolism. Chem Biol Interact. 1993;89:1–34. doi: 10.1016/0009-2797(93)03203-7. [DOI] [PubMed] [Google Scholar]
- (69).Penning TM, Burczynski ME, Hung CF, McCoull KD, Palackal NT, Tsuruda LS. Dihydrodiol dehydrogenases and polycyclic aromatic hydrocarbon activation: Generation of reactive and redox active o-quinones. Chem. Res. Toxicol. 1999;12:1–18. doi: 10.1021/tx980143n. [DOI] [PubMed] [Google Scholar]
- (70).Xue W, Warshawsky D. Metabolic activation of polycyclic and heterocyclic aromatic hydrocarbons and DNA damage: a review. Toxicol. Appl. Pharmacol. 2005;206:73–93. doi: 10.1016/j.taap.2004.11.006. [DOI] [PubMed] [Google Scholar]
- (71).Zhang F, Bolton JL. Synthesis of the equine estrogen metabolites 2-hydroxyequilin and 2-hydroxyequilenin. Chem. Res. Toxicol. 1999;12:200–203. doi: 10.1021/tx980189g. [DOI] [PubMed] [Google Scholar]
- (72).Bolton JL, Trush MA, Penning TM, Dryhurst G, Monks TJ. Role of quinones in toxicology. Chem. Res. Toxicol. 2000;13:135–160. doi: 10.1021/tx9902082. [DOI] [PubMed] [Google Scholar]
- (73).Roy D, Liehr JG. Estrogen, DNA damage and mutations. Mutat. Res. 1999;424:107–115. doi: 10.1016/s0027-5107(99)00012-3. [DOI] [PubMed] [Google Scholar]
- (74).Nutter LM, Ngo EO, Abul-Hajj YJ. Characterization of DNA damage induced by 3,4-estrone-o-quinone in human cells. J. Biol. Chem. 1991;266:16380–16386. [PubMed] [Google Scholar]
- (75).Lavigne JA, Goodman JE, Fonong T, Odwin S, He P, Roberts DW, Yager JD. The Effects of Catechol-O-Methyltransferase Inhibition on Estrogen Metabolite and Oxidative DNA Damage Levels in Estradiol-treated MCF-7 Cells. Cancer Res. 2001;61:7488–7494. [PubMed] [Google Scholar]
- (76).Rajapakse N, Butterworth M, Kortenkamp A. Detection of DNA strand breaks and oxidized DNA bases at the single-cell level resulting from exposure to estradiol and hydroxylated metabolites. Environ. Mol. Mutagen. 2005;45:397–404. doi: 10.1002/em.20104. [DOI] [PubMed] [Google Scholar]
- (77).Li JJ, Gonzalez A, Banerjee S, Banerjee SK, Li SA. Estrogen carcinogenesis in the hamster kidney: role of cytotoxicity and cell proliferation. Environ. Health Perspect. 1993;5:259–264. doi: 10.1289/ehp.93101s5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Banerjee SK, Banerjee S, Li SA, Li JJ. Induction of chromosome aberrations in Syrian hamster renal cortical cells by various estrogens. Mutat. Res. 1994;311:191–197. doi: 10.1016/0027-5107(94)90176-7. [DOI] [PubMed] [Google Scholar]
- (79).Chen ZH, Na HK, Hurh YJ, Surh YJ. 4-Hydroxyestradiol induces oxidative stress and apoptosis in human mammary epithelial cells: possible protection by NF-kappaB and ERK/MAPK. Toxicol. Appl. Pharmacol. 2005;208:46–56. doi: 10.1016/j.taap.2005.01.010. [DOI] [PubMed] [Google Scholar]
- (80).Zhang F, Swanson SM, van Breemen RB, Liu X, Yang Y, Gu C, Bolton JL. Equine estrogen metabolite 4-hydroxyequilenin induces DNA damage in the rat mammary tissues: formation of single-strand breaks, apurinic sites, stable adducts, and oxidized bases. Chem. Res. Toxicol. 2001;14:1654–1659. doi: 10.1021/tx010158c. [DOI] [PubMed] [Google Scholar]
- (81).Ozcagli E, Sardas S, Biri A. Assessment of DNA damage in postmenopausal women under hormone replacement therapy. Maturitas. 2005;51:280–285. doi: 10.1016/j.maturitas.2004.08.010. [DOI] [PubMed] [Google Scholar]
- (82).Malins DC, Polissar NL, Gunselman SJ. Progession of human breast cancers to the metastatic state is linked to hydroxyl radical-induced DNA damage. Proc. Natl. Acad. Sci. USA. 1996;93:2557–2563. doi: 10.1073/pnas.93.6.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Malins DC, Anderson KM, Jaruga P, Ramsey CR, Gilman NK, Green VM, Rostad SW, Emerman JT, Dizdaroglu M. Oxidative changes in the DNA of stroma and epithelium from the female breast: potential implications for breast cancer. Cell Cycle. 2006;5:1629–1632. doi: 10.4161/cc.5.15.3098. [DOI] [PubMed] [Google Scholar]
- (84).Karihtala P, Soini Y. Reactive oxygen species and antioxidant mechanisms in human tissues and their relation to malignancies. Apmis. 2007;115:81–103. doi: 10.1111/j.1600-0463.2007.apm_514.x. [DOI] [PubMed] [Google Scholar]
- (85).Liehr JG. Role of DNA adducts in hormonal carcinogenesis. Regul. Toxicol. Pharmacol. 2000;32:276–282. doi: 10.1006/rtph.2000.1432. [DOI] [PubMed] [Google Scholar]
- (86).Russo J, Russo IH. Genotoxicity of steroidal estrogens. Trends Endocrinol. Metab. 2004;15:211–214. doi: 10.1016/j.tem.2004.05.007. [DOI] [PubMed] [Google Scholar]
- (87).Chakravarti D, Mailander PC, Li KM, Higginbotham S, Zhang HL, Gross ML, Meza JL, Cavalieri EL, Rogan EG. Evidence that a burst of DNA depurination in SENCAR mouse skin induces error-prone repair and forms mutations in the H-ras gene. Oncogene. 2001;20:7945–7953. doi: 10.1038/sj.onc.1204969. [DOI] [PubMed] [Google Scholar]
- (88).Saeed M, Rogan E, Fernandez SV, Sheriff F, Russo J, Cavalieri E. Formation of depurinating N3Adenine and N7Guanine adducts by MCF-10F cells cultured in the presence of 4-hydroxyestradiol. Int. J. Cancer. 2007;120:1821–1824. doi: 10.1002/ijc.22399. [DOI] [PubMed] [Google Scholar]
- (89).Zahid M, Kohli E, Saeed M, Rogan E, Cavalieri E. The greater reactivity of estradiol-3,4-quinone vs estradiol-2,3-quinone with DNA in the formation of depurinating adducts: Implications for tumor-initiating activity. Chem. Res. Toxicol. 2006;19:164–172. doi: 10.1021/tx050229y. [DOI] [PubMed] [Google Scholar]
- (90).Saeed M, Zahid M, Gunselman SJ, Rogan E, Cavalieri E. Slow loss of deoxyribose from the N7deoxyguanosine adducts of estradiol-3,4-quinone and hexestrol-3′,4′-quinone. Implications for mutagenic activity. Steroids. 2005;70:29–35. doi: 10.1016/j.steroids.2004.09.011. [DOI] [PubMed] [Google Scholar]
- (91).Debrauwer L, Rathahao E, Jouanin I, Paris A, Clodic G, Molines H, Convert O, Fournier F, Tabet JC. Investigation of the regio- and stereo-selectivity of deoxyguanosine linkage to deuterated 2-hydroxyestradiol by using liquid chromatography/ESI-ion trap mass spectrometry. J. Am. Soc. Mass Spectrom. 2003;14:364–372. doi: 10.1016/S1044-0305(03)00066-7. [DOI] [PubMed] [Google Scholar]
- (92).Terashima I, Suzuki N, Shibutani S. Mutagenic properties of estrogen quinone-derived DNA adducts in simian kidney cells. Biochemistry. 2001;40:166–172. doi: 10.1021/bi002273c. [DOI] [PubMed] [Google Scholar]
- (93).Hayashi N, Hasegawa K, Barrett JC, Tsutsui T. Estrogen-induced cell transformation and DNA adduct formation in cultured Syrian hamster embryo cells. Mol. Carcinogenesis. 1996;16:149–156. doi: 10.1002/(SICI)1098-2744(199607)16:3<149::AID-MC5>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- (94).Embrechts J, Lemiere F, Dongen WV, Esmans EL, Buytaert P, van Marck E, Kockx M, Makar A. Detection of estrogen DNA-adducts in human breast tumor tissue and healthy tissue by combined nano LC-nano ES tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2003;14:482–491. doi: 10.1016/S1044-0305(03)00130-2. [DOI] [PubMed] [Google Scholar]
- (95).Bolton JL. Quinoids, quinoid radicals, and phenoxyl radicals formed from estrogens and antiestrogens. Toxicology. 2002;177:55–65. doi: 10.1016/s0300-483x(02)00195-6. [DOI] [PubMed] [Google Scholar]
- (96).Ding S, Shapiro R, Geacintov NE, Broyde S. Conformations of stereoisomeric base adducts to 4-hydroxyequilenin. Chem. Res. Toxicol. 2003;16:695–707. doi: 10.1021/tx0340246. [DOI] [PubMed] [Google Scholar]
- (97).Yasui M, Laxmi YR, Ananthoju SR, Suzuki N, Kim SY, Shibutani S. Translesion synthesis past equine estrogen-derived 2′-deoxyadenosine DNA adducts by human DNA polymerases eta and kappa. Biochemistry. 2006;45:6187–6194. doi: 10.1021/bi0525324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Ding S, Shapiro R, Geacintov NE, Broyde S. 4-hydroxyequilenin-adenine lesions in DNA duplexes: stereochemistry, damage site, and structure. Biochemistry. 2007;46:182–191. doi: 10.1021/bi061652o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Kolbanovskiy A, Kuzmin V, Shastry A, Kolbanovskaya M, Chen D, Chang M, Bolton JL, Geacintov NE. Base selectivity and effects of sequence and DNA secondary structure on the formation of covalent adducts derived from the equine estrogen metabolite 4-hydroxyequilenin. Chem. Res. Toxicol. 2005;18:1737–1747. doi: 10.1021/tx050190x. [DOI] [PubMed] [Google Scholar]
- (100).Chang M, Shin YG, van Breemen RB, Blond SY, Bolton JL. Structural and functional consequences of inactivation of human glutathione S-transferase P1-1 mediated by the catechol metabolite of equine estrogens, 4-hydroxyequilenin. Biochemistry. 2001;40:4811–4820. doi: 10.1021/bi002513o. [DOI] [PubMed] [Google Scholar]
- (101).Abel EL, Lyon RP, Bammler TK, Verlinde CL, Lau SS, Monks TJ, Eaton DL. Estradiol metabolites as isoform-specific inhibitors of human glutathione S-transferases. Chem. Biol. Interact. 2004;151:21–32. doi: 10.1016/j.cbi.2004.10.006. [DOI] [PubMed] [Google Scholar]
- (102).Bianco NR, Perry G, Smith MA, Templeton DJ, Montano MM. Functional implications of antiestrogen induction of quinone reductase: inhibition of estrogen-induced deoxyribonucleic acid damage. Mol. Endocrinol. 2003;17:1344–1355. doi: 10.1210/me.2002-0382. [DOI] [PubMed] [Google Scholar]
- (103).Cassidy PB, Edes K, Nelson CC, Parsawar K, Fitzpatrick FA, Moos PJ. Thioredoxin reductase is required for the inactivation of tumor suppressor p53 and for apoptosis induced by endogenous electrophiles. Carcinogenesis. 2006;27:2538–2549. doi: 10.1093/carcin/bgl111. [DOI] [PubMed] [Google Scholar]
- (104).Ronai Z. Deciphering the mammalian stress response - a stressful task. Oncogene. 1999;18:6084–6086. doi: 10.1038/sj.onc.1203175. [DOI] [PubMed] [Google Scholar]
- (105).Davis W, Jr., Ronai Z, Tew KD. Cellular thiols and reactive oxygen species in drug-induced apoptosis. J Pharmacol Exp Ther. 2001;296:1–6. [PubMed] [Google Scholar]
- (106).Adler V, Yin Z, Tew KD, Ronai Z. Role of redox potential and reactive oxygen species in stress signaling. Oncogene. 1999;18:6104–6111. doi: 10.1038/sj.onc.1203128. [DOI] [PubMed] [Google Scholar]
- (107).Stevens JL, Liu H, Halleck M, Bowes RC, Chen QM, van de Water B. Linking gene expression to mechanisms of toxicity. Toxicol. Lett. 2000;112-113:479–486. doi: 10.1016/s0378-4274(99)00200-3. [DOI] [PubMed] [Google Scholar]
- (108).Moran LK, Gutteridge JM, Quinlan GJ. Thiols in cellular redox signalling and control. Curr. Med. Chem. 2001;8:763–772. doi: 10.2174/0929867013372904. [DOI] [PubMed] [Google Scholar]
- (109).Sarmanova J, Susova S, Gut I, Mrhalova M, Kodet R, Adamek J, Roth Z, Soucek P. Breast cancer: role of polymorphisms in biotransformation enzymes. Eur. J. Hum. Genet. 2004;12:848–854. doi: 10.1038/sj.ejhg.5201249. [DOI] [PubMed] [Google Scholar]
- (110).Sanchez RI, Mesia-Vela S, Kauffman FC. Induction of NAD(P)H quinone oxidoreductase and glutathione S-transferase activities in livers of female August-Copenhagen Irish rats treated chronically with estradiol: comparison with the Sprague-Dawley rat. J. Steroid Biochem. Mol. Biol. 2003;87:199–206. doi: 10.1016/j.jsbmb.2003.08.007. [DOI] [PubMed] [Google Scholar]
- (111).Lee JM, Anderson PC, Padgitt JK, Hanson JM, Waters CM, Johnson JA. Nrf2, not the estrogen receptor, mediates catechol estrogen-induced activation of the antioxidant responsive element. Biochim. Biophys. Acta. 2003;1629:92–101. doi: 10.1016/j.bbaexp.2003.08.006. [DOI] [PubMed] [Google Scholar]
- (112).Husbeck B, Powis G. The redox protein thioredoxin-1 regulates the constitutive and inducible expression of the estrogen metabolizing cytochromes P450 1B1 and 1A1 in MCF-7 human breast cancer cells. Carcinogenesis. 2002;23:1625–1630. doi: 10.1093/carcin/23.10.1625. [DOI] [PubMed] [Google Scholar]
- (113).Yao J, Chang M, Li Y, Pisha E, Liu X, Yao D, Elguindi EC, Blond SY, Bolton JL. Inhibition of cellular enzymes by equine catechol estrogens in human breast cancer cells: specificity for glutathione S-transferase P1-1. Chem. Res. Toxicol. 2002;15:935–942. doi: 10.1021/tx020018i. [DOI] [PubMed] [Google Scholar]
- (114).Baldwin MA, Benz CC. Redox control of zinc finger proteins. Methods Enzymol. 2002;353:54–69. doi: 10.1016/s0076-6879(02)53036-6. [DOI] [PubMed] [Google Scholar]
- (115).Wang LH, Yang XY, Zhang X, Mihalic K, Fan YX, Xiao W, Howard OM, Appella E, Maynard AT, Farrar WL. Suppression of breast cancer by chemical modulation of vulnerable zinc fingers in estrogen receptor. Nat. Med. 2004;10:40–47. doi: 10.1038/nm969. [DOI] [PubMed] [Google Scholar]
- (116).Jung J, Ishida K, Nishihara T. Anti-estrogenic activity of fifty chemicals evaluated by in vitro assays. Life Sci. 2004;74:3065–3074. doi: 10.1016/j.lfs.2003.10.030. [DOI] [PubMed] [Google Scholar]
- (117).Atsriku C, Scott GK, Benz CC, Baldwin MA. Reactivity of zinc finger cysteines: chemical modifications within labile zinc fingers in estrogen receptor. J. Am. Soc. Mass Spectrom. 2005;16:2017–2026. doi: 10.1016/j.jasms.2005.08.009. [DOI] [PubMed] [Google Scholar]
- (118).Li JJ, Li SA, Gustafsson JA, Nandi S, Sekely LI. Springer-Verlag; New York: 1996. [Google Scholar]
- (119).Mitra K, Marquis JC, Hillier SM, Rye PT, Zayas B, Lee AS, Essigmann JM, Croy RG. A rationally designed genotoxin that selectively destroys estrogen receptor-positive breast cancer cells. J. Am. Chem. Soc. 2002;124:1862–1863. doi: 10.1021/ja017344p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Sharma U, Marquis JC, Nicole Dinaut A, Hillier SM, Fedeles B, Rye PT, Essigmann JM, Croy RG. Design, synthesis, and evaluation of estradiol-linked genotoxicants as anti-cancer agents. Bioorg. Med. Chem. Lett. 2004;14:3829–3833. doi: 10.1016/j.bmcl.2004.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Lareef MH, Garber J, Russo PA, Russo IH, Heulings R, Russo J. The estrogen antagonist ICI-182-780 does not inhibit the transformation phenotypes induced by 17-beta-estradiol and 4-OH estradiol in human breast epithelial cells. Int. J. Oncol. 2005;26:423–429. [PubMed] [Google Scholar]
- (122).Russo J, Hasan Lareef M, Balogh G, Guo S, Russo IH. Estrogen and its metabolites are carcinogenic agents in human breast epithelial cells. J. Steroid Biochem. Mol. Biol. 2003;87:1–25. doi: 10.1016/s0960-0760(03)00390-x. [DOI] [PubMed] [Google Scholar]
- (123).Cuendet M, Liu X, Pisha E, Li Y, Yao J, Yu L, Bolton JL. Equine estrogen metabolite 4-hydroxyequilenin induces anchorage-independent growth of human mammary epithelial MCF-10A cells: differential gene expression. Mutat. Res. 2004;550:109–121. doi: 10.1016/j.mrfmmm.2004.02.005. [DOI] [PubMed] [Google Scholar]