

Abstract
Escherichia coli K1 and Group B Streptococcus (GBS) are the most common bacteria that cause meningitis during the neonatal period. Complement, the first line of defence in the host, acts on these bacteria to opsonize with various components of complement for subsequent presentation to phagocytes. To counteract these opsonization effects, E. coli and GBS bind to the complement regulators C4 binding protein and Factor H, respectively. Nonetheless, the deposition of complement components on these two bacteria from neonatal serum and their effect on the host cell interaction is unclear. Here we demonstrated that the deposition of complement proteins from adult serum prevented the invasion of E. coli into human brain microvascular endothelial cells, whereas the invasion of GBS was enhanced. In contrast, treatment with cord serum had no effect on the invasion of both these bacteria. We also examined the effect of the deposited complement proteins on phagocytosis using THP-1 cells and THP-1 cells differentiated into macrophages. Escherichia coli treated with adult serum neither attached nor entered these cells, whereas GBS was phagocytosed and survived efficiently. We further demonstrate that the inhibitory effect of complement proteins is the result of the bound complement inhibitors C4b-binding protein, in the case of E. coli, and Factor H, in the case of GBS. Taken together, these results suggest that E. coli and GBS utilize contrasting mechanisms of complement-mediated interactions with their target cells for successful establishment of disease.
Keywords: bacteria, complement, endothelial cells, invasion, meningitis
Introduction
Escherichia coli K1 (E. coli) and Group B Streptococcus Type III (GBS) are the most common Gram-negative and Gram-positive bacteria, respectively, that cause meningitis in neonates. Despite advances in antimicrobial therapy and supportive care, the mortality and morbidity rates associated with this disease have not changed over the last three decades. This poor outcome is because of our inability to develop new modes of prevention and is primarily the result of inadequate knowledge of the pathogenesis and pathophysiology of neonatal meningitis. These bacteria infect neonates either by vertical transmission (transplacental) or horizontal transmission (nosocomial infections); they survive and multiply to reach a high degree of bacteraemia, which is subsequently required for the traversal across the blood–brain barrier. Although it is hypothesized that the lack of protective antibodies against the bacteria and the immature immune system of infants are major risk factors for the incidence of meningitis, it is unclear how the bacteria survive in the neonatal blood to reach the high degree of bacteremia.
To survive in the host, bacteria have to first overcome the complement system, the first-line of defence against invading pathogens. The complement system has three pathways of activation, the classical, alternative and lectin pathways; all three pathways result in C3 activation and merge into a common pathway that results in the formation of the membrane attack complex (MAC), or C5b-9.1–5 The classical pathway is primarily activated through the binding of C1q to antibody–antigen complexes. The lectin pathway is initiated when the mannose-binding lectin binds to certain carbohydrate residues on bacteria or viruses, and the alternative pathway is initiated when spontaneously activated C3 binds to activating surfaces such as those of certain bacteria or viruses. However, several bacteria have devised strategies to capture complement inhibitors and so inactivate the complement.6–8 Earlier studies from our laboratory demonstrated that the outer membrane protein A (OmpA) of E. coli K1 binds C4b-binding protein (C4BP), the major inhibitor of the classical pathway, and inactivates both C3b and C4b to avoid serum bactericidal activity.9,10 On the other hand, the binding of Factor H (FH), an inhibitor of the alternative pathway of complement, had little effect on the survival of E. coli. Notably, E. coli enters and survives in the absence or presence of antibody opsonization in macrophages;11 however, it is not clear whether deposited complement contributes to increased phagocytosis and/or survival. While GBS are known to be resistant to the MAC, opsonization of GBS with the complement fragments C3b and iC3b promotes efficient phagocytosis and killing of bacteria. In the absence of type III capsular specific antibody in neonates, GBS opsonophagocytosis occurs via the lectin pathway of complement.12–15 It has been hypothesized that GBS survives phagocytosis by binding to FH via the sialic acid residues present on its surface and by inactivating bound complement.16 In support of this, recent studies have shown that FH binds to GBS; however, its role in resisting phagocytosis is not clearly known (manuscript in preparation). Thus, these two bacteria utilize contrasting mechanisms for survival in serum. Since these bacteria are prevalent pathogens in the neonatal population, especially in low-birth-weight infants, and complement activation is the primary line of innate defence, we studied the relative quantities and deposition of complement proteins on these bacteria using whole cord blood and serum in comparison with adult blood and serum. The effect of serum complement deposition on the phagocytosis and interaction with human brain microvascular endothelial cells (HBMEC) was also evaluated to obtain insights into whether bound complement enhances the virulence of these strains.
Materials and methods
Bacterial strains and growth conditions
OmpA+E. coli K1 was a rifampin-resistant mutant of E. coli K1 strain RS 218 (serotype O18:K1:H7) and was isolated from the cerebrospinal fluid of a neonate with meningitis. It invades HBMEC in a cell culture model.17 OmpA–E. coli K1 was a non-invasive derivative of the OmpA+E. coli K1, in which the ompA gene was disrupted and expressed no OmpA.18,19 OmpA+E. coli K1 was cultured in brain–heart infusion broth with 12·5 μg/ml rifampicin while OmpA–E. coli K1 was cultured in 12·5 μg/ml rifampicin and 12·5 μg/ml tetracycline. GBS type III strain COH-1 (wt-GBS) is a tetracycline-resistant clinical isolate from an infected patient and is highly encapsulated.20 The acapsular GBS (acap-GBS) was a transposon mutant of wt-GBS and does not produce a capsule.21 Both wt-GBS and acap-GBS were a kind gift from Prof. Craig Rubens at the Children's Hospital and Regional Medical Center, Seattle, WA. All the bacteria were grown overnight in appropriate broth containing respective antibiotics and were washed three times in phosphate-buffered saline (PBS) by centrifugation before being used in experiments.
Antibodies and other reagents
The purification of human FH and C4BP-PS (C4BP in high-affinity complex with Protein S) from serum, and recombinant C4BP-α chain (essentially the same as C4BP-PS but devoid of protein S) were described previously.22,23 Polyclonal antibodies to C1q, C3, C5, C9, C4BP and FH were obtained from Calbiochem (San Diego, CA). Secondary antibodies, raised in rabbit and goat, coupled to horseradish peroxidase (HRP) were obtained from Bio-Rad (Hercules, CA) and Calbiochem, respectively.
Serum and blood survival studies
Adult and cord blood was collected in citrate buffer according to the policies of the Children's Hospital Los Angeles. To obtain serum, 10 ml blood was replenished with 120 μl 100 mm CaCl2 and allowed to stand at room temperature for 1 hr followed by overnight at 4°. The serum bactericidal assays were performed as described previously.10 Briefly, the bacterial suspensions [104–106 colony-forming units (CFU)] were incubated with freshly prepared serum diluted to 40% in gelatin–veronal buffer with calcium (Sigma, St Louis, MO). Aliquots of 10 μl were removed at various times, serially diluted in PBS and then the dilutions were plated on sheep blood agar. The plates were incubated at 37° overnight and the colonies were counted. Heat-inactivated serum was obtained by incubating serum in a water bath for 30 min at 56°. To perform bactericidal assays using whole blood, 120 μl 100 mm CaCl2 in the presence of heparin (2 U/ml) or hirudin (10 U/ml) was added to 10 ml citrated blood and aliquots of blood were incubated with bacteria for varying periods. The experiments were repeated at least five times in triplicates.
Analysis of deposited complement proteins on E. coliand GBS by flow cytometry
Briefly, 108 cells were incubated in 50 μl adult serum (AS) or cord serum (CS) for the indicated periods. The bacteria were washed three times with PBS and then the sample was split into five parts. Each part was incubated with one antibody against C3, C5, C9, FH, or C4BP (1 : 1000 dilution) for 30 min on ice. The bacterial pellets were then incubated with the respective fluorescein isothiocyanate-conjugated secondary antibodies (1 : 500 dilution) for 30 min at room temperature, washed and resuspended in PBS. Flow cytometry was carried out on a Becton-Dickinson Flow analyser (University of Southern California FACS core facility; Becton-Dickinson, San Jose, CA) using cell quest software and approximately 10 000 gated events were recorded.
Determination of FH and C4BP levels in serum samples by enzyme-linked immunosorbent assay (ELISA)
Various concentrations of serum (1 : 500, 1 : 1000, and 1 : 2000 dilutions in PBS) were incubated in a 96-well ELISA plate either overnight at 4° or for 2 hr at room temperature. Purified FH or C4BP were used to generate standard curves. The plate was washed with PBS and incubated for 1 hr with 1% bovine serum albumin (BSA) in PBS. Subsequently, the plate was incubated with anti-FH or anti-C4BP antibodies (1 : 5000 dilution in 1% BSA) for 1 hr at room temperature, washed, and incubated with respective secondary antibodies coupled to HRP (1 : 15 000 dilution) for 1 hr. After washing the plate with PBS three times, the colour was developed with TMB Microwell peroxidase substrate (KPL, Gaithersburg, MD) and read at 450 nm on an ELISA reader (Molecular Devices Corp., Sunnyvale, CA).
Binding analysis of FH and C4BP from AS or CS to GBS and E. coli
To study differences in binding of FH and C4BP from AS and CS by GBS and E. coli, the bacteria (108 CFU) were incubated with adult or cord serum (40 μl in 250 μl reaction volume). The bacteria were washed in PBS three times by centrifugation, boiled in sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE) sample buffer, and equal volumes of the supernatants were separated by SDS–PAGE. The resolved proteins were then transferred to nitrocellulose membranes, stained briefly with Ponceu and scanned; the various bands were analysed by densitometry to determine the equality of protein loading. The blots were then incubated with specific human anti-FH or anti-C4BP antibodies (1 : 2000 dilution) and subsequently with specific HRP-conjugated secondary antibodies (1 : 10 000 dilution). The blots were further developed using chemiluminescence. Purified FH and recombinant C4BP-α were loaded on the gels as controls.
Bacterial invasion assays
To check for a differential effect of opsonization on invasion, GBS and E. coli were incubated in either AS or CS and then tested for their capabilities for invading HBMEC. Briefly, HBMEC were cultured to 90–95% confluence, washed three times with RPMI-1640 and then incubated with GBS or E. coli. Invasion assays were performed by a modification of the method of Tang et al.17 Approximately 107 bacteria were incubated with 40% serum, either heat-inactivated or normal, for 15 min, washed, and then added to confluent HBMEC monolayers at a multiplicity of infection (MOI) of 1 : 100 (cell : bacteria ratio) in 500 μl experimental medium containing M199/ Ham F12 (1 : 1 volume/volume) with 5% heat-inactivated fetal bovine serum, 2 mm l-glutamine and 1 mm sodium pyruvate. The plates were incubated for 90 min at 37° in 5% CO2. The monolayers were washed and incubated in the experimental medium containing gentamycin (100 μg/ml) for 1 hr to kill extracellular bacteria. The monolayers were washed once again and lysed with 0·5% Triton X-100 in saline. The internalized bacteria released into Triton X-100 were then diluted in PBS and enumerated on blood agar plates. To determine total cell-associated bacteria in the case of HBMEC, the gentamycin step was omitted in the above described experiments and performed simultaneously. Similar assays were performed using THP-1 (a human monocytic cell line) and THP-1 differentiated into macrophages with 20 μm phorbol 12-myristate 13-acetate, which are designated as THP-DM.
Statistical analysis
Comparison between groups was performed using paired Student's t-test for statistical analysis. P-values lower than 0·05 were considered significant.
Results
Survival of E. coli and GBS in human adult and cord serum
Previous studies from our laboratory demonstrated that OmpA+E. coli could efficiently mediate resistance to bactericidal activity of adult human serum.10 However, it is not clear whether newborn serum is able to kill these bacteria. Hence, we analysed the survival of OmpA+E. coli (E44) in CS and compared it to survival in AS by incubating 106 CFU/ml of the bacteria in 40% serum for various time-points. Bacteria treated with heat-inactivated serum were used as a control. The number of surviving bacteria was assessed by plating different dilutions on sheep blood agar plates. OmpA–E. coli (E91), which is sensitive to AS, was used as a negative control. We observed that there was an ∼ 40% decrease in the number of OmpA+E. coli surviving within 1 hr of incubation in AS while OmpA–E. coli were readily killed (Fig. 1a,b). However, both OmpA+ and OmpA–E. coli survived in CS and multiplied to approximately 120% at 1 hr postincubation. GBS are known to be resistant to lysis by serum because of the presence of a well-developed capsule and so we also included an acap-GBS in these studies. In agreement with this concept, wt-GBS survived efficiently in AS, whereas the acap-GBS showed a decrease of approximately 40% after 1 hr when incubated in AS (Fig. 1c,d). Both strains of GBS survived and multiplied in CS. Heat-inactivated serum is inefficient in killing either E. coli K1 or GBS strains even after 4 hr of incubation (data not shown).
Figure 1.
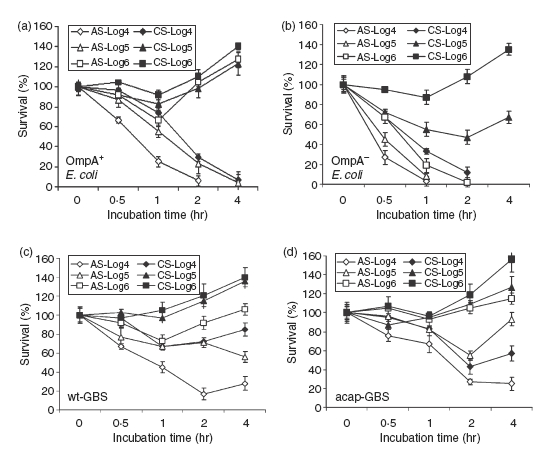
Effect of inoculum sizes on the bactericidal activity of cord serum (CS) and adult serum (AS). Various inoculum sizes of OmpA+ (E44), OmpA–Escherichia coli (E91), wild-type Group B Streptococci (wt-GBS), or acapsular GBS (acap-GBS) ranging from 104 to 106 colony-forming units were subjected to serum bactericidal assay for the times indicated. Aliquots of the bacteria in serum were then serially diluted and plated on blood agar. The experiments were performed in triplicate, at least three times with the data representing relative survival with the bacterial survival at 0 min set as 100%. Error bars represent ± SD.
In addition, the serum survival assays were also performed using lower inocula of bacteria to examine the effectiveness of serum killing. The data suggested that OmpA+E. coli was killed at 104 and 105 CFU/ml in AS while it survived and multiplied at a dose of 106 (Fig. 1a). However, OmpA+E. coli survived in CS even at an inoculum size of 105 CFU/ml, demonstrating a 10-fold difference in survival between AS and CS (Fig. 1b). OmpA–E. coli, on the other hand, did not survive in AS even at 106 CFU/ml but survived in CS at inocula of 105 and 106 CFU/ml (Fig. 1b). Regardless, OmpA+E. coli at an inoculum of 104 CFU/ml was susceptible to killing in CS between 2 and 4 hr incubation, suggesting that at a higher bacterial load the complement components might be completely exhausted in CS more quickly when compared to AS. Similar results were also obtained with GBS (Fig. 1c,d). Both OmpA+E. coli and wt-GBS had multiplied significantly by 4 hr postinoculation.
Analysis of E. coli and GBS survival in human cord or adult blood
We next assessed the survival of the wild-type and mutant strains of E. coli and GBS in adult blood (AB) and cord blood (CB) to examine the comparative bactericidal potential of whole blood. Since heparin is known to affect complement, it may influence the results. Low concentrations (up to 2 U/ml) of heparin slightly enhance, whereas high concentrations (20 U/ml) inhibit, complement activation.24,25 In contrast, hirudin or its analogue lepirudin have no effect on the complement activation. Therefore, we used 2 U/ml heparin or 10 U/ml hirudin in the whole blood assays – concentrations that are sufficient to avoid clotting of the blood for up to 5 hr at 37°. Under the conditions employed in our experiments, both heparin-treated and hirudin-treated blood showed similar bactericidal activities on E. coli K1 and GBS within a 5–10% standard error. Therefore, we presented the results obtained from heparin. Similar to the data obtained for sera, OmpA+E. coli was readily killed at inocula of 104 and 105 CFU/ml in AB, whereas at higher inocula, up to 70% of the bacteria survived, after which they multiplied to 130% by 4 hr postincubation (Fig. 2a). In contrast, both 105 and 106 CFU/ml inocula survived up to 50% in CB and multiplied at later time-points. Interestingly, the inoculum of 104 CFU/ml was readily killed even in CB. However, all the inoculum sizes of OmpA–E. coli were killed in both AB and CB, except the 106 CFU/ml inoculum, which showed 60% survival in CB at 1 hr postinoculation (Fig. 2b). Since AS killed the bacteria at a similar level to AB, we assumed that phagocytosis may not play a significant role in killing or clearing the bacteria. However, it is possible that OmpA+E. coli hide in phagocytes to avoid complement attack, as was demonstrated previously.11 With respect to GBS survival in blood, wt-GBS survived in both CB and AB at an inoculum size of 106 CFU/ml (Fig 2c). On the other hand, AB was capable of completely killing the acap-GBS at all inoculum sizes (Fig 2d) while the acap-GBS survived in CB at 106 CFU/ml, indicating that phagocytosis of the bacteria was less efficient in CB than AB.
Figure 2.
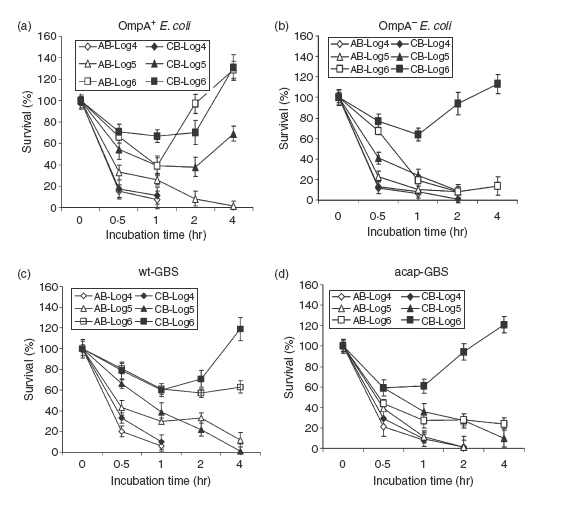
Escherichia coli K1 and Group B Streptococcus (GBS) survival in cord blood (CB) and adult blood (AB). OmpA+E. coli (E44), OmpA–E. coli (E91), wild-type GBS (wt-GBS), or acapsular GBS (acap-GBS) was subjected to bactericidal activity using whole blood for the periods indicated. Aliquots of the bacteria taken at each time-point were then serially diluted and plated on blood agar. The experiments were performed at least four times in duplicate. The data was expressed as per cent survival considering the survival at 0 min as 100% and the error bars represent ± SD.
Analysis of deposition of complement components on bacteria by flow cytometry
Since the killing of E. coli has been partially attributed to complement in AS, the survival of the highly serum-susceptible OmpA–E. coli in CS suggested that the complement system of CS was unable to kill the bacteria. This could be the result of inefficiency of complement to deposit terminal MAC to induce bacteriolysis. We decided to first quantitatively analyse the deposition of complement components C3b and C9 on OmpA+ and OmpA–E. coli after incubating the bacteria in serum. Both components are critical to the formation of MAC, which is responsible for bacterial lysis. Bacteria were analysed by flow cytometry after incubation in serum, as described earlier using specific antibodies to C3 (recognizes also C3b) and C9. Bacteria incubated with neither serum nor antibodies were chosen as controls. Bacteria were scored positive for complement binding if they were more fluorescent than those treated without the primary antibody. Forward-scatter and side-scatter analysis suggested cell membrane distortion upon serum exposure; however, this did not differ between OmpA+ and OmpA–E. coli. Percentages of cells in the population of the positive gate were plotted on a graph. Flow cytometry analysis showed that C3b deposition on OmpA–E. coli from adult serum reached a maximum value (mean shift in frequency) of 70% within 5 min, whereas it was slower and reached similar values in 15 min in the case of OmpA+E. coli (Fig 3a). In contrast, C3b deposited on both OmpA+ and OmpA–E. coli incubated with CS revealed lower quantities up to 10 min. However, the deposition levels were similar between AS and CS by 30 min postincubation. Approximately 70–80% of C9 was detected on OmpA–E. coli within 15 min, while the amount of C9 formed on OmpA+E. coli was approximately 40%. Interestingly, both the strains demonstrated only 10% deposition of C9 when opsonized in CS (Fig 3c). These results suggest that CS is less efficient at activating complement when compared to AS.
Figure 3.
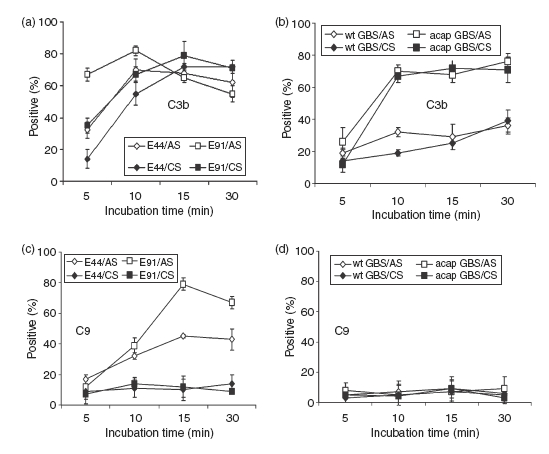
Analysis of C3b and C9 deposited on bacteria incubated in serum. The amounts of C3b and C9 deposited on OmpA+E. coli (E44), OmpA–Escherichia coli (E91), wild-type Group B Streptococci (wt-GBS), or acapsular GBS (acap-GBS) incubated in cord serum (CS) and adult serum (AS) were analysed by flow cytometry using specific fluorescence-labelled antibodies against C3 and C9. The percentages of cells in the population in the positive gate from at least three experiments were taken and presented as means ± SD.
As mentioned earlier, GBS is resistant to complement killing because of the presence of a capsule. However, it was shown earlier that streptococci opsonized with complement components mediate phagocytosis for subsequent killing.26–29 Therefore, we examined the deposition of C3b and C9 on GBS also. The C3b deposition on wt-GBS was comparatively lower than that of acap-GBS when incubated in AS. Approximately 30% C3b was observed on the wt-GBS at 10 min, compared to ∼ 75% on acap-GBS, which did not increase with time (Fig. 3b). Of note, the deposition of C3b on wt-GBS incubated with CS was similar to that of AS, suggesting that capsule indeed inhibits the deposition of complement. Acap-GBS incubated with CS, however, showed similar levels of C3b deposition as that incubated in AS, indicating that C3 component of complement is not deficient in CS. The C9 deposition on both wt-GBS and acap-GBS incubated in AS and CS reached only 5–10% even 30 min postincubation (Fig. 3d).
Determination of FH and C4BP concentrations in AS and CS; and their binding to E. coli and GBS
Several studies have previously demonstrated the presence of low levels of C9 in cord serum compared to adult serum.30–32 Therefore, it is possible that the low amounts of C9 deposited on both E. coli as well as GBS when incubated in CS compared to AS are the result of lack of C9 in CS. On par with reported values, estimation of C9 levels in the serum samples used in the study revealed that several CS samples contain approximately 10% of adult C9 levels (data not shown). Given the fact that E. coli and GBS bind C4BP and FH respectively to subsequently evade complement attack, the concentrations of these two proteins were also determined by ELISA. As shown in Fig. 4a, FH levels ranged from 700 to 825 μg/ml in AS samples whereas CS samples were only 15–40% of AS levels. On the other hand, C4BP concentrations in most of the CS samples were less than 20% of adult levels (20–100 μg/ml in CS versus 350–450 μg/ml in AS). These results suggest that despite the presence of significantly lower levels of FH and C4BP in some CS samples, the bacteria still survive efficiently and this is the result of the lack of terminal complement component C9.
Figure 4.
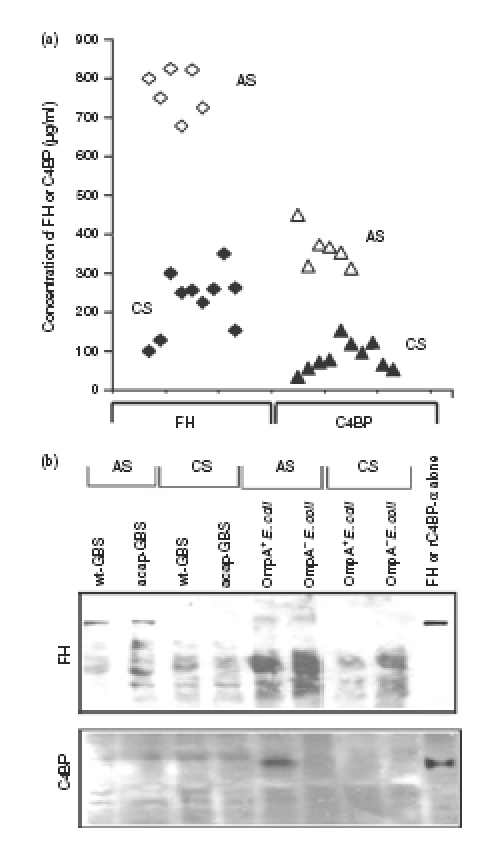
Effect of the concentrations of Factor H (FH) or C4-binding protein (C4BP) in serum on their binding to Escherichia coli and Group B Streptococcus (GBS). (a) The amounts of FH and C4BP in various serum samples were determined by enzyme-linked immunosorbent assay. Each point represents an individual sample. (b) OmpA+E. coli (E44), OmpA–E. coli (E91), wild-type GBS (wt-GBS), or acapsular GBS (acap-GBS) were incubated in 40% cord serum (CS) or adult serum (AS) for 1 hr. The bacteria were then washed three times in phosphate-buffered saline, boiled in sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE) sample buffer, and the supernatants were separated by SDS–PAGE. Purified FH and rC4BP-α were also loaded as controls. The proteins were transferred to nitrocellulose and immunoblotted with antibodies to FH or C4BP.
Next, we examined the binding of FH or C4BP to E. coli and GBS after incubating with CS or AS. Equal numbers of bacteria incubated with either AS or CS for 1 hr were washed and boiled in SDS–PAGE buffer. The supernatants were resolved on SDS–polyacrylamide gels and then immunoblotted with antibodies against either FH or C4BP. The data revealed that OmpA+E. coli bound significantly greater quantities of C4BP when incubated in AS but not in CS (Fig. 4b). In contrast, OmpA–E. coli, GBS or acap-GBS did not bind C4BP from either AS or CS. Similarly, both wt-GBS and acap-GBS were observed to bind much higher levels of FH when incubated in AS than CS. The binding of FH to GBS strains was significantly greater from AS when compared to E. coli. No binding of FH from CS was observed with either of the strains (Fig. 4b). Other proteins that reacted with anti-FH antibody also bound both strains and could be variants of FH, such as FHL-1. These results suggest that the acquisition of either FH or C4BP by these bacteria was significantly lower in CS compared to AS.
Effect of AS or CS incubation on the invasion of HBMEC by E. coli and GBS
Deposition of complement proteins on bacteria often aids their adherence to host cells for successful colonization. For example, acquisition of FH/FHL-1 by group A streptococci might enhance the binding of the bacteria to host cells and increase the rate of invasion in host cells and similar was observed for C4BP and Candida albicans.33,34 Both E. coli and GBS bound higher levels of FH and C4BP from AS compared to CS, we examined whether differential opsonization of the bacteria with these complement regulators had any effect on the invasion of the E. coli and GBS into HBMEC, which are the target cells for both these bacteria while crossing the blood–brain barrier in neonatal meningitis. Both E. coli and GBS (107 CFU) were incubated for 15 min in 40% AS or CS at 37°, washed and then added to confluent HBMEC monolayers. Both binding and invasion assays were performed as described earlier.10 In these experiments, the percentage invasion was calculated based on the inoculum size of the bacteria after treatment with either AS or CS. As shown in Fig. 5a, OmpA+E. coli treated with CS invaded HBMEC with similar success to that of control cells to which 5% heat-inactivated newborn calf serum had been added. In contrast, treatment of OmpA+E. coli with AS significantly inhibited both binding and invasion, suggesting that some component in AS was inhibiting these processes. OmpA–E. coli treated with either CS or AS showed no invasion into HBMEC although the binding levels were similar to those of OmpA+E. coli. Incubation of OmpA+E. coli with heat-inactivated serum showed similar binding or invasion of the bacteria as in controls, suggesting that complement components might be responsible for the inhibitory effect. Our previous studies demonstrated that OmpA+E. coli binds C4BP in greater quantities than OmpA–E. coli, indicating that C4BP might be preventing OmpA interaction with the HBMEC receptor. To examine this, OmpA+E. coli were incubated with purified C4BP-PS (most C4BP in blood is complexed with Protein S) or FH for 15 min at room temperature, washed, and then added to HBMEC monolayers. In addition, to rule out the role of Protein S (PS), recombinant C4BP-α chain (rC4BP) was also included in these experiments. The results indicated that OmpA+E. coli-bound C4BP-PS or rC4BP prevented the entry of the bacteria into HBMEC by ∼ 60%. The FH had no effect on the invasion, although the binding was increased by ∼ 20%.
Figure 5.
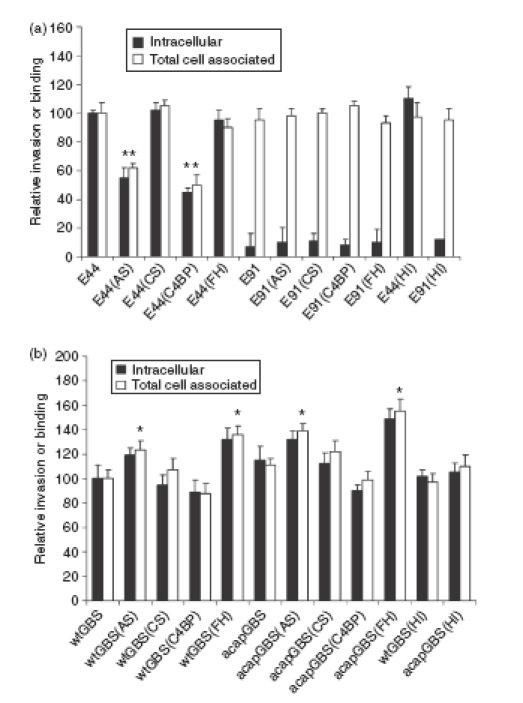
Binding to and invasion of human brain microvascular endothelial cells (HBMEC) by Escherichia coli and group B streptococci (GBS) incubated in adult serum (AS), cord serum (CS), purified Factor H (FH), or C4-binding protein (C4BP). (a) OmpA+E. coli (E44) or OmpA–E. coli (E91) and (b) wild-type GBS (wt-GBS) or acapsular GBS (acap-GBS) were incubated in 40% cord serum (CS) or adult serum (AS), purified Factor H (FH) or C4-binding protein (C4bp) (25 μg/ml) for 15 min, washed, and added to HBMEC monolayers at a multiplicity of infection of 1 : 100 (cell to bacteria ratio). Binding and invasion assays were performed as described in the Materials and methods. Heat-inactivated (HI) serum was used as a negative control. The experiments were repeated three times in triplicate with similar results. The data are presented as means ± SD. The invasion of the bacteria treated with serum was considered significantly enhanced or reduced when compared to untreated controls. *P < 0·02 by two-tailed t-test; **P < 0·05.
In contrast, wt-GBS incubated with AS demonstrated an increase of approximately 40% more invasion, while wt-GBS grown with CS did not show a significant difference from untreated wt-GBS (Fig. 5b). Notably, acap-GBS showed approximately 10% higher invasion than wt-GBS, either untreated or treated with AS. There was no significant increase in invasion by acap-GBS opsonized with CS over control acap-GBS. Since it has been shown that GBS binds to FH, we anticipated that FH might participate in enhancing the invasion of GBS. Therefore, we treated GBS with purified FH, washed them and then incubated with HBMEC. We also used C4BP-PS and rC4BP as controls. The wt-GBS treated with FH showed increased invasion, whereas C4BP-PS-treated or rC4BP-treated cells revealed no significant difference in their invasion. As seen with serum-treated bacteria, acap-GBS showed higher invasion rates treated with FH when compared to wt-GBS. Taken together these data suggest that deposition of complement inhibitors FH and C4BP on E. coli and GBS, respectively, affects differentially their interaction with HBMEC.
Effect of E. coli-bound and GBS-bound complement proteins on phagocytosis of the bacteria in vitro
Our previous studies revealed that E. coli enters both monocytes and macrophages in the absence of opsonization and survives intracellularly.11 However, it is not clear whether complement proteins deposited on the bacteria contribute to binding and entry into phagocytic cells. Therefore, OmpA+ and OmpA–E. coli were incubated with AS and CS for 15 min, washed, and then incubated with THP-1 cells (a human monocytic cell line) for 1·5 hr at 37°. In addition, THP-1 cells differentiated into macrophages (THP-DM) were also used for phagocytosis assays. Our studies revealed that only 50% of OmpA+E. coli treated with AS survived in both THP-1 and THP-DM cells when compared to the untreated bacteria (Fig. 6a). In contrast, the bacteria treated with CS entered the cells with 125% higher frequency than untreated OmpA+E. coli. The OmpA–E. coli could not survive in these cells irrespective of the treatment. To examine whether the FH or C4BP had any effect on the phagocytosis of E. coli, as in the case of endothelial cell invasion, OmpA+ and OmpA–E. coli were treated with FH or C4BP-PS or rC4BP for 15 min at room temperature, washed and incubated with THP-1 and THP-DM cells. The data revealed that C4BP-treated OmpA+E. coli had 50% lower survival than the untreated bacteria in these cells. In contrast, FH had no effect on the survival of OmpA+E. coli. Since THP-1 cells do not attach to the flask, the binding of untreated and serum-treated OmpA+E. coli was studied using only THP-DM cells and the results revealed that binding of bacteria treated with AS, C4BP-PS or rC4BP was 50% lower compared to that of untreated bacteria (data not shown). OmpA–E. coli showed no significant increase in the binding to these cells. These results suggest that C4BP might be inhibiting the interaction of OmpA+E. coli with the receptors present on THP-1 or THP-DM cells and that C4BP-α chain alone is sufficient to exert the inhibitory effect.
Figure 6.
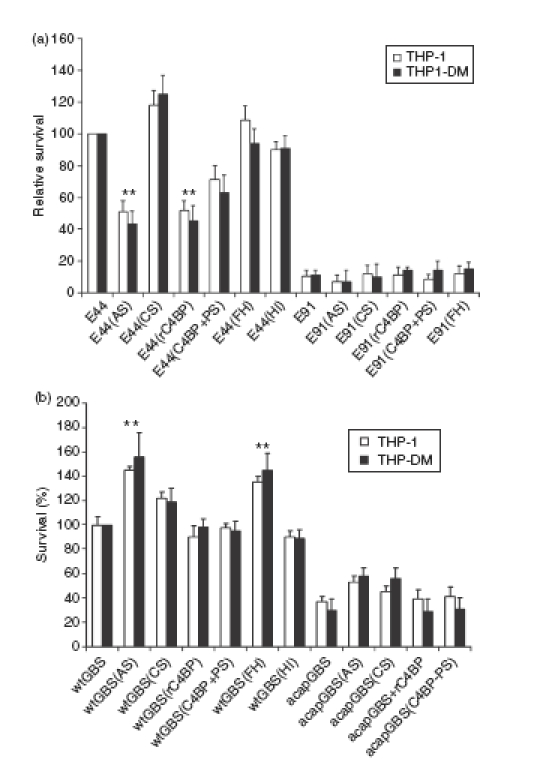
Effect of serum, Factor H (FH) and C4-binding protein (C4BP) on the entry of Escherichia coli and Group B Streptococcus (GBS) into THP-1 cells or THP-DM. E. coli (a) or GBS (b) were incubated with 40% adult serum (AS) or cord serum (CS), purified Factor H (FH) or C4-binding protein (rC4BP or C4BP-PS) (25 μg/ml) for 15 min, washed, and then further incubated with either THP1 cell suspension or THP-DM monolayers at a multiplicity of infection of 1 : 10. Heat-inactivated (HI) serum was also included as a negative control. After incubation, the cells were washed and further incubated with gentamicin-containing medium (100 mg/ml) for 1 hr to kill extracellular bacteria. Numbers of internalized bacteria were determined by plating on blood agar and expressed as per cent invasion with respect to control bacteria (incubated with neither sera nor purified proteins). Experiments were conducted at least three times and the data are presented as means ± SD. The survival of the bacteria, treated with serum or purified proteins, was considered significantly increased or decreased when compared to untreated controls. **P < 0·05 by two-tailed t-test.
Similarly, GBS treated with CS and AS were added to THP1 and THP-DM cells and the entry of GBS was analysed as described. The data revealed that these cells phagocytosed wt-GBS incubated with AS with 1·4-fold higher frequency than wt-GBS treated with CS (Fig. 6b). In contrast, the uptake of acap-GBS was significantly lower than control and acap-GBS opsonized in AS. To examine the effect of FH and C4BP binding on phagocytosis by these cells, the bacteria were treated with purified FH, C4BP-PS or rC4BP before invasion assays. wt-GBS incubated with FH showed 40% higher survival in both these cells compared to the bacteria treated with C4BP-PS or rC4BP, suggesting that FH binding to wt-GBS enhances the chances of survival in addition to the protection given by the capsule. Although acap-GBS bound greater quantities of FH, it could not survive well in these phagocytes, probably because of lack of capsule. The binding of acap-GBS treated with FH to THP-DM was similar to that of acap-GBS without treatment (data not shown), suggesting that the bacteria were killed very efficiently by macrophages.
Discussion
Infections with E. coli K1 and GBS continue to threaten the neonatal population because they cause meningitis, a disease with a high mortality rate. The circulating bacteria must survive and reach a high degree of bacteraemia for subsequent crossing of the blood–brain barrier, suggesting that these bacteria withstand the host defence mechanisms. As innate and adaptive immune responses are critical for controlling infections in humans, newborns are considered to be immunocompromised because of their underdeveloped immune system. However, it is not clear why only certain populations of neonates are at high risk for developing these bacterial infections. In addition, the reasons for the requirement for a high degree of bacteraemia are also unknown. Complement is one of the key components of the innate immune defence that promotes phagocytosis and supports local inflammatory responses against pathogens. The present study was undertaken to evaluate whether the complement proteins of newborns contribute to lower phagocytosis and/or interaction with HBMEC.
Previous studies from our laboratory demonstrated that OmpA+E. coli survive efficiently in AS, whereas OmpA–E. coli were killed.10 In addition, C4BP was shown to interact with OmpA of E. coli and to mediate the degradation of C3b and C4b, leading to evasion of serum bactericidal activity.9,10 We show here that both OmpA+ and OmpA–E. coli survive equally well in CS, which is attributed to the deposition of lower amounts of C9 on these bacteria despite the presence of equal amounts of C3b. Of note, the CS killed both bacteria at an inoculum of 104 CFU/ml but was inefficient in killing higher inocula, suggesting that overload of bacteria could exhaust the complement. In addition, C9, which is necessary for the formation of MAC, was present in only small quantities in several cord serum samples and this could also contribute to the inefficiency of CS in killing the bacteria. The C9 deficiency of cord serum is in agreement with several previous studies in which the investigators have shown that neonatal serum is significantly deficient in C9 (260 + 50 μg/ml in adults versus < 42 μg/ml in neonates,30). In addition, Lassiter et al.31,35 demonstrated that deficiency of C9 component is associated with diminished deposition of C9 on E. coli and killing of the bacteria in neonatal rats. In AB, OmpA–E. coli could not survive at any inoculum size, whereas it survived efficiently at 106 CFU/ml in CB. OmpA+E. coli is capable of surviving in both adult and cord blood at inoculum sizes higher than 104 CFU/ml, indicating that the bacteria are also able to resist phagocytosis. These results match our previous data demonstrating that OmpA+E. coli enter and survive in peripheral blood monocytes and macrophages.11 It is well known that neonatal neutrophils have functional deficiencies when compared to adult cells.36 Nonetheless, the function of monocytes and macrophages is relatively normal, indicating that these cells might be responsible for the majority of the phagocytosis in neonates as observed in our studies at lower bacterial inocula.
On the other hand, GBS is known to be serum-resistant because of its thick capsule. However, Rooijakkers et al.37 showed that the Gram-positive Staphylococcus aureus does accumulate the MAC on the capsule. We demonstrated in this study that GBS strains are mildly affected by the MAC, as seen by the slight decrease of survival after 15–30 min in AS, but they recover and multiply over longer periods of incubation. Such multiplication of GBS (including the acapsular mutant) is more pronounced in CS than in AS. This difference between adult and cord samples might explain why newborn children are prone to severe invasive infections by these bacteria. It has been hypothesized that type III capsular polysaccharide inhibits the alternative pathway activation by preventing C3b deposition on the surface of GBS.16 Therefore, complement activation might be of major importance for the opsonization of GBS with low amounts of capsule. In agreement with this concept, our studies also revealed deposition of significantly greater amounts of C3b on acap-GBS compared to wt-GBS from both AS and CS. The deposition of C9 was similar on both these strains and significantly lower than the deposition of C9 on E. coli strains. One explanation for the reduced deposition of C9 is that the binding of FH leads to rapid degradation of the deposited C3b and thus does not allow the deposition of downstream complement molecules in AS. However, in the case of CS, the amounts of C9 are significantly lower than in AS and with the FH action the result is lower deposition.
Phagocytosis of bacteria requires the interaction of specific receptors on the phagocyte surface with bound opsonins.38 The initial events in phagocytosis are the recognition and engulfment of bacteria by phagocytes; however, the phagocytotic capacity differs significantly between various cells. Interestingly, our studies demonstrated that treatment of OmpA+E. coli with CS significantly enhances bacterial survival in THP-1 or THP-DM cells when compared with untreated cells. In contrast, treatment with AS significantly prevented the binding to and entry into THP-DM, suggesting that some component(s) in AS is inhibiting the interaction of OmpA+E. coli with these cells. Since the binding of C3b to E. coli is similar between AS and CS, complement receptors might not be involved in the reduction of binding to and entry of THP-DM cells. Our unpublished studies revealed that OmpA binds to Fcγ receptor I, irrespective of opsonization with either complement or antibody. The same site is also involved in the binding to Ecgp, a gp96-like OmpA receptor on endothelial cells.39 Therefore, the presence of lower levels of C4BP in CS allows these bacteria to interact with macrophages, to enter and multiply so as to reach a high degree of bacteraemia. In contrast, OmpA+E. coli-bound C4BP from AS prevents their entry into macrophages, thereby preventing the finding of a niche for its own multiplication. On a par with this concept, purified C4BP-treated OmpA+E. coli, but not FH-treated, could not bind to and enter the THP-DM cells in our assays. A compelling observation of these studies is that the incubation of OmpA+E. coli with AS or purified C4BP significantly inhibited the entry of the bacteria into HBMEC. Neither CS nor FH showed any effect on E. coli invasion of HBMEC. These observations indicate for the first time that the lower than threshold levels of C4BP might be a risk factor for E. coli infection in certain neonates. The GBS is known to interact with neutrophils via CR3 receptors; however, binding of FH might contribute to the resistance to phagocytosis as in the case of group A streptococci.40,41 The GBS treated with AS entered THP-1 or THP-DM cells and survived efficiently in them compared to control bacteria, which were treated with heat-inactivated fetal calf serum. Treatment with CS also led to increased survival by the bacteria. Since the deposition of C3b on GBS treated with CS is significantly greater than the C3b levels in GBS treated with AS, the survival could be the result of GBS-bound FH activity. Our data using purified FH confirmed that FH might be contributing to the survival of GBS in these phagocytes.
We propose that the presence of significantly lower levels of C4BP opens up a window of opportunity for E. coli to interact with the OmpA receptor (Ecgp) on HBMEC, allowing them to cross the blood–brain barrier. Supporting this concept, treatment with either AS or recombinant C4BP considerably reduced the invasion of OmpA+E. coli into HBMEC. No effect on the bacterial invasion of HBMEC has been observed with CS or FH. Therefore, C4BP binding to OmpA offers a survival advantage to E. coli in serum; however, it also presents a disadvantage by preventing the interaction of E. coli with macrophages, which it would enter and hide in for subsequent multiplication. In addition, C4BP binding to E. coli also prevents the interaction of the bacteria with Ecgp on HBMEC for subsequent invasion. On the contrary, the FH bound to GBS enhances the invasion of HBMEC, whereas treatment with C4BP did not show any effect. Thus, binding of FH to GBS enhances the virulence by both increasing its resistance to serum bactericidal activity and by enhancing the interaction with HBMEC. Taken together, these data indicate that E. coli and GBS use different strategies to evade host complement and to interact with HBMEC. In the case of E. coli, identification of C4BP binding sites and the molecular mechanisms involved in the interaction will provide clues for developing therapeutic strategies. In addition, establishing the C4BP levels that are protective to E. coli infections would provide a reference point for identifying at-risk neonates. In the case of GBS, it is not clear how FH contributes to enhanced survival in phagocytes to increase the invasion of HBMEC. It is possible that FH interacts with specific receptors that induce either survival or invasive mechanisms in these cells.
Acknowledgments
This work was supported by National Institutes of Health grant AI40567 (N.V.P.) and by the Swedish Research Council (A.B.).
References
- 1.Kirschfink M, Mollnes TE. Modern complement analysis. Clin Diagn Lab Immunol. 2003;10:982–9. doi: 10.1128/CDLI.10.6.982-989.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gadjeva M, Thiel S, Jensenius JC. The mannan-binding-lectin pathway of the innate immune response. Curr Opin Immunol. 2000;13:74–8. doi: 10.1016/s0952-7915(00)00185-0. [DOI] [PubMed] [Google Scholar]
- 3.Gewurz H, Pickering RJ, Muschel LH, Mergenhagen SE, Good RA. Complement-dependent biological functions in complement deficiency in man. Lancet. 1966;13:356–60. doi: 10.1016/s0140-6736(66)92658-4. [DOI] [PubMed] [Google Scholar]
- 4.Bladen HA, Gewurz H, Mergenhagen SE. Interactions of the complement system with the surface and endotoxic lipopolysaccharide of Veillonella alcalescens. J Exp Med. 1967;125:767–86. doi: 10.1084/jem.125.5.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marcus RL, Shin HS, Mayer MM. An alternate complement pathway: C-3 cleaving activity, not due to C42a, on endotoxic lipopolysaccharide after treatment with guinea pig serum; relation to properdin. Proc Natl Acad Sci USA. 1971;68:1351–4. doi: 10.1073/pnas.68.6.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dave S, Brooks-Walter A, Pangburn MK, McDaniel LS. PspC, a pneumococcal surface protein, binds human factor H. Infect Immun. 2001;69:3435–7. doi: 10.1128/IAI.69.5.3435-3437.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lindahl G, Sjobring U, Johnsson E. Human complement regulators: a major target for pathogenic microorganisms. Curr Opin Immunol. 2000;12:44–51. doi: 10.1016/s0952-7915(99)00049-7. [DOI] [PubMed] [Google Scholar]
- 8.Horstmann RD, Sievertsen HJ, Knobloch J, Fischetti VA. Antiphagocytic activity of streptococcal M protein: selective binding of complement control protein factor H. Proc Natl Acad Sci USA. 1988;85:1657–61. doi: 10.1073/pnas.85.5.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prasadarao NV, Blom AM, Villoutreix BO, Linsangan LC. A novel interaction of outer membrane protein A with C4b binding protein mediates serum-resistance of Escherichia coli K1. J Immunol. 2002;169:6352–60. doi: 10.4049/jimmunol.169.11.6352. [DOI] [PubMed] [Google Scholar]
- 10.Wooster DG, Maruvada R, Blom AM, Prasadarao NV. Logarithmic phase Escherichia coli K1 efficiently avoids serum killing by promoting C4bp-mediated C3b and C4b degradation. Immunology. 2006;117:482–93. doi: 10.1111/j.1365-2567.2006.02323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sukumaran SK, Shimada H, Prasadarao NV. Entry and intracellular replication of Escherichia coli K1 in macrophages require expression of outer membrane protein A. Infect Immun. 2003;71:5951–61. doi: 10.1128/IAI.71.10.5951-5961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aoyagi Y, Adderson EE, Min JG, Matsushita M, Fujita T, Takahashi S, Okuwaki Y, Bohnsack JF. Role of l-ficolin/mannose-binding lectin-associated serine protease complexes in the opsonophagocytosis of type III group B streptococci. J Immunol. 2005;174:418–25. doi: 10.4049/jimmunol.174.1.418. [DOI] [PubMed] [Google Scholar]
- 13.Baker CJ, Edwards MS, Kasper DL. Role of antibody to native type III polysaccharide of group B Streptococcus in infant infection. Pediatrics. 1981;68:544–9. [PubMed] [Google Scholar]
- 14.Edwards MS, Nicholson-Weller A, Baker CJ, Kasper DL. The role of specific antibody in alternative complement pathway-mediated opsonophagocytosis of type III, group B Streptococcus. J Exp Med. 1980;151:1275–87. doi: 10.1084/jem.151.5.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takahashi S, Aoyagi Y, Adderson EE, Okuwaki Y, Bohnsack JF. Capsular sialic acid limits C5a production on type III group B streptococci. Infect Immun. 1999;67:1866–70. doi: 10.1128/iai.67.4.1866-1870.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marques MB, Kasper DL, Pangburn MK, Wessels MR. Prevention of C3 deposition by capsular polysaccharide is a virulence mechanism of type III group B streptococci. Infect Immun. 1992;60:3986–93. doi: 10.1128/iai.60.10.3986-3993.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prasadarao NV, Wass CA, Weiser JN, Stins MF, Huang SH, Kim KS. Outer membrane protein A of Escherichia coli contributes to invasion of brain microvascular endothelial cells. Infect Immun. 1996;64:146–53. doi: 10.1128/iai.64.1.146-153.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bremer E, Silhavy TJ, Maldener M, Cole ST. Isolation and characterization of mutants deleted for the sulA-ompA region of the E. coli K-12 chromosome. FEMS Microbiol Lett. 1986;33:173–8. [Google Scholar]
- 19.Weiser JN, Gotschlich EC. Outer membrane protein A (OmpA) contributes to serum resistance and pathogenicity of Escherichia coli K1. Infect Immun. 1991;59:2252–8. doi: 10.1128/iai.59.7.2252-2258.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rubens CE, Raff HV, Jackson JC, Chi EY, Bielitzki JT, Hillier SL. Pathophysiology and histopathology of group B Streptococcal sepsis in Macaca nemestrina primates induced after intraamniotic inoculation: evidence for bacterial cellular invasion. J Infect Dis. 1991;164:320–30. doi: 10.1093/infdis/164.2.320. [DOI] [PubMed] [Google Scholar]
- 21.Rubens CE, Heggen LM, Haft RF, Wessels MR. Identification of cpsD, a gene essential for type III capsule expression in group B streptococci. Mol Microbiol. 1993;8:843–55. doi: 10.1111/j.1365-2958.1993.tb01631.x. [DOI] [PubMed] [Google Scholar]
- 22.Abdullah M, Davies RJ, Hill JA. The application of DNA-cellulose chromatography in the isolation of immunoglobulin M and complement component C4b-binding protein from human serum. J Chromatogr. 1985;25:129–36. doi: 10.1016/s0021-9673(01)95476-7. [DOI] [PubMed] [Google Scholar]
- 23.Holme ER, Qi M, Ahmed AE, Veitch J, Auda G, Whaley K. Purification and characterization of RHP (factor H) and study of its interactions with the first component of complement. Mol Immunol. 1992;29:957–64. doi: 10.1016/0161-5890(92)90134-j. [DOI] [PubMed] [Google Scholar]
- 24.Lappegard KT, Fung M, Bergseth G, Riesenfeld J, Lambris JD, Videm V, Mollnes TE. Effect of complement inhibition and heparin coating on artificial surface-induced leukocyte and platelet activation. Ann Thorac Surg. 2004;77:932–41. doi: 10.1016/S0003-4975(03)01519-4. [DOI] [PubMed] [Google Scholar]
- 25.Mollnes TE, Brekke OL, Fung M, et al. Essential role of the C5a receptor in E coli-induced oxidative burst and phagocytosis revealed by a novel lepirudin-based human whole blood model of inflammation. Blood. 2002;100:1869–77. [PubMed] [Google Scholar]
- 26.Shyur SD, Raff HV, Bohnsack JF, Kelsey DK, Hill HR. Comparison of the opsonic and complement triggering activity of human monoclonal IgG1 and IgM antibody against group B streptococci. J Immunol. 1992;148:1879–84. [PubMed] [Google Scholar]
- 27.Levy NJ, Kasper DL. Antibody-independent and -dependent opsonization of group B streptococcus requires the first component of complement C1. Infect Immun. 1985;49:19–24. doi: 10.1128/iai.49.1.19-24.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Payne NR, Concepcion NF, Anthony BF. Opsonic effect of jacalin and human immunoglobulin A on type II group B streptococci. Infect Immun. 1990;58:3663–70. doi: 10.1128/iai.58.11.3663-3670.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rainard P. Assessment by a fluorochrome microassay of phagocytic killing of group B streptococci adherent to glass. J Immunol Methods. 1986;94:113–8. doi: 10.1016/0022-1759(86)90222-x. [DOI] [PubMed] [Google Scholar]
- 30.Lassiter HA, Watson SW, Seifring ML, Tanner JE. Complement factor 9 deficiency in serum of human neonates. J Infect Dis. 1992;166:53–7. doi: 10.1093/infdis/166.1.53. [DOI] [PubMed] [Google Scholar]
- 31.Lassiter HA, Walz BM, Wilson JL, et al. The administration of complement component C9 enhances the survival of neonatal rats with Escherichia coli sepsis. Pediatr Res. 1997;42:128–36. doi: 10.1203/00006450-199707000-00020. [DOI] [PubMed] [Google Scholar]
- 32.Winkelstein JA, Kurlandsky LE, Swift AJ. Defective activation of the third component of complement in the sera of newborn infants. Pediatr Res. 1979;13:1093–6. doi: 10.1203/00006450-197910000-00001. [DOI] [PubMed] [Google Scholar]
- 33.Pandiripally V, Wei L, Skerka C, Zipfel PF, Cue D. Recruitment of complement factor H-like protein 1 promotes intracellular invasion by group A streptococci. Infect Immun. 2003;71:7119–28. doi: 10.1128/IAI.71.12.7119-7128.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meri T, Blom AM, Hartmann A, Lenk D, Meri S, Zipfel PF. The hyphal and yeast forms of Candida albicans bind the complement regulator C4b-binding protein. Infect Immun. 2004;72:6633–41. doi: 10.1128/IAI.72.11.6633-6641.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lassiter HA, Wilson JL, Feldhoff RC, Hoffpauir JM, Klueber KM. Supplemental complement component C9 enhances the capacity of neonatal serum to kill multiple isolates of pathogenic Escherichia coli. Pediatr Res. 1994;35:389–96. [PubMed] [Google Scholar]
- 36.Nupponen I, Venge P, Pohjavuori M, Lassus P, Andersson S. Phagocyte activation in preterm infants following premature rupture of the membranes or chorioamnionitis. Acta Paediatr. 2000;89:1207–12. doi: 10.1080/080352500750027583. [DOI] [PubMed] [Google Scholar]
- 37.Rooijakkers SH, Ruyken M, Roos A, et al. Immune evasion by a staphylococcal complement inhibitor that acts on C3 convertases. Nat Immunol. 2005;6:920–7. doi: 10.1038/ni1235. [DOI] [PubMed] [Google Scholar]
- 38.Smith CL, Baker CJ, Anderson DC, Edwards MS. Role of complement receptors in opsonophagocytosis of group B streptococci by adult and neonatal neutrophils. J Infect Dis. 1990;162:489–95. doi: 10.1093/infdis/162.2.489. [DOI] [PubMed] [Google Scholar]
- 39.Prasadarao NV. Identification of Escherichia coli outer membrane protein A receptor on human brain microvascular endothelial cells. Infect Immun. 2002;70:4556–63. doi: 10.1128/IAI.70.8.4556-4563.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kotarsky H, Hellwage J, Johnsson E, Skerka C, Svensson HG, Lindahl G, Sjobring U, Zipfel PF. Identification of a domain in human factor H and factor H-like protein-1 required for the interaction with streptococcal M proteins. J Immunol. 1998;160:3349–54. [PubMed] [Google Scholar]
- 41.Blackmore TK, Fischetti VA, Sadlon TA, Ward HM, Gordon DL. M protein of the group A Streptococcus binds to the seventh short consensus repeat of human complement factor H. Infect Immun. 1998;66:1427–31. doi: 10.1128/iai.66.4.1427-1431.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]