

Abstract
Estimating individual genome-wide autozygosity is important both in the identification of recessive disease variants via homozygosity mapping and in the investigation of the effects of genome-wide homozygosity on traits of biomedical importance. Approaches have tended to involve either single-point estimates or rather complex multipoint methods of inferring individual autozygosity, all on the basis of limited marker data. Now, with the availability of high-density genome scans, a multipoint, observational method of estimating individual autozygosity is possible. Using data from a 300,000 SNP panel in 2618 individuals from two isolated and two more-cosmopolitan populations of European origin, we explore the potential of estimating individual autozygosity from data on runs of homozygosity (ROHs). Termed Froh, this is defined as the proportion of the autosomal genome in runs of homozygosity above a specified length. Mean Froh distinguishes clearly between subpopulations classified in terms of grandparental endogamy and population size. With the use of good pedigree data for one of the populations (Orkney), Froh was found to correlate strongly with the inbreeding coefficient estimated from pedigrees (r = 0.86). Using pedigrees to identify individuals with no shared maternal and paternal ancestors in five, and probably at least ten, generations, we show that ROHs measuring up to 4 Mb are common in demonstrably outbred individuals. Given the stochastic variation in ROH number, length, and location and the fact that ROHs are important whether ancient or recent in origin, approaches such as this will provide a more useful description of genomic autozygosity than has hitherto been possible.
Introduction
In plant and animal genetics, the detrimental effects of parental relatedness on fitness have long been recognized.1 The mechanism of these effects is thought to be increased levels of homozygosity for deleterious recessive alleles, although overdominance might also play a role.2
In human populations in which consanguinity is customary or population size and isolation result in elevated levels of background parental relatedness, evidence has been reported of several effects, including an increased risk of monogenic disorders,3–5an increased risk of complex diseases involving recessive variants with intermediate or large effect sizes,6–9 and genome-wide effects on disease traits such as blood pressure10–17and LDL cholesterol.15 These are consistent with a causal role for many recessive variants with individually small effects scattered throughout the genome.
Central to any investigation of the effects of parental relatedness on the health of offspring is the need for a reliable and accurate method of quantifying this phenomenon at an individual level. The first method proposed was the inbreeding coefficient, F, defined as the probability of inheriting two identical-by-descent (IBD) alleles at an autosomal locus or, equivalently, the average proportion of the autosomal genome that is inherited IBD.18 This is estimated with Wright's path method,19 which calculates an individual's probability of inheriting two IBD alleles, given a specified pedigree and given that an allele present in a parent is transmitted to a specified offspring with a probability of 0.5. Before the availability of marker data from high-density genome scans, researchers had no option but to use this approach, despite the fact that, even where pedigrees are known and accurate, it has two major disadvantages.20
First, meiosis is a highly random process. Whereas on average, half of the DNA making up a gamete is maternally derived and half is paternally derived, there is a high degree of stochastic variance about this average.21,22 As a consequence, grandchildren vary in the proportion of DNA they inherit from each of their four grandparents, and although the mean F coefficient of the offspring of first cousins is 0.0625, the standard deviation is 0.0243.20 This variance increases with each meiosis (i.e., each degree of cousinship), so it is perfectly possible for the offspring of third cousins to be more autozygous (homozygous by descent) than the offspring of second cousins. Because the F coefficient (denoted here as Fped to distinguish it from genomic estimates of autozygosity) is derived on the basis of this expectation, it is, therefore, only a very approximate estimate of individual genome-wide autozygosity.
Second, Fped estimates the proportion of an individual's genome that is IBD, relative to that of a poorly characterized founder generation. This generation is usually fairly recent, and, moreover, the founders are presumed to be unrelated, when in fact, members of historical populations were often related several times over through multiple lines of descent. As a result, this approach fails to capture the effects of distant parental relationships and, therefore, underestimates autozygosity, particularly in small, isolated populations or in populations with a long tradition of consanguineous marriage.23,24
With the increasing availability of high-density genome-scan data, interest has grown in exploring whether a more reliable and accurate estimate of autozygosity might be derived on the basis of genomic marker data. Much of the impetus for this comes from those searching for specific disease genes via homozygosity mapping, rather than from a general interest in the health effects of parental relatedness. Since the 1980s, many autosomal-recessive genes underlying monogenic human diseases have been identified with homozygosity mapping, which exploits the fact that regions flanking the disease gene will be identical by descent (IBD) in people with the disease whose parents are related to each other.25 Botstein and Risch identified nearly 200 studies, published between 1995 and 2003, that used homozygosity mapping in consanguineous families to identify rare recessive disease genes.26 Homozygosity mapping requires an estimate of the proportion of the genome that is autozygous for each affected individual, on the basis of which a LOD score for linkage to a specified locus is computed. Accurate estimation of autozygosity is crucial: underestimation results in an inflated LOD score and, thus, false evidence for linkage,27,28 and overestimation results in false negatives.
Quantification of individual autozygosity is also of interest to those investigating recessive effects in complex-disease genetics. Several studies in consanguineous or small, isolated populations with above average levels of parental relatedness have found evidence for a genome-wide effect of homozygosity on coronary heart disease,29–31 cancer,29,32–34 blood pressure,10–17 and LDL cholesterol.15 These findings are consistent with studies suggesting that the variants associated with increased risk of common complex disease are more likely to be rare than to be common in the population;35,36 are more likely to be distributed abundantly rather than sparsely across the genome,37 and are more likely to be recessive than to be dominant.38 Further empirical development of this idea has, however, been hampered by the inadequacy of available measures of autozygosity.
Here, we describe a multipoint, observational approach to estimating autozygosity from genomic data that exploits the fact that autozygous genotypes are not evenly distributed throughout the genome but are distributed in runs or tracts (Figure 1). This idea was first suggested by Broman and Weber, who proposed identifying autozygous segments from runs of consecutive homozygous markers.39 Can runs of homozygosity (ROHs), observable from high-density genome-scan data, be used for a reliable and accurate estimate of autozygosity at both the individual level and the population level? How do individuals with different ancestry, characterized in terms of population size, endogamy, and parental relatedness, differ in terms of ROHs? At a population level, do ROHs reflect differences in population isolation?
Figure 1.
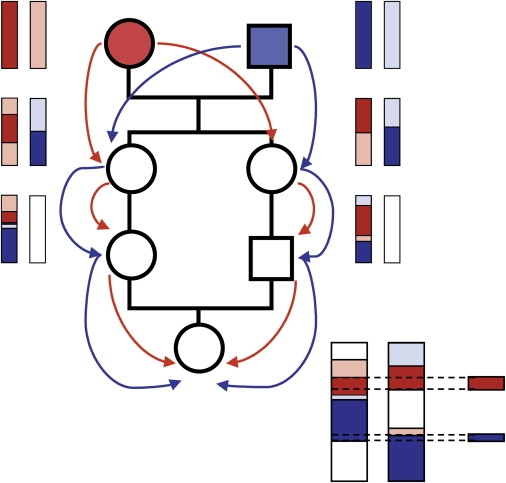
Pedigree of the Offspring of First Cousins
An example chromosome is illustrated. The female common ancestor is red. The chromosome inherited from one of her parents is colored red, and the chromosome inherited from her other parent is colored pink. The male common ancestor is blue. The chromosome inherited from one of his parents is colored dark blue, and the chromosome inherited from his other parent is colored light blue. The second generation are sisters. They share around 50% of their chromosomes IBD. The segments colored red and pink are segments inherited from their mother, and the segments colored dark and light blue are segments inherited from their father. The third generation are first cousins. In each case, the second (white) chromosome derives from their fathers (not shown), the red and pink segments are inherited from their maternal grandmother, and the dark and light blue segments are inherited from their maternal grandfather. The offspring of these first cousins has segments inherited from both founders on both copies of the chromosome. Where the same segments have been passed down both sides of the pedigree, the offspring of first cousins has extended identical-by-descent tracts or runs of homozygosity.
This paper has three objectives. First, it uses various measures derived from ROHs to compare four European populations: two isolated island populations and two more-cosmopolitan populations. The key study population is the Scottish isolate of Orkney, a remote archipelago off the north coast of Scotland. Three additional populations are used for comparison: a representative Scottish comparison population,40 an isolate population from a Dalmatian island in Croatia,15 and the HapMap CEU (northwest-European-derived population from Utah, USA) founders from the Centre d'Étude du Polymorphisme Humain (CEPH).41 Second, with the use of high-quality pedigree information available for the Orkney population, correlations are reported between Fped and a genome-wide autozygosity measure derived from ROHs (Froh). Finally, this study assesses the utility of Froh as a measure of autozygosity.
Subjects and Methods
Study Populations
The Orkney Complex Disease Study (ORCADES) is an ongoing, family-based, cross-sectional study that seeks to identify genetic factors influencing cardiovascular and other disease risk in the population isolate of the Orkney Isles in northern Scotland. The North Isles of Orkney, the focus of this study, consist of a subgroup of ten inhabited islands with census populations varying from ∼30 to ∼600 people on each island. Although transport links have steadily improved between the North Isles and the rest of Orkney, the geographical position of these islands, coupled with weather and sea conditions, means that even today they are isolated and that they would have been considerably more so in the past.
Although consanguinity is not the cultural norm in Orkney—indeed, there is evidence of consanguinity avoidance during the twentieth century42—two key factors make the North Isles population ideal for this type of study. First, the North Isles have experienced a period of severe population decline over the last 150 years, fueled by high emigration and low fertility. The population fell from an estimated peak of 7700 in the 1860s to 2217 by 2001. Second, endogamous marriage was widespread during the nineteenth century and into the twentieth centuries.43 Therefore, despite consanguinity avoidance, the combined effects of steep population decline and endogamy have led to inflated levels of parental relatedness in the current population.
ORCADES received ethical approval from the appropriate research ethics committees in 2004. Data collection was carried out in Orkney between 2005 and 2007. Informed consent and blood samples were provided by 1019 Orcadian volunteers who had at least one grandparent from the North Isles of Orkney.
A Scottish comparison population was derived from the controls of the Scottish Colon Cancer Study (SOCCS).40 This consists of 984 subjects, not known to have colon cancer, matched by residential postal area and age to a series of incident cases of colorectal cancer. Subjects were resident throughout Scotland, with dates of birth ranging from 1921 to 1983.
The Dalmatian sample consists of 849 Croatian individuals, aged 18–93, sampled from the population of one island.15 Both the SOCCS and the Croatian projects were approved by the relevant ethics committees.
The CEU sample consists of 60 unrelated individuals from Utah, USA, of northwest-European ancestry, collected by the CEPH in 1980.41
Genotyping
Genotyping procedures for the Scottish,40 Dalmatian,44 and CEU45 samples are described elsewhere. All were genotyped on the Illumina Infinium HumanHap300v2 platform (Illumina, San Diego, CA, USA). After extraction of genomic DNA from whole blood with the use of Nucleon kits (Tepnel, Manchester, UK), 758 Orcadian samples were genotyped, according to the manufacturer's instructions, on the Illumina Infinium HumanHap300v2 platform. Analysis of the raw data was done via BeadStudio software, with the recommended parameters for the Infinium assay, with the use of the genotype-cluster files provided by Illumina.
Individuals with less than 95% call rate were removed, as were SNPs with more than 10% missing genotypes. SNPs failing Hardy-Weinberg equilibrium at a threshold of 0.0001 were removed. IBD sharing between all first- and second-degree relative pairs was assessed with the Genome program in PLINK,46 and individuals falling outside expected ranges were removed from the study. Sex checking was performed with PLINK, and individuals with discordant pedigree and genomic data were removed. On completion of data-cleaning and quality-control procedures, 725 individuals and 316,364 autosomal SNPs remained. The male-to-female ratio of study participants is 0.86. The mean year of birth is 1952, varying from 1909 to 1988.
A consensus SNP panel was then created, with use of only those markers that satisfied these quality control criteria in all four populations, leaving a final sample of 289,738 autosomal SNPs and 2618 individuals (60 from CEU, 725 from Orkney, 849 from the Dalmatian island, and 984 from Scotland).
Fped Estimates
The pedigrees of all individuals in the ORCADES sample were traced back for as many generations as possible in all ancestral lineages, with the use of official birth, marriage, death, and census records held by the General Register Office for Scotland in Edinburgh. Fped was calculated for each individual via Wright's path method.19
Limited pedigree information is available for the Dalmatian-isolate data set, and this is too incomplete for an estimate of Fped. It was, however, possible to analyze these data with the use of grandparental-endogamy levels.
No pedigree information is available for the Scotland data set; however, we analyzed data according to the rurality of subjects' residential address47 in order to determine whether there is any evidence for an association between remote rurality and autozygosity in Scotland.
Runs of Homozygosity
ROHs were identified via the Runs of Homozygosity program implemented in PLINK version 1.0.46 This slides a moving window of 5000 kb (minimum 50 SNPs) across the genome to detect long contiguous runs of homozygous genotypes. An occasional genotyping error or missing genotype occurring in an otherwise-unbroken homozygous segment could result in the underestimation of ROHs. To address this, the program allows one heterozygous and five missing calls per window.
A threshold was set for the minimum length (kb) needed for a tract to qualify as homozygous. Because strong linkage disequilibrium (LD), typically extending up to about 100 kb, is common throughout the genome,48–51 short tracts of homozygosity are very prevalent. For exclusion of these short and very common ROHs that occur in all individuals in all populations, the minimum length for an ROH was set at 500 kb. All empirical studies have identified a few very long stretches of LD, measuring up to several hundred kb in length,49 which could result in the occurrence of longer ROHs in outbred individuals. Such ROHs will not be excluded by this methodology; however, the purpose here is not to identify only those ROHs that result from parental relatedness but to identify all ROHs and then relate these to pedigree and population data for an assessment of the extent to which these result from parental relatedness and population isolation.
We set a threshold for the minimum number of SNPs constituting a ROH in order to ensure that these are true ROHs—i.e., that between the first SNP and the last SNP the entire unobserved stretch of the chromosome is homozygous. With, for example, only three consecutive homozygous genotypes, there would be a very high probability that these three could be homozygous by chance alone and that the intervening, unobserved chromosomal stretches could be heterozygous. We have deliberately not taken LD into account here. By using a minimum-length cutoff of 500 kb, most shorter ROHs resulting from LD will be eliminated; however, some longer stretches will remain. This is intentional: we are interested in identifying and quantifying these common ROHs, whatever their origin. We used allele frequencies for a random sample of chromosomal segments across the entire autosomes to estimate the mean probability of finding 10, 25, and 50 consecutive homozygous SNPs by chance alone in each population. On this basis, the minimum number of contiguous homozygous SNPs constituting a ROH was set at 25 (p < 0.0001 in each of the four populations). Two additional parameters were added for ensuring that estimates of F were not artificially inflated by apparently homozygous tracts in sparsely covered genomic regions: tracts with a mean tract density > 50 kb/SNP were excluded, and the maximum gap between two consecutive homozygous SNPs was set at 100 kb.
For exclusion of the possibility that apparent ROHs are in fact regions of hemizygous deletion, an analysis of deletions was carried out in the Orkney data set. An Objective Bayes' Hidden Markov model, as employed in QuantiSNP v. 1.0, was used for identification of heterozygous deletions with a sliding window of 2 Mb over the genome and 25 iterations. All of the samples were corrected for genomic GC content prior to copy-number inference as a means of ensuring that the variation of the observed log2 R ratio is not attributed to the region-specific GC content.52 We included in the downstream analysis all heterozygous deletions with an estimated Bayes' factor ≥ 10 to ensure a low false-negative rate, as reported in Colella et al., 2007.53 A custom Perl script was developed for comparison of the identified heterozygous deletions and ROHs.
All deletions overlapping with ROHs were identified. When deletions covered the entire length of the ROH or when less than 0.5 Mb of the tract remained after the deletion was taken account of, the ROH was removed from the analysis. Because the Dalmatian, CEU, and Scotland data sets were uncorrected for deletions, uncorrected Orkney data are shown when there are population comparisons. Analyses using only the Orkney data set use data corrected for deletions.
Froh Estimates
A genomic measure of individual autozygosity (Froh) was derived, defined as the proportion of the autosomal genome in runs of homozygosity above a specified length threshold:
in which ∑Lroh is the total length of all of an individual's ROHs above a specified minimum length and Lauto is the length of the autosomal genome covered by SNPs, excluding the centromeres. The centromeres are excluded because they are long genomic stretches devoid of SNPs and their inclusion might inflate estimates of autozygosity if both flanking SNPs are homozygous. The length of the autosomal genome covered by our consensus panel of SNPs is 2,673,768 kb. We show individual and population mean values of Froh for a range of different ROH-length thresholds.
Statistical Analysis
For statistical analyses, the Orkney population was split into endogamous Orcadians, defined as those with at least three grandparents born in Orkney, on the same island, typically ∼10 km2 in size and with a population of 50–500 (n = 390); mixed Orcadians, defined as those with at least three grandparents born in Orkney but on different islands in the archipelago—i.e., from an area over 500 km2 with a population of ∼20,000 (n = 286); and half Orcadians, defined as those with one pair of Orcadian-born and one pair of Scottish-mainland-born grandparents (n = 49). Although pedigree information is not available for an assessment of whether the parents of half-Orcadian subjects are related beyond five generations in the past, it is reasonable to assume that they are likely to be unrelated for at least 10–12 generations. It is known that there was major Scottish immigration to Orkney in the 15th and 16th centuries, before10–12 generations ago. Although Scottish immigration has certainly occurred sporadically since then, rates have been low. An analysis of the area of origin of the Scottish parents of our half-Orcadian subjects shows that they came from all over Scotland: we found no evidence for strong Orcadian connections with any specific Scottish settlement, which might increase the chances of parental relatedness in this group. Furthermore, the surnames of the ancestors of the Orcadian parents of this group were markedly different from those of the ancestors of the non-Orcadian Scottish parents.
The Dalmatian population was split into endogamous Dalmatians, defined as those with all four grandparents born in the same village—i.e., from a 1 km2 area, with a population of < 2000 (n = 431); mixed Dalmatian, defined as those with all four grandparents born on the same island but not in the same village—i.e., from a 90 km2 area with a population of 3600 (n = 221); and Croatian, defined as residents of the island with grandparents born elsewhere in Croatia (n = 197). The CEU and Scottish populations were not subdivided.
All calculations were performed with SPSS and Excel software. The proportions of each subpopulation with ROHs measuring less than 1, 1.5, and 2 Mb were calculated. All subjects in all subpopulations had ROHs shorter than 1.5 Mb. Subpopulations start to become differentiated from each other for ROHs > 1.5 Mb, with the effects of endogamy on ROHs starting to emerge above this threshold. Unless otherwise specified, all analyses exploring the effects of endogamy and parental relatedness on ROHs therefore define a ROH as measuring ≥ 1.5 Mb.
Subpopulation means were calculated for the total length of ROHs per individual. The number of ROHs was plotted against the total length of ROHs, per individual, for each subpopulation.
The correlation between Fped and Froh was calculated with the use of a subset of 249 individuals, from the Orkney sample, who satisfied the condition of having at least two grandparents on the same side of the family born in Orkney and no grandparents born outside of Scotland and who were either the offspring of consanguineous parents (parents related as 2nd cousins or closer) or those for whom it was possible to establish pedigrees for at least six generations in all Orcadian ancestral lineages or five generations in non-Orcadian ancestral lineages.
Correlations were also calculated between Froh, Fped, and two other measures: multilocus heterozgyosity (MLH), which is defined as the proportion of markers that are heterozygous,54 and the measure of autozygosity implemented in PLINK, termed here Fplink, which estimates autozygosity from genotype frequencies, giving more weight to rare alleles.46
Prevalence and Genomic Location of ROHs in Different Subpopulations
Next, we explored the hypothesis that ROHs in outbred individuals tend to cluster in the same genomic locations, whereas those present in the offspring of related parents tend to be more randomly distributed across the autosomes. We compared the location of ROHs in three groups: the half-Orcadian group, consisting of all half Orcadians with at least one ROH measuring ≥ 1.5 Mb (n = 46); an offspring-of-cousins group, which was constructed by consideration of all individuals from the Orkney sample with parents related as 3rd cousins or closer and the selection of those 20 with the greatest total length of ROHs; and a control population derived from our cross-sectional sample from Scotland. Because some individuals in the Scottish sample have long ROHs that could be indicative of parental relatedness, we restricted the control sample to those with no more than eight ROHs, totaling no more than 17 Mb: the maximum values in the half-Orcadian group, the members of which are known to be the offspring of unrelated parents. There were 943 individuals in the control group. ROHs measuring at least 1.5 Mb in all three groups were compared. Control-group ROHs overlapping by at least 0.5 Mb with ROHs in either Orcadian group were counted. The number of control overlaps per ROH (and per Mb of ROH) in the half-Orcadian group was compared with that in the offspring-of-cousins group.
We then investigated whether ROHs in half Orcadians occurred in regions of lower-than-average recombination. Based on sex-averaged mean recombination rates per Mb, derived from the deCODE genetic map, we used the UCSC Genome Browser (March 2006)55 to calculate the mean recombination rate of all complete Mb of ROH in our half-Orcadian sample.
Results
Copy-Number Variation
We detected 224 deletions that overlapped with ROHs (median length of deletion 995 kb). Overlapping deletions were detected in 57 individuals (7.6% of sample). After removal of these overlaps from the sample and removal of the entire affected ROH if less than 0.5 Mb remained, ROH statistics were recalculated. There was no significant difference between results before and after correction for deletion for the mean total length of ROHs (correcting for deletions reduced this by less than 0.3% in the sample as a whole) or the mean number of ROHs (reduced by 0.02%). Furthermore, no significant differences were found when data were analyzed by subpopulation and when different length parameters were used for defining ROHs. This provides strong evidence that the ROHs identified are true homozygous tracts and not hemizgyous deletions.
Urban versus Rural Analysis of Scottish Sample
No difference was found in the mean total length of ROHs between those living in rural areas and those in urban areas of Scotland, regardless of whether the analysis used a dichotomous classification or a more-detailed, eight-category classification, from large urban to remote rural (data not shown). Data were also analyzed for a subset (n = 426) of the sample with information on grandparental country of birth. On average, those with four Scottish-born grandparents (n = 254) had a slightly greater sum of ROHs than did those with at least one grandparent born outside of Scotland, but differences were not significant (data not shown). The Scottish sample was, therefore, not split into subpopulations for further analyses.
Effect of Stochastic Variation on Individual Autozygosity
On average, the difference in the total length of ROHs between full sibling pairs was 10.3 Mb. However, the distribution is skewed, with half of all individuals having less than 5 Mb difference yet some 7% differing by more than 30 Mb. The greatest difference between sibling pairs was 91 Mb, or 3.4% of the autosomes (paternity was confirmed from patterns of genomic sharing in all cases).
Effects of Population Isolation and Endogamy on Length and Number of ROHs
The proportions of subpopulations with ROHs of a given length are shown in Figure 2. All individuals in all populations have ROHs measuring less than 1.5 Mb. If we consider the populations as a whole, on average, a significantly greater proportion of the autosomes of Orcadians are in ROHs measuring 0.5–1.5 Mb (77.7 Mb) than is the case for either the Dalmatian (73.2 Mb), the Scottish (75.8 Mb), or the CEU (74.1 Mb) populations. There are no significant differences between groups within populations, however, which suggests that this reflects population differences in genetic diversity or LD of ancient origin rather than effects of more recent endogamy or population isolation.
Figure 2.
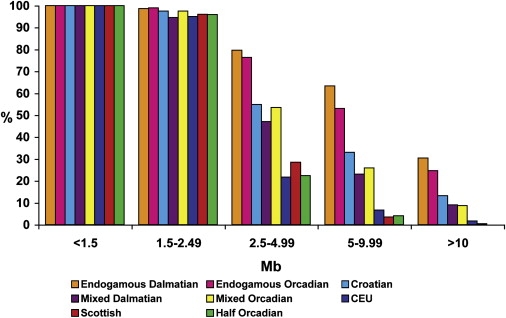
Proportion of Subpopulations with One or More ROHs of a Given Length
The proportion of individuals with one or more ROHs of up to 0.5–1.49, 1.5–2.49, 2.5–4.99, and 5–9.99 Mb in length, or over 10 Mb in length, is plotted for each of the eight population groups defined in the Statistical Analysis section of Subjects and Methods.
For ROHs above 1.5 Mb, three distinct groupings, which are clearly related to endogamy and isolation, emerge: a greater proportion of the endogamous Dalmatian and Orcadian samples than of the other samples have long ROHs (28% have ROHs > 10 Mb); only a small proportion of the CEU, Scottish, and half-Orcadian samples have long ROHs (0.5% > 10 Mb), and the proportion of Croatian and mixed Dalmatian and Orcadian samples with long ROHs falls in between (10% > 10 Mb).
Forty-nine individuals had no ROHs longer than 1.5 Mb. This number included at least one individual from each subpopulation, although they were predominantly half-Orcadian, Scottish, and CEU samples. The shortest sum of ROHs across all of the samples was found in a Scottish individual, who had ROHs longer than 0.5 Mb covering only 1.5% of the autosomes (39 Mb). This compares with a mean of 3.5% across all of the populations (93 Mb).
The number of ROHs longer than 1.5 Mb per individual, plotted against the total length of those ROHs, is shown for each group in Figure 3. The half-Orcadian group is used as a reference, because we know that these individuals are the offspring of unrelated parents. Reference lines are shown on all graphs for the maximum number of ROHs, the maximum total length of ROHs, and the line of best fit for the half-Orcadian group. Compared with the half-Orcadian group, all other groups have a greater variance in the number and sum of ROHs and contain individuals with more and longer ROHs. Again, the same three groupings are apparent. Data points for the half-Orcadian, Scottish, and CEU samples are generally narrowly distributed along both axes, indicating that these individuals have few, relatively short ROHs. The two endogamous samples are much more widely spread along both axes, reflecting the presence of many, much longer ROHs. The Croatian, mixed Orcadian, and mixed Dalmatian groups are intermediate, reflecting the fact that these less carefully specified groups are probably made up of individuals with a mixture of ancestries, from the outbred to the very endogamous. The percentage of each group with more and longer ROHs than the maximum for the half Orcadians was calculated. Again, the Scottish (5%) and CEU (8%) groups differed least and the endogamous Dalmatians (64%) and Orcadians (54%) differed most from the half Orcadians. The Croatians (33%), mixed Dalmatians (26%), and mixed Orcadians (23%) were intermediate.
Figure 3.
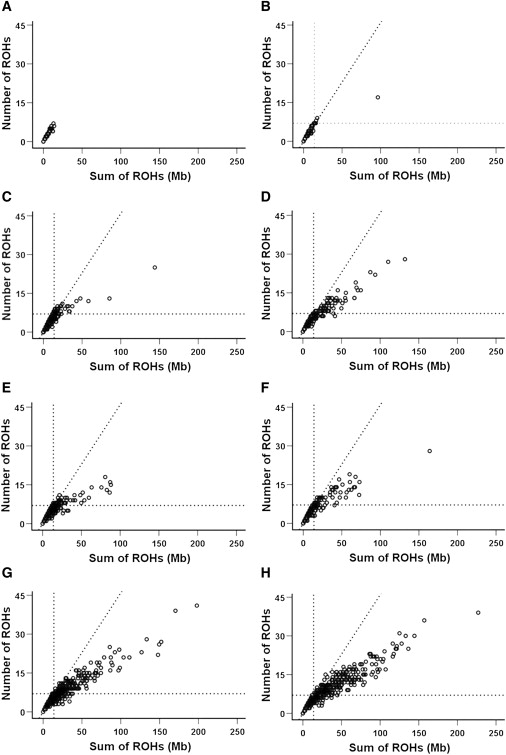
Number of ROHs Compared to Total Length of ROHs
(A) Half Orcadian, (B) CEU, (C) Scottish, (D) Croatian, (E) Mixed Orcadian, (F) Mixed Dalmatian, (G) Endogamous Orcadian, and (H) Endogamous Dalmatian.
The effect of different degrees of parental relatedness on the sum and number of ROHs is shown in Figure 4 for the 249 individuals in the Orkney sample with good pedigree information. Although a trend for increasing number and total length of ROHs is evident from the half-Orcadian through the mixed to the endogamous and offspring-or-cousins subgroups, there is considerable overlap between groups.
Figure 4.
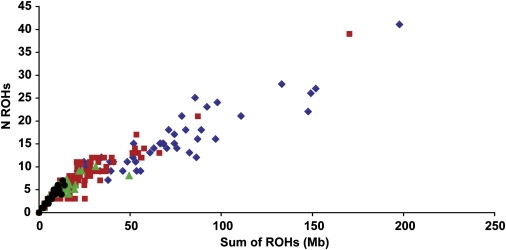
Effect of Endogamy on Sum and Number of ROHs
Offspring of 1st or 2nd cousins are shown in blue, endogamous Orcadians who are not the offspring of 1st or 2nd cousins are shown in red, mixed Orcadians are shown in green, and half Orcadians are shown in black.
Comparison of Fped and Froh
A subset of 249 Orcadian individuals with complete and reliable pedigree data were used to compare Fped and Froh. The mean (standard error) Fped of the sample is 0.0038 (0.0005), approximately equivalent to a parental relationship of third cousins. Mean Fped values for Orcadian subpopulations are shown in Table 1. These vary from 0.02, for the offspring of 1st or 2nd cousins, to 0.0002 (equivalent to a parental relationship of 5th cousins) in the mixed Orcadian group. Mean Fped values are compared with mean Froh values for a range of minimum-length thresholds. The mean value of Froh 5 (i.e., with a minimum-length threshold of 5 Mb) is closest to that of Fped, whereas Froh 0.5 (i.e., with a minimum-length threshold of 0.5 Mb) is an order of magnitude higher. This suggests that a shared maternal and paternal ancestor in the preceding six generations results predominantly in ROHs longer than 5 Mb. It is clear from the half-Orcadian group, whose parents do not share a common ancestor for at least six generations and probably at least 10–12 generations, that ROHs measuring less than 3 or 4 Mb are not uncommon in the absence of parental relatedness. On average, these individuals have over 3% (84 Mb) of their autosomes in ROHs over 0.5 Mb long and 0.2% (almost 6 Mb) in ROHs longer than 1.5 Mb.
Table 1.
Mean Values of Fped and Froh for Orkney Subpopulations
| Orkney Subpopulation | N | Mean (SE) Fped | Equivalent Parental Cousin Relationship (Single Loop) | Mean (SE) Froh 0.5 | Mean (SE) Froh 1.5 | Mean (SE) Froh 5 |
|---|---|---|---|---|---|---|
| Offspring of 1st or 2nd cousins | 42 | 0.0182 (0.0014) | 2nd cousin | 0.0569 (0.0024) | 0.0271 (0.0022) | 0.0169 (0.0017) |
| Endogamous Orcadian | 114 | 0.0015 (0.0004) | 3rd – 4th cousin | 0.0379 (0.0008) | 0.0087 (0.0007) | 0.003 (0.0004) |
| Mixed Orcadian | 44 | 0.0002 (0.0001) | 5th cousin | 0.033 (0.0006) | 0.0046 (0.0005) | 0.0012 (0.0004) |
| Half Orcadian | 49 | 0 | None | 0.0315 (0.0004) | 0.0021 (0.0002) | 0.0001 (0.00007) |
| Total | 249 | 0.0038 (0.0005) | 3rd cousin | 0.039 (0.0008) | 0.0098 (0.0007) | 0.0045 (0.0005) |
Correlation between Froh, Fped, Fplink, and MLH
We used the total sample to examine correlations between different genetic estimates of autozygosity or homozygosity. Because MLH is in fact a measure of heterozygosity, we have used 1 − MLH in our calculations. Allele frequencies for Fplink were estimated by naive counting in all individuals, as implemented in PLINK. Fplink and 1 − MLH are highly correlated (r = 0.94). Froh 1.5 is more highly correlated with 1 − MLH (r = 0.80) than with Fplink (r = 0.74).
We used a subset of the Orcadian sample (n = 249) to estimate correlations with Fped. Froh 1.5 was most highly correlated with Fped (r = 0.86; 95% confidence interval 0.83–0.89). Correlations between Fped and Froh 1.5 were significantly higher than both the correlation between Fplink and Fped (r = 0.77; 0.72–0.82) and that between 1 − MLH and Fped (r = 0.76; 0.71–0.82). Froh 1.5 was slightly, but not significantly, more strongly correlated with Fped than was either Froh 0.5 or Froh 5.
Correlations between Fped and Froh 0.5, Froh1.5, and Froh 5 are shown in Figure 5. For each value of Fped there is a range of values for Froh, reflecting stochastic variation in ancestral recombination, the existence of multiple distant parental relationships undetectable with the use of pedigrees, and possible pedigree misspecifications. The closer the parental relationship, the greater the variance in the autozygosity of offspring. This is clear from the wide distribution of Froh values in the endogamous group compared to the mixed Orcadian group. Although as we have shown, ROHs shorter than around 1.5 Mb do not appear to reflect differences in recent ancestral endogamy, data from the half-Orcadian sample illustrate that the prevalence of these shorter ROHs clearly varies between individuals. Use of a minimum-ROH-length threshold of 5 Mb might better reflect the effects of parental relatedness on autozygosity; however, it also obscures a great deal of individual genetic variation of more ancient origin. This is illustrated by the regression lines on each panel: the y intercept gives the value of Froh when Fped = 0. This is a measure of the proportion of the autosomes in ROHs not captured by Fped. Thus, 0.034 of the autosomes are in ROHs longer than 0.5 Mb but are not captured by Fped. The equivalent figures are 0.0053 for ROHs longer than 1.5 Mb and 0.0014 for ROHs longer than 5 Mb. This clearly shows that Fped fails to account for autozygosity of ancient origin.
Figure 5.
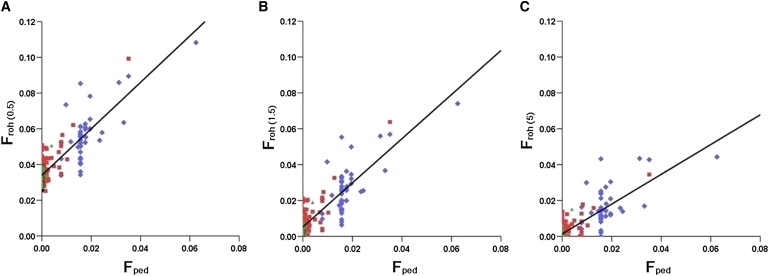
Correlation between Fped and Froh in Orkney Sample
Correlations, with regression lines, are shown for three different minimum-ROH-length thresholds. (A) shows the correlation between Fped and Froh 0.5, (B) shows the correlation between Fped and Froh 1.5, and (C) shows the correlation between Fped and Froh 5. For colors and details of subgroups, see Figure 4 legend. N = 249.
Mean Froh by Subpopulation
Mean Froh and the mean total length of ROHs for each subpopulation are shown for a range of minimum ROH lengths in Figure 6. This figure again shows the effect on Froh, in all populations, of changing the ROH-length cutoff point. The same three distinct groupings emerge for ROHs longer than 1.5 Mb, although when shorter ROHs are included, the picture is less clear. With 1.5 Mb used as the minimum length, endogamous Dalmatians have a mean Froh of 0.013 (35 Mb), endogamous Orcadians 0.011 (28 Mb), Croatians 0.007 (18 Mb), mixed Dalmatians 0.006 (15 Mb), mixed Orcadians 0.005 (14 Mb), CEU 0.003 (8 Mb), Scottish 0.003 (7 Mb), and half Orcadians 0.002 (6 Mb). With a 5 Mb threshold, the same relationship between groups is seen, but values for all groups are reduced (to 17 Mb in endogamous Dalmatians and 0.3 Mb in half Orcadians).
Figure 6.
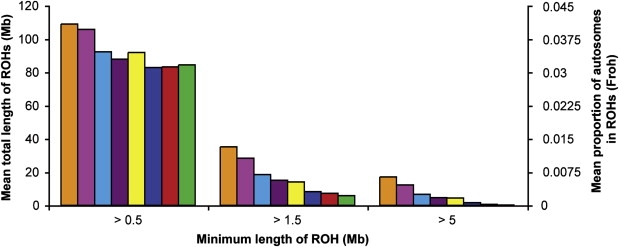
Mean Total Length of ROHs over a Range of Minimum Tract Lengths
The average total length of ROHs per individual, calculated from ROHs above 0.5, 1.5 and 5 Mb, is plotted for each of the eight population groups defined in the Statistical Analysis section of Subjects and Methods. For colors, see Figure 2 legend.
Comparison of ROHs in the Offspring of Unrelated Parents and the Offspring of Cousins
We next investigated whether ROHs found in half Orcadians are more common than those found in the offspring of related parents. We defined “common” as overlapping by at least 0.5 Mb with ROHs found in a subset of the Scottish sample. The number of ROHs measuring ≥ 1.5 Mb was 143 in the half-Orcadian sample, 3159 in the Scottish control sample, and 382 in the offspring-of-cousins sample. Results are summarized in Table 2. On average, each half-Orcadian ROH overlapped with more than twice as many controls as did ROHs in the offspring-of-cousins group. Only 12.6% of half-Orcadian ROHs, but almost a third of ROHs in the offspring-of-cousins group, did not overlap with any controls. We also looked at the mean number of overlaps per Mb of ROH in the two samples in order to correct for the fact that ROHs in the offspring-of-cousins group tend to be longer. There were more than three times as many control overlaps per Mb of ROH in the half-Orcadian group than there were in the offspring-of-cousins group. If we consider only those ROHs measuring > 5 Mb in the offspring-of-cousins sample (i.e., those that are most likely to result from recent shared parental ancestry), the mean number of overlaps per Mb was only1.4 (SD 2.0).
Table 2.
Overlaps between ROHs Found in Orcadians and Those Found in a Scottish Control Sample
| Half Orcadian | Offspring of Cousins | |
|---|---|---|
| Number of individuals | 46 | 20 |
| Number of ROHs ≥ 1.5 Mb | 143 | 382 |
| Mean (SE) number of control overlaps per ROH | 20.5 (22.5) | 9.6 (16.0) |
| Maximum number of controls overlapping with a ROH | 123 | 123 |
| Percentage of ROHs overlapping with no controls | 12.6 | 29 |
| Mean (SE) number of control overlaps per Mb of ROH | 10.9 (11.8) | 3 (6.3) |
Data on chromosome 1 for ten individuals in the half-Orcadian group (shown in blue) and seven individuals in the offspring-of-cousins group (shown in red) are illustrated by way of example in Figure 7. These are all of the individuals in the sample with ROHs on chromosome 1, except that data for only one individual per sibship is shown. This removed six individuals from the offspring-of-cousins group but none from the half-Orcadian group. The numbers shown below each colored segment are the numbers of ROHs in the control sample overlapping with the illustrated ROH. It is clear that although there is a tendency for ROHs from both groups to cluster in certain chromosomal regions, the longer ROHs in the offspring-of-cousins group are more randomly distributed along the chromosome.
Figure 7.
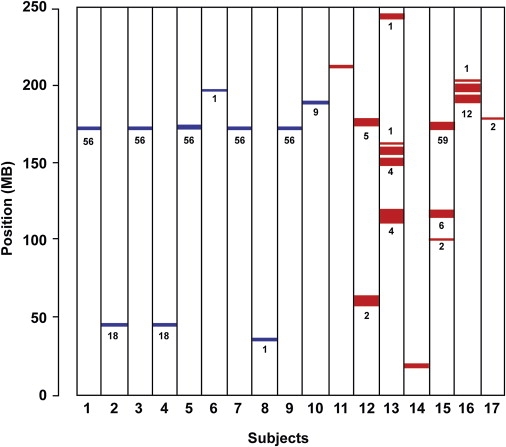
Size and Location of ROHs on Chromosome 1, Comparing Half Orcadians and Offspring of Cousins
ROHs measuring ≥ 1.5 Mb in ten half Orcadians are shown in blue, and those of seven offspring of 1st–3rd cousins are shown in red. The numbers shown below each colored segment are the numbers of overlapping ROHs in the Scottish control sample.
Next, we identified all ROHs in the half-Orcadian group that overlapped by at least 0.5 Mb with common ROHs identified by Lencz.56 In a sample of 322 non-Hispanic European Americans, Lencz identified 339 ROHs present in at least ten subjects. Of the 143 half-Orcadian ROHs, 57% overlapped with Lencz et al.'s list. Only 7% (ten ROHs) overlapped with neither Lencz et al.'s list nor our control group.
Finally, we investigated whether the ROHs in half Orcadians were found in areas of lower-than-average recombination. The mean recombination rate for the regions where half-Orcadian ROHs are located is 0.52 of the mean genome-wide recombination rate. For common ROHs (i.e., half-Orcadian ROHs that overlap with ROHs in the control group), this figure was 0.38 of the genome-wide mean.
Discussion
Our findings are consistent with a number of recent observational studies using high-density genome-scan data, which have suggested that ROHs longer than 1 Mb are more common in outbred individuals than previously thought.39,56–60
We have quantified this phenomenon by describing the number and length of ROHs in individuals who are known to have no common maternal and paternal ancestor in at least five generations (and probably 10–12 generations). Our analysis of copy-number variation in the Orkney sample is consistent with studies that have shown that observed ROHs are true homozygous tracts and not deletions or other chromosomal abnormalities.39,45,57,60 Heterozygous deletions are not easily differentiated from ROHs, because the employed algorithm uses the B allele frequency as one of its input parameters to infer CNV status. Therefore, homozygosity at consecutive SNPs increases the posterior probability of being called a heterozygous deletion. In other words, this is a very robust estimation of the prevalence of ROHs in the Orkney sample, which to some extent overcorrects for heterozygous deletions. Other studies have suggested that ROHs cluster in regions of the genome where recombination rates are low,57–60 and our data support this. The picture of genome-wide homozygosity now emerging is that short stretches, measuring tens of kb and indicative of ancient LD patterns, are common, covering up to one third of the genome.45 At the other end of the spectrum, very long ROHs, measuring tens of Mb, are the signature of parental relatedness. In between, ROHs might result from recent parental relatedness or might be autozygous segments of much older pedigree that have occurred because of the chance inheritance through both parents of extended haplotypes that are at a high frequency in the general population, possibly because they convey or conveyed some selective advantage.56 The Phase II HapMap study estimates that ROHs measuring in excess of around 100 kb constitute 13%–14% of the genome in Europeans.45 Lencz et al.56 give a similar estimate. The findings of our study are not directly comparable, given that we have not examined ROHs shorter than 500 kb; however, we have shown (Figure 2) that ROHs measuring between 500 and 1500 kb were present in all individuals in all the subpopulations that we studied, totaling on average 75 Mb per individual (2%–3% of the autosomes). The fact that we found small but significant differences among our four populations in the mean sum of these short ROHs but no significant differences within populations (e.g., between endogamous Orcadians and half Orcadians) lends support to the view that population differences in the prevalence of ROHs shorter than around 1.5 Mb reflect LD patterns of ancient origin rather than the effects of more recent endogamy.
We have demonstrated clearly that data on ROHs measuring more than 1.5 Mb accurately reflect differences in population isolation, as measured by grandparental endogamy (Figures 2, 3, and 6). Furthermore, characterizing populations in terms of ROHs allows us to situate those with unknown degrees of isolation along a spectrum. For example, beyond knowing that the Scottish sample is broadly representative of the general Scottish population, we have no information on the precise birthplace of participants' grandparents. Data on ROHs would suggest that endogamy and consanguinity are uncommon, although not unheard of, in the recent ancestry of modern Scots. The 36 (4%) outliers in Scottish sample with ROHs suggestive of parental relatedness (total ROHs ≥ 5 Mb) were no more likely to live in rural or island locations than in urban locations. This is unsurprising: Scotland is a small, largely urbanized country with high population mobility. There are, however, small, remote island communities off the west and north coasts of Scotland that have been shown to have greater LD and lower haplotype diversity than mainland urban and rural Scottish populations,61 consistent with lower effective population sizes, isolation, and genetic drift. Orkney is one such isolated community; however, as we show, even within such small populations, there is a great diversity of ancestry, from the tightly endogamous to the completely outbred. Our data show that having at least three grandparents from within a 2–3 mile radius (as is the case in the North Isles of Orkney and the Dalmatian villages) is associated with considerably more and longer ROHs than is merely coming from Orkney or a Dalmatian island. The distribution of ROHs in the CEU sample, which is widely used as a northwest-European reference population, does indeed appear to be very similar in this respect to that in the Scottish sample. Consistent with other studies,45 we identify one outlier (NA12874), who is likely to be the offspring of consanguineous parents. The Dalmatian subsample of the offspring of Croatian settlers is more autozygous by various ROH-based measures than the mixed-Dalmatian and mixed-Orcadian subgroups, suggesting that these settlers came from fairly small, semi-isolated communities where endogamy was not uncommon.
We found that Froh is strongly correlated with Fped, significantly more so than the other two measures investigated. Perfect correlation is not expected, largely because of the deficiencies of Fped. This is particularly the case in isolated populations, where multiple distant parental relationships, undetectable with only a few generations of pedigree information, inflate autozygosity, such that the offspring of distant cousins can be almost as autozygous as the offspring of first cousins.24 The individual with the second highest Froh in the Orkney sample, for example, is the offspring of a couple whose closest relationship is that of 3rd cousins but who are multiply related at least 24 different ways in the last eight generations alone. We illustrate the deficiencies of Fped in Figure 5, in which the y intercept of the regression line is an indication of the autozygosity captured by Froh but not by Fped. Although it is unlikely that any approach could accurately identify the precise nature of distant parental cousin relationships for individuals with such complex pedigrees as those found in our Orkney sample, Froh can accurately rule out the possibility that an individual is the offspring of first cousins: during preliminary data analysis, before all pedigree relationships had been verified by checking of inferred IBD sharing among first-degree relatives, a sibling pair, putatively the offspring of first cousins, was identified as having Froh values significantly lower than predicted from pedigree. Upon checking of inferred IBD sharing among pairs of their genotyped relatives, an ancestral false paternity was identified that explained this anomaly.
A key objective of this research was to explore whether ROHs could be used for derivation of a measure of individual autozygosity. Before the advent of dense genome scans, the approach to estimating autozygosity from genetic-marker data was invariably inferential. We propose a very different, observational approach. Termed Froh, this is defined as the proportion of the autosomal genome in ROHs above a specified length threshold. Our purpose here is not to develop a fully fledged statistical methodology tested against the alternatives—further work is needed to refine the methodology, particularly in relation to the most appropriate length threshold for defining ROHs—but, rather, to outline a broad approach and highlight issues for future consideration. Equally, a detailed evaluation of alternative methods is beyond the scope of this paper; however, we have made some preliminary comparisons with two of the measures, Fplink and multilocal heterozygosity (MLH). Both correlate strongly with Froh. Whereas 1 − MLH is a measure of genome-wide homozygosity54 with no attempt to distinguish loci that are homozygous because of IBD and loci that are homozygous by chance, Fplink46 uses expected genome heterozygosity to control for homozygosity by chance. Carothers et al.20 have proposed another measure of autozygosity, which uses locus-specific heterozygosity to give more weight to polymorphic loci that are homozygous. Unlike our approach, all three methods are single-point approaches and do not exploit the nature of autozygosity that comes in runs or tracts. Another drawback of Fplink and the method proposed by Carothers et al. is that they require estimation of population allele frequencies, a nontrivial problem in many populations.62 Leutenegger et al.22 have also proposed a multipoint approach to autozygosity inference. Their method uses a hidden Markov model that requires that markers are in linkage equilibrium. Hence, it is computationally more complex to deal with extremely dense SNP maps, because LD needs to be taken into account or a subset of SNPs in low LD needs to be selected. Both of these are subject to ongoing research. The method is, on the other hand, very well suited for dense microsatellite maps or mixed microsatellite-SNP maps.28
Froh differs from all these approaches in that it is based on the assumption that ROHs are a signature of autozygosity (Figure 1), which might be the result of recent parental relatedness but equally might be of much more ancient origin. This is clearly illustrated by our half-Orcadian population, whose parents are known to be unrelated and who, therefore, have inherited no IBD alleles for at least five and probably 10–12 generations. We show, however, that on average, half Orcadians have a total of 6 Mb worth of ROHs measuring longer than 1.5 Mb (0.2% of the autosomes). In the two nonisolate populations studied, the comparable statistics are 7.25 Mb (0.3% of autosomes), in the Scottish population, and 8.3 Mb (0.3%), in the CEU population (Figure 6).
Consistent with the findings of other studies,56,59 we have shown that these shorter ROHs are almost invariably common but not universal in the population, occurring in both a Scottish control group (Figure 7) and an outbred non-Hispanic European American population.56 Common ROHs are a source of individual genetic variation that might play a causal role in common complex disease and that, therefore, merit further exploration as risk factors in their own right.56 We feel that it is also entirely appropriate to count them in our Froh statistic for the purposes of investigating the effect of genome-wide homozygosity on quantitative disease or disease-related traits. For this purpose, we suggest a minimum-length threshold of 0.5 Mb, because this is the limit of resolution possible with a 300,000-SNP genome-wide scan and is also considerably longer than most stretches of LD.48–51 There is, though, clearly potential for exploration of the prevalence and distribution of even-shorter ROHs with the use of data sets with more densely spaced markers.
When the research aim is to use homozygosity mapping to identify the variants causing rare recessive diseases, Froh can be modified in order to reflect only the effects of recent parental relatedness. Our analysis of the genomic location of ROHs shows that many of the most common ROHs are equally present in the offspring of both related and unrelated parents (see Table 2 and Figure 7). We propose that Froh could be modified by identification of such common ROHs and removal of them from both the numerator and the denominator, thus reducing the risk of false negatives. An alternative approach would be to set a higher minimum-length threshold, for example, 5 Mb (see Table 1 and Figure 5), but this would have the effect of underestimating the effects of recent parental relatedness by failure to count any shorter ROHs of recent origin, while still not totally eliminating longer, common ROHs.
We have shown here that ROHs measuring 1.5 Mb and longer can be used to distinguish between populations with different histories of isolation. ROHs also distinguish effectively between individuals with different degrees of parental relatedness in their ancestry. This approach is simple, observational, and based on sound theoretical justification. Although our study is based on Illumina data, this method is generally applicable, and we see no reason why it could not be used with data generated on other platforms. With some refinement, Froh has potential as a measure of individual genome-wide autozygosity for comparison to phenotype. The essential challenge in any attempt to estimate individual autozygosity from genomic data is to set a limit distinguishing autozygous from merely homozygous genotypes. Single-point methodologies based on estimation of population allele frequencies implicitly use time as a limit but face the serious drawback of requiring allele-frequency data for a founder or reference population. Our multipoint approach, which exploits the potential of ROHs as a measure of autozygosity, uses ROH length as a limit. Here, we have described how Froh is affected by the length threshold used and by the inclusion of common ROHs. The next challenge is to establish the optimum-length threshold and determine to what extent Froh should be modified with reference to the prevalence of common ROHs. These issues are the subject of ongoing research, involving the simulation of high-density genotype data by gene dropping fully phased Hap300 data down representative pedigrees. Work is also in progress to apply this approach to data sets from highly consanguineous populations and, in particular, to investigate whether the Froh length cutoff used here is universally applicable. Common, shorter ROHs also merit further investigation as a risk factor in common complex disease and will have utility in narrowing down genomic regions in the search for functional genetic variants.56 The availability of denser genome-wide scans with 1 million or more SNPs will facilitate more reliable identification and enumeration of shorter ROHs, and the use of these large data sets in different populations will improve understanding of the frequency of common ROHs and how these differ among populations.
Web Resources
The URLs for data presented herein are as follows:
UCSC Genome Browser, http://genome.ucsc.edu
Acknowledgments
We thank the people of Orkney; Lorraine Anderson and the research nurse team in Orkney; Rosa Bisset, Kay Lindsay, Gail Crosbie, and the administrative team at Public Health Sciences, University of Edinburgh; Ruby McMenemy, Sam Harcus, George Gray, Orkney Library and Archive, and the Orkney Family History Society for help with reconstructing pedigrees; Graeme Grimes, Colin Semple, Sarfraz Mohammed, Dave du Feu, Craig Nicol, and Lesley McGoohan for IT support; and Evi Theodoratu for advice relating to the Scottish data set. ORCADES is supported by the Chief Scientist Office of the Scottish Executive, the MRC Human Genetics Unit, the Royal Society, the Edinburgh Wellcome Trust Clinical Research Facility, and the European Union Framework Program 6. Ruth McQuillan is supported by a University of Edinburgh College of Medicine and Veterinary Medicine Ph.D. studentship. Rehab Abdel-Rahman is supported by a fund from the Supreme Council of Egyptian Universities. Igor Rudan is supported by grant no. 108-1080315-0302, Branka Janicijevic by grant no. 196-1962766-2763, Nina Smolej-Narancic by grant no. 196-1962766-2747, and Pavao Rudan by grant no. 196-1962766-2751 of the Croatian Ministry of Science, Education and Sport.
References
- 1.Keller L., Waller D. Inbreeding effects in wild populations. Trends Ecol. Evol. 2002;17:230–241. [Google Scholar]
- 2.Charlesworth B., Charlesworth D. The genetic basis of inbreeding depression. Genet. Res. 1999;74:329–340. doi: 10.1017/s0016672399004152. [DOI] [PubMed] [Google Scholar]
- 3.Bittles A.H. Consanguineous marriage and childhood health. Dev. Med. Child Neurol. 2003;45:571–576. doi: 10.1017/s001216220300104x. [DOI] [PubMed] [Google Scholar]
- 4.Khlat M., Khoury M. Inbreeding and diseases: demographic, genetic, and epidemiologic perspectives. Epidemiol. Rev. 1991;13:28–41. doi: 10.1093/oxfordjournals.epirev.a036072. [DOI] [PubMed] [Google Scholar]
- 5.Modell B., Darr A. Science and society: genetic counselling and customary consanguineous marriage. Nat. Rev. Genet. 2002;3:225–229. doi: 10.1038/nrg754. [DOI] [PubMed] [Google Scholar]
- 6.Bonifati V., Rizzu P., van Baren M.J., Schaap O., Breedveld G.J., Krieger E., Dekker M.C., Squitieri F., Ibanez P., Joosse M. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- 7.van Duijn C.M., Dekker M.C., Bonifati V., Galjaard R.J., Houwing-Duistermaat J.J., Snijders P.J., Testers L., Breedveld G.J., Horstink M., Sandkuijl L.A. Park7, a novel locus for autosomal recessive early-onset parkinsonism, on chromosome 1p36. Am. J. Hum. Genet. 2001;69:629–634. doi: 10.1086/322996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ewald H., Wikman F.P., Teruel B.M., Buttenschon H.N., Torralba M., Als T.D., El Daoud A., Flint T.J., Jorgensen T.H., Blanco L. A genome-wide search for risk genes using homozygosity mapping and microarrays with 1,494 single-nucleotide polymorphisms in 22 eastern Cuban families with bipolar disorder. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2005;133:25–30. doi: 10.1002/ajmg.b.30106. [DOI] [PubMed] [Google Scholar]
- 9.Mani A., Meraji S.M., Houshyar R., Radhakrishnan J., Mani A., Ahangar M., Rezaie T.M., Taghavinejad M.A., Broumand B., Zhao H. Finding genetic contributions to sporadic disease: a recessive locus at 12q24 commonly contributes to patent ductus arteriosus. Proc. Natl. Acad. Sci. USA. 2002;99:15054–15059. doi: 10.1073/pnas.192582999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rudan I., Smolej-Narancic N., Campbell H., Carothers A., Wright A., Janicijevic B., Rudan P. Inbreeding and the genetic complexity of human hypertension. Genetics. 2003;163:1011–1021. doi: 10.1093/genetics/163.3.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krieger H. Inbreeding effects on metrical traits in Northeastern Brazil. Am. J. Hum. Genet. 1969;21:537–546. [PMC free article] [PubMed] [Google Scholar]
- 12.Martin A.O., Kurczynski T.W., Steinberg A.G. Familial studies of medical and anthropometric variables in a human isolate. Am. J. Hum. Genet. 1973;25:581–593. [PMC free article] [PubMed] [Google Scholar]
- 13.Hurwich B.J., Nubani N. Blood pressures in a highly inbred community–Abu Ghosh, Israel. 1. Original survey. Isr. J. Med. Sci. 1978;14:962–969. [PubMed] [Google Scholar]
- 14.Halberstein R.A. Blood pressure in the Caribbean. Hum. Biol. 1999;71:659–684. [PubMed] [Google Scholar]
- 15.Campbell H., Carothers A.D., Rudan I., Hayward C., Biloglav Z., Barac L., Pericic M., Janicijevic B., Smolej-Narancic N., Polasek O. Effects of genome-wide heterozygosity on a range of biomedically relevant human quantitative traits. Hum. Mol. Genet. 2007;16:233–241. doi: 10.1093/hmg/ddl473. [DOI] [PubMed] [Google Scholar]
- 16.Saleh E.A., Mahfouz A.A., Tayel K.Y., Naguib M.K., Bin-al-Shaikh N.M. Hypertension and its determinants among primary-school children in Kuwait: an epidemiological study. East. Mediterr. Health J. 2000;6:333–337. [PubMed] [Google Scholar]
- 17.Badaruddoza Inbreeding effects on metrical phenotypes among north Indian children. Collegium Antropologicum. 2004;28(Suppl(2)):311–319. [PubMed] [Google Scholar]
- 18.Hartl D., Clark A.G. Sinauer Associates; Sunderland, MA: 1997. Principles of Population Genetics. [Google Scholar]
- 19.Wright S. Coefficients of inbreeding and relationship. Am. Nat. 1922;56:330–338. [Google Scholar]
- 20.Carothers A.D., Rudan I., Kolcic I., Polasek O., Hayward C., Wright A.F., Campbell H., Teague P., Hastie N.D., Weber J.L. Estimating human inbreeding coefficients: comparison of genealogical and marker heterozygosity approaches. Ann. Hum. Genet. 2006;70:666–676. doi: 10.1111/j.1469-1809.2006.00263.x. [DOI] [PubMed] [Google Scholar]
- 21.Stam P. The distribution of the fraction of the genome identical by descent in finite random mating populations. Genet. Res. 1980;35:131–155. [Google Scholar]
- 22.Leutenegger A.L., Prum B., Genin E., Verny C., Lemainque A., Clerget-Darpoux F., Thompson E.A. Estimation of the inbreeding coefficient through use of genomic data. Am. J. Hum. Genet. 2003;73:516–523. doi: 10.1086/378207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woods C.G., Valente E.M., Bond J., Roberts E. A new method for autozygosity mapping using single nucleotide polymorphisms (SNPs) and EXCLUDEAR. J. Med. Genet. 2004;41:e101. doi: 10.1136/jmg.2003.016873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu F., Elefante S., van Duijn C.M., Aulchenko Y.S. Ignoring distant genealogic loops leads to false-positives in homozygosity mapping. Ann. Hum. Genet. 2006;70:965–970. doi: 10.1111/j.1469-1809.2006.00279.x. [DOI] [PubMed] [Google Scholar]
- 25.Smith C. The detection of linkage in human genetics. J. Royal Stat. Soc. B. 1953;15:153–192. [Google Scholar]
- 26.Botstein D., Risch N. Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease. Nat. Genet. 2003;33(Suppl):228–237. doi: 10.1038/ng1090. [DOI] [PubMed] [Google Scholar]
- 27.Miano M.G., Jacobson S.G., Carothers A., Hanson I., Teague P., Lovell J., Cideciyan A.V., Haider N., Stone E.M., Sheffield V.C. Pitfalls in homozygosity mapping. Am. J. Hum. Genet. 2000;67:1348–1351. doi: 10.1016/s0002-9297(07)62966-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leutenegger A.L., Labalme A., Genin E., Toutain A., Steichen E., Clerget-Darpoux F., Edery P. Using genomic inbreeding coefficient estimates for homozygosity mapping of rare recessive traits: application to Taybi-Linder syndrome. Am. J. Hum. Genet. 2006;79:62–66. doi: 10.1086/504640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shami S.A., Qaisar R., Bittles A.H. Consanguinity and adult morbidity in Pakistan. Lancet. 1991;338:954. doi: 10.1016/0140-6736(91)91828-i. [DOI] [PubMed] [Google Scholar]
- 30.Puzyrev V.P., Lemza S.V., Nazarenko L.P., Panphilov V.I. Influence of genetic and demographic factors on etiology and pathogenesis of chronic disease in north Siberian aborigines. Arctic Med. Res. 1992;51:136–142. [PubMed] [Google Scholar]
- 31.Ismail J., Jafar T.H., Jafary F.H., White F., Faruqui A.M., Chaturvedi N. Risk factors for non-fatal myocardial infarction in young South Asian adults. Heart. 2004;90:259–263. doi: 10.1136/hrt.2003.013631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simpson J.L., Martin A.O., Elias S., Sarto G.E., Dunn J.K. Cancers of the breast and female genital system: search for recessive genetic factors through analysis of human isolate. Am. J. Obstet. Gynecol. 1981;141:629–636. doi: 10.1016/s0002-9378(15)33302-0. [DOI] [PubMed] [Google Scholar]
- 33.Lebel R.R., Gallagher W.B. Wisconsin consanguinity studies. II: Familial adenocarcinomatosis. Am. J. Med. Genet. 1989;33:1–6. doi: 10.1002/ajmg.1320330102. [DOI] [PubMed] [Google Scholar]
- 34.Rudan I. Inbreeding and cancer incidence in human isolates. Hum. Biol. 1999;71:173–187. [PubMed] [Google Scholar]
- 35.Carlson C.S., Eberle M.A., Kruglyak L., Nickerson D.A. Mapping complex disease loci in whole-genome association studies. Nature. 2004;429:446–452. doi: 10.1038/nature02623. [DOI] [PubMed] [Google Scholar]
- 36.Freimer N., Sabatti C. The use of pedigree, sib-pair and association studies of common diseases for genetic mapping and epidemiology. Nat. Genet. 2004;36:1045–1051. doi: 10.1038/ng1433. [DOI] [PubMed] [Google Scholar]
- 37.Wright A., Charlesworth B., Rudan I., Carothers A., Campbell H. A polygenic basis for late-onset disease. Trends Genet. 2003;19:97–106. doi: 10.1016/s0168-9525(02)00033-1. [DOI] [PubMed] [Google Scholar]
- 38.Reich D.E., Lander E.S. On the allelic spectrum of human disease. Trends Genet. 2001;17:502–510. doi: 10.1016/s0168-9525(01)02410-6. [DOI] [PubMed] [Google Scholar]
- 39.Broman K.W., Weber J.L. Long homozygous chromosomal segments in reference families from the centre d'Etude du polymorphisme humain. Am. J. Hum. Genet. 1999;65:1493–1500. doi: 10.1086/302661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tenesa A., Farrington S.M., Prendergast J.G., Porteous M.E., Walker M., Haq N., Barnetson R.A., Theodoratou E., Cetnarskyj R., Cartwright N. Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nat. Genet. 2008;40:631–637. doi: 10.1038/ng.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 42.Brennan E.R., Relethford J.H. Temporal variation in the mating structure of Sanday, Orkney Islands. Ann. Hum. Biol. 1983;10:265–280. doi: 10.1080/03014468300006431. [DOI] [PubMed] [Google Scholar]
- 43.Boyce A.J., Holdsworth V.M.L., Brothwell D. Demographic and genetic studies in the Orkney islands. In: Roberts D.F., Sunderland E., editors. Genetic Variation in Britain. Taylor and Francis; London: 1973. [Google Scholar]
- 44.Vitart V., Rudan I., Hayward C., Gray N.K., Floyd J., Palmer C.N., Knott S.A., Kolcic I., Polasek O., Graessler J. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat. Genet. 2008;40:437–442. doi: 10.1038/ng.106. [DOI] [PubMed] [Google Scholar]
- 45.Frazer K.A., Ballinger D.G., Cox D.R., Hinds D.A., Stuve L.L., Gibbs R.A., Belmont J.W., Boudreau A., Hardenbol P., Leal S.M. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Department, R.A., ed. (2004). Scottish Executive Urban Rural Classification 2003–2004, E.a.
- 48.Abecasis G.R., Ghosh D., Nichols T.E. Linkage disequilibrium: ancient history drives the new genetics. Hum. Hered. 2005;59:118–124. doi: 10.1159/000085226. [DOI] [PubMed] [Google Scholar]
- 49.Wall J.D., Pritchard J.K. Haplotype blocks and linkage disequilibrium in the human genome. Nat. Rev. Genet. 2003;4:587–597. doi: 10.1038/nrg1123. [DOI] [PubMed] [Google Scholar]
- 50.Abecasis G.R., Noguchi E., Heinzmann A., Traherne J.A., Bhattacharyya S., Leaves N.I., Anderson G.G., Zhang Y., Lench N.J., Carey A. Extent and distribution of linkage disequilibrium in three genomic regions. Am. J. Hum. Genet. 2001;68:191–197. doi: 10.1086/316944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reich D.E., Cargill M., Bolk S., Ireland J., Sabeti P.C., Richter D.J., Lavery T., Kouyoumjian R., Farhadian S.F., Ward R. Linkage disequilibrium in the human genome. Nature. 2001;411:199–204. doi: 10.1038/35075590. [DOI] [PubMed] [Google Scholar]
- 52.Marioni J.C., Thorne N.P., Valsesia A., Fitzgerald T., Redon R., Fiegler H., Andrews T.D., Stranger B.E., Lynch A.G., Dermitzakis E.T. Breaking the waves: improved detection of copy number variation from microarray-based comparative genomic hybridization. Genome Biol. 2007;8:R228. doi: 10.1186/gb-2007-8-10-r228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Colella S., Yau C., Taylor J.M., Mirza G., Butler H., Clouston P., Bassett A.S., Seller A., Holmes C.C., Ragoussis J. QuantiSNP: an Objective Bayes Hidden-Markov Model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 2007;35:2013–2025. doi: 10.1093/nar/gkm076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Charpentier M., Setchell J.M., Prugnolle F., Knapp L.A., Wickings E.J., Peignot P., Hossaert-McKey M. Genetic diversity and reproductive success in mandrills (Mandrillus sphinx) Proc. Natl. Acad. Sci. USA. 2005;102:16723–16728. doi: 10.1073/pnas.0507205102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lencz T., Lambert C., DeRosse P., Burdick K.E., Morgan T.V., Kane J.M., Kucherlapati R., Malhotra A.K. Runs of homozygosity reveal highly penetrant recessive loci in schizophrenia. Proc. Natl. Acad. Sci. USA. 2007;104:19942–19947. doi: 10.1073/pnas.0710021104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Simon-Sanchez J., Scholz S., Fung H.C., Matarin M., Hernandez D., Gibbs J.R., Britton A., de Vrieze F.W., Peckham E., Gwinn-Hardy K. Genome-wide SNP assay reveals structural genomic variation, extended homozygosity and cell-line induced alterations in normal individuals. Hum. Mol. Genet. 2007;16:1–14. doi: 10.1093/hmg/ddl436. [DOI] [PubMed] [Google Scholar]
- 58.Gibson J., Morton N.E., Collins A. Extended tracts of homozygosity in outbred human populations. Hum. Mol. Genet. 2006;15:789–795. doi: 10.1093/hmg/ddi493. [DOI] [PubMed] [Google Scholar]
- 59.Curtis D., Vine A.E., Knight J. Study of regions of extended homozygosity provides a powerful method to explore haplotype structure of human populations. Ann. Hum. Genet. 2008;72:261–278. doi: 10.1111/j.1469-1809.2007.00411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li L.H., Ho S.F., Chen C.H., Wei C.Y., Wong W.C., Li L.Y., Hung S.I., Chung W.H., Pan W.H., Lee M.T. Long contiguous stretches of homozygosity in the human genome. Hum. Mutat. 2006;27:1115–1121. doi: 10.1002/humu.20399. [DOI] [PubMed] [Google Scholar]
- 61.Vitart V., Carothers A.D., Hayward C., Teague P., Hastie N.D., Campbell H., Wright A.F. Increased level of linkage disequilibrium in rural compared with urban communities: a factor to consider in association-study design. Am. J. Hum. Genet. 2005;76:763–772. doi: 10.1086/429840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hoffman J.I., Boyd I.L., Amos W. Exploring the relationship between parental relatedness and male reproductive success in the Antarctic fur seal Arctocephalus gazella. Evolution Int. J. Org. Evolution. 2004;58:2087–2099. doi: 10.1111/j.0014-3820.2004.tb00492.x. [DOI] [PubMed] [Google Scholar]