

Abstract
All reported mutations in ALAS2, which encodes the rate-regulating enzyme of erythroid heme biosynthesis, cause X-linked sideroblastic anemia. We describe eight families with ALAS2 deletions, either c.1706-1709 delAGTG (p.E569GfsX24) or c.1699-1700 delAT (p.M567EfsX2), resulting in frameshifts that lead to replacement or deletion of the 19–20 C-terminal residues of the enzyme. Prokaryotic expression studies show that both mutations markedly increase ALAS2 activity. These gain-of-function mutations cause a previously unrecognized form of porphyria, X-linked dominant protoporphyria, characterized biochemically by a high proportion of zinc-protoporphyrin in erythrocytes, in which a mismatch between protoporphyrin production and the heme requirement of differentiating erythroid cells leads to overproduction of protoporphyrin in amounts sufficient to cause photosensitivity and liver disease.
Main Text
Each of the seven inherited porphyrias results from a partial deficiency of an enzyme of heme biosynthesis. Mutations that cause porphyria have been identified in all the genes of the heme biosynthetic pathway except ALAS1 and ALAS2, which encode the ubiquitously expressed (ALAS1) and erythroid-specific (ALAS2) isoforms of mitochondrial 5-aminolevulinate synthase (ALAS) (EC 2.3.1.37), the initial, rate-regulating enzyme of the pathway.1 ALAS2 is essential for hemoglobin formation by erythroid cells, and ALAS1 cannot replace this function. No mutations have been identified in ALAS1, but pathogenic mutations in the 14.4 kb, 11-exon-containing ALAS2 cause X-linked hereditary sideroblastic anemia (XLSA [MIM 301300]) with iron overload.2
Erythropoietic protoporphyria (EPP [MIM 177000]) is an inherited disorder caused by partial mitochondrial deficiency of ferrochelatase (FECH) (EC 4.99.1.1), the terminal enzyme of heme biosynthesis. Accumulation of protoporphyrin IX in erythrocytes and other tissues leads to lifelong photosensitivity and, in about 2% of patients, severe liver disease.3 Most patients have autosomal-dominant EPP (dEPP), in which clinical expression normally requires coinheritance of an FECH mutation that abolishes or markedly reduces FECH activity trans to a hypomorphic FECH IVS3-48C allele carried by about 11% of western Europeans.4 About 4% of families have autosomal-recessive EPP.4 However, mutational analysis fails to detect FECH mutations in about 7% of EPP families, of which about 3% are homozygous for the wild-type FECH IVS3-48T allele,5 suggesting possible involvement of another locus.
Within this subgroup of families with mutation-negative EPP, we studied eight families in which at least one individual had acute photosensitivity clinically indistinguishable from that of dEPP. These families were identified through referral to specialist porphyria centers or during surveys of EPP in the UK6 or South Africa7 and were of western European (four families), Jewish, north African, Indo-Asian, or Sudanese (one family each) ancestry. Our study was conducted in accord with the World Medical Association Declaration of Helsinki ethical principles for medical research involving human subjects and its subsequent amendments. All patients or their parents gave informed consent to investigation. Prior ethical approval was obtained for patient surveys.6,7
Genomic DNA was extracted from whole blood. For sequencing of ALAS2, FECH, and SLC25A37 (MIM 610387) (GenBank accession numbers: human ALAS2 [NM000032.3], FECH [NM000140], and SLC25A37 [NM0166112] cDNAs and ALAS2 [NT011630] and SLC25A37 [NT023666] genes), all exons and their flanking sequences were amplified by polymerase chain reaction (PCR) (primers and conditions are available from the authors). PCR-amplified double-stranded DNA was purified from agarose gels with the QIAquick gel extraction kit (QIAGEN, Cawley, UK) before being cycle sequenced with fluorescent ddNTPs (BigDye) and an ABI Prism 3130XL Genetic Analyzer (PE Biosystems, Warrington, UK). We confirmed the presence or absence of mutations by sequencing both strands. Genotyping with FECH intragenic single-nucleotide polymorphisms (SNPs),4 and microsatellite markers for FECH7 and ALAS2 was performed on the ABI PRISM 3100 automated sequencer. The ALAS2 microsatellite markers (16AG at position 54992283, 17GT at position 55066050, and 23AC at position 55535957) were identified at the UCSC genome bioinformatics site (Santa Cruz, CA; see Web Resources). Results were analyzed with the ABI PRISM GeneMapper software version 3.0. Erythrocyte porphyrins were measured as previously described.7,8 The percentage of zinc protoporphyrin was calculated from flourescence emission spectra of ethanol8 or acetone9 extracts of erythrocyte haemolystaes. FECH activity was measured as described4 or indirectly from the amount of protoporphyrin formed from 5-aminolevulinate in the presence and absence of Fe2+.10 Differences between quantitative variables were assessed with the Mann-Whitney test, and those between proportions were assessed with Fisher's exact test.
We differentiated patients in these eight families from others with FECH mutation-negative EPP by showing that the percentage of erythrocyte protoporphyrin present as its zinc chelate (19%–65%, median 44%) was markedly greater than in patients with dEPP (4%–13%, median 8%). Erythrocyte protoporphyrin concentrations were also higher in our patients, in whom they were increased 24-fold (range: 6- to 103-fold) (Table 1) compared with 14-fold (range: 4- to 44-fold) in 171 patients with dEPP (p < 0.001). In one patient with iron deficiency, erythrocyte protoporphyrin increased markedly (101-fold) but then decreased as iron stores were replenished (Figure 1). Lymphocyte FECH activity, measured in ten patients, ranged from 74%–106% (median 85%) of the mean normal value, indicating that protoporphyrin accumulation was not caused by FECH deficiency resulting from a mutation of the ubiquitously expressed FECH gene. We further eliminated involvement of FECH by using intragenic SNPs4 or microsatellite markers7 to show that protoporphyrin accumulation did not segregate with FECH haplotypes in two families; other families were uninformative or not tested. Because abnormal expression of mRNA for mitoferrin has been implicated in the pathogenesis of a similar form of protoporphyria (Shaw et al., Blood 108, ASH Annual Meeting Abstracts, 6a), we sequenced all exons of SLC25A37 and their flanking sequences in all eight probands but were unable to identify any disease-specific mutation.
Table 1.
Erythrocyte Porphyrins, Hematological Measurements, and Serum Iron Indices in Patients with X-Linked Dominant Protoporphyria and Their Unaffected Relatives
| XLDPP |
Unaffected Relatives |
|||
|---|---|---|---|---|
| Male | Female | Male | Female | |
| Total protoporphyrin (fold increase) | 27, 11–103 (7) | 24, 6–64 (16) | Less than 1.0 | |
| Zinc protoporphyrin (% total) | 45, 19–58 (7) | 46, 30–65 (15) | Less than 80 | |
| Hemoglobin (g/dl) | 14.3, 12.2–17.5 (5) | 13.0, 12.4–14.1 (8) | 14.2, 13.1–14.7 (8) | 12.5, 11.9–14.2 (5) |
| MCV (fl) | 87, 78–92 (13) | 88, 84–96 (13) | ||
| Ferritin (μg/liter) | 24, 10–104 (5)∗ | 56, 21–154 (6) | 102, 41–231 (8) | 51, 33–91 (5) |
| Transferrin (g/liter) | 2.87, 1.93–3.81 (9) | 2.27, 1.59–2.84 (13) | ||
| Transferrin saturation (%) | 16, 3–39 (11)∗ | 26, 22–35 (13) | ||
| Transferrin receptor-1 (mg/liter) | 1.44, 1.24–2.59 (10)∗∗ | 1.16, 0.84–1.66 (13) | ||
Measurements are medians and ranges. ∗p = 0.02 versus unaffected male relatives; ∗∗p = 0.04 versus unaffected relatives; differences between other groups are not significant. Total porphyrin is expressed as n-fold increase (times upper limit of normal); for 17 patients with XLDPP in whom total erythrocyte porphyrin was measured by the same method,8 the median concentration was 51.2 μmol/liter, range 20.1–195.6 μmol/liter (reference range: less than 1.7 μmol/liter). Values in italics include male and female subjects.
Figure 1.
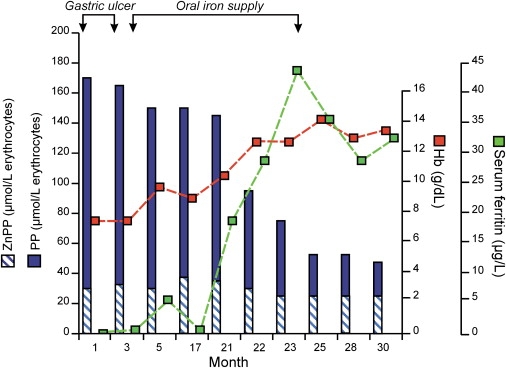
Treating Iron Deficiency Decreases Erythrocyte Protoporphyrin Concentrations
Iron deficiency caused by bleeding from a gastric ulcer was treated with omeprazole and oral iron.
Parent-child transmission of overt disease is uncommon in EPP.11 Our families were unusual in showing an apparent dominant pattern of inheritance with an absence of father-son transmission, which suggested X-linkage (Figure 2). We therefore investigated two candidate genes that are located on the X chromosome and are involved in heme formation, GATA1 (MIM 305371) (data not shown) and ALAS2. Protoporphyrin accumulation segregated with an X chromosome haplotype defined by microsatellite markers around ALAS2 in three families (data not shown). Sequencing of genomic DNA identified two different deletions (c.1706–1709 delAGTG in six families; c.1699–1700 delAT in two families) in ALAS2 exon 11, an exon that is present in all ALAS2 transcripts.12 The frameshifts produced by these deletions lead to predicted alterations of the 19–20 C-terminal amino acids of ALAS2 (Figures 3A and 3B); either deletion (delAT) or replacement by a 23 residue sequence (delAGTG) that extends the enzyme by 4 amino acids and alters the predicted secondary structure (Supplemental Data; Figure 1). These mutations segregated with photosensitivity (LOD score 7.8) and were absent from 129 unrelated EPP patients (106 dEPP; 23 FECH-mutation-negative EPP) and 100 normal chromosomes. The delAGTG mutation occurred on five different haplotypes, indicating that it has arisen on at least five separate occasions, whereas the two delAT families, both from southwest England, had the same background haplotype and may come from a single extended family (data not shown). These findings show that these deletions in ALAS2 cause a previously unrecognized X-linked dominant protoporphyria (XLDPP) that, in contrast to dEPP and other autosomal-dominant porphyrias,1 has close to 100% penetrance (Figure 2).
Figure 2.
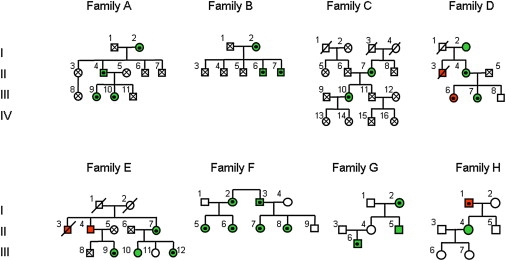
Pedigrees of Eight Families with FECH-Mutation-Negative Protoporphyria
Green circles and squares represent patients with photosensitivity. Red circles and squares represent patients with photosensitivity and clinically overt liver disease. Protoporphyric liver disease was confirmed at autopsy or by needle biopsy in all these patients except patient I1 (family H), for whom a diagnosis has not been established. Patient II3 (family E) has been reported previously.23 Clinical information was not obtainable for patients C I, 3 and 4 or E I, 1 and 2.
Black dots within circles or squares indicate individuals in whom either the delAGTG (families A–E, H) or delAT (families F and G) ALAS2 mutations were identified. Crosses within circles or squares indicate individuals in whom sequencing excluded the presence of an ALAS2 mutation. The absence of a black dot or cross indicates an individual from whom a DNA sample was not available for analysis. The LOD score for linkage between photosensitivity and the ALAS2 mutation was calculated for families A–E.
Figure 3.
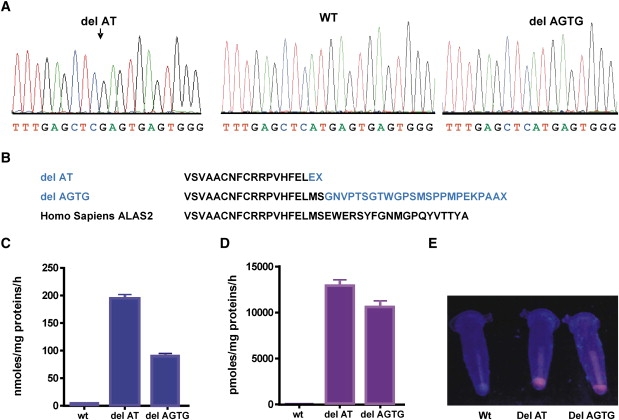
C-Terminal Deletions in ALAS2 Cause X-Linked Dominant Protoporphyria
(A) Sequence analysis of genomic DNA from male patients showing deletions in the ALAS2 gene.
(B) Predicted effects of deletions on ALAS2 C-terminal sequences.
(C–E) Prokaryotic expression of wild-type and mutant ALAS2 enzymes: Rates of formation of ALA (C) and porphyrin (D) by bacterial lysates; means and ranges for three experiments are shown. (E) Porphyrin fluorescence (UVA light) in bacterial pellets.
All previously described mutations in ALAS2 have caused XLSA (Human Gene Mutation Database), including a missense mutation (S568G) in the region lost or replaced in our patients.13 Previously reported frameshift or other null ALAS2 mutations causing XLSA have been embryonically lethal in males.2 In our families, both sexes were affected, and patients had neither anemia nor iron overload (Table 1). Instead, there was some evidence of diminished iron stores, particularly in males (Table 1). Similar abnormalities have been observed in dEPP14 and might result from accumulation of protoporphyrin rather than FECH deficiency.15 Five (17%) patients had overt liver disease, suggesting that XLPP, like autosomal recessive EPP3,4, carries a higher risk of liver disease than dEPP. Liver disease was more common in males (p = 0.008), and one obligate carrier was asymptomatic (Figure 2; family G, II4), but otherwise we found no evidence that X inactivation led to milder disease in females. Erythrocyte protoporphyrin concentrations in photosensitive patients were not significantly different between the sexes (Table 1). These data show that disruption of the C-terminal region of ALAS2 leads to the production of protoporphyrin in excess of the amount required for hemoglobinization and in quantities sufficient to cause photosensitivity and liver damage, in spite of normal FECH activity; this is a situation unique in human disease.
To investigate the effect of the mutants on ALAS2 activity, we expressed both mutant enzymes in Escherichia coli. PCR-amplified cDNAs for the delAT and delAGTG mutations were introduced into pMALc2-AE2 (ALAS2 WT)16 by site-directed mutagenesis with the QuickChange Site Directed Mutagenesis Kit (Stratagene, La Jolla, CA) and the following oligonucleotides: delAT sense 5′-ACACTTTGAGCTCGAGTGAGTGGGAACG-3′ and delAGTG sense 5′-CACTTTGAGCTCATGAGTGGG AACGTTCCTACTTGC-3′ and their complementary antisense oligonucleotides. We confirmed the sequences of the resulting clones for the entire coding region, and we sequenced each mutated ALAS2 cDNA to ensure that only the desired mutation had been introduced and that the remainder of the sequence was correct. Expression constructs were transfected into Escherichia coli BL21 (Invitrogen), and overnight cultures were grown in LB (Lennox L Broth Base, Invitrogen) media with 100 mg/ml ampicillin (PANPHARMA).The next day, 20 ml cultures in LB/ampicillin media were initiated with the overnight cultures and grown to 1.2 A600 units. Induction with 0.1 mM isopropyl β-D-thiogalactopyranoside was performed in LB/ampicillin media for 4 hr at 22°C. Cells were pelleted at 2500 rpm for 10 min. The recombinant bacteria were grown, and ALAS2 activities of controls and mutant enzymes were determined in bacteria lysates as previously described but with minor modifications.16 Enzyme activities were expressed in pmol of 5-aminolevulinate (ALA) and porphyrin/hr/mg protein at 37°C.
These expression studies showed that both deletions markedly increase ALAS2 activity and that some of the ALA that is produced is further metabolised to porphyrin (Figures 3C–3E). E. coli BL21 transformed with mutant plasmids accumulated porphyrin (mutant, 5000 nmol/g protein; wild-type, less than 8 nmol/g) without the addition of substrates for ALAS2. These findings of a gain of function strongly suggest that protoporphyrin and its zinc chelate accumulates in XLDPP because the rate of ALA formation is increased to such an extent that insertion of Fe2+ into PP by FECH becomes rate limiting for heme synthesis. Gain-of-function mutations have not previously been identified in genes of the heme biosynthetic pathway1 but, as in our families, characteristically cause dominant disorders.
Excretion of ALA and other protoporphyrin precursors is normal in XLDPP (data not shown), indicating that most of the ALA produced by erythroid cells is metabolized to protoporphyrin. Some is used for hemoglobin synthesis, but the fate of the rest is uncertain. Because FECH activity in erythroid cells exceeds that required for hemoglobin synthesis,2 some may be converted to free heme and exported from the cytoplasm.17 However, the accumulation of zinc protoporphyrin in XLDPP, indicating that FECH is using its alternative metal substrate, suggests that formation of excess heme can be prevented by lack of available iron. The phenotype of iron deficiency in XLDPP (Figure 1) closely resembles that of Ireb2−/− mice, in which deletion of iron-regulatory protein 2 (IRP2) leads to overexpression of ALAS2, erythroblast iron deficiency, and microcytic anemia.18 In the patient whose clinical course is shown in Figure 1, iron repletion decreased protoporphyrin accumulation and corrected the anemia. However, zinc protoporphyrin did not decrease, as it does in uncomplicated iron deficiency.19 This might indicate that the synthesis of zinc protoporphyrin becomes limited by intra-mitochondrial availability of Zn2+ when the protoporphyrin pool is greatly expanded.18 These findings are consistent with the hypothesis that the regulatory system that enables efficient utilization of iron for heme synthesis during erythroid differentiation20 allows matching of erythroblast iron uptake to intra-mitochondrial heme synthesis to be maintained even when excess protoporphyrin is present. The mechanism by which this is achieved is unknown but might involve regulation of transferrin receptor-1 expression in erythroblasts through heme-mediated degradation of IRP2.21
The 26 C-terminal amino acids of ALAS2 are highly conserved and have diverged from ALAS1 (Figure 4), which suggests that this sequence might have an important, but unknown, erythroid-specific function. During erythropoiesis, tight coordination of substrate supply to FECH normally prevents accumulation of toxic amounts of protoporphyrin. Coordination is largely achieved through iron-dependent post-transcriptional regulation of synthesis of ALAS2.2 This system fails in XLDPP because the mutations that we have described greatly increase ALAS2 activity. A possible mechanism might be stabilization against degradation or an intrinsic increase in specific activity. This C-terminal sequence is not present in ALASRc, the only ALAS for which the crystal structure has been reported.22 Sequence similarities between ALASRc and human ALAS222 suggest that it is not directly involved in pyridoxal 5-phospate-dependent catalysis. Our findings indicate that it modulates enzyme activity, but the mechanism of this effect remains to be determined. The discovery of gain-of-function mutations in ALAS2 identifies a previously undefined type of human porphyria, provides new information about the regulation of substrate supply for heme synthesis during erythroid differentiation, and identifies a potential tool for increasing erythroid heme synthesis in experimental systems.
Figure 4.
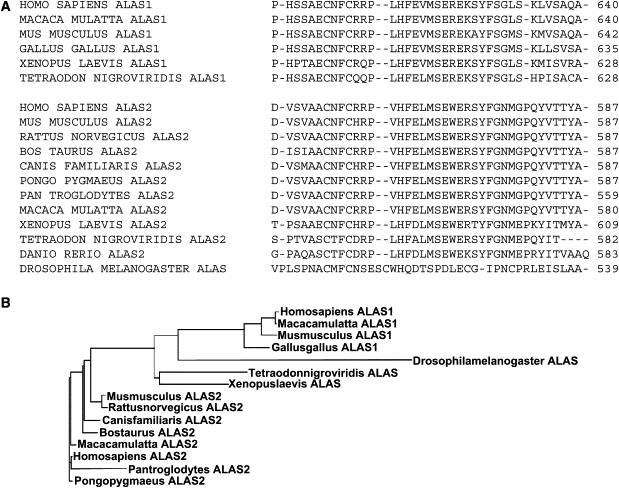
Comparison of C-terminal Sequences of ALAS enzymes
(A) Alignment of ALAS C-terminal sequences.
(B) Phylogenetic tree showing relationships between ALAS genes constructed with ClustalW.
Acknowledgments
We thank the family members for their cooperation; Yves Nordmann for his clinical contribution; David Bishop, Mount Sinai School of Medicine, New York, for kindly providing the bacterial expression vector containing the normal ALAS2 cDNA sequence; Jerôme Fagart (INSERM U773) for the secondary-structure predictions; and Vasco Pereira Da Silva, Sylvie Simonin, Anne Marie Robreau Fraolini, Nicola Mason, Jacqueline Woolf, and Brandon Davidson for expert laboratory assistance. This work was supported by Agence Nationale pour la Recherche-Groupement d'Interet Scientifiqe (ANR-GIS) Maladies Rares, reference ANR-07-MRAR-008-01 (C.B.); the British Skin Foundation (S.D.W); Royal College of Physicians (S.A.H.); a Dorothy Hodgkin Royal Society Fellowship (Y.M.); WellChild, Cheltenham, UK and the Children's Liver Disease Foundation, Birmingham,UK (G.M.V); and the South African Medical Research Council (A.V.C, P.N.M., and M.P.).
Supplemental Data
Web Resources
URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbio.nlm.nih.gov/Omim/ (for EPP, XLSA)
Human Gene Mutation Database (HGMD), http://archive.uwcm.ac.uk/uwcm/mg/hgmd0.html/
For ALAS2 microsatellite markers, http://www.genome.ucsc.edu/
Protein sequence comparisons, ClustalW, http://www.ebi.ac.uk/clustalW/
Network protein sequence analysis, http://pbil.univ-lyon1.fr/pf_bioinfo/rubrique9.html
References
- 1.Sassa S. Modern diagnosis and management of the porphyrias. Br. J. Haematol. 2006;135:281–292. doi: 10.1111/j.1365-2141.2006.06289.x. [DOI] [PubMed] [Google Scholar]
- 2.Bottomley S.S. Sideroblastic anemias. In: Lukens J.N., Rogers G.M., Paraskevas F., Glader B.E., editors. Wintrobe's Clinical Hematology, J.P. Greer J. Foerster. Lippincott Williams & Wilkins; Philadelphia: 2004. pp. 1012–1033. [Google Scholar]
- 3.Cox T.M. Erythropoietic protoporphyria. In: Kadish K.M., Smith K.M., Guilard R., editors. The Porphyrin Handbook, Vol 14, Medical aspects of porphyrias. Academic Press; Amsterdam: 2003. pp. 121–150. [Google Scholar]
- 4.Gouya L., Martin-Schmitt C., Robreau A.M., Austerlitz F., Da Silva V., Brun P., Simonin S., Lyoumi S., Grandchamp B., Beaumont C. Contribution of a common single-nucleotide polymorphism to the genetic predisposition for erythropoietic protoporphyria. Am. J. Hum. Genet. 2006;78:2–14. doi: 10.1086/498620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whatley S.D., Mason N.G., Holme S.A., Anstey A.V., Elder G.H., Badminton M.N. Gene dosage analysis identifies large deletions of the FECH gene in 10% of UK families with erythropoietic protoporphyria. J. Invest. Dermatol. 2007;127:2790–2794. doi: 10.1038/sj.jid.5700924. [DOI] [PubMed] [Google Scholar]
- 6.Holme S.A., Anstey A.V., Finlay A.Y., Elder G.H., Badminton M.N. Erythropoietic protoporphyria in the U.K.: Clinical features and effect on quality of life. Br. J. Dermatol. 2006;155:574–581. doi: 10.1111/j.1365-2133.2006.07472.x. [DOI] [PubMed] [Google Scholar]
- 7.Parker M., Corrigall A.V., Hift R.J., Meissner P.N. Molecular characterisation of erythropoietic protoporphyria in South Africa. Br. J. Dermatol. 2008;159:182–191. doi: 10.1111/j.1365-2133.2008.08580.x. [DOI] [PubMed] [Google Scholar]
- 8.Deacon A.C., Elder G.H. Front line tests for the investigation of suspected porphyria. J. Clin. Pathol. 2001;54:500–507. doi: 10.1136/jcp.54.7.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hart D., Piomelli S. Simultaneous quantitation of zinc protoporphyrin and free protoporphyrin in erythrocytes by acetone extraction. Clin. Chem. 1981;27:220–222. [PubMed] [Google Scholar]
- 10.Tovey J.A., Elder G.H. Ferrochelatase activity in human lymphocytes: Effect of storage on haem formation. Ann. Clin. Biochem. 1990;27:80–81. doi: 10.1177/000456329002700118. [DOI] [PubMed] [Google Scholar]
- 11.Went L.N., Klasen E.C. Genetic aspects of erythropoietic protoporphyria. Ann. Hum. Genet. 1984;48:105–117. doi: 10.1111/j.1469-1809.1984.tb01006.x. [DOI] [PubMed] [Google Scholar]
- 12.Cox T.C., Sadlon T.J., Schwarz Q.P., Matthews C.S., Wise P.D., Cox L.L., Bottomley S.S., May B.K. The major splice variant of human 5-aminolevulinate synthase-2 contributes significantly to erythroid heme biosynthesis. Int. J. Biochem. Cell Biol. 2004;36:281–295. doi: 10.1016/s1357-2725(03)00246-2. [DOI] [PubMed] [Google Scholar]
- 13.Harigae H., Furuyama K., Kimura A., Neriishi K., Tahara N., Kondo M., Hayashi N., Yamamoto M., Sassa S., Sasaki T. A novel mutation of the erythroid-specific delta-aminolaevulinate synthase gene in a patient with X-linked sideroblastic anaemia. Br. J. Haematol. 1999;106:175–177. doi: 10.1046/j.1365-2141.1999.01479.x. [DOI] [PubMed] [Google Scholar]
- 14.Holme S.A., Worwood M., Anstey A.V., Elder G.H., Badminton M.N. Erythropoiesis and iron metabolism in dominant erythropoietic protoporphyria. Blood. 2007;110:4108–4110. doi: 10.1182/blood-2007-04-088120. [DOI] [PubMed] [Google Scholar]
- 15.Lyoumi S., Abitbol M., Andrieu V., Henin D., Robert E., Schmitt C., Gouya L., de Verneuil H., Deybach J.C., Montagutelli X. Increased plasma transferrin, altered body iron distribution and microcytic hypochromic anemia in ferrochelatase deficient mice. Blood. 2007;109:811–818. doi: 10.1182/blood-2006-04-014142. [DOI] [PubMed] [Google Scholar]
- 16.Cotter P.D., Rucknagel D.L., Bishop D.F. X-linked sideroblastic anemia: Identification of the mutation in the erythroid-specific delta-aminolevulinate synthase gene (ALAS2) in the original family described by Cooley. Blood. 1994;84:3915–3924. [PubMed] [Google Scholar]
- 17.Keel S.B., Doty R.T., Yang Z., Quigley J.G., Chen J., Knoblaugh S., Kingsley P.D., De Domenico I., Vaughn M.B., Kaplan J. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science. 2008;319:825–828. doi: 10.1126/science.1151133. [DOI] [PubMed] [Google Scholar]
- 18.Cooperman S.S., Meyron-Holtz E.G., Olivierre-Wilson H., Ghosh M.C., McConnell J.P., Rouault T.A. Microcytic anemia, erythropoietic protoporphyria, and neurodegeneration in mice with targeted deletion of iron-regulatory protein 2. Blood. 2005;106:1084–1089. doi: 10.1182/blood-2004-12-4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Labbé R.F., Rettmer R.L. Zinc protoporphyrin: A product of iron-deficient erythropoiesis. Semin. Hematol. 1989;26:40–46. [PubMed] [Google Scholar]
- 20.Schranzhofer M., Schrifrer M., Cabrera J.A., Kopp K., Chiba P., Beug H., Mullner E.W. Remodelling the regulation of iron metabolism during erythroid differentiation to ensure efficient heme biosynthesis. Blood. 2006;107:4159–4167. doi: 10.1182/blood-2005-05-1809. [DOI] [PubMed] [Google Scholar]
- 21.Ishikawa H., Kato M., Hori H., Ishimori K., Kirisako T., Tokunaga F., Iwai K. Involvement of heme regulatory motif in heme-mediated ubiquitinisation and degradation of IRP2. Mol. Cell. 2005;19:171–181. doi: 10.1016/j.molcel.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 22.Astner I., Schulze J.O., van den Heuvel J., Jahn D., Schubert W.D., Heinz D.W. Crystal structure of 5-aminolevulinate synthase, the first enzyme of heme biosynthesis, and its link to XLSA in humans. EMBO J. 2005;24:3166–3177. doi: 10.1038/sj.emboj.7600792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eales L., Day R.S., Pimstone N.R. Protoporphyrin (proto)-determined hepatopathty in a South African Jewish family. Ann. Clin. Res. 1978;10:205–213. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.