

Abstract
In two siblings we found a mitochondrial encephalomyopathy, characterized by developmental delay, hemiplegia, convulsions, asymmetrical brain atrophy, and low cytochrome c oxidase (COX) activity in skeletal muscle. The disease locus was identified on chromosome 2 by homozygosity mapping; candidate genes were prioritized for their known or predicted mitochondrial localization and then sequenced in probands and controls. A homozygous nonsense mutation in the KIAA0971 gene segregated with the disease in the proband family. The corresponding protein is known as fas activated serine-threonine kinase domain 2, FASTKD2. Confocal immunofluorescence colocalized a tagged recombinant FASTKD2 protein with mitochondrial markers, and membrane-potential-dependent in vitro mitochondrial import was demonstrated in isolated mitochondria. In staurosporine-induced-apoptosis experiments, decreased nuclear fragmentation was detected in treated mutant versus control fibroblasts. In conclusion, we found a loss-of-function mutation in a gene segregating with a peculiar mitochondrial encephalomyopathy associated with COX deficiency in skeletal muscle. The corresponding protein is localized in the mitochondrial inner compartment. Preliminary data indicate that FASTKD2 plays a role in mitochondrial apoptosis.
Main Text
Mitochondrial disorders are clinical phenotypes associated with abnormalities of the terminal component of aerobic energy metabolism, i.e., oxidative phosphorylation (OXPHOS), a complex pathway linking cellular respiration to adenosine triphosphate (ATP) synthesis. OXPHOS is carried out in the inner mitochondrial membrane by the mitochondrial respiratory chain (MRC). The MRC is composed of a series of multiheteromeric enzymes resulting from a dual genetic contribution, i.e., the nuclear and the mitochondrial genomes. Whereas the 13 MRC proteins encoded by mtDNA are all essential for OXPHOS, they make up only a tiny fraction of the total number of proteins involved in a functional MRC, most of which are in fact nucleus encoded and imported into mitochondria. As a consequence, mitochondrial disease can be due to mutations in either mtDNA or nuclear DNA genes functionally related to MRC and OXPHOS.1 In fact, a huge number of pathogenic mutations of mtDNA have been building up in the last two decades, in association with a wide spectrum of clinical presentations. Albeit at a lesser rate, substantial progress has also been made in the characterization of nuclear OXPHOS-related genes responsible for mitochondrial syndromes.2
In addition to approximately 80 MRC structural subunits, OXPHOS nuclear genes encode a remarkable number of factors that control the formation, turnover, and activity of MRC, including mtDNA biogenesis and expression, and their adaptation to tissue- and development-specific needs.3–5 The size of the mitochondrial proteome is not precisely known, but the most recent studies have set it up to more than 1400 entries.6,7 Each of these genes and proteins may be considered as a candidate for disease. Therefore, it is not surprising that in spite of the tremendous expansion of scientific and medical knowledge in the field, the genetic etiology of many mitochondrial clinical syndromes still remains elusive, the mechanisms leading to disease are poorly understood, and the very function of several factors involved in mitochondrial disorders is not, or is at best partially, known. Approximately 60% of biochemically defined OXPHOS disorders still fail to get diagnosed at the molecular level. To identify additional nuclear genes that cause mitochondrial disease phenotypes, we have embarked on a systematic, genome-wide linkage-analysis project based on homozygosity mapping of consanguineous multiplex families characterized by mitochondrial recessive traits.
The family reported in this paper (Figure 1) is composed of first-cousin parents of Bedouin origin, with an offspring of seven siblings, two of whom are affected by a peculiar, early-onset encephalomyopathy.
Figure 1.
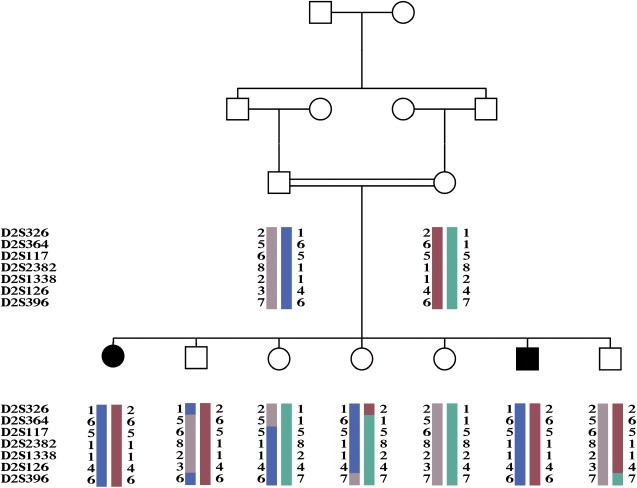
Pedigree and Haplotypes
Filled symbols indicate affected individuals. Each haplotype is indicated by a distinct color.
Patient II-1, a female, was born at term after an uneventful pregnancy by caesarean section due to nonprogression of labor. Birth weight was 3800 grams. Bilateral congenital hip dislocation was managed by abduction splinting. Marked irritability and inconsolable cry were noted since birth. Nonetheless, social and motor development was reported as normal until 7 months of age. At that time, she suffered from febrile illness and developed refractory generalized tonic-clonic convulsions. Because atypical pneumonia was suspected, she was treated with oral erythromycin. Brain MRI examination disclosed generalized symmetric atrophy (Figure 2A). In the following years, her epileptic disease persisted, the psychomotor development was markedly delayed, and left side hemiplegia ensued, with facial-nerve involvement. Brain CT scan performed at 5 yr of age revealed severe atrophic changes on the right hemisphere (Figure 2B). Because of fixed contractures, the hip and ankle tendons were surgically released at 7 yr of age. Echocardiography, abdominal ultrasound, blood count, and liver and renal functions were repeatedly normal and plasma lactate was mildly increased (2.4–3.2 mM in different samples; normal values [n.v.] < 1.8 mM). At 14 yr she obeyed simple commands in two languages, could recognize colors, and had a 20 word vocabulary. She could sit herself up and move herself relatively smoothly along the floor while sitting, but was never able to stand or walk. Her hearing was intact but the eyesight was impaired because of bilateral optic atrophy. She had left spastic hemiparesis; on the right side the muscle tone was decreased with reduced strength (3/5). Myoclonic and gelastic seizures were partly controlled by carbamazepine, valproate, and clonazepam. The EEG revealed bilateral epileptic activity, which was more prominent over the left hemisphere. Biochemical assays were first performed in isolated mitochondria at Hadassah-Hebrew University Medical Center in Jerusalem, Israel. As shown in Table 1, the specific activity of cytochrome c oxidase (COX) was reduced to 21% of the control mean (3.1 nmoles/min/mg versus a control mean of 14.8 ± 3.6). The activities of the other MRC complexes, as well as that of the pyruvate dehydrogenase complex (PDHc), were within the control range. The respiratory-chain activities were measured 2 yr later in the muscle homogenate by a second laboratory, at the Istituto Neurologico “Carlo Besta” in Milan, Italy. Again, the activity of COX, normalized to that of citrate synthase (CS), was severely reduced, to 14% of the control mean, while the activities of complex II and III were in the lower limit of the normal or just below it (Table S1 available online). The activity of complex I could not be measured because of the scarcity of the material. In spite of the isolated COX defect detected in muscle homogenate, no gross abnormality of COX and succinate dehydrogenase (SDH) histochemical reactions were reported in muscle, and the trichrome Gomori stain was normal. The activities of all MRC complexes were normal in cultured skin fibroblasts (Table S1).
Figure 2.
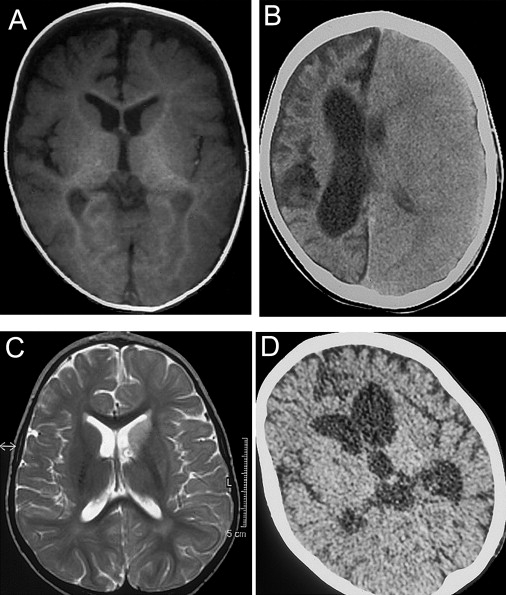
Brain Imaging
(A) Magnetic resonance imaging (MRI) T1-weighted sequence on a transverse section of supratentorial brain of patient II-1 at disease onset (7 months of age).
(B) Computerized tomography (CT) scan of the brain in patient II-1 at 5 yr.
(C) MRI T2-weighted sequence on a transverse section of supratentorial brain in patient II-6 at disease onset (1 yr of age).
(D) CT scan of the same brain section in patient II-1 at 30 months.
Table 1.
Biochemical Assays on Pt II-1 Muscle Mitochondria
| Enzyme | Specific Activity | Pt II-1 | Control Mean ± SD | % |
|---|---|---|---|---|
| Citrate Synthase | μmol/min/mg | 1.4 | 2.12 ± 0.37 | 66 |
| Succinate-ubiquinone reductase | nmol/min/ng | 56 | 77 ± 15 | 73 |
| Succinate dehydrogenase | nmol/min/ng | 206 | 327 ± 52 | 63 |
| NADH-ferricyanide reductase | μmol/min/mg | 2.7 | 4.65 ± 1.02 | 58 |
| NADH-cyt. c oxidoreductase | nmol/min/mg | 445 | 586 ± 186 | 76 |
| Succinate-cyt. c oxidoreductase | nmol/min/mg | 251 | 333 ± 100 | 75 |
| Cytochrome c oxidase | μmol/min/mg | 3.1 | 14.8 ± 3.6 | 21 |
| Mg ATPase with DNP | pmol/min/mg | 888 | 721 ± 203 | 123 |
| PDHc | nmol/min/mg | 81 | 80 ± 24 | 101 |
Patient II-6, the 6th child of the couple, was born at term, with a birth weight of 4150 gr. His early development was uneventful, and at 10 months he could sit himself up and crawl freely. After a febrile gastroenteritis at 1 yr of age, the patient experienced subacute neurological deterioration with muscle hypotonia and extrapyramidal movements, mainly on the left limbs. Brain MRI disclosed increased signal intensities on the left nucleus caudatus, globus pallidus, and crus cerebri. The lateral ventricles and the basal cysternae were of normal size (Figure 2C). A convulsive disorder, first noted at around one year, became refractory to treatment at the third year of life. The patient went into prolonged periods of status epilepticus; the EEG disclosed flattening of activity over the right hemisphere and triphasic peaked waves over the left hemisphere. A brain CT scan at 30 months revealed generalized and white-matter atrophy, more pronounced on the left basal ganglia, with bilateral dilatation of the ventricles and basal cysternae (Figure 2D). At four years, he was bed-ridden with neither communication nor any voluntary activity. There was bilateral optic atrophy and strabismus. His general muscle tone was decreased with hyperreflexia and dystonic posturing. The gag reflex was weak and he was fed via gastrostome. He had two episodes of deep-vein thrombosis at sites of central-line catheters, but blood coagulation screen was negative. Echocardiography, abdominal ultrasound, blood count, blood gases, liver and renal function tests, urinary organic acids, plasma very-long-chain fatty acids, plasma lactate, and amino acid levels were normal. CSF lactate was increased to 3.8 mM (n.v. < 1.8 mM), and the activity of COX in his lymphocytes was reported as markedly decreased. No other biological material is available for this patient. The parents gave informed consent to a biochemical and genetic screening according to the Ethical Committee of Hadassah University Hospital, Jerusalem, Israel. Blood samples were collected from parents and all seven siblings of the family; DNA was extracted by standard methods. However, 2 yr ago we lost contact with the family; therefore, we are currently unable to give an updated follow-up of the probands.
Depletion of mtDNA and mtDNA large-scale rearrangements were ruled out in skeletal muscle of patient II-1 by real-time PCR8 and long-PCR analysis,9 respectively. Pathogenic mtDNA point mutations were also excluded by sequence analysis of the entire mtDNA molecule10 from skeletal muscle of the same patient.
A nuclear-genome-wide search was performed with the ABI PRISM Linkage Mapping Set v.2.5 (Perkin Elmer, USA), characterized by over 375 markers that define a 10 cM resolution human index map. PCR reactions with fluorescently labeled primers were run under the conditions suggested by the supplier. An aliquot of each PCR reaction was run on an ABI PRISM 3100 DNA sequencer, and results were processed by GENEMAPPER software. Statistical analysis was performed on the basis of an autosomal-recessive disease with complete penetrance. Multipoint parametric linkage analysis was performed with SIMWALK2 2.91.11 The linkage analysis in the family gave a pairwise lodmax score of 2.428 on D2S1338. A region with a LOD score of ∼2 is located between recombinant markers D2S326 and D2S396 on chromosome 2q31-q36 (Figure S1, available online). No other genomic region gave positive lod scores by linkage analysis, suggesting that the 2q31-q36 contained the responsible gene.
Haplotype reconstruction revealed a homozygous condition caused by identity-by-descent alleles in the affected individuals (Figure 1). Because of their biochemical and clinical features, we assumed that the responsible protein was likely to be involved in mitochondrial metabolism and be targeted to mitochondria.
The D2S326-D2S396 interval contains 382 gene entries, some of which encode proteins that are known to target to mitochondria. Other genes within this interval encode unknown polypeptides that score high when analyzed with dedicated prediction softwares for mitochondrial targeting, including MitoProt, TargetP, and PSORT-II. Sequencing of coding regions of 14 such genes (Table S2) failed to reveal disease-segregating mutations, with the exception of a homozygous missense mutation in the gene encoding BCS1L, an assembly factor of MRC complex III (CIII).12 The mutation predicts a p.D210N amino acid change in a conserved region of the BCS1L protein. However, the presence of an isolated defect of complex IV (CIV) rather than CIII in the muscle of patient II-1, the absence of an assembly defect of CIII in either muscle or fibroblasts (Figures S2), and the detection of the p.D210N mutation in one out of 240 chromosomes from Italian control samples cast doubts about its pathogenic role. We then sequenced the exons and the intron-exon junctions of an anonymous gene entry (KIAA0971; primer sequences are available upon request) encoding a protein with a high score for mitochondrial targeting. The KIAA0971 cDNA (NM_014929) corresponds to a gene spanning 27 kb and organized in 12 exons. The open reading frame (ORF) of KIAA0971 predicts an amino acid sequence that contains a Fas-activated serine-threonine (FAST) kinase domain (NP_055744) and is therefore known as FASTKD2, for FAST kinase domain protein 2 (MIM ∗606965). However, the human FASTKD2 N terminus contains two potential translation-initiation methionine residues, at position +1 and +17 (M1, M17); both M1 and the sequence between M1 and K16 are absent in all of the nonhuman species in which FASTKD2 is present, i.e., mammals and birds, including the closest living human relatives, e.g., Pan troglodytes and Pongo pygmaeus, whereas M17 and the rest of the protein sequence are both conserved (Figure S3A). For instance, in both Pan troglodytes and Pongo pygmaeus, the potential predicted sequence corresponding to M1–M17 is identical to the human sequence, except that isoleucine is present at the apparent amino acid position 1 instead of methionine, whereas the methionine at position 17 is conserved (Figure S3B). Taken together, these observations strongly suggest that in humans as in other species, the “true” FASTKD2 protein starts from M17 rather than from M1. Moreover, when mitochondrial-targeting prediction softwares are used, the sequence starting at the alleged M1 residue scores relatively high for mitochondrial targeting, but that from M17 scores much higher (Mitoprot 72% versus 91%; TargetP 43% versus 76%). Therefore, we considered the sequence starting from M17 as the bona fide physiological human KIAA0971-encoding protein; it is hereafter referred to as FASTKD2 and consists of 694 amino acid residues, instead of the potentially translatable 710 residues. According to the nucleotide numeration of NM_014929, and considering the ATG encoding M17 as the translation initiation codon, we found a homozygous c.1246C→T nucleotide transition in both patients, whereas the two parents were heterozygous (Figures 3A). The c.1246C→T change determines the conversion of a CGA triplet, encoding R416, into a TGA termination codon. This is by definition a deleterious change (R416X) predicting the synthesis of a prematurely truncated protein, missing the 278/694 C-terminal amino acid residues (Figure 3B). The mutation was absent in 240 control alleles. The missing portion of the mutant KIAA0971 protein sequence contains two predicted transmembrane domains (403–421 and 482–500 amino acid residues), as well as the FAST kinase domain. Whereas the function of FASTKD2 is presently unknown, previous work showed FAST to be anchored to the outer face of the outer mitochondrial membrane, from where it plays an inhibitory role in the activation of the Fas-dependent apoptotic cascade.13 In order to establish experimentally the subcellular localization of FASTKD2, a sequence encoding a hemagglutinin (HA) tag of the influenza virus was added in frame at the 3′ end of the KIAA0971 ORF encoding FASTKD2, and the modified cDNA was inserted into pCDNA3.2 (Invitrogen), a mammalian expression vector. This construct was then transfected by electroporation in COS7 cells and HeLa cells. After 24 hr, cells were incubated with a monoclonal anti-HA and a polyclonal anti-mtSSB (mitochondrial single-strand-DNA binding protein) antibody. Immunofluorescence images were acquired with a confocal microscope (Biorad). As shown in Figure 4, the FASTKD2HA-specific immunofluorescence pattern obtained with the anti-HA antibody coincided with that of mtSSB, a mitochondrion-specific protein. This result demonstrates that FASTKD2HA targets to mitochondria. Mitochondria are double-membrane organelles; therefore, mitochondrial proteins can be localized in the outer mitochondrial membrane (OMM), in the mitochondrial intermembrane (MIM) space, or in the inner mitochondrial compartment (IMC), consisting of the inner mitochondrial membrane (IMM) and the mitochondrial matrix (MM). Translocation into the IMC is an energy-dependent process requiring both ATP and a mitochondrial membrane potential (ΔΨ).14 For demonstration of active mitochondrial import into the IMC, [35]S-radiolabeled in vitro-translated FASTKD2, obtained with the TNT Quick Coupled Transcription/Translation Systems (Promega), was incubated for 45 min at 37°C with freshly prepared HeLa-cell mitochondria energized by the addition of ATP in the presence or absence of 20 μM valinomycin.15
Figure 3.
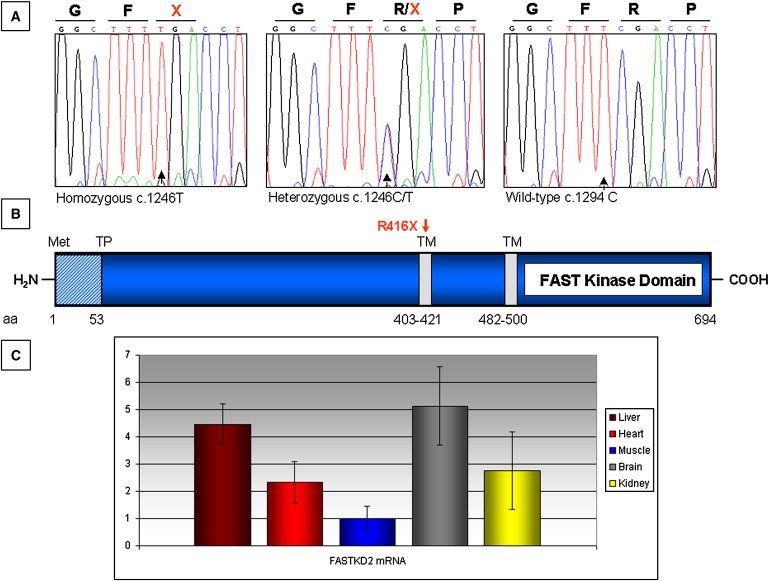
FASTKD2 Features
(A) Sequence analysis of exon 7 of KIAA0971 showing the c.1246C→T change in patient II-1 (homozygous T), her father (heterozygous C/T), and a control (homozygous C).
(B) Schematic structure of FASTKD2 protein. TP indicates a predicted cleavage site; TM indicates two putative transmembrane domains. A red arrow indicates the position of the R416X mutation.
(C) Quantitative real-time PCR of FASTKD2 mRNA relative to GAPDH mRNA in different mouse tissues. The relative amount of FASTKD2/GAPDH mRNA in muscle was taken as reference. Errors bars indicate the standard deviation (± SD).
Figure 4.
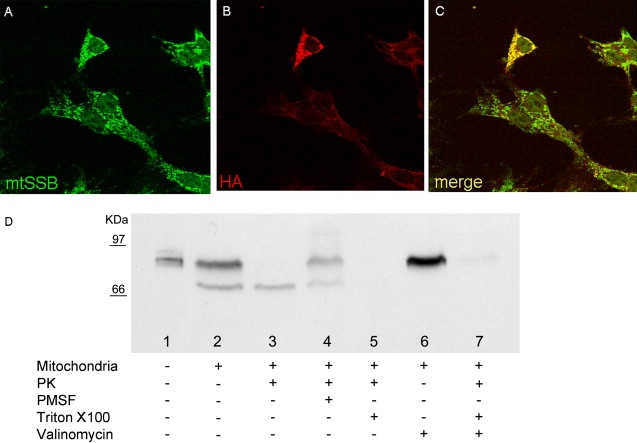
Immunofluorescence and In Vitro Import Assay
(A) Immunofluorescence specific to mtSSB.
(B) Immunofluorescence specific to FASTKD2HA.
(C) Merge. All images were taken in COS7 cells 24 hr after FASTKD2HA transfection.
(D) Import assay on isolated HeLa cell mitochondria. Lane 1, in vitro-translated FASTKD2; lane 2, FASTKD2 + mitochondria; lane 3, FASTKD2 + mitochondria + PK; lane 4, FASTKD2 + mitochondria + PK + PMSF; lane 5, FASTKD2 + mitochondria FASTKD2 + PK + Triton X-100; lane 6, FASTKD2 + mitochondria + valinomycin; and lane 7, FASTKD2 + mitochondria + valinomycin + PK.
After incubation, some samples were treated with proteinase K (PK), with or without the previous addition of PMSF, an inhibitor of PK, and/or Triton X-100, a detergent that disrupts membranes. All samples were loaded into SDS-PAGE; after fixation in 10% acetic acid and 25% isopropanol, the gel was washed for 20 min in Amplify reagent (Amersham) and layered onto a phosphorimaging screen (Biorad). After overnight exposure, autoradiography was carried out in a Molecular Imager apparatus (Biorad).
Upon exposure to freshly prepared energized mitochondria, two radiolabeled bands were visualized (Figure 4D), a larger band corresponding to the full-length in vitro-translated FASTKD2 and a shorter one corresponding to a mature protein species lacking an N-terminal mitochondrial target sequence. A potential cleavage site of the 79 kDa FASTKD2 precursor protein by the mitochondrial matrix protease (MMP) is present between M52 and Q53 and predicts the mature imported protein from the FASTKD2 precursor to be composed of 642 amino acid residues for a MW of approximately 73 kDa. The two bands shown in lane 2 correspond nicely to the size of either FASTKD2 protein species. Treatment with PK determined the complete digestion of the upper band in intact mitochondria, whereas a substantial amount of the lower band was left intact, as expected to occur for a protein species imported into, and protected by, the IMC. This digestion is specific and can be blocked by the addition of PMSF. After treatment of the mitochondrial membranes with Triton X-100, exposure to PK determined the complete digestion of both protein species, again indicating that the shorter polypeptide was located inside the IMC of the organelle. As reported for other mitochondrial IMC proteins, the import process was dependent on the presence of a mitochondrial proton-motive force (ΔΨ), because it was blocked by treatment of the mitochondrial suspension with valinomycin, an ionophore drug that dissipates ΔΨ. These results demonstrate that FASTKD2 is indeed imported into the IMC through the ΔΨ-dependent TOM-TIM import machinery, which targets proteins to either the IMM or the MM.16 The presence of two predicted transmembrane domains suggests that FASTKD2 is anchored to the IMM, and the full protection from PK digestion indicates that it may project toward the IMC.
Electronic data from arrays available in public databases (Genecards) shows that FASTKD2 is ubiquitously expressed. To measure FASTKD2 gene expression experimentally, we created specific primers for the mouse ortholog (NM_172422). Then we extracted RNA from different mouse tissues, and first-strand cDNA was synthesized by standard methods. We analyzed by real-time PCR the abundance of the FASTKD2 transcript, normalized via the GAPDH transcript as reference. FASTKD2 transcript was expressed ubiquitously, but in variable amounts from tissue to tissue, with skeletal muscle < heart ≤ kidney < liver ≤ brain (Figure 3C).
Because nuclear genes coding for assembly factors of COX, such as SURF-1, are known to cause CIV deficiency, a role for FASTKD2 in COX assembly was evaluated by blue-native gel electrophoresis (BNGE)-western-blot analysis.17 However, we failed to demonstrate abnormality in the assembly status and abundance of COX by using an anti-COX1 antibody complex, in muscle of patient II-1 (Figure S2). Therefore, whether the COX defect is a direct and specific consequence of, or is rather part of a more general mitochondrial dysfunction induced by, the abolishment of FASTKD2 remains an open issue for future investigation.
The presence of a FAST domain in FASTKD2 and the involvement of FAST in the apoptotic process prompted us to evaluate nuclear fragmentation, taken as an index of apoptotic response,18 in mutant versus control fibroblasts incubated with 1 mM staurosporine for 3 hr (Figure 5A). Staurosporine, a potent protein kinase C inhibitor, is known to induce apoptosis through a mitochondria-mediated pathway19 and to cause oxidative stress through mitochondrion-generated ROS in several cells.20 As shown in Figure 5B, the percentage of fragmented nuclei in mutant cells was significantly lower than that of control cells (Chi-square value = 0.003). This result suggests a specific role of FASTKD2 in the regulation of mitochondrial apoptosis, because it does not seem to depend upon the activity of the respiratory-chain complexes that were all in the normal range in mutant fibroblasts. In support of this hypothesis stands the observation that overexpression of recombinant FASTKD2HA in HeLa or COS7 cells consistently led to shrinkage of the cell body, followed by nuclear fragmentation and eventually apoptotic death (Figure 5C). As a consequence, we were unable to generate stably overexpressing FASTKD2HA cell lines. Whether this drastic effect is due to a specific function of FASTKD2, or to a generic toxic effect due to damage of the inner mitochondrial membrane, remains to be elucidated.
Figure 5.
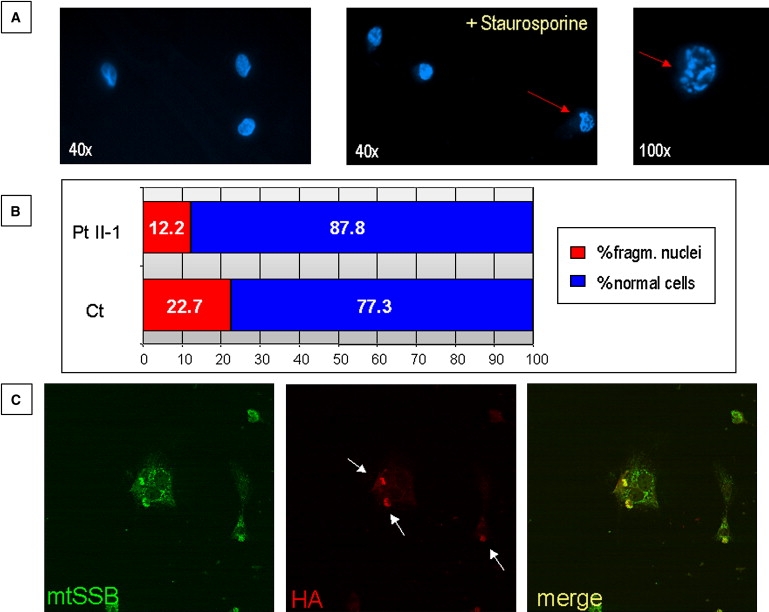
Staurosporine-Induced Apoptosis
(A) Hoechst staining of fibroblasts before and after staurosporine treatment. Red arrows indicate cells with fragmented nucleus.
(B) Quantification of cells with fragmented nucleus in control (Ct) and patient (Pt II-1) fibroblasts.
(C) Immunofluorescence images of COS7 cells 48 hr after FASTKD2HA transfection.
The clinical and MRI findings of the FASTKD2-associated disease are rather unusual for mitochondrial disorders. Combination of hemiplegia and epilepsy may be reminiscent of mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS, MIM #540000),21 but other features were missing, including RRF and lactic acidosis. Other signs were peculiar of this syndrome and resembled some key features of Rasmussen's encephalopathy, an allegedly immunological condition characterized, like our probands, by infantile onset, intractable epilepsy, emispheric atrophy with dilatation of the ipsilateral ventricle system, and hemiparesis with a slowly progressive, relentless course.22 However, we failed to identify mutations in the KIAA0971 sequence in five consecutive patients with Rasmussen's encephalopathy, suggesting that the KIAA0971-associated syndrome is a genetic condition that phenocopies but does not account for sporadic Rasmussen's encephalopathy.
In conclusion, we found a previously unreported gene mutated in a peculiar mitochondrial COX-defective encephalomyopathy. We demonstrated that the responsible protein, FASTKD2, is localized in the IMC, where it may play a still-unknown role in apoptosis. Although FASTKD2 is not directly involved in COX assembly, its ablation is indeed associated with COX deficiency in skeletal muscle. The elucidation of the function of FASTKD2 is made difficult by the likely rarity of the clinical condition linked to its disruption (to our knowledge, this is the first and so far only family with a FASTKD2 mutation) and by the difficulty to establish cell lines stably expressing recombinant FASTKD2HA. In addition, FASTKD2 seems to be present only in mammals and birds, whereas no orthologs can be found in other animals, plants, and lower eukaryotes. This prevents the possibility to create models in user-friendly organisms such as yeast, worm, or fly. The restriction of FASTKD2 to relatively recent classes of animals, notably those endowed with homeothermy, may suggest a role for FASTKD2 in the finely-tuned regulation of heath production by mitochondria. The use of siRNA in cell culture, and the creation of a murine knockout model for the FASTKD2 gene, which is under way in our laboratory, will help clarify the function of this new, elusive component of the IMC and give a mechanistic interpretation to the mitochondrial syndrome affecting humans.
Acknowledgments
This work was supported by the Pierfranco and Luisa Mariani Foundation, Fondazione Telethon-Italy grant number GGP07019, the Italian Ministry of University and Research (FIRB 2003 - project RBLA038RMA), the Italian Ministry of Health RF2006 ex 56/05/21, Marie Curie intra-European fellowship (FP6-2005-Mobility-5) number 040140-MAD, and MITOCIRCLE and EUMITOCOMBAT (LSHM-CT-2004-503116) network grants from the European Union framework program 6.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ensembl, http://www.ensembl.org
GeneCards, http://www.genecards.org/index.shtml
Mitoprot, http://ihg2.helmholtz-muenchen.de/ihg/mitoprot.html
NCBI database, http://www.ncbi.nlm.nih.gov
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Zeviani M., Di Donato S. Mitochondrial disorders. Brain. 2004;127:2153–2172. doi: 10.1093/brain/awh259. [DOI] [PubMed] [Google Scholar]
- 2.Rötig A., Lebon S., Zinovieva E., Mollet J., Sarzi E., Bonnefont J.P., Munnich A. Molecular diagnostics of mitochondrial disorders. Biochim. Biophys. Acta. 2004;1659:129–135. doi: 10.1016/j.bbabio.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 3.Zeviani M. The expanding spectrum of nuclear gene mutations in mitochondrial disorders. Semin. Cell Dev. Biol. 2001;12:407–416. doi: 10.1006/scdb.2001.0278. [DOI] [PubMed] [Google Scholar]
- 4.Spinazzola A., Zeviani M. Disorders of nuclear-mitochondrial intergenomic communication. Biosci. Rep. 2007;27:39–51. doi: 10.1007/s10540-007-9036-1. [DOI] [PubMed] [Google Scholar]
- 5.Zeviani M., Carelli V. Mitochondrial disorders. Curr. Opin. Neurol. 2007;20:564–571. doi: 10.1097/WCO.0b013e3282ef58cd. [DOI] [PubMed] [Google Scholar]
- 6.Calvo S., Jain M., Xie X., Sheth S.A., Chang B., Goldberger O.A., Spinazzola A., Zeviani M., Carr S.A., Mootha V.K. Systematic identification of human mitochondrial disease genes through integrative genomics. Nat. Genet. 2006;38:576–582. doi: 10.1038/ng1776. [DOI] [PubMed] [Google Scholar]
- 7.Pagliarini D.J., Calvo S.E., Chang B., Sheth S.A., Vafai S.B., Ong S.E., Walford G.A., Sugiana C., Boneh A., Chen W.K. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spinazzola A., Viscomi C., Fernandez-Vizarra E., Carrara F., D'Adamo P., Calvo S., Marsano R.M., Donnini C., Weiher H., Strisciuglio P. MPV17 encodes an inner mitochondrial memmbrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat. Genet. 2006;38:570–575. doi: 10.1038/ng1765. [DOI] [PubMed] [Google Scholar]
- 9.Coulter-Mackie M.B., Applegarth D.A., Toone J., Gagnier L. A protocol for detection of mitochondrial DNA deletions: Characterization of a novel deletion. Clin. Biochem. 1998;31:627–632. doi: 10.1016/s0009-9120(98)00074-5. [DOI] [PubMed] [Google Scholar]
- 10.Bugiani M., Invernizzi F., Alberio S., Briem E., Lamantea E., Carrara F., Moroni I., Farina L., Spada M., Donati M.A. Clinical and molecular findings in children with complex I deficiency. Biochim. Biophys. Acta. 2004;1659:136–147. doi: 10.1016/j.bbabio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 11.Sobel E., Sengul H., Weeks D.E. Multipoint estimation of identity-by-descent probabilities at arbitrary positions among marker loci on general pedigrees. Hum. Hered. 2001;52:121–131. doi: 10.1159/000053366. [DOI] [PubMed] [Google Scholar]
- 12.Petruzzella V., Tiranti V., Fernandez P., Ianna P., Carrozzo R., Zeviani M. Identification and characterization of human cDNAs specific to BCS1, PET112, SCO1, COX15, and COX11, five genes involved in the formation and function of the mitochondrial respiratory chain. Genomics. 1998;54:494–504. doi: 10.1006/geno.1998.5580. [DOI] [PubMed] [Google Scholar]
- 13.Li W., Simarro M., Kedersha N., Anderson P. FAST is a survival protein that senses mitochondrial stress and modulates TIA-1-regulated changes in protein expression. Mol. Cell. Biol. 2004;24:10718–10732. doi: 10.1128/MCB.24.24.10718-10732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neupert W. Protein import into mitochondria. Annu. Rev. Biochem. 1997;66:863–917. doi: 10.1146/annurev.biochem.66.1.863. [DOI] [PubMed] [Google Scholar]
- 15.Fernandez-Vizarra E., Lopez-Perez M.J., Enriquez J.A. Isolation of biogenetically competent mitochondria from mammalian tissues and cultured cells. Methods. 2002;26:292–297. doi: 10.1016/S1046-2023(02)00034-8. [DOI] [PubMed] [Google Scholar]
- 16.Wiedemann N., Frazier A.E., Pfanner N. The protein import machinery of mitochondria. J. Biol. Chem. 2004;279:14473–14476. doi: 10.1074/jbc.R400003200. [DOI] [PubMed] [Google Scholar]
- 17.Nijtmans L.G., Henderson N.S., Holt I.J. Blue native electrophoresis to study mitochondrial and other protein complexes. Methods. 2002;26:327–334. doi: 10.1016/S1046-2023(02)00038-5. [DOI] [PubMed] [Google Scholar]
- 18.Zhivotosky B., Orrenius S. Assessment of apoptosis and necrosis by DNA fragmentation and morphological criteria. In: Bonifacino J.S., Dasso M., Hardford J.B., Lippincott-Schwartz J., Yamada K.M., editors. Current Protocols in Cell Biology. John Wiley & Sons; New York: 2001. Chapter 18, Unit 18.3. [DOI] [PubMed] [Google Scholar]
- 19.Yang J., Liu X., Bhalla K., Kim C.N., Ibrado A.M., Cai J., Peng T.I., Jones D.P., Wang X. Prevention of apoptosis by Bcl-2: Release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 20.Cai J., Jones D.P. Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. J. Biol. Chem. 1998;273:11401–11404. doi: 10.1074/jbc.273.19.11401. [DOI] [PubMed] [Google Scholar]
- 21.Pavlakis S.G., Phillips P.C., DiMauro S., De Vivo D.C., Rowland L.P. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: A distinctive clinical syndrome. Ann. Neurol. 1984;16:481–488. doi: 10.1002/ana.410160409. [DOI] [PubMed] [Google Scholar]
- 22.Bien C.G., Granata T., Antozzi C., Cross J.H., Dulac O., Kurthen M., Lassmann H., Mantegazza R., Villemure J.G., Spreafico R., Elger C.E. Pathogenesis, diagnosis and treatment of Rasmussen encephalitis: A European consensus statement. Brain. 2005;128:454–471. doi: 10.1093/brain/awh415. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.