

Abstract
Technological and scientific advances, stemming in large part from the Human Genome and HapMap projects, have made large-scale, genome-wide investigations feasible and cost effective. These advances have the potential to dramatically impact drug discovery and development by identifying genetic factors that contribute to variation in disease risk as well as drug pharmacokinetics, treatment efficacy, and adverse drug reactions. In spite of the technological advancements, successful application in biomedical research would be limited without access to suitable sample collections. To facilitate exploratory genetics research, we have assembled a DNA resource from a large number of subjects participating in multiple studies throughout the world. This growing resource was initially genotyped with a commercially available genome-wide 500,000 single-nucleotide polymorphism panel. This project includes nearly 6,000 subjects of African-American, East Asian, South Asian, Mexican, and European origin. Seven informative axes of variation identified via principal-component analysis (PCA) of these data confirm the overall integrity of the data and highlight important features of the genetic structure of diverse populations. The potential value of such extensively genotyped collections is illustrated by selection of genetically matched population controls in a genome-wide analysis of abacavir-associated hypersensitivity reaction. We find that matching based on country of origin, identity-by-state distance, and multidimensional PCA do similarly well to control the type I error rate. The genotype and demographic data from this reference sample are freely available through the NCBI database of Genotypes and Phenotypes (dbGaP).
Introduction
Our capacity to measure human genetic variation and apply it to address scientific questions related to evolution,1 population structure,2,3 and interindividual phenotypic variation4 is expanding at an increasing rate. At least as important as the technologies to measure genetic variation is the availability of suitable samples and their descriptive data. In the past, the resources to conduct large-scale genetic investigations have been restricted to a relatively small number of well-funded academic and commercial groups, limiting the access to the raw data. However, recent changes in attitudes in the scientific community, on ethical review boards, and at funding agencies are leading to greater openness in sharing genetic data with the intent to improve opportunities for discovery through their creative use and careful integration.5,6
In 2005, GlaxoSmithKline initiated the Population Reference Sample (POPRES) project with the goal of bringing together a DNA sample set that would be extensively genotyped in order to support a variety of efforts related to pharmacogenetics research. We found that the application of pharmacogenetics research associated with drug development could be hampered by (1) lack of readily available population controls for adequately powered study designs, (2) high costs of conducting highly exploratory genome-wide studies, (3) extended study timelines that may not meet clinical development needs, and (4) lack of samples representative of the multinational patient populations from which the prevalence of pharmacogenetically relevant polymorphisms can be estimated. The POPRES project was carried out to begin addressing these issues, with the further objective of making the resulting genotypic and demographic data publicly available to help drive development in the broader genetics research community.
There are many projects, especially in pharmacogenetics, wherein the sample collection is focused on the acquisition of cases. One important example is the identification and collection of cases with adverse drug reactions (ADRs) through postmarketing surveillance. In these situations, the acquisition or selection of a suitable set of controls can add a substantial burden to the experimental process. Having a large collection of DNA samples previously scrutinized and genetically characterized would facilitate the search for genetic risk factors. This is particularly true if the case samples to be matched with controls are not of northern European origin, which is the background of most genome-wide studies published to date and publicly available. The availability of key demographic, phenotypic, and clinical data for the selected subjects would enhance their application.
Investigations into genetic risk factors underlying ADRs are highly exploratory, because there is generally little a priori evidence to support a genetic hypothesis. The availability of population controls with existing genotype data that could be matched to the cases substantially lowers the cost and time to conduct this research and could facilitate exploratory efforts. For ADRs that have relatively low frequency, there is little power lost in the use of population controls versus drug-treated, clinically matched controls.7 A large resource of genotyped controls would also allow for more careful matching of what can be genetically diverse cases to controls on the basis of their patterns of genetic variation.8
Many pharmacogenetic studies utilize samples collected in clinical trials, which are becoming increasingly global and diverse in their origin.9 Therefore, in addition to the value of genome-wide genotype data for exploratory scans, the availability of DNA for the subjects included in the POPRES initiative allows for measurement of variants that are of particular interest to pharmacogenetic research, as well as estimation of their population-specific relative frequencies. This can be useful for predicting population-specific ADR risks or possible variability in drug response. Furthermore, population genetic studies of more diverse samples, such as POPRES, provide important information about the similarity or differentiation of these populations,10 informing future study designs and interpretation.
The availability of a densely genotyped population reference sample will increase opportunities for many areas of genetics research, by us and others, by providing a well-characterized, readily available set of samples representative of the populations of interest from which to draw controls and estimate population parameters of interest. Furthermore, such resources will foster development of statistical methods and analysis strategies and provide a resource for innovative population-genetics research. In this paper, we describe the collections currently comprising 5,886 POPRES subjects, genotyping and analysis methods used in preparing the data being provided to the public domain, and selected data-analysis results. Lastly, we present an example application matching controls to a small set of ADR cases.
Material and Methods
The subjects included in the POPRES initiative are derived from ten collections. Each collection is briefly described. Where available, see accompanying references for further collection details. All subjects included in this study were either collected in an anonymous fashion or have been multiply coded by the collecting institution as well as the POPRES data managers (see 11 for definitions).
UCSF African Americans
African American subjects were recruited across the United States to serve as controls for studies of multiple sclerosis (MS) genetic susceptibility conducted at the University of California, San Francisco.12 In general, individuals were invited to participate in the study by the probands and constitute primarily spouses or friends of MS patients. In addition to the ability to give consent and willingness to participate, inclusion criteria included male and female gender, age of more than 16 years, no personal or familial history of MS, and no history of autoimmunity. Exclusion criteria included chronic diseases and recreational drug use. All study participants were self-reported African Americans, but European ancestry was documented on the basis of genotyping results of 186 informative single-nucleotide polymorphisms (SNPs).13
Healthy Japanese Controls
Participants were recruited through the James Lance GlaxoSmithKline Medicines Research Unit in Sydney, Australia. Eligibility criteria included self-described Japanese ethnic background, age of more than 20 years, and freedom from chronic disease. Blood samples were collected in an anonymous fashion, i.e., no identifiers were associated with the biological sample that could associate it back with the participant. Sex is the only personal information recorded for each subject.
Healthy Taiwanese Controls
Participants were recruited through the Tri-Service General Hospital in Taipei, Taiwan. Eligibility criteria included self-described ethnicity as Han Chinese, age of at least 20 years, and freedom from chronic disease. Blood samples were collected in an anonymous fashion. Sex is the only personal information recorded for each subject.
Healthy Mexican Controls
Participants were recruited through a hospital-based clinic in Guadalajara, Mexico. Eligibility criteria included self-described ethnicity as Mexican or Hispanic, age of at least 18 years, and freedom from chronic disease. Blood samples were collected in an anonymous fashion. Sex is the only personal information recorded for each subject.
Healthy Caucasian Controls
Participants were recruited through (1) the Royal Adelaide Hospital in Adelaide, Australia; (2) Duke University, North Carolina, USA; and (3) the University of Ottawa Heart Institute, Ottawa, Canada. Inclusion criteria included self-described ethnicity as Caucasian, age of at least 18 years, and healthiness. Here, healthy individuals are those who are free from clinical cardiac, pulmonary, gastrointestinal, hepatic, renal, hematological, neurological, and psychiatric disease as determined by history, physical examination, or screening investigations. Blood samples were collected in an anonymous fashion. Sex is the only personal information recorded for each subject.
London Life Sciences Population Study
The LOLIPOP study is a population-based study of Indian Asians and European whites, aged 35–75 years, identified from the lists of 58 general practitioners in West London.14 To date, 938 northern Europeans and 431 Indian Asians from this collection are included in POPRES. Although extensive cardiovascular-related phenotypic data were collected on these participants, the POPRES database only includes nonidentifying demographic information: age at collection, self-identified race and/or ethnicity, and country of birth.
CoLaus Study, Lausanne, Switzerland
This is a population-based study of European subjects drawn from Lausanne, Switzerland, through the Centre Hospitalier Universitaire Vaudois (CHUV) University Hospital.15 From this collection, 2,809 subjects were included in POPRES. Although extensive phenotypic data were collected on these participants, the POPRES database only includes nonidentifying demographic information: age at collection, self-identified race and/or ethnicity, native language, country of birth, and parental and grandparental countries of birth.
Duke Healthy Volunteers
Healthy volunteers were recruited from the Duke and North Carolina State University campuses. Volunteers were to be aged between 18 and 90 years and have no known cognitive impairments. All races and ethnicities were included. Five hundred and eighty-six subjects from this collection were included in POPRES. Only nonidentifying personal demographic information was made available, including and limited to age at collection, self-identified race and/or ethnicity, and sex.
Informed Consent and Ethical Approval
All participants in the component studies that contributed to POPRES provided written informed consent for the use of their DNA in genetic studies. The informed-consent form was different for each study, some providing more explicit descriptions of the variety of ways that genotype data derived from the sample may be used than others. Informed consents are available through the dbGaP submission. The informed consents of the Healthy Caucasian Controls collections were the most extensive. Given the anonymized nature of the collection, these samples were included in POPRES without need for further ethical review. Specific ethical review board approval for the controlled release of deidentified genotype data was sought for the Healthy Taiwanese Controls, Healthy Japanese Controls, Healthy Mexican Controls, CoLaus, and Duke collections. All were granted, with the exception of the Healthy Taiwanese Controls, which will not be publicly released. The nature of the original consent and ethical review board approval for the LOLIPOP collection was sufficient for the current usage.
Genotyping
Genotyping was performed on the Affymetrix (Mountain View, CA) GeneChip 500K Array Set with the published protocol for 96-well-plate format. Samples were genotyped in nine batches over a period of 19 months (Table 1) with a 2%–3% sample duplicate rate to help assess genotype data quality. The CoLaus and LOLIPOP collections were genotyped in multiple batches. All other collections were typed within a single batch. Batch information for each subject is available with the genotype data.
Table 1.
Summary of the Collections Included in the POPRES Study
| Region | Africa | East Asia | South Asia | Latin America | Europe | Mix | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Study | UCSF African American | Japanese | Taiwanese | LOLIPOP | Mexican | USA | Canadian | Australian | LOLIPOP | CoLaus | Duke |
| Collection Site | United States | Sydney, Australia | Taiwan | London, England | Guadalajara | North Carolina | Ottawa | Adelaide | London, England | Lausanne, Switzerland | North Carolina |
| Collection Type | Healthy | Healthy | Healthy | Population | Healthy | Healthy | Healthy | Healthy | Population | Population | Healthy |
| Sample Size | 436 | 106 | 174 | 431 | 205 | 27 | 105 | 69 | 938 | 2809 | 586 |
| 500K, Initial QC | 346 | 73 | 109 | 360 | 149 | 27 | 105 | 69 | 598 | 2509 | 490 |
| 500K, Final QC | 346 | 73 | 108 | 359 | 112 | 27 | 105 | 69 | 481 | 2507 | - |
| Genotyping batcha | 9 | 1 | 1 | 7 | 1 | 2 | 2 | 2 | 3, 7 | 4, 5, 6 | 8 |
| Age (min/med/max) | 18/45/81 | >20 | ≥20 | 35/50/74 | ≥18 | ≥18 | ≥18 | ≥18 | 23/54/75 | 35/52/75 | 18/22/79 |
| Sex (F:M) | 279:157 | 62:44 | 84:90 | 121:310 | 93:112 | 18:9 | 63:42 | 47:22 | 213:724b | 1508:1301 | 331:255 |
| 500K, Initial QC | 223:123 | 44:29 | 48:61 | 103:257 | 69:80 | 18:9 | 63:42 | 47:22 | 184:414 | 1350:1159 | 285:205 |
| 500K, Final QC | 223:123 | 44:29 | 47:61 | 103:256 | 46:66 | 18:9 | 63:42 | 47:22 | 180:301 | 1348:1159 | - |
| Call Rate (per SNP) | |||||||||||
| Median | 0.99 | 0.99 | 0.98 | 0.99 | 0.98 | 1.00 | 0.98 | 0.99 | 0.99 | 0.99 | - |
| 95th %ile | 0.94 | 0.85 | 0.87 | 0.91 | 0.87 | 0.85 | 0.89 | 0.87 | 0.92 | 0.93 | - |
Batch defined by month that genotyping completed: 1, Nov. 2005; 2, Mar. 2006; 3, Aug. 2006; 4, Sep. 2006; 5, Nov. 2006; 6, Dec. 2006; 7, Jan. 2007; 8, Mar. 2007; and 9, May 2007.
One subject was missing sex information and failed genotyping (i.e., sex could not be inferred).
The dynamic model (DM) genotype-calling algorithm uses perfect-match and mismatch probe intensities to call genotypes for individual arrays. DM was used to measure raw experiment quality. Individual arrays that failed to achieve a 90% DM call rate (at p = 0.26) were generally reattempted in genotyping by rehybridization. Duplicate concordance for the StyI arrays was distinctly lower than that for the NspI arrays on four plates in batch 7 genotyping of the LOLIPOP collection. The samples on these four plates were regenotyped on the StyI array with fresh DNA aliquots and with the Affymetrix protocol performed in its entirety.
A series of identity checks was performed. Samples were removed if reported gender was inconsistent with X-linked genotypes. Samples with no reported gender were left in the dataset, and their gender was inferred from the genetic data. In addition to the 500K genotyping, a subset of 88 SNPs was typed with the single base chain extension (SBCE) assay16 for all subjects (43 on NspI, and 45 on StyI). The SBCE genotypes were compared with those called by DM on the 500K SNP panel. Samples less than 90% concordant between the SBCE data and the Affymetrix 500K SNP panel data on a single array were removed from the dataset.
Final genotype calling was performed with the Bayesian robust linear model with Mahalanobis distance classifier algorithm (BRLMM). Only arrays passing an 85% DM call-rate threshold were input into BRLMM. BRLMM is a clustering algorithm that requires batches of arrays to make calls. Arrays were batched together for BRLMM by plate, with a minimum batch size of fifty. Affymetrix Power Tools v1.4 was used to run BRLMM, with the maximum confidence threshold set to 0.3. Defaults were used for all other parameters. Any inconsistent genotypes for duplicated samples were removed. Samples were considered successfully genotyped if they passed identity checks and achieved a minimum 95% BRLMM call rate on both arrays after removal of inconsistent genotypes.
There are 500,566 unique markers included in the genotyping array. A set of 3,247 markers identified as mapping to multiple sites on the genome were excluded, leaving 497,625 for subsequent analysis.
Quality Control
Genome-wide genotyping with an Affymetrix 500K SNP panel was attempted for all subjects over an 18 month period of time. Two rounds of initial quality control were performed. The first included standard checks. Only subjects with call rates greater than 95% for both NspI and StyI chips and confirmed genotype-sex concordance were retained. Relatedness among subjects was evaluated on the basis of identity-by-descent estimates. This identified 48 closely related subjects, primarily from the Mexican cohort, that were subsequently excluded. For the LOLIPOP collection, it was determined after genotyping that some subjects received for the POPRES initiative were not a random sample of the larger LOLIPOP collection. Rather, it consisted of subjects that had been collected early in the project, which had an initial focus on recruiting cardiovascular-disease-related patients. A subset of subjects were subsequently selected for inclusion in POPRES with a 6% coronary heart disease (CHD) rate that brought CHD-related endpoints in the dataset in line with LOLIPOP overall. This resulted in the removal of 125 subjects. Preliminary principal-component analysis (PCA, see below) within Europeans identified 111 subjects from the European LOLIPOP sample on two genotyping plates strongly correlated with scores on the second component, suggesting a problem with genotype data quality. These subjects were excluded. Two additional subjects were excluded because they had highly negative inbreeding F scores, which were calculated with PLINK.17 The F scores were twice the magnitude of all other samples, indicating potential contamination. A total of 4,835 subjects (82%) passed this first round of checks. The second round of quality control included further PCA to identify subjects with data quality concerns or misreported genetic ancestry. Four thousand, one hundred, and eighty-seven subjects (72%) passed the second round of checks. We note that the Duke data were not available during these further quality-control measures and are not included in subsequent analyses. However, the collection is described herein and the genotype data are publicly available.
With the set of subjects that passed both initial rounds of quality control, we carried out a series of more stringent quality-control steps in an effort to further reduce the likelihood of genotyping errors that could negatively influence genetic studies using these data. First, to overcome concerns that the small batch sizes used to cluster and call genotypes in the original data set could bias the results (e.g., 4), a high-performance computing system was used to apply BRLMM to the entire set of files, including data from sample duplicates, for the NspI and StyI chips separately. We refer to the genotypes generated by this combined calling strategy as “pooled” genotypes and those produced in small groups of samples as “batched” genotypes. The quality of the pooled versus batched genotype calls were assessed by comparison of the sample duplicate concordance and call rates of each (Figure S1 available online). We found that with the BRLMM quality threshold of 0.3, the batched genotypes resulted in higher duplicate concordance than the pooled calls (99.66% versus 99.56%) as well as higher call rates (97.66% versus 95.12%). For this reason, we relied on the batched calls for all reported analyses.
We then evaluated the influence of the BRLMM quality threshold on duplicate concordance and its relationship to genotype call rate (Figure S1). As expected, duplicate concordance increased and call rate decreased as the quality threshold decreased from 0.5 toward zero. On the basis of the improvement in heterozygote concordance we observed (0.98 to 0.99) by decreasing the quality threshold from the initial value of 0.3 to 0.2 with only a modest corresponding decrease in call rates (0.96 to 0.93), we selected the 0.2 threshold for this more restricted data set.
We then excluded 54,191 SNPs (10.8%) that had three or more discrepancies between the batched and pooled calls or that exhibited a batch call rate below 90% (Table S1). These pruned SNPs showed lower average duplicate-chip concordance rates (96.6% versus 99.8%) and higher levels of Hardy-Weinberg disequilibrium (20% versus 5% of SNPs with heterozygosity levels above the p < 0.001 threshold). The remaining SNPs have an average call rate of 97.7%, and an analysis of individuals for which duplicate chips were run shows a concordance rate of 99.8%. Our selection of a 90% threshold contrasts with the 95% call rate applied in most other studies using the Affymetrix 500K panel. However, because we use a more stringent confidence threshold (0.2 versus the BRLMM default value of 0.5), we achieve higher genotype quality (duplicate concordance) with lower call rates (see Figure S1).
Principal-Component Analysis
Principal-component analysis was conducted with the smartpca software18 and default settings with no outlier removal. Analysis was carried out after the removal of some apparently related individuals (high identity-by-descent estimates), and individuals were identified as outliers in preliminary PCA runs based on regional subsets of the data (e.g., Europe, East Asia, etc). Furthermore, because of the large overrepresentation of UK and Swiss individuals, we randomly selected a subset of 200 UK and 125 French-speaking Swiss subjects. This resulted in a sample of 3,082 POPRES subjects. As a reference, and to provide data from Africans in the analysis, we included genotype data (release 23) on the same subset of SNPs from 207 unrelated subjects from the four core HapMap samples19: Yorubans from Ibadan, Nigeria; Japanese from the Tokyo area; Chinese from Beijing; and Centre d'Etude du Polymorphism Humain (CEPH) Europeans from Utah (CEU). To reduce the linkage disequilibrium between markers, we first used the PLINK software to remove all markers with genotypic r2 greater than 0.8, calculated in sliding windows 50 SNPs wide, shifted and recalculated every five SNPs. This process reduced the number of SNPs analyzed to 286,930.
Previous studies have shown that regions with structural variation such as inversions can strongly influence PCA results.4,20 We found from previous work (data not shown) that plots showing the per-SNP correlation between individual genotype scores (0, 1, or 2) and individual PC coordinates are a useful diagnostic for identifying PCs that might be influenced by long-range LD regions. For instance, in the initial analysis of European samples,21 known inversions on chromosomes 8p23 and 17q21 appear as peaks in the correlation plots for some of the lower PCs (e.g., PC 3). (Alternatively, we could have plotted the absolute values or the square of SNP loadings from the PCA, but here we used the correlation-based approach because much of this work was done before the release of recent versions of smartpca that provide the SNP loadings.) The only strong peaks in the correlation plots within the top seven PCs were for the approximately north-to-south European principal component, which exhibited two large peaks, with p values of association as extreme as 10−40 to 10−100. One of these peaks located at 134.6–137.6 Mb on chromosome 2 centered on the LCT gene (136.4–136.5 Mb). The other peak on chromosome 6 at 29.1–32.8 Mb contained the major histocompatibility complex (MHC) complex, including the HLA-A, -B, -C, -DR, and -DQ genes.
To assess whether such aberrant regions might influence the PCA results and obscure genome-wide patterns, we performed a second PCA analysis where we first removed SNPs from regions surrounding putative peaks of correlation. Although none of the other first seven PCs, aside from PC 5, showed a strong peak of correlated markers, we conservatively removed all SNPs within 2 Mb of a marker highly correlated with any of the first ten principal components. We defined the threshold for calling highly correlated SNPs as being within the top 0.2% of r2 values for correlations of markers against the given principal component. This process excluded over half of the markers (including the lactase and MHC regions mentioned above), leaving 226,211 SNPs for the subsequent analysis, and resulted in a final set of 143,893 SNPs after excluding markers by the procedure based on the sliding-window-based pruning step described above. Using this more stringent set of SNPs, we reran PCA on the same set of individuals and found that, aside from negation of the eigenvectors, the PCs revealed the same structure as when the full set of markers was used, and the first seven principal components had a correlation greater than 0.98 between the two runs. This suggests that the initial PCA was capturing genome-wide patterns of variation rather than patterns localized to specific sets of markers, and the peaks of correlation observed were simply particular sets of markers that happened to be correlated with the population structure (such as in the case of the lactase gene with PC 5, the roughly north-to-south European PC). Although the results were similar between the two runs, we present the results from the second of the two PCA runs.
Case-Control Matching and Genome-wide Analysis
We performed four different methods of case-control matching to assess their impact on type I error rates in an example motivated by the search for major genetic risk factors for adverse drug reactions. Twenty-two HIV-positive patients of European origin with clinically diagnosed abacavir-associated hypersensitivity reaction were genotyped with the Affymetrix 500K SNP panel as previously described.7 One case was dropped because of very low genotyping efficiency (<85%). Ten controls were matched to each case by four methods: (1) continental origin, selecting Europeans from the United Kingdom, (2) country of sampling or country of birth (if available), (3) minimizing pairwise identity by state (IBS) distance, and (4) minimizing pairwise distance among selected principal components.
Continent of origin matching was carried out with POPRES subjects of self-identified European origin who were collected in, or reported to have ancestry from, England or the United Kingdom. Country matching was carried out by selection of sex-matched controls from the same country of origin as the cases (Table S2). When there were excess numbers of controls available, ten were randomly selected for each case. Controls from adjoining countries were selected when there were insufficient numbers of controls available from the case countries. IBS matching was carried out by estimation of the pairwise IBS distance from each case to each POPRES subject that satisfied the quality-control criteria described above. IBS estimation was carried out with PLINK v1.01,17 excluding 58,089 SNPs found within genomic regions highly correlated with the scores from the top four PCs in a European-only analysis (as described above), 61,275 SNPs missing more than 5% of genotypes, and 96,880 SNPs with minor-allele frequencies less than 5%. For each case, the ten POPRES subjects with the shortest IBS distance to the case were selected as controls. PCA matching was carried out with PCA scores. PCA, excluding 58,089 SNPs described above, was carried out on the combined cases and subset of POPRES defined as European origin, with analysis limited to 200 subjects per country and principal-component scores assigned to all eligible controls. Inspection of the resulting eigenvalues led to the selection of the first four components for genetic matching. Prior to matching, eigenscores were rescaled to reflect their relative importance by multiplication of each eigenscore by the square root of the corresponding eigenvalue. Pairwise Euclidean distances were then estimated between each case and all POPRES subjects. Ten controls were selected for each case; they were randomly selected controls within the 2.5th percentile of the multivariate distance distribution, with care not to allow the reuse of controls among cases.
For each of the four selections of controls, genome-wide association analysis was carried out with Fisher's exact test, as described previously.7 SNPs were excluded from analysis if they were missing mapping position, had genotyping efficiency less than 90%, had minor-allele frequency less than 1%, or had deviations of genotype frequencies from Hardy-Weinberg expectations that were highly significant (p value < 10−7) in controls. We also excluded 26 SNPs identified in a previous study to have highly erroneous genotype calls within the 21 cases.7 Comparisons across analyses were carried out on a final set of 393,699 SNPs that passed the QC in all four case-control samples.
Public Data Availability
The subject-level data described in this study are available via the dbGaP archive sponsored by the National Center for Biotechnology Information (see Web Resources) pending acceptance of a standard Data Use Certification and endorsement by the requesting investigator's institution. Data include the demographic variables listed in the following section, PCA scores, and genotype data described herein.
Results
Sample and Data Overview
The POPRES study includes DNA samples from 5,886 subjects derived from ten constituent collections (Table 1; the LOLIPOP study is divided between subjects of Indian Asian and European origin). Based on the inclusion criteria and recruiting methods, these collections are broadly described as either population samples or healthy subjects (see Methods for collection-specific details). Basic demographic data available for all subjects includes sex, country of collection, and self-described racial background. Additional information available for some collections includes age at collection, state or city of collection, country of birth, parental country birthplaces, grandparental country birthplaces, and native language. Complete demographic summaries of each collection are provided in the Supplemental Results and subject-level details are available via controlled access in a public repository (see Web Resources). All participants were at least 18 years of age at time of recruitment. The sex ratio varies widely among studies.
The distribution of minor-allele frequencies by collection is presented in Figure 1. The frequency distributions are very consistent among the five European collections as well as between the two East Asian collections. However, the distributions differ substantially among the five major geographic regions represented by these collections. East Asia shows the highest proportion of low frequency SNPs (22% of SNPs less than 0.01 frequency), followed by Europe (15%), South Asia (13%), Mexico (10%), and lastly African American (1.9%). These frequency distributions differ markedly from those observed in the resequenced ENCODE regions of the HapMap project,19 wherein Europeans showed an increase in low frequency SNPs compared to East Asians and levels comparable to Africans. These differences reflect the biased nature of the SNPs included on the genotyping array.22
Figure 1.
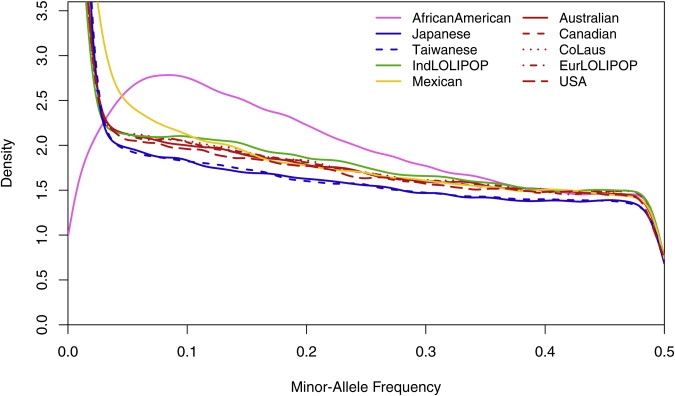
Distribution of Minor-Allele Frequency by Collection
Colors and line types for the densities of each collection are shown within the figure.
The distribution within African Americans is most distinct. There is a large proportion of SNPs with frequencies between 0.05 and 0.2, which is consistent with the African HapMap ENCODE and Affymetrix 500K SNP data (Figure S2). However, the African Americans have a very small proportion of low frequency and monomorphic SNPs compared to the other continental groups and compared to HapMap Africans (Figure S2). This does not reflect the underlying SNP frequency distribution in African Americans,1 but rather the influence of African and European admixture of African Americans with the SNPs in this panel. Although 15% of these SNPs have minor-allele frequencies less than 0.01 in Europeans and 11% in YRI, only 1.6% of them have minor-allele frequencies less than 0.01 in both. This smaller proportion of low frequency SNPs suggests that this panel would be more informative for studies in African Americans, compared to Africans.
Analysis of Population Structure
We performed a principal-component analysis on the genotype data to investigate the main axes of variation present in this sample. PCA makes inferences solely on the basis of the genotype data without inclusion of any other information; hence, the analysis results reflect the clustering within those data. The results of the PCA with the POPRES and HapMap data combined exhibit the anticipated structure first of clustering continents and next of regions within continents (Figure 2 and Figures S3 and S4). As expected, the first principal component (PC 1) distinguishes Africans from non-Africans. The next three principal components also characterize continental regions: PC 2 distinguishes East Asians from Africans and Europeans, with South Asians and Mexicans at intermediate values; PC 3 distinguishes South Asians from East Asians; and PC 4 distinguishes Mexicans from non-Mexicans.
Figure 2.
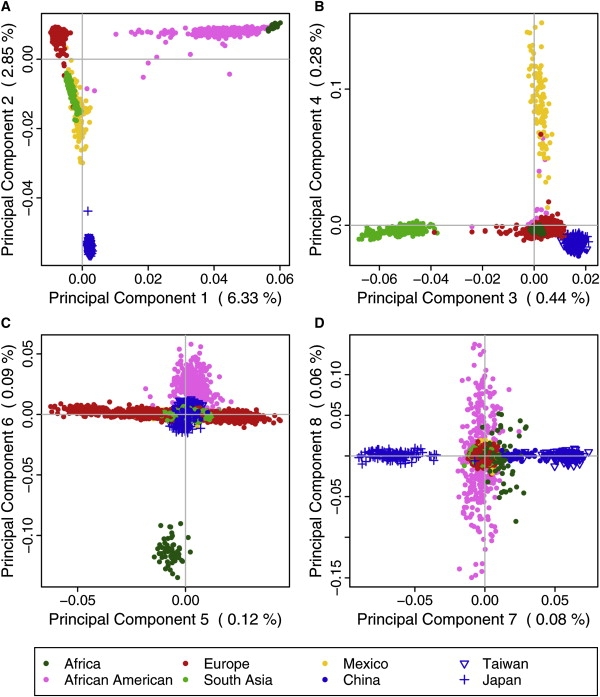
Genetic Structure Illustrated through Scatter Plots of Consecutive Principal Components
Subject scores are colored by continental and/or ethnic origin (see legend). East Asian populations are indicated by varying point types. Percent of variation explained by each component is given in parentheses on each axis label.
The subsequent principal components mark within-continent variation. PC 5 reveals a north-to-south cline within Europeans (Figure 3), consistent with existing studies of European substructure.20,23,24 The majority of Europeans sampled from North America and Australia are most similar to northern Europeans, with modest numbers of outlier observations. The CEU sample had the highest median scores on this component, followed by Australia and USA (collected in North Carolina), then by Canada, having a median more similar to central than to northern Europe. PC 6 distinguishes the African Americans from the HapMap Africans. Interpretation of the asymmetrical distributions of the Africans and African Americans along the European north-south cline in Figure 2C suggests that the Africans are slightly more similar to southern Europeans, whereas the African Americans lie slightly shifted to the right and on average appear more like northern Europeans on this principal component. This may be partially due to northern European admixture in African Americans. However, caution should be used in this interpretation, because the Africans and African Americans are slightly more similar to their respective subpopulations of Europeans only on genotypes that distinguish southern from northern Europeans, and this similarity is not necessarily true of overall genotype relatedness.
Figure 3.
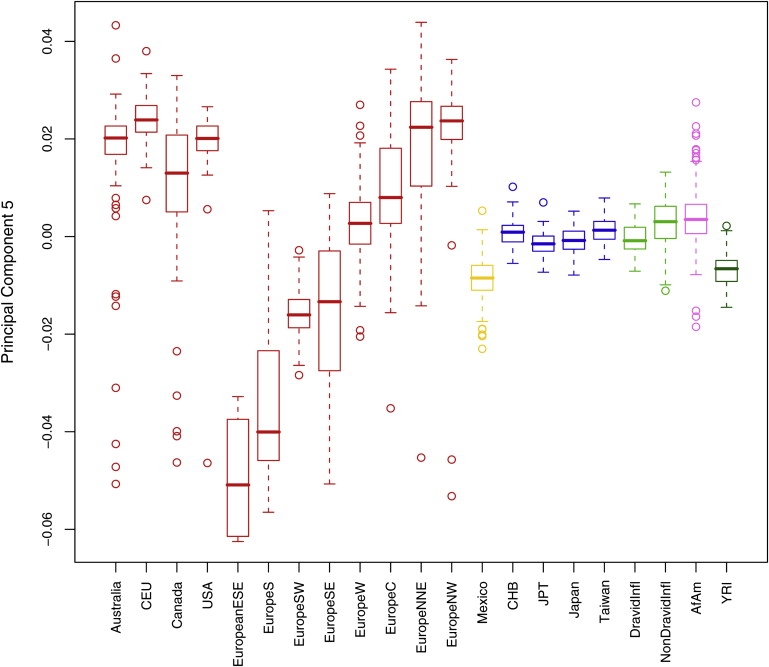
Distribution of Subject-Level Principal Component 5 Scores by Reported Ancestry
Each box and whisker indicates the median (heavy line), interquartile range (IQR, box), and minimum and maximum observations (whiskers). Whiskers are truncated at the last observation within 1.5 times the IQR from the edge of the box, with outliers shown individually. Plots for the remaining principal components are available in Figure S2, available online.
Principal component 7 (Figure 2D) separates the three East Asian populations: Japan (left), HapMap CHB (center right), and Taiwan (far right). Note that the Africans, unlike African Americans or other continents, appear more similar to the Chinese than Japanese on the PC that distinguishes East Asian substructure. We do not show further results because PC 8 and subsequent PCs display substructure within Africans and African Americans, but do not correspond to any known geographic or population structure among individuals. The first two PCs explain a total of 9.2% of the genetic variation within this sample. The remaining five PCs, though clearly informative, only explain an additional 1.0% combined. Loadings for the first seven PCs are included in Table S3. HapMap subject scores are available in Table S4, and POPRES subject scores are available with the subject-specific data through dbGaP.
Case-Control Matching
One of the primary motives in the development of the POPRES resource was to provide a source of pregenotyped population samples that could be drawn on as needed as a comparator (i.e., control) group for association studies of adverse drug reactions. The rationale for this approach and its implications on statistical power for ADR genetics research have been considered elsewhere.7 In that previous work, we argued that use of population controls required that they be matched appropriately to the cases. Given such a resource, there are multiple ways in which cases and controls could be matched. Here, we extend our previous work with 21 clinically diagnosed abacavir-associated hypersensitivity reaction (ABC HSR) cases7 by comparing four strategies for matching them to these POPRES controls: (1) matched by continental origin by selecting northern Europeans from the United Kingdom, (2) matched by reported country or region of birth, (3) minimizing pairwise identity-by-state (IBS) distances between cases and controls (Figure S5), and (4) minimizing distances between cases and controls on the basis of multivariate PCA scores (Figures S6 and S7). For each method, controls were matched to this small sample of cases in a 10:1 ratio.
The results of each genome-wide association analysis, using controls selected as described above, are summarized in Figure S8. All four methods identify the known MHC region (tagging HLA-B∗5701) among the top 20 associated SNPs, with PCA matching yielding the lowest p value and highest rank (p value = 2.1 × 10−6, rank = 2), followed by UK (4.2 × 10−6, 8), country (7.6 × 10−6, 5), and IBS (2.9 × 10−5, 16) matching. A comparison of the ranking among the top 100 SNPs from each analysis showed that the country- and IBS-matching methods were the most concordant (ρ = 0.58). Country- and PCA-matching methods were the least concordant (ρ = 0.03). The remaining pairwise comparisons were only modestly correlated (ρ < 0.15).
With a single realization of each matching algorithm, it is not possible to assess the impact of the matching on the power to identify the known effect of the HLA-B∗5701 allele. However, with nearly 400,000 SNPs for which the null hypothesis of no association is true, we can reasonably assess the effect of each matching algorithm on the type I error rate. The proportion of tests with p values falling below a range of significance thresholds, shown in Figure 4, is very similar among the country (genomic control λ = 1.00 for allelic test), IBS (λ = 1.00), and PCA (λ = 1.00) matching methods and falls close to the expected proportion at each level. In contrast, the analysis that only drew from population controls in the UK (λ = 1.13) resulted in a significant excess of low p values at all levels below 0.1, roughly doubling the numbers observed with the other matching methods. Whereas all four control matching procedures resulted in relatively low p values for the known association, the UK controls (i.e., matching only by continent) suffered from an increase in the false-positive rate, even with this small number of cases. Figure S8 shows that relatively small p values are observed across the genome and vary substantially across control selections.
Figure 4.
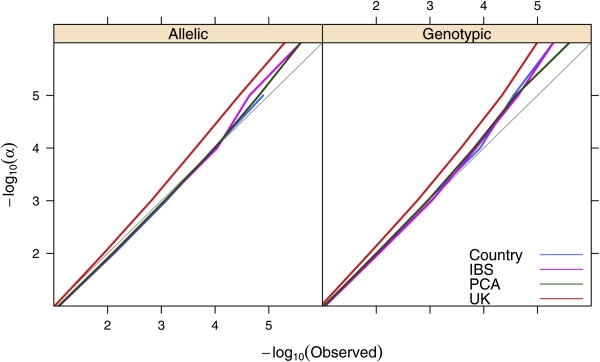
P-Plot Comparing Observed versus Expected Proportion of Associations over a Range of Significance Thresholds
Separate lines are presented for each of the four control matching strategies. Results of the allelic exact test are shown on the left and genotypic exact tests on the right. A light gray line corresponds to unity.
Discussion
We have brought together DNA from nearly 6,000 subjects participating in ten studies with ancestry from five major geographic regions and dozens of countries as a resource for genetics research. Genotype data from a genome-wide panel of 500,000 SNPs attempted on nearly all participant samples were carefully evaluated to yield a set of subjects and markers with high data quality that may be appropriate for a range of applications. These data are freely available for legitimate research purposes through the public dbGaP website.
Principal-component analysis of these data illustrates the overall data quality, in terms of both the genotypes and the labels of subject origins. The seven highly informative principal components provided a high degree of discrimination among African, East Asian, South Asian, European, and Mexican ancestry. They also illustrated finer differentiation in the separation of Africans and African Americans and differentiation between the three Asian populations of Japan, mainland China, and Taiwan, and they highlighted genetic gradients within African Americans, Mexicans, and Europeans. These results provided ample opportunities to identify subjects with ancestry labels that do not match their genetic background. Very few subjects demonstrated PC score patterns that deviated noticeably from the majority of their groups. The score information (available via dbGaP) may be used in future applications to re-label subjects or to exclude them from further analyses.
The potential impact of cases and controls that are poorly matched for their genetic background on the type I error rates of association studies is well understood (e.g., 25). Most studies of unrelated subjects attempt to control for this through careful study design and sampling (e.g., 26), statistical correction (e.g., 27), or measurement and correction of sample structure by use of PCA or related methods (e.g., 28,29). Alternatively, sets of healthy or population controls that have been genotyped for compatible genome-wide panels can be queried for controls that genetically match the genotyped cases,8,30 recently illustrated for genome-wide genotype data.8 In the limited application presented here with 21 subjects with abacavir-associated hypersensitivity reaction, we found that matching controls to cases on the basis of country of origin, minimizing pairwise IBS distances, and minimizing distances among the top principal components were similarly effective in controlling type I error. The latter two genotype-based methods would clearly be preferred when there is uncertainty about genetic background of the cases or controls, or when the populations sampled are admixed or otherwise genetically heterogeneous. It is important to note that with such a small number of cases included in this example application, there is insufficient power for subtle population or genotype-quality-dependent differences between the cases and controls to be detected. An analysis with a larger number of cases and controls could highlight limitations in the sample-matching schemes or in the POPRES data that were not readily apparent in this example.
Most studies that include whole-genome genotype data do not have need for external sources of controls for key analyses, and even with ∼5,000 subjects genotyped, the power of this resource to investigate common disease genetics is limited, particularly for non-European populations. Nevertheless, the data published herein should prove useful for characterizing the genetic background of study participants, particularly for small sample sizes or poorly characterized sample collections. POPRES genotype data may be included with study genotype data to conduct analyses of population structure. The Affymetrix 500K SNP panel shares a reasonably large number of SNPs with other popular SNP panels, including Illumina 1M (138,143) and Affymetrix 6.0 (469,874), which in many cases will be sufficient for inferring patterns of population structure. Subject scores may also be computed directly from the SNP loadings published herein (Table S3). The legitimacy of this approach is most obvious for genotype data derived from the Affymetrix 500K and 6.0 SNP panels. However, it should be possible to derive informative subject scores with this approach from the subset of SNPs that overlap with the Illumina panels, though the accuracy of this approach has not been assessed. Beyond the global patterns of variation observed in the analyses included in this report, finer-scale structure may also be investigated in subsets of the POPRES data, such as within Europeans.21
As described, nearly all of the subjects currently included in POPRES have been genotyped with the Affymetrix 500K SNP panel. The choice to standardize on this panel was largely influenced by the timing of the project. Since the time this project was initiated, genome-wide genotyping panels from multiple vendors have expanded and improved in quality. Although there is no expectation that the entire POPRES collection will be genotyped on another genome-wide panel, selected subsets will be genotyped with newer panels as required to support ongoing research, and much of these data will eventually be deposited to dbGaP. This includes existing data on the Illumina (San Diego, CA) 550K and 1M panels typed on ∼500 POPRES subjects of European origin. Developments around use of representative patterns of haplotype structure to impute unmeasured genotypes may also be employed with this and similar resources to make the results from the Affymetrix 500K panel compatible with other panels.31–33
In developing this resource, we considered several alternative designs. The first objective is to use this collection as a resource for generating contrast (control) groups for pharmacogenetic studies. In the context of studying the occurrence of an ADR, the controls would ideally match the cases for disease status, treatment, duration of treatment, age, gender, and any other disease- or ADR-related clinical characteristics so that associated markers can be inferred to be causally related. However, developing a general resource applicable to a diversity of diseases and relevant to a number of drugs (approved or in development) would probably require extremely large samples and be difficult, if not impossible, to ascertain. When the outcome under study is relatively rare (prevalence < 10%), as many ADRs are, an alternative to having treatment-matched patients is having patients matched for disease status but unknown for their propensity for an adverse event given the lack of treatment. Because the outcome is rare, a relatively small percentage of the controls would have had the adverse event, if they had been treated. This more feasible design would result in little loss of power to detect even modest genetic effects. Even so, unless the number of relevant diseases is very small and foreseeable, even large collection sizes will be limited once study-specific strata are considered.
With these limitations, we considered that a collection representative of the populations from which the cases were sampled without regard to disease status would be the most feasible design. A population sample design would result in disease frequencies in proportions similar to those of the population at large. For rare outcomes, the frequency of those genetically predisposed to the outcome of interest would be low, resulting in a small loss of power to identify predisposing factors. In this design, the disease status and outcome of interest are likely to be confounded, requiring further investigation to disentangle the relevance of each result.
It is often of interest to estimate the frequencies of alleles associated with a pharmacogenetic response. One can estimate these frequencies in the population of affected individuals (i.e., patients) or in the population at large. Although estimates in patients are more representative of the intent to treat population, having an appropriate sample for a large number of diseases is not feasible. Estimating the genetic parameters in the population at large will only be limiting if the genetic variant, or one in linkage disequilibrium with it, plays an important role in both the disease susceptibility and the pharmacogenetic response under investigation. This may be expected to occur when the variations with pharmacogenetic impact are located within the drug target. In such cases, caution should be exercised in the interpretation of results.
The range and value of genetic studies possible with such a resource rests largely on the quality, quantity, and sampling of the data available. The public release of the POPRES resource will have immediate opportunities to impact a variety of studies and contribute to the growing body of data that will further many areas of human genetics research. We support the public access to these data for appropriate research uses and encourage the further development of such resources for the benefit of the scientific community.
Supplemental Data
Supplemental Data include eight figures, four tables, and a summary of demographic variables available for each collection and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Each box and whisker indicates the median (heavy line), interquartile range (IQR, box), and minimum and maximum observations (whiskers). Whiskers are truncated at 1.5 times the IQR, with outliers shown individually.
Web Resources
The URL for data presented herein is as follows:
Acknowledgments
We thank Clive E. Bowman, Michael Klotsman, Ann Marie McNeill, David P. Yarnall, Ross Haggart, Steve Haneline, Kelley Johansson, Devon Kelly, Devi Smart, Sarah Tate, Jill Ratchford, and Mike Lawson at GlaxoSmithKline for their many contributions to the development of the POPRES resource. We thank Arlene Hughes and Bill Spreen at GlaxoSmithKline for their work on pharmacogenetics research into abacavir-associated hypersensitivity reaction. We gratefully acknowledge Yolande Barreau, Anne-Lise Bastian, Binasa Ramic, Martine Moranville, Martine Baumer, Marcy Sagette, Jeanne Ecoffey, and Sylvie Mermoud for their roles in the CoLaus data collection. The CoLaus study was supported by research grants from GlaxoSmithKline and from the Faculty of Biology and Medicine of Lausanne, Switzerland. We thank Anna C. Need for providing access to the Duke healthy-controls collection. We thank Robin Lincoln at UCSF for expert specimen management. Recruitment of the UCSF African American samples was funded by grants from the National Institutes of Health (RO1 NS046297) and National Multiple Sclerosis Society (RG3060C8). All genotyping for this study was funded by GlaxoSmithKline, of which several authors are employees.
References
- 1.Lohmueller K.E., Indap A.R., Schmidt S., Boyko A.R., Hernandez R.D., Hubisz M.J., Sninsky J.J., White T.J., Sunyaev S.R., Nielsen R. Proportionally more deleterious genetic variation in European than in African populations. Nature. 2008;451:994–997. doi: 10.1038/nature06611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jakobsson M., Scholz S.W., Scheet P., Gibbs J.R., VanLiere J.M., Fung H.C., Szpiech Z.A., Degnan J.H., Wang K., Guerreiro R. Genotype, haplotype and copy-number variation in worldwide human populations. Nature. 2008;451:998–1003. doi: 10.1038/nature06742. [DOI] [PubMed] [Google Scholar]
- 3.Li J.Z., Absher D.M., Tang H., Southwick A.M., Casto A.M., Ramachandran S., Cann H.M., Barsh G.S., Feldman M., Cavalli-Sforza L.L. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- 4.Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manolio T.A., Rodriguez L.L., Brooks L., Abecasis G., Ballinger D., Daly M., Donnelly P., Faraone S.V., Frazer K., Gabriel S. New models of collaboration in genome-wide association studies: The Genetic Association Information Network. Nat. Genet. 2007;39:1045–1051. doi: 10.1038/ng2127. [DOI] [PubMed] [Google Scholar]
- 6.Mailman M.D., Feolo M., Jin Y., Kimura M., Tryka K., Bagoutdinov R., Hao L., Kiang A., Paschall J., Phan L. The NCBI dbGaP database of genotypes and phenotypes. Nat. Genet. 2007;39:1181–1186. doi: 10.1038/ng1007-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nelson M.R., Bacanu S.A., Mosteller M., Li L., Bowman C.E., Roses A.D., Lai E.H., Ehm M.G. Genome-wide approaches to identify pharmacogenetic contributions to adverse drug reactions. Pharmacogenomics J. 2008 doi: 10.1038/tpj.2008.4. in press. Published online February 26, 2008. [DOI] [PubMed] [Google Scholar]
- 8.Luca D., Ringquist S., Klei L., Lee A.B., Gieger C., Wichmann H.E., Schreiber S., Krawczak M., Lu Y., Styche A. On the use of general control samples for genome-wide association studies: Genetic matching highlights causal variants. Am. J. Hum. Genet. 2008;82:453–463. doi: 10.1016/j.ajhg.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thiers F.A., Sinskey A.J., Berndt E.R. Trends in the globalization of clinical trials. Nat. Rev. Drug Discov. 2008;7:13–14. [Google Scholar]
- 10.Rosenberg N.A., Pritchard J.K., Weber J.L., Cann H.M., Kidd K.K., Zhivotovsky L.A., Feldman M.W. Genetic structure of human populations. Science. 2002;298:2381–2385. doi: 10.1126/science.1078311. [DOI] [PubMed] [Google Scholar]
- 11.International Conference on Harmonization E15 Definitions for genomic biomarkers, pharmacogenomics, pharmacogenetics, genomic data and sample coding categories. Federal Register. 2008;78:19074–19076. [PubMed] [Google Scholar]
- 12.Oksenberg J.R., Barcellos L.F., Cree B.A., Baranzini S.E., Bugawan T.L., Khan O., Lincoln R.R., Swerdlin A., Mignot E., Lin L. Mapping multiple sclerosis susceptibility to the HLA-DR locus in African Americans. Am. J. Hum. Genet. 2004;74:160–167. doi: 10.1086/380997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patterson N., Hattangadi N., Lane B., Lohmueller K.E., Hafler D.A., Oksenberg J.R., Hauser S.L., Smith M.W., O'Brien S.J., Altshuler D. Methods for high-density admixture mapping of disease genes. Am. J. Hum. Genet. 2004;74:979–1000. doi: 10.1086/420871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kooner J.S., Chambers J.C., Aguilar-Salinas C.A., Hinds D.A., Hyde C.L., Warnes G.R., Gomez Perez F.J., Frazer K.A., Elliott P., Scott J. Genome-wide scan identifies variation in MLXIPL associated with plasma triglycerides. Nat. Genet. 2008;40:149–151. doi: 10.1038/ng.2007.61. [DOI] [PubMed] [Google Scholar]
- 15.Firmann M., Mayor V., Vidal P.M., Bochud M., Pecoud A., Hayoz D., Paccaud F., Preisig M., Song K.S., Yuan X. The CoLaus study: A population-based study to investigate the epidemiology and genetic determinants of cardiovascular risk factors and metabolic syndrome. BMC Cardiovasc. Disord. 2008;8:6. doi: 10.1186/1471-2261-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen J., Iannone M.A., Li M.S., Taylor J.D., Rivers P., Nelsen A.J., Slentz-Kesler K.A., Roses A., Weiner M.P. A microsphere-based assay for multiplexed single nucleotide polymorphism analysis using single base chain extension. Genome Res. 2000;10:549–557. doi: 10.1101/gr.10.4.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., De Bakker P.I., Daly M.J. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patterson N., Price A.L., Reich D. Population structure and Eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.The International HapMap Consortium A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tian C., Plenge R.M., Ransom M., Lee A., Villoslada P., Selmi C., Klareskog L., Pulver A.E., Qi L., Gregersen P.K. Analysis and application of European genetic substructure using 300 K SNP information. PLoS Genet. 2008;4:e4. doi: 10.1371/journal.pgen.0040004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Novembre J., Johnson T., Bryc K., Kutalik Z., Boyko A., Auton A., Indap A., King K.S., Bergmann S., Nelson M.R. Genes mirror geography within Europe. Nature. 2008 doi: 10.1038/nature07331. in press. Published online August 28, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clark A.G., Hubisz M.J., Bustamante C.D., Williamson S.H., Nielsen R. Ascertainment bias in studies of human genome-wide polymorphism. Genome Res. 2005;15:1496–1502. doi: 10.1101/gr.4107905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Novembre J., Stephens M. Interpreting principal component analyses of spatial population genetic variation. Nat. Genet. 2008;40:646–649. doi: 10.1038/ng.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bauchet M., McEvoy B., Pearson L.N., Quillen E.E., Sarkisian T., Hovhannesyan K., Deka R., Bradley D.G., Shriver M.D. Measuring European population stratification with microarray genotype data. Am. J. Hum. Genet. 2007;80:948–956. doi: 10.1086/513477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lander E.S., Schork N.J. Genetic dissection of complex traits. Science. 1994;265:2037–2048. doi: 10.1126/science.8091226. [DOI] [PubMed] [Google Scholar]
- 26.McCarthy M.I., Abecasis G.R., Cardon L.R., Goldstein D.B., Little J., Ioannidis J.P., Hirschhorn J.N. Genome-wide association studies for complex traits: Consensus, uncertainty and challenges. Nat. Rev. Genet. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 27.Devlin B., Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 28.Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 29.Yu J., Pressoir G., Briggs W.H., Vroh B., Yamasaki I., Doebley M., Mc J.F., Mullen M.D., Gaut B.S., Nielsen D.M. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006;38:203–208. doi: 10.1038/ng1702. [DOI] [PubMed] [Google Scholar]
- 30.Hinds D.A., Stokowski R.P., Patil N., Konvicka K., Kershenobich D., Cox D.R., Ballinger D.G. Matching strategies for genetic association studies in structured populations. Am. J. Hum. Genet. 2004;74:317–325. doi: 10.1086/381716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marchini J., Howie B., Myers S., McVean G., Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 32.Browning S.R., Browning B.L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 2007;81:1084–1097. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scott L.J., Mohlke K.L., Bonnycastle L.L., Willer C.J., Li Y., Duren W.L., Erdos M.R., Stringham H.M., Chines P.S., Jackson A.U. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Each box and whisker indicates the median (heavy line), interquartile range (IQR, box), and minimum and maximum observations (whiskers). Whiskers are truncated at 1.5 times the IQR, with outliers shown individually.