

Abstract
Down syndrome (DS) is the most common cause of mental retardation. Many neural phenotypes are shared between DS individuals and DS mouse models; however, the common underlying molecular pathogenetic mechanisms remain unclear. Using a transchromosomic model of DS, we show that a 30%–60% reduced expression of Nrsf/Rest (a key regulator of pluripotency and neuronal differentiation) is an alteration that persists in trisomy 21 from undifferentiated embryonic stem (ES) cells to adult brain and is reproducible across several DS models. Using partially trisomic ES cells, we map this effect to a three-gene segment of HSA21, containing DYRK1A. We independently identify the same locus as the most significant eQTL controlling REST expression in the human genome. We show that specifically silencing the third copy of DYRK1A rescues Rest levels, and we demonstrate altered Rest expression in response to inhibition of DYRK1A expression or kinase activity, and in a transgenic Dyrk1A mouse. We reveal that undifferentiated trisomy 21 ES cells show DYRK1A-dose-sensitive reductions in levels of some pluripotency regulators, causing premature expression of transcription factors driving early endodermal and mesodermal differentiation, partially overlapping recently reported downstream effects of Rest +/−. They produce embryoid bodies with elevated levels of the primitive endoderm progenitor marker Gata4 and a strongly reduced neuroectodermal progenitor compartment. Our results suggest that DYRK1A-mediated deregulation of REST is a very early pathological consequence of trisomy 21 with potential to disturb the development of all embryonic lineages, warranting closer research into its contribution to DS pathology and new rationales for therapeutic approaches.
Introduction
Down syndrome (DS [MIM 190685]) is a complex condition characterized by many phenotypic features, including mental retardation, smaller brain size, reduced numbers of neurons, reduced dendritic spine density and plasticity, and early-Alzheimer's-disease-like neurodegeneration.1,2 Mouse models for DS also display behavioral and cognitive defects, synaptic plasticity defects and long-term potentiation (LTP) deficit in the hippocampus, and reduced hippocampal and cerebellar neuron numbers.3–8 However, despite these similarities, causative mechanisms common to human and mouse DS systems remain to be elucidated.
Cultured fetal DS brain cell-derived neurospheres were found to have decreased transcript levels of neuron-restrictive silencer factor (NRSF or REST [MIM 600571]) and downstream targets such as SCG109[MIM 600621]. REST modulates expression of genes encoding fundamental neuronal functions including ion channels, synaptic proteins, and neurotransmitter receptors.10–15 It is essential both for the repression of these genes in non-neuronal tissues10 and for the orchestrated activation of transcription of these genes during neuronal differentiation, acting as a silencer or as a transcription activator.12–14,16–18 Coordinated activation of transcription of REST targets is both necessary and sufficient for the transition from pluripotent embryonic stem (ES) cells to neural progenitor cells (NPCs)14 and onward to mature neurons,13,14 and the REST pathway has been implicated in an inherited form of mental retardation.19 Using a transchromosomic mouse model of DS, we show here that a reduced expression of Rest is an alteration that persists from undifferentiated ES cells to the adult brain and is reproducible across several DS models. We map the region capable of affecting Rest levels in both mouse and human cells to the DYRK1A [MIM 600855] locus and demonstrate the sensitivity of Rest levels to the dose and kinase activity of DYRK1A. Recently, Rest +/− cells were found to have reduced levels of key pluripotency regulators (Oct4 [MIM 164177], Nanog [MIM 607937] and Sox2 [MIM 184429]), shedding new light on the function of Rest in the regulation of ES cell pluripotency and self-renewal.20 We demonstrate that trisomy 21 ES cells share certain aspects of the aberrant control of pluripotency and early differentiation reported in Rest +/− cells,20 suggesting that DYRK1A-mediated deregulation of REST could be an important potential contributor to a variety of DS phenotypes.
Material and Methods
Material
All general reagents and tissue-culture media were from Sigma (Dorset, UK) unless otherwise stated, and all primers were supplied by Invitrogen (Paisley, UK). Transchromosomic ES cells 47-1, 40-2, and 46-1 were derived from D3 ES cells by the introduction of all or parts of HSA21 via microcell-mediated chromosome transfer.21 Mapping data for 40-2 and 46-1 have been previously published,21 and they were verified and refined to the single-gene resolution in the current study (data available upon request). Antibodies were as follows: anti-REST from Upstate, anti-GAPDH from Invitrogen (ZYMED laboratories), and anti-PTEN from Abcam. Anti-β-actin, anti-calnexin, and anti-β tubulin isotype III antibodies were from Sigma. Sequences of primers and probes used for quantitative RT-PCR and linkage analysis are available in Table S3, available online. EGCG (epigallocatechin gallate) was from Sigma. The Dual-Luciferase Reporter Assay System was from Promega.
Mice
Tc1 mice backcrossed to C57BL/6J (2–3 generations) and Ts1Cje mice backcrossed to C57BL/6J (> 10 generations) were maintained at the National Institute for Medical Research in accordance with UK Home Office regulations.8 The TgDyrk1A mice were maintained on C57BL6/SJL background in the facility of the Genes and Disease Program, Center for Genomic Regulation. Whole brain hemispheres from 6- to 11-month-old adult mice were used. In all comparisons, sex-matched littermates were used.
ES Cell Culture and Differentiation
Pluripotent D3 and 47-1 ES cells were seeded onto embryonic-fibroblast feeder layers and were maintained in DMEM with 15% fetal-calf serum (Hyclone), 2 mM glutamine, 1 × nonessential amino acids (Invitrogen), 50 U/ml penicillin, 50 μg/ml streptomycin, 1:150,000 β-mercaptoethanol, and 103 U/ml LIF-ESGRO (Chemicon). For the first passage, transchromosomic cells were cultured in ES medium with 500 μg/ml G418 for ensuring retention of HSA21. Cells were cultured without G418 for a second passage, for minimization of differences in culture conditions between wild-type (WT) and transchromosomic cells, and without G418 or feeders for the final passage before extraction, for minimization of background from feeder cells. Transchromosomic cells were verified by FISH as retaining HSA21 in > 90% of cells, with the use of human Cot1 probe as described previously.21 For differentiation into NPCs, D3 and 47-1 cells were treated as described.22 In brief, cells were cultured in the absence of LIF to form embryoid bodies, which were then treated with 5 μM RA for four days before dissociation22 and replating onto poly-D-lysine- and laminin-coated dishes in Neurobasal N2 medium (Invitrogen).
Immunofluorescence was carried out as described.14 Images were captured with the use of a Q550 Imaging Workstation with DM5000 microscope and Qwin v3.2 software (Leica Microsystems [UK], Bucks). For cell counts and neurite characterization, 10–15 different images of each of the D3 and 47-1 cell lines from independent neural-differentiation experiments were collected by automated unbiased random sampling (total of 30 images per cell line). Mature neurons, identified by immunofluorescent staining, were counted blindly by two researchers and expressed relative to DAPI-staining nuclei. Similar numbers of D3 and 47-1 cells were counted (ranging from 400–3000 cells per experiment).
Microarray Analysis
RNA was extracted from four cultures of each cell line in undifferentiated state with the use of RNEasy mini spin columns with on-column Dnase-1 digestion (QIAGEN, Crawley, UK) and labeled according to the standard Affymetrix protocol before hybridization to MG-U74Av2 microarrays (Affymetrix UK, High Wycombe, UK). Images were scaled to a target intensity of 500, and all samples were verified as having Gapdh [MIM 138400] and β-actin [MIM 102630] 3′/5′ ratios less than 3 and at least 40% of probe sets called present (range 40.3–51.1). All array data have been deposited in the MIAMExpress database, under the experiment number ‘E-MEXP-654’.
Differentially expressed genes were identified with two different search strategies. In the first, absolute and comparative data were exported from Affymetrix MicroArray Suite (MAS) software into a microarray-analysis program based on Filemaker Pro 5 (Filemaker), developed by G.D. A search was carried out for probe sets called “increased” or “decreased” by MAS (change p value < 0.05) in all comparisons between 47-1 and D3 cells, with the highest signal being called “present” in each case. In the second method, absolute data were imported into Genespring v6.1 (Silicon Genetics, CA, USA), normalized to the 50th percentile of each array, and normalized to the median for each probe set. The data were then filtered for removal of all probe sets that were called present in fewer than four samples and those that changed fewer than two times between the two cell lines. ANOVA was applied to the remaining probe sets, with the Benjamini-Hochberg multiple-testing correction.
Quantitative RT-PCR
RNA from mouse brains was extracted with RNABee (Tel-Test, Texas, USA), according to the manufacturer's protocol, followed by DNase-1 digestion (Roche) and cDNA synthesis with Superscript II reverse transcriptase (Invitrogen). RNA from D3 and 47-1 cells was extracted with the use of the RNeasy Plus Mini Kit (QIAGEN). Quantitative RT-PCR was carried out via an Applied Biosystems 7700 Sequence Detector v1.7 and SYBR green or Taqman PCR mix, according to the manufacturer's protocol (Applied Biosystems, Warrington, UK). All transcripts were measured in duplicate against standard curves relative to Gapdh.
RNAi Silencing
Knockdown of DYRK1A, Ttc3 [MIM 602259], Dscr3 [MIM 605298], Setd4, and Cbr1[MIM 114830] was achieved by transfection of RNAi oligonucleotides with Lipofectamine 2000 (Invitrogen), according to the manufacturer's specific protocol for D3 cells with minor modifications. In brief, ES cells were trypsinized, pelleted by centrifugation, and resuspended in ES cell medium. Then, 5 × 105 cells were seeded into previously gelatinized 12-well plates, and a transfection mixture containing 4 μg of Lipofectamine-2000 and 100 pmol of the specific sequence (or scrambled sequence) of RNAi reagents in Opti-MEM I Reduced Serum Medium was added to each sample. Samples were incubated for 24 hr before RNA extraction was performed. RNAi oligonucleotide sequences are available in Table S4.
Luciferase Assay
Mouse Rest transcript NM_011263 promoter sequence (1013 bp) was inserted into the PGL-3 basic vector, upstream of the firefly Luciferase reporter gene. Rest-PGL-3 was cotransected with the pRL-CMV expression vector into D3 cells with Lipofectamine 2000 (Invitrogen), according to the manufacturer's specific protocol for D3 cells with minor modifications. Luciferase activity was measured with the Dual-Luciferase Reporter assay system (Promega). Renilla luciferase activity was used for standardization of transfection efficiency.
Protein Analysis
For Western blotting, D3 and 47-1 ES cells were solubilized in 30 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM NaF, 1 μg/ml leupeptin, and 5 KU/ml aprotinin containing 1% Triton X-100. The lysate was clarified by centrifugation at 435,000 × g max for 30 min at 4°C. Immunoblot analyses were performed as described previously.23 Western blotting of brain homogenates was performed as described previously.24
Human-Genome Linkage Analysis
EBV-transformed cell lines of 135 individuals from ten CEPH (Centre d'Etude du Polymorphisme Humain) pedigrees were obtained from the Coriell cell repositories. Cell culture and RNA extractions were performed as described previously.25 Gene-expression levels of REST and four normalization genes (AGPAT1 [MIM 603099], B2M [MIM 109700], EEF1A1 [MIM 130590] and UBE2D2 [MIM 602962]) were measured by Taqman qRT-PCR with six replicates per gene per sample, and expression values were median normalized with q-base software, as described previously.26 Normalized gene-expression values were used for performing quantitative multipoint genome-wide linkage analysis with the Merlin package with the –VC option and default parameters.
Results
Decrease in Rest Level Is an Early and Persistent Phenotype of Trisomy 21
We sought to examine transcripts altered by trisomy 21 in pluripotent, undifferentiated mouse ES cells. For this purpose, we compared the transchromosomic mouse ES cell line 47-1, which was engineered to contain a whole HSA21 on an otherwise euploid mouse genome,21 with parental D3 cells using Affymetrix MG-U74Av2 mouse arrays (n = 4). Unsupervised hierarchical clustering successfully segregated 47-1 from D3, indicating a global perturbation of transcription by trisomy 21 (Figure 1A). Rest was found among the eight most significantly decreased mouse transcripts (Figure S1 and Tables S1 and S2). Given that Rest had previously been found reduced only in DS fetal brain cells,9 its apparent reduction in pluripotent ES cells would provide a new insight; we decided to investigate this further. Rest suppression in 47-1 cells was verified by quantitative RT-PCR (Figure 1B). Both alternatively spliced forms of the transcript (Rest 1 and Rest 4) were significantly decreased (Figure 1C; see Table S3 for all primer sequences), suggesting that suppression occurs at the level of transcription rather than alternative splicing. Suppression of Rest protein was verified by Western blotting of whole cell lysates, in which a single 200 kD band was seen (Figure 1). Rest protein level was highly significantly reduced, by > 40%, irrespective of the protein used for normalization (Gapdh, Calnexin, Pten, or all three; Figure 1D). In order to prove that Rest suppression is not simply a clone- or system-specific artifact, we measured Rest transcript levels in an independently derived transchromosomic ES cell line carrying a smaller portion of HSA21 (40-2)21 and in adult brains from two independent mouse models of DS: (1) transchromosomic Tc1 mice, which model a range of features of DS, including changes in behavior, synaptic plasticity, cerebellar neuronal number, congenital heart defects, and skeletal malformations;8 and (2) Ts1Cje mice, which are trisomic for a segment of mouse chromosome 16 carrying mouse orthologs of 85 HSA21 genes5 and display DS neurological phenotypes similar to those of Tc1 mice.5–7 All three systems showed a 34%–41% reduction of Rest transcript levels compared to WT (Figures 1E–1G). Other tissues of the DS mouse models also showed a decrease in Rest and altered dose of some of the genes containing NSRE elements27 that Rest binds to (examples shown in Figure S2).Therefore, Rest suppression persists from ES cells to the adult brain, it is reproducible regardless of differences in genetic background and the species of origin of the extra chromosome, and, importantly, it is a phenotype shared between several DS mouse models (Figure 1F, Figure S2) and human DS.9
Figure 1.
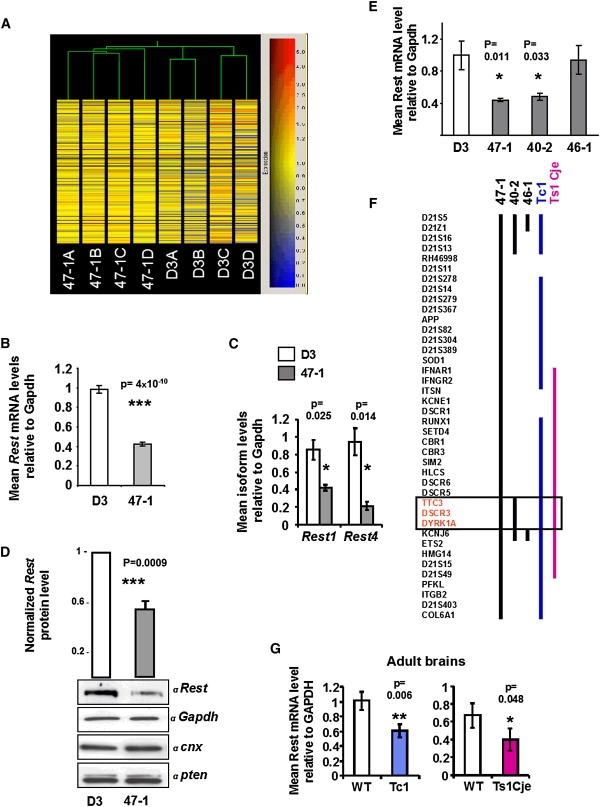
Transcript Profiling and Analysis of Rest Levels in Down Syndrome Model Systems
(A) Unsupervised clustering of transchromosomic (+HSA21) 47-1 and normal control D3 mouse ES cell lines. RNA samples from four cultures of each cell line were analyzed independently on Affymetrix MG-U74Av2 arrays, and hierarchical clustering based on Spearman correlation was performed, after elimination of all probe sets called absent across all chips.
(B) qRT-PCR measurement of the level of Rest transcript in undifferentiated ES cells (n = 9 independent cultures).
(C) qRT-PCR analysis of alternative forms of the Rest transcript (Rest-1 and Rest-4) in undifferentiated ES cells (n = 4).
(D) Western blot of Rest protein expression in undifferentiated ES cells. Bars show densitometric intensity of the Rest band, normalized against each of the three normalizing protein bands (shown below) and averaged across values obtained from three independent cell cultures for each normalization.
(E) qRT-PCR analysis of Rest in partially trisomic transchromosomic ES lines 40-2 and 46-1, compared with D3 and 47-1 (n = 4).
(F) Map showing regions of HSA21 that are trisomic (black bar indicates HSA21 fragments [mapping data based on refs. 8,21, Table S5, and unpublished data]; red bar indicates equivalent mouse chromosome 16 segment5) in trisomy 21 models. The box delineates the minimal trisomic region correlating with Rest suppression.
(G) qRT-PCR analysis of Rest in adult brains of Tc1 mice and their WT littermates (n = 5) and in adult brains of Ts1Cje mice and their WT littermates (n = 6). In all graphs, means and standard errors are shown, and statistical significance by Student's t test is indicated by one (p < 0.05), two (p < 0.01), or three (p < 0.001) asterisks.
Rest-Level Control Maps to DYRK1A Locus in Mouse ES Cells and Human Lymphoblastoid Lines
Combining results from the different model systems yielded a minimal trisomic region sufficient to cause Rest suppression, mapped to a ∼2 Mb HSA21 interval. In order to dissect this region further, we used another ES cell line (46-1), from the same panel as 40-2, which is not trisomic for three genes in the candidate region. Because this cell line showed no significant reduction in Rest levels (Figure 1E), the minimal candidate region for Rest reduction could be mapped to only three genes: TTC3, DSCR3, and DYRK1A (Figure 1F).
As an alternative, independent approach to identifying loci that regulate REST-gene expression, we undertook a human genome-wide eQTL analysis.25,28,29 We measured variation of REST transcript levels in human lymphoblastoid cell lines of 135 individuals from ten three-generation CEPH families. Interindividual differences in REST expression were observed (variance ratio = 4.7), with a significant proportion of this variability having a genetic component (heritability = 0.64; p = 3E-6). We performed genome-wide quantitative linkage analysis using a set of 2688 autosomal SNPs30 distributed throughout the human genome. Interestingly, the highest LOD score (LOD = 3.81; p = 1.4E-5; Figure 2A) mapped to a 3 Mb genomic region on HSA21 that overlaps with the minimal region responsible for Rest suppression in mouse models of DS (Figure 2B; highlighted square).
Figure 2.
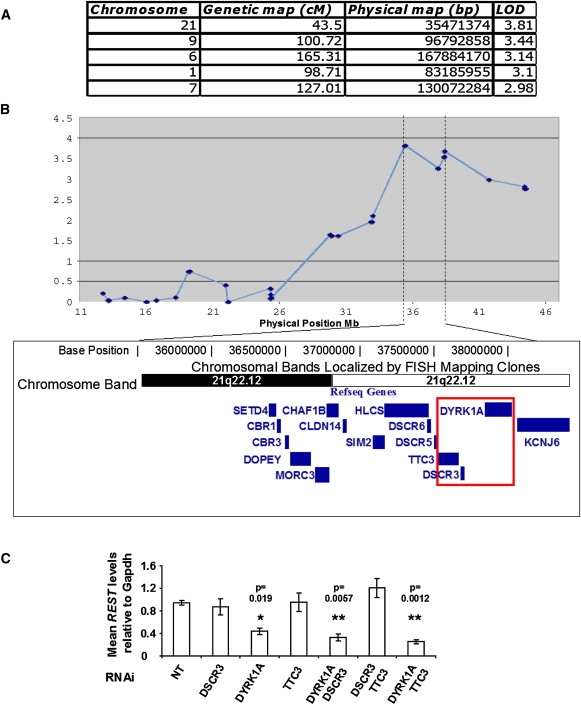
Rest Levels Are Controlled by the DYRK1A Genomic Locus in Human Cells and Are Sensitive to DYRK1A Levels in Normal Mouse Cells
(A) Table showing most significant human genome-wide eQTLs for REST expression levels. The four columns show the chromosome where the peak is located, the genetic map position of the SNP marker with the highest LOD score, its physical position according to the hg17 assembly, and the corresponding LOD score, respectively.
(B) Results of multipoint REST eQTL analysis of HSA21. Dotted lines show the interval of most significant linkage genome-wide and the corresponding annotated gene content derived from the UCSC genome browser. The highlighted box indicates overlap with the common trisomic region identified by segmental models in Figure 1F.
(C) Individual and combination gene-by-gene dissection of the candidate overlap region with the use of RNAi silencing in normal mouse E14 ES cells. The RNAi targets are indicated along the horizontal axis, and the vertical bars show the qRT-PCR levels for Rest (n = 3 independent transfection experiments). The specificity and efficiency of silencing is shown in Figure S3. Data are shown normalized to control samples transfected with a nontargeting “scrambled” RNAi sequence. Means and standard errors are shown, and statistical significance by Student's t test is indicated by one (p < 0.05) or two (p < 0.01) asterisks.
DYRK1A Dosage Imbalance Perturbs Rest Levels
These data, taken together, allowed us to hypothesize that one or more of the three genes in the minimal candidate region (TTC3, DSCR3, or DYRK1A) would control Rest transcript levels in mice and humans. In order to test this hypothesis, we used RNAi oligonucleotides to specifically silence the three genes, individually or in pairs, in normal, pluripotent, undifferentiated mouse E14 ES cells (for sequences of all siRNA reagents, see Table S4; silencing effectiveness was measured by quantitative RT-PCR; see Figure S3). Rest mRNA levels specifically responded only to the dose of Dyrk1A and not to the other two genes (Figure 2C). Interestingly, the level of Rest was reduced when the Dyrk1a transcript was suppressed (Figure 2C). We then used human-specific DYRK1A RNAi oligonucleotides (targeting the 3′UTR) to silence only the products of the third copy of the DYRK1A gene in transchromosomic 47-1 ES cells. This approach only partially succeeded in suppressing human DYRK1A mRNA (∼0.5-fold; Figure 3A), with no significant effect on mouse Dyrk1A (not shown). This correction was sufficient for rescuing Rest levels to within the range of normal (D3 control) values (Figure 3A). This provides strong evidence that Rest dysregulation is mediated by DYRK1A. The effect of the selective DYRK1A-kinase inhibitor, epigallocatechin gallate (EGCG),31 on Rest expression was then assessed. Short-term culture with the inhibitor slightly reduced Rest levels in D3 cells but had little effect on trisomic 47-1 cells (Figure 3B). Longer treatment significantly reduced Rest levels in both cell lines as compared with untreated cells (Figure 3B), in concordance with the effect of complete RNAi silencing of DYRK1A (Figure 2C, Figure S3). We also observed an inhibitory effect of EGCG on the Rest-promoter activity in undifferentiated ES cells (Figure 3C). Lastly, we studied Rest in brains from adult Dyrk1A transgenic (TgDyrk1A) mice, which display several DS-related neural phenotypes and show a 1.94-fold increase in Dyrk1A protein levels.24 A significant (∼30%) reduction of Rest mRNA was observed (Figure 3D), demonstrating that the increased Dyrk1A gene dosage is sufficient to cause the suppression of Rest, to an extent similar to that observed in brains of DS mouse models.
Figure 3.
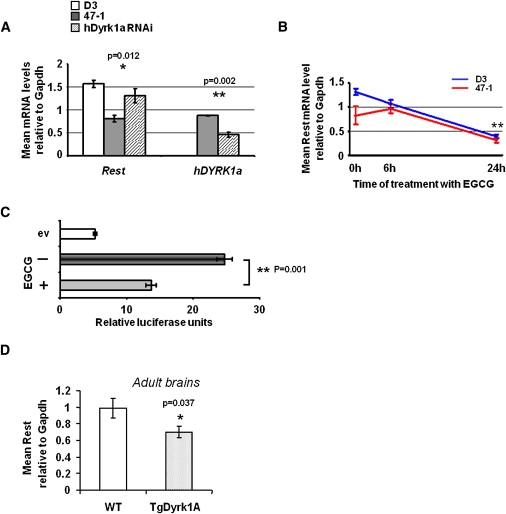
Trisomy of DYRK1A Reduces Rest mRNA Levels in Mouse Models of DS
(A) qRT-PCR analysis of Rest and human DYRK1A levels in undifferentiated mouse ES cells: D3 (open bars), 47-1 (filled bars), and 47-1 transfected with RNAi specifically targeting human DYRK1A mRNA in the 3′UTR (striped bars) (n = 3 independent transfection experiments). The data are shown relative to control samples transfected with nontargeting “scrambled” RNAi sequence.
(B) qRT-PCR analysis of Rest levels in undifferentiated mouse D3 and 47-1 ES cells (blue symbols and red symbols, respectively) treated with the DYRK1A-kinase inhibitor, green-tea compound EGCG (10 μM), for 0, 6, or 24 hr.
(C) Undifferentiated D3 mouse ES cells were transfected with a construct containing 1013 bp of mouse Rest promoter sequence cloned upstream of a firefly luciferase reporter gene and were then treated (+) or not treated (−) with 10 μM EGCG for 24 hr (ev: cells transfected with empty vector, containing the luciferase gene without any promoter). Horizontal bars represent arbitrary luminescence units. Firefly luminescence was normalized against Renilla luciferase activity for taking into account transfection efficiency (n = 3 independent transfection experiments).
(D) qRT-PCR analysis of Rest in adult brains of TgDyrk1A mice and WT littermates (n = 5). Means and standard errors are shown, and statistical significance by Student's t test is indicated by one (p < 0.05) or two (p < 0.01) asterisks.
These data suggest that Rest expression is very sensitive to the level of Dyrk1A, with both over- and underexpression of Dyrk1A resulting in Rest suppression. The DYRK1A-inhibitor data implicate DYRK1A phosphorylation in the mechanism behind this effect. Our data cannot exclude the possibility that other HSA21 genes cooperate with DYRK1A in modulating the Rest levels. The potential contribution of several other genes from the eQTL region was considered: DOPEY2, which was implicated in cerebellar morphogenesis,32 is not expressed at the blastocyst stage (Unigene database), whereas Cbr1, recently highlighted as a strong potential candidate for the generation of DS phenotypes,33 and Setd4 were both ruled out by the demonstration that their respective RNAi knockdowns had no effect on Rest levels in mouse ES cells (see Figure S4). Much more detailed analysis would have to be carried out for the examination of additional contributory effects of all other HSA21 genes.
DYRK1A-Rest Deregulation Disturbs Pluripotency and Embryonic Stem Cell Fate
Our data show that the level of Rest is approximately halved in undifferentiated trisomy 21 ES cells (Figure 1). It was recently reported that Rest +/− cells show reduced levels of key regulators of pluripotency, Oct4, Nanog, and Sox2, resulting in aberrantly premature expression of differentiation-driving transcription factors (TFs).20 The differences in the levels of Oct4, Nanog, and Sox2 in our microarray data were not above the rigorous significance cutoff thresholds. However, when their expression was more sensitively tested by qRT-PCR on a larger number of independent cultures (n = 9), a result partially overlapping with that of Rest +/− cells20 was obtained: Oct4 levels were not significantly changed, but Nanog and Sox2 were both significantly reduced in trisomy 21 ES cells (Figure 4A). Next, we measured the levels of several TF drivers of embryonic-layer-specific differentiation (downstream targets of Oct4, Nanog, and Sox2) that were increased in Rest +/− cells.20 We found that the TF drivers of endoderm (Gata4 [MIM 600576], Gata6 [MIM 601656], and Foxa2 [MIM 600288]) and mesoderm (Snai1 [MIM 604238] and Pitx2 [MIM 601542]) were all aberrantly increased in undifferentiated trisomy 21 ES cells, whereas TF drivers of ectoderm (Fgf5 [MIM 165190]) were unchanged (Figure 4B, Tables S1 and S2). We then demonstrated that the reduced levels of Nanog and Sox2 in 47-1 cells could be partially restored (though still not reaching the levels in D3 cells; not shown) by human-specific DYRK1A RNAi transfection (Figure 4C), similar to Rest (Figure 3A). The partial knockdown of human DYRK1A in this experiment did not significantly alter the levels of lineage-specific TFs (not shown), probably because it would take a stronger and more lasting knockdown to stimulate the cascade of events in the reassembly of the complexes repressing the transcription of these TFs once they had been derepressed. We then investigated whether the drastic reduction in Rest caused by a 24 hr incubation with DYRK1A-kinase inhibitor in normal mouse D3 cells had any effects on the pluripotency-regulating network. This treatment reduced the level of Rest in D3 cells by 3.5-fold (Figure 3B), and it was also sufficient to trigger a reduction in the levels of Nanog and Sox2 and a premature increase in the expression of endodermal and mesodermal TFs Foxa2, Gata4, and Snai1 (Figure 4D). Taken together, these data show that the pluripotency-regulating network is disturbed in trisomy 21 ES cells in a specific way, which is similar in part to the disturbance reported for the heterozygous knockout of Rest,20 and that this deregulation is partially sensitive to the dose and enzymatic activity of DYRK1A.
Figure 4.
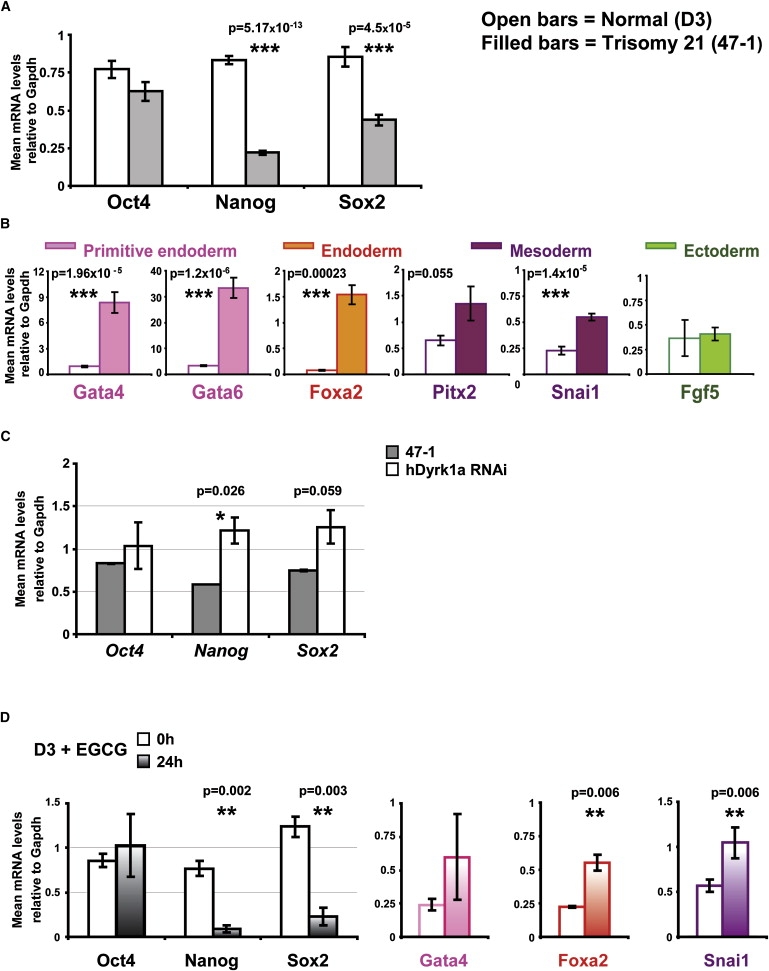
Trisomy 21-Caused Perturbation of the Regulatory Network Maintaining Pluripotency in Undifferentiated ES Cells Is Sensitive to DYRK1A Activity
(A) qRT-PCR measurements of the mRNA levels of key regulators of pluripotency in undifferentiated D3 (open bars) and 47-1 (filled bars) mouse ES cells (n = 9).
(B) qRT-PCR measurements of the mRNA levels of selected differentiation-driving TFs that are known downstream targets of the regulators of pluripotency. TFs driving specific embryonic-layer lineages are color coded, as per labeled color symbols. D3 (open bars) and 47-1 (filled bars) (n = 9).
(C) qRT-PCR analysis of Sox2 and Nanog levels in undifferentiated mouse ES cells: 47-1 (filled bars) and 47-1 transfected with RNAi specifically targeting human DYRK1A mRNA in the 3′UTR (open bars) (n = 3 independent transfection experiments). The data are shown relative to the control samples transfected with nontargeting “scrambled” RNAi sequence.
(D) qRT-PCR analysis of the levels of regulators of pluripotency and selected lineage-specific TFs in undifferentiated normal mouse D3 cells treated with the DYRK1A-kinase inhibitor, green-tea compound EGCG (10 μM), for 0 hr (open symbols) or 24 hr (reverse striped symbols). TFs driving specific embryonic-layer lineages are color coded, as per labeled color symbols.
In all graphs, means and standard errors are shown, and statistical significance by Student's t test is indicated by one (p < 0.05), two (p < 0.01), or three (p < 0.001) asterisks.
When we allowed the ES cells to differentiate into embryoid bodies (EBs), the trisomy 21 EBs (47-1) showed a significantly higher level of the primitive endoderm marker (Gata4) and severely reduced levels of neuroectodermal markers (Nestin [MIM 600915], Tubb3 [MIM 602661], Map2 [MIM 157130]), compared with the normal EBs (D3) (Figure 5A), suggesting a skewed ratio of early layer-specific progenitor cells in favor of primitive endoderm at the expense of neuroectodermal progenitors. This was further investigated by replating the dissociated EB onto N2 medium, allowing the differentiation of neuroectodermal progenitors into neurons. Trisomy 21 cells (47-1) produced a significantly reduced number of neurons per total number of DAPI-staining nuclei and lower relative levels of Tubb3 and Map2 mRNA in the same cell population (Figures 5B and 5B′). There was also a trend toward an increase in abnormal branching in 47-1-derived neurons, compared with D3 neurons (Figure 5C), quantified as an altered ratio of secondary to primary neurites (see Figure S5). These data reproduce the main features of the neurogenesis defect previously reported for human DS fetal neurospheres.9
Figure 5.
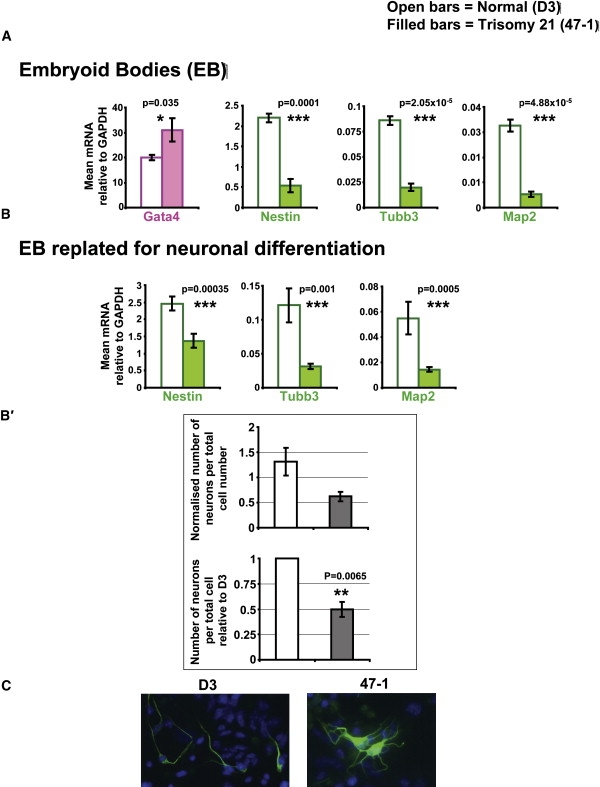
Trisomy 21 ES Cells Give Rise to Embryoid Bodies with a Disturbed Composition of Lineage-Specific Progenitors
(A) qRT-PCR measurements of the mRNA levels of selected lineage-specific markers in EBs derived by culturing D3 (open bars) and 47-1 (filled bars) ES cells (n = 8 experiments) in the absence of LIF, as well as in the presence of retinoic acid. Markers of specific embryonic-layer lineages are color coded, as in Figure 4.
(B and B′) Measurement of the progress of in vitro neurogenesis of EBs from Figure 5A replated onto Neurobasal N2 medium for promoting the differentation of neural progenitors into neurons. (B) mRNA levels of Nestin, Map2, and Tubb3, measured by qRT-PCR (n = 7 experiments). (B′) Proportion of cells positive for immunofluorescent staining with a neuronal marker, relative to the number of DAPI-staining nuclei (n = 2 experiments). Lower graph shows values normalized to WT D3 levels.
(C) Representative immunofluorescence images of Tubb3-positive (green) cells derived from EBs of D3 and 47-1 cells after differentiation for 48 hr in Neurobasal N2 medium, with DAPI (blue) nuclear counterstain. See Figure S5 for quantitative analysis of neurite branching. In all graphs, means and standard errors are shown, and statistical significance by Student's t test is indicated by one (p < 0.05), two (p < 0.01), or three asterisks (p < 0.001).
Discussion
Dual-specificity tyrosine-phosphorylated and -regulated kinase, DYRK1A, is a well-characterized HSA21 gene and ortholog of the Drosophila minibrain (mnb) gene, whose mutation causes abnormal spacing of neuroblasts and reduced production of neuronal progeny in Drosophila.34 Dosage imbalance of DYRK1A together with DSCR1 (MIM 602917) (also on HSA21) has been reported to cause a dysregulation of the NFATc (MIM 602699) pathway in DS.35 This mechanism is most likely independent of the Rest dysregulation we report here, given that DSCR1 is not trisomic in two systems showing Rest reduction in our experiments (40-2 and TC1) and that the Rest reduction we observe developmentally precedes the changes in NFATc. We report a downregulation of Rest mRNA in adult brains of three different DS mouse models—Tc1, Ts1Cje, and TgDyrk1A—compared to their WT littermates (Figures 1G and 3D). Interestingly, all three of these mouse models and a fourth mouse model that is trisomic for four HSA21 genes, including DYRK1A (Tg152f7), share learning and memory deficits, motor defects, including hyperactivity and other behavioral changes, and LTP deficits in the hippocampus or other signs of hippocampal dysfunction.4–8,24 This comparison between the yeast artificial chromosome (YAC)-transgenic, gene-transgenic, and segmental-trisomy models should be taken with a degree of caution, because even though there is a hippocampal dysfunction in both models, the reasons behind each could be different. Also, the impact of Dyrk1A overexpression could be different in different genetic backgrounds within disomic or trisomic contexts and in different brain regions. Our data are partly in contradiction with a recently generated 33-gene trisomy model of DS, Ts1Rhr, that shows no hippocampal-volume change and no electrophysiological and behavioral defects associated with hippocampal functions despite having three copies of Dyrk1A.36 However, given that (1) crossing the Ms1RhR reciprocal-deletion model to the Ts65DN mouse model rescues most of the latter model's cognitive phenotypes36 and that (2) TgDyrk1A mouse models show compelling phenotypes,4,24 it is likely that additional factors, such as strain-specific modifier genes, might account for the apparent contradiction. Additional studies on the Ts1RhR mouse model are clearly necessary to explain these discrepancies. Interestingly, monosomy of this 33-gene segment (mouse model Ms1RhR) produced striking reductions in hippocampal volume,36 comparable to the severe reduction in brain size and morphology in Dyrk1A+/− mice37 and consistent with the fact that truncation of DYRK1A in humans causes microcephaly.38 This result is compatible with our observation that downregulation of Dyrk1A below physiological (disomic) levels drastically reduces Rest level (Figure 2C, Figure 3B), thereby deregulating early differentiation and decreasing the relative proportion of neuroectodermal progenitors. In support of this explanation, an early developmental function for Rest has also been demonstrated in other models: Rest-knockout mice exhibit early embryonic lethality,39 and RNAi strategies in Xenopus revealed that a reduced transcriptional dose of Rest causes incorrect patterning of the ectoderm and abnormal neurogenesis.40 The observation that both overdose and inhibition of Dyrk1A have the same consequence of reducing the Rest transcript level is intriguing. Several different hypotheses could explain this phenomenon. It is plausible that an imbalance could occur in a multiprotein complex regulating REST transcription, subcomponents of which could be DYRK1A targets. Such mechanisms have been previously demonstrated in yeast, in which under- or overexpression of single subunits of a multiprotein complex reduced the fitness of the complex as a whole with an equal outcome.41 We also identify the DYRK1A locus on 21q22 as the most significant eQTL for the control of Rest level in human lymphoblastoid lines (LBLs) (Figures 2A and 2B). Interestingly, this locus, as well as the next two most likely eQTLs (9q22 and 6q27; Figure 2A), form three of the 12 loci genome-wide, with the suggestive linkage to Alzheimer's disease (AD, [MIM 104300]), in a study of 437 families.42 It might be interesting to examine the roles of DYRK1A and REST in aging and survival of neurons, as well as in pathogenesis of AD.
DYRK1A displays a dynamic spatio-temporal pattern of expression during mouse brain development.43 Its expression in preneurogenic and neurogenic progenitors43 places it at the critical point when a sudden change in the level of Rest transcription takes place,14 after the proteolytic degradation of Rest triggers the exit from ES to preneurogenic progenitor. The transient burst of DYRK1A expression in the asymmetric neuronal-progenitor cell division fits with its potential role as the trigger for neuro-differentiation commitment.43,44 Our data suggest that one of the mechanisms by which DYRK1A could exert this triggering role could be through precipitating local spatio-temporal changes in the level of Rest. Repertoire of downstream target genes regulated by REST is critically dependent on the level of REST,14,27,45 regulation and maintenance of which is not fully understood. Differentiation is triggered by an orchestrated reduction in Rest levels, which is mediated both by ubiquitin-dependent proteolytic degradation and by control of transcription.14,46 Forced partial knockout of Rest destabilizes a complex network of miRNAs and proteins responsible for the maintenance of self-renewal and pluripotency of both human and mouse ES cells.20,47,48 This produced a premature and aberrant expression of differentiation-driving TFs of all embryonic lineages in undifferentiated ES cells.20 Our data (Tables S1 and S2, Figures 4A and 4B) show that the halving of Rest dose by trisomy 21 reproduces many of these perturbations (decreased levels of Nanog and Sox2 and increased levels of Gata4, Gata6, Foxa2, Snai1, and Pitx2), but not all of them (Oct4 and Fgf5 levels are unchanged). The reasons for these differences are unclear and require additional investigation, but they are likely to result from the trisomic contribution of other HSA21 elements. Nevertheless, the reduction in Rest has the potential to significantly perturb the pluripotency network in trisomy 21 ES cells, and this perturbance seems to be partially sensitive to the dose and kinase activity of DYRK1A (Figures 4C and 4D). DYRK1A is localized in the cell nuclei and/or cytoplasm, depending on the specific cell type, and some of its phosphorylation targets include histone subunits and TFs.49–51 The potential contribution of other HSA21 genes to this deregulation remains to be investigated. We cannot rule out the possibility that DYRK1A-kinase activity affects other regions in the Rest promoter, besides affecting transcription from the promoter region, shown in Figure 3C (though we ruled out any significant contribution from several other regions, totaling 1393 bp of the Rest promoter; data available on request). We also cannot rule out the possibility that DYRK1A-kinase activity affects other unknown targets within the complex miRNA-protein-regulatory network, whose back- and forward-feeding loops control levels of Rest, Oct4, Nanog, and Sox2.20,47,48
ES cells with premature expression of differentiation-driving TFs have been reported to give rise to EBs with increased lineage-specific progenitors.20 To test if this phenomenon could be reproduced in trisomy 21 ES cells, we differentiated the 47-1 and D3 cells for 8 days in the absence of LIF, as well as in the presence of retinoic acid. Consistent with the premature expression of TFs observed in the trisomy 21 ES cells, their differentiation into EBs resulted in a skewed ratio of lineage progenitor markers: compared with normal (D3) cells, the trisomic (47-1) cells produced EBs with higher levels of Gata4 (a marker of extra-embryonic endoderm and heart development) and reduced markers of the EB neuroectodermal compartment (nestin, Tubb3, and Map-2) (Figure 5A). Neurogenesis starting from such EBs was retarded (Figure 5B), consistent with the prediction that the EBs contained fewer neuronal progenitor cells relative to other progenitors. Two other reports support these observations: (1) we have recently shown that trisomy 21 ES cells ectopically form teratomas displaying the full spectrum of differentiated tissues in vivo but with a strongly reduced neuronal cell fate compared with euploid controls,52 and (2) Bahn et al. have shown that human fetal-brain neurospheres display a severely reduced ability to form neurons, compared to neurospheres grown from age-matched euploid feti.9 The DS neurospheres also produced neurons with abnormally branched neurites,9 a result reproduced by our transchromosomic ES cells (Figure 5C and Figure S5) and by partial REST siRNA knockdown in a differentiating NPC cell line.53 Taken together, these data suggest that the DYRK1A-REST perturbation has the potential to significantly contribute to the development of defects in neuronal number and morphology in DS. The premature reduction in REST levels could skew cell-fate decisions to give rise to a relative depletion in the number of neuronal progenitors, and the same reduction in REST levels could cause an aberrantly accelerated differentiation of each progenitor, producing abnormally highly branched neurons.
In summary, we have demonstrated that Rest levels are disturbed at a very early developmental stage in trisomy 21 ES cells (preceding ES cell differentiation) and shown this disturbance to be dose-sensitive to the level and activity of DYRK1A. Our results suggest that this deregulation has the potential to disturb the development of all embryonic lineages, warranting more detailed research into its contribution to all aspects of DS pathology and rationales for novel therapeutic approaches.
Supplemental Data
Supplemental Data include five figures and five tables and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
MIAMEXPRESS Database: http://www.ebi.ac.uk/microarray-as/ae/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
UCSC Genome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway
Accession Number
The experiment accession number in the MIAMExpress database for the microarray data reported in this paper is E-MEXP-654.
Acknowledgments
This work was supported by the AnEUploidy grant from Framework Programme 6 from the EU Commission and by the Specialist Programme Grant 06003 from the Leukaemia Research Fund-UK. We also thank the following for their support: Barts and The London Charitable Foundation, Fondation Jerome Lejeune, Swiss National Science Foundation and Center of Excellence “Frontiers in Genetics”, “Childcare” Foundation, National Institute of Child Health and Human Development, Spanish Ministry of Education and Science, Medical Research Council UK, and The Wellcome Trust. We thank Philip Cohen (University of Dundee) for advice.
References
- 1.Epstein C. Down Syndrome. In: Scriver C.R.B.A., Sly W.S., Valle D., editors. The metabolic and molecular bases of inherited disease. McGraw-Hill; New York: 2001. pp. 1223–1256. [Google Scholar]
- 2.Antonarakis S.E., Lyle R., Dermitzakis E.T., Reymond A., Deutsch S. Chromosome 21 and down syndrome: from genomics to pathophysiology. Nat. Rev. Genet. 2004;5:725–738. doi: 10.1038/nrg1448. [DOI] [PubMed] [Google Scholar]
- 3.Reeves R.H., Irving N.G., Moran T.H., Wohn A., Kitt C., Sisodia S.S., Schmidt C., Bronson R.T., Davisson M.T. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat. Genet. 1995;11:177–184. doi: 10.1038/ng1095-177. [DOI] [PubMed] [Google Scholar]
- 4.Smith D.J., Stevens M.E., Sudanagunta S.P., Bronson R.T., Makhinson M., Watabe A.M., O'Dell T.J., Fung J., Weier H.U., Cheng J.F. Functional screening of 2 Mb of human chromosome 21q22.2 in transgenic mice implicates minibrain in learning defects associated with Down syndrome. Nat. Genet. 1997;16:28–36. doi: 10.1038/ng0597-28. [DOI] [PubMed] [Google Scholar]
- 5.Sago H., Carlson E.J., Smith D.J., Kilbridge J., Rubin E.M., Mobley W.C., Epstein C.J., Huang T.T. Ts1Cje, a partial trisomy 16 mouse model for Down syndrome, exhibits learning and behavioral abnormalities. Proc. Natl. Acad. Sci. USA. 1998;95:6256–6261. doi: 10.1073/pnas.95.11.6256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olson L.E., Roper R.J., Baxter L.L., Carlson E.J., Epstein C.J., Reeves R.H. Down syndrome mouse models Ts65Dn, Ts1Cje, and Ms1Cje/Ts65Dn exhibit variable severity of cerebellar phenotypes. Dev. Dyn. 2004;230:581–589. doi: 10.1002/dvdy.20079. [DOI] [PubMed] [Google Scholar]
- 7.Siarey R.J., Villar A.J., Epstein C.J., Galdzicki Z. Abnormal synaptic plasticity in the Ts1Cje segmental trisomy 16 mouse model of Down syndrome. Neuropharmacology. 2005;49:122–128. doi: 10.1016/j.neuropharm.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 8.O'Doherty A., Ruf S., Mulligan C., Hildreth V., Errington M.L., Cooke S., Sesay A., Modino S., Vanes L., Hernandez D. An aneuploid mouse strain carrying human chromosome 21 with down syndrome phenotypes. Science. 2005;309:2033–2037. doi: 10.1126/science.1114535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bahn S., Mimmack M., Ryan M., Caldwell M.A., Jauniaux E., Starkey M., Svendsen C.N., Emson P. Neuronal target genes of the neuron-restrictive silencer factor in neurospheres derived from fetuses with Down's syndrome: a gene expression study. Lancet. 2002;359:310–315. doi: 10.1016/S0140-6736(02)07497-4. [DOI] [PubMed] [Google Scholar]
- 10.Chong J.A., Tapia-Ramirez J., Kim S., Toledo-Aral J.J., Zheng Y., Boutros M.C., Altshuller Y.M., Frohman M.A., Kraner S.D., Mandel G. REST: a mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell. 1995;80:949–957. doi: 10.1016/0092-8674(95)90298-8. [DOI] [PubMed] [Google Scholar]
- 11.Schoenherr C.J., Anderson D.J. The neuron-restrictive silencer factor (NRSF): a coordinate repressor of multiple neuron-specific genes. Science. 1995;267:1360–1363. doi: 10.1126/science.7871435. [DOI] [PubMed] [Google Scholar]
- 12.Sun Y.M., Greenway D.J., Johnson R., Street M., Belyaev N.D., Deuchars J., Bee T., Wilde S., Buckley N.J. Distinct Profiles of REST Interactions with Its Target Genes at Different Stages of Neuronal Development. Mol. Biol. Cell. 2005;16:5630–5638. doi: 10.1091/mbc.E05-07-0687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuwabara T., Hsieh J., Nakashima K., Taira K., Gage F.H. A small modulatory dsRNA specifies the fate of adult neural stem cells. Cell. 2004;116:779–793. doi: 10.1016/s0092-8674(04)00248-x. [DOI] [PubMed] [Google Scholar]
- 14.Ballas N., Grunseich C., Lu D.D., Speh J.C., Mandel G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell. 2005;121:645–657. doi: 10.1016/j.cell.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 15.Bruce A.W., Donaldson I.J., Wood I.C., Yerbury S.A., Sadowski M.I., Chapman M., Gottgens B., Buckley N.J. Genome-wide analysis of repressor element 1 silencing transcription factor/neuron-restrictive silencing factor (REST/NRSF) target genes. Proc. Natl. Acad. Sci. USA. 2004;101:10458–10463. doi: 10.1073/pnas.0401827101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bessis A., Champtiaux N., Chatelin L., Changeux J.P. The neuron-restrictive silencer element: a dual enhancer/silencer crucial for patterned expression of a nicotinic receptor gene in the brain. Proc. Natl. Acad. Sci. USA. 1997;94:5906–5911. doi: 10.1073/pnas.94.11.5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoo J., Jeong M.J., Lee S.S., Lee K.I., Kwon B.M., Kim D.S., Park Y.M., Han M.Y. The neuron restrictive silencer factor can act as an activator for dynamin I gene promoter activity in neuronal cells. Biochem. Biophys. Res. Commun. 2001;283:928–932. doi: 10.1006/bbrc.2001.4857. [DOI] [PubMed] [Google Scholar]
- 18.Su X., Kameoka S., Lentz S., Majumder S. Activation of REST/NRSF target genes in neural stem cells is sufficient to cause neuronal differentiation. Mol. Cell. Biol. 2004;24:8018–8025. doi: 10.1128/MCB.24.18.8018-8025.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tahiliani M., Mei P., Fang R., Leonor T., Rutenberg M., Shimizu F., Li J., Rao A., Shi Y. The histone H3K4 demethylase SMCX links REST target genes to X-linked mental retardation. Nature. 2007;447:601–605. doi: 10.1038/nature05823. [DOI] [PubMed] [Google Scholar]
- 20.Singh S.K., Kagalwala M.N., Parker-Thornburg J., Adams H., Majumder S. REST maintains self-renewal and pluripotency of embryonic stem cells. Nature. 2008;453:223–227. doi: 10.1038/nature06863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hernandez D., Mee P.J., Martin J.E., Tybulewicz V.L., Fisher E.M. Transchromosomal mouse embryonic stem cell lines and chimeric mice that contain freely segregating segments of human chromosome 21. Hum. Mol. Genet. 1999;8:923–933. doi: 10.1093/hmg/8.5.923. [DOI] [PubMed] [Google Scholar]
- 22.Bibel M., Richter J., Schrenk K., Tucker K.L., Staiger V., Korte M., Goetz M., Barde Y.A. Differentiation of mouse embryonic stem cells into a defined neuronal lineage. Nat. Neurosci. 2004;7:1003–1009. doi: 10.1038/nn1301. [DOI] [PubMed] [Google Scholar]
- 23.Delom F., Emadali A., Cocolakis E., Lebrun J.J., Nantel A., Chevet E. Calnexin-dependent regulation of tunicamycin-induced apoptosis in breast carcinoma MCF-7 cells. Cell Death Differ. 2007;14:586–596. doi: 10.1038/sj.cdd.4402012. [DOI] [PubMed] [Google Scholar]
- 24.Altafaj X., Dierssen M., Baamonde C., Marti E., Visa J., Guimera J., Oset M., Gonzalez J.R., Florez J., Fillat C. Neurodevelopmental delay, motor abnormalities and cognitive deficits in transgenic mice overexpressing Dyrk1A (minibrain), a murine model of Down's syndrome. Hum. Mol. Genet. 2001;10:1915–1923. doi: 10.1093/hmg/10.18.1915. [DOI] [PubMed] [Google Scholar]
- 25.Deutsch S., Lyle R., Dermitzakis E.T., Attar H., Subahmanyan L., Gehrig C., Parand L., Gagnebin M., Rougemont J., Jongeneel C.V. Gene expression variation and expression quantitative trait (eQTL) mapping of human chromosome 21 genes. Hum. Mol. Genet. 2005;14:3741–3749. doi: 10.1093/hmg/ddi404. [DOI] [PubMed] [Google Scholar]
- 26.Lyle R., Gehrig C., Neergaard-Henrichsen C., Deutsch S., Antonarakis S.E. Gene expression from the aneuploid chromosome in a trisomy mouse model of down syndrome. Genome Res. 2004;14:1268–1274. doi: 10.1101/gr.2090904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Otto S.J., McCorkle S.R., Hover J., Conaco C., Han J.J., Impey S., Yochum G.S., Dunn J.J., Goodman R.H., Mandel G. A new binding motif for the transcriptional repressor REST uncovers large gene networks devoted to neuronal functions. J. Neurosci. 2007;27:6729–6739. doi: 10.1523/JNEUROSCI.0091-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morley M., Molony C.M., Weber T.M., Devlin J.L., Ewens K.G., Spielman R.S., Cheung V.G. Genetic analysis of genome-wide variation in human gene expression. Nature. 2004;430:743–747. doi: 10.1038/nature02797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schadt E.E., Lamb J., Yang X., Zhu J., Edwards S., Guhathakurta D., Sieberts S.K., Monks S., Reitman M., Zhang C. An integrative genomics approach to infer causal associations between gene expression and disease. Nat. Genet. 2005;37:710–717. doi: 10.1038/ng1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deloukas P., Schuler G.D., Gyapay G., Beasley E.M., Soderlund C., Rodriguez-Tome P., Hui L., Matise T.C., McKusick K.B., Beckmann J.S. A physical map of 30,000 human genes. Science. 1998;282:744–746. doi: 10.1126/science.282.5389.744. [DOI] [PubMed] [Google Scholar]
- 31.Bain J., McLauchlan H., Elliott M., Cohen P. The specificities of protein kinase inhibitors: an update. Biochem. J. 2003;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rachidi M., Lopes C., Costantine M., Delabar J.M. C21orf5, a new member of Dopey family involved in morphogenesis, could participate in neurological alterations and mental retardation in Down syndrome. DNA Res. 2005;12:203–210. doi: 10.1093/dnares/dsi004. [DOI] [PubMed] [Google Scholar]
- 33.Sultan M., Piccini I., Balzereit D., Herwig R., Saran N.G., Lehrach H., Reeves R.H., Yaspo M.L. Gene expression variation in Down's syndrome mice allows prioritization of candidate genes. Genome Biol. 2007;8:R91. doi: 10.1186/gb-2007-8-5-r91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tejedor F., Zhu X.R., Kaltenbach E., Ackermann A., Baumann A., Canal I., Heisenberg M., Fischbach K.F., Pongs O. minibrain: a new protein kinase family involved in postembryonic neurogenesis in Drosophila. Neuron. 1995;14:287–301. doi: 10.1016/0896-6273(95)90286-4. [DOI] [PubMed] [Google Scholar]
- 35.Arron J.R., Winslow M.M., Polleri A., Chang C.P., Wu H., Gao X., Neilson J.R., Chen L., Heit J.J., Kim S.K. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature. 2006;441:595–600. doi: 10.1038/nature04678. [DOI] [PubMed] [Google Scholar]
- 36.Olson L.E., Roper R.J., Sengstaken C.L., Peterson E.A., Aquino V., Galdzicki Z., Siarey R., Pletnikov M., Moran T.H., Reeves R.H. Trisomy for the Down syndrome “critical region” is necessary but not sufficient for brain phenotypes of trisomic mice. Hum. Mol. Genet. 2007;16:774–782. doi: 10.1093/hmg/ddm022. [DOI] [PubMed] [Google Scholar]
- 37.Fotaki V., Dierssen M., Alcantara S., Martinez S., Marti E., Casas C., Visa J., Soriano E., Estivill X., Arbones M.L. Dyrk1A haploinsufficiency affects viability and causes developmental delay and abnormal brain morphology in mice. Mol. Cell. Biol. 2002;22:6636–6647. doi: 10.1128/MCB.22.18.6636-6647.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moller R.S., Kubart S., Hoeltzenbein M., Heye B., Vogel I., Hansen C.P., Menzel C., Ullmann R., Tommerup N., Ropers H.H. Truncation of the Down syndrome candidate gene DYRK1A in two unrelated patients with microcephaly. Am. J. Hum. Genet. 2008;82:1165–1170. doi: 10.1016/j.ajhg.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen Z.F., Paquette A.J., Anderson D.J. NRSF/REST is required in vivo for repression of multiple neuronal target genes during embryogenesis. Nat. Genet. 1998;20:136–142. doi: 10.1038/2431. [DOI] [PubMed] [Google Scholar]
- 40.Olguin P., Oteiza P., Gamboa E., Gomez-Skarmeta J.L., Kukuljan M. RE-1 silencer of transcription/neural restrictive silencer factor modulates ectodermal patterning during Xenopus development. J. Neurosci. 2006;26:2820–2829. doi: 10.1523/JNEUROSCI.5037-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Papp B., Pal C., Hurst L.D. Dosage sensitivity and the evolution of gene families in yeast. Nature. 2003;424:194–197. doi: 10.1038/nature01771. [DOI] [PubMed] [Google Scholar]
- 42.Blacker D., Bertram L., Saunders A.J., Moscarillo T.J., Albert M.S., Wiener H., Perry R.T., Collins J.S., Harrell L.E., Go R.C. Results of a high-resolution genome screen of 437 Alzheimer's disease families. Hum. Mol. Genet. 2003;12:23–32. doi: 10.1093/hmg/ddg007. [DOI] [PubMed] [Google Scholar]
- 43.Hammerle B., Elizalde C., Tejedor F.J. The spatio-temporal and subcellular expression of the candidate Down syndrome gene Mnb/Dyrk1A in the developing mouse brain suggests distinct sequential roles in neuronal development. Eur. J. Neurosci. 2008;27:1061–1074. doi: 10.1111/j.1460-9568.2008.06092.x. [DOI] [PubMed] [Google Scholar]
- 44.Hammerle B., Vera-Samper E., Speicher S., Arencibia R., Martinez S., Tejedor F.J. Mnb/Dyrk1A is transiently expressed and asymmetrically segregated in neural progenitor cells at the transition to neurogenic divisions. Dev. Biol. 2002;246:259–273. doi: 10.1006/dbio.2002.0675. [DOI] [PubMed] [Google Scholar]
- 45.Greenway D.J., Street M., Jeffries A., Buckley N.J. RE1 Silencing transcription factor maintains a repressive chromatin environment in embryonic hippocampal neural stem cells. Stem Cells. 2007;25:354–363. doi: 10.1634/stemcells.2006-0207. [DOI] [PubMed] [Google Scholar]
- 46.Westbrook T.F., Hu G., Ang X.L., Mulligan P., Pavlova N.N., Liang A., Leng Y., Maehr R., Shi Y., Harper J.W. SCFbeta-TRCP controls oncogenic transformation and neural differentiation through REST degradation. Nature. 2008;452:370–374. doi: 10.1038/nature06780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boyer L.A., Lee T.I., Cole M.F., Johnstone S.E., Levine S.S., Zucker J.P., Guenther M.G., Kumar R.M., Murray H.L., Jenner R.G. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim J., Chu J., Shen X., Wang J., Orkin S.H. An extended transcriptional network for pluripotency of embryonic stem cells. Cell. 2008;132:1049–1061. doi: 10.1016/j.cell.2008.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Himpel S., Tegge W., Frank R., Leder S., Joost H.G., Becker W. Specificity determinants of substrate recognition by the protein kinase DYRK1A. J. Biol. Chem. 2000;275:2431–2438. doi: 10.1074/jbc.275.4.2431. [DOI] [PubMed] [Google Scholar]
- 50.Woods Y.L., Rena G., Morrice N., Barthel A., Becker W., Guo S., Unterman T.G., Cohen P. The kinase DYRK1A phosphorylates the transcription factor FKHR at Ser329 in vitro, a novel in vivo phosphorylation site. Biochem. J. 2001;355:597–607. doi: 10.1042/bj3550597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Galceran J., de Graaf K., Tejedor F.J., Becker W. The MNB/DYRK1A protein kinase: genetic and biochemical properties. J. Neural Transm. Suppl. 2003;67:139–148. doi: 10.1007/978-3-7091-6721-2_12. [DOI] [PubMed] [Google Scholar]
- 52.Mensah A., Mulligan C., Linehan J., Ruf S., O'Doherty A., Grygalewicz B., Shipley J., Groet J., Tybulewicz V., Fisher E. An additional human chromosome 21 causes suppression of neural fate of pluripotent mouse embryonic stem cells in a teratoma model. BMC Dev. Biol. 2007;7:131. doi: 10.1186/1471-213X-7-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lepagnol-Bestel A.M., Maussion G., Ramoz N., Moalic J.M., Gorwood P., Simonneau M. Nrsf silencing induces molecular and subcellular changes linked to neuronal plasticity. Neuroreport. 2007;18:441–446. doi: 10.1097/WNR.0b013e328011dc81. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.