

Abstract
Following up on our previous study, we conducted a genome-wide analysis of admixture for two Uyghur population samples (HGDP-UG and PanAsia-UG), collected from the northern and southern regions of Xinjiang in China, respectively. Both HGDP-UG and PanAsia-UG showed a substantial admixture of East-Asian (EAS) and European (EUR) ancestries, with an empirical estimation of ancestry contribution of 53:47 (EAS:EUR) and 48:52 for HGDP-UG and PanAsia-UG, respectively. The effective admixture time under a model with a single pulse of admixture was estimated as 110 generations and 129 generations, or admixture events occurred about 2200 and 2580 years ago for HGDP-UG and PanAsia-UG, respectively, assuming an average of 20 yr per generation. Despite Uyghurs' earlier history compared to other admixture populations, admixture mapping, holds promise for this population, because of its large size and its mixture of ancestry from different continents. We screened multiple databases and identified a genome-wide single-nucleotide polymorphism panel that can distinguish EAS and EUR ancestry of chromosomal segments in Uyghurs. The panel contains 8150 ancestry-informative markers (AIMs) showing large frequency differences between EAS and EUR populations (FST > 0.25, mean FST = 0.43) but small frequency differences (7999 AIMs validated) within both populations (FST < 0.05, mean FST < 0.01). We evaluated the effectiveness of this admixture map for localizing disease genes in two Uyghur populations. To our knowledge, our map constitutes the first practical resource for admixture mapping in Uyghurs, and it will enable studies of diseases showing differences in genetic risk between EUR and EAS populations.
Introduction
Xinjiang, a territory located at the far west of China and crossed by the Silk Road, is an important pathway connecting East Asia with Central Asia and Europe. About half of total population in Xinjiang are Uyghurs (> 9.4 million), who demonstrate an array of mixed European and Asian anthropological features.1 Genetic studies have found that extant Uyghur populations represent an admixture of eastern and western Eurasian mtDNA2 and Y-chromosome3 lineages. By analyzing the single-nucleotide polymorphism (SNP) data of chromosome 21 in a Uyghur population sampled from southern Xinjiang, we showed, in a recent study, that both East-Asian (EAS) and European (EUR) ancestries contribute to the current gene pool of Uyghur populations, with 60% EUR ancestry and 40% EAS ancestry.4 It was believed that more interaction occurred recently between the Han Chinese and Uyghur populations in northern Xinjiang, and the EUR ancestry contribution was estimated as only 30%5 or 36.3%6 in some previous studies. However, there were also much higher estimations (> 50%) when both classical markers7 and mtDNA8 were used; especially, sequences of 55% European mtDNA were found in Uyghurs sampled in the easternmost section of Kazakhstan, which is only 18 km from the boundary with northern Xinjiang,8 and those Uyghurs were known to have emigrated from Xinjiang. According to historical records, the ancestors of Uyghur (Gaoche) can be traced back to the Chidi and the Dingling in the third century B.C. A full analysis of the genetic structure of modern Uyghurs of Xinjiang would shed light on the understanding of human migratory history and of the admixture mechanisms of Europeans and East Asians.
Apart from their interesting genetic history, Uyghurs have potential utility for the mapping of genes underlying diseases. The mixed ancestries of chromosomal segments in Uyghur genomes provide an experiment of nature that can be exploited for the localization of genes. This idea was theoretically proposed two decades ago by Chakraborty et al.;9 i.e., the information about linkage that is generated by admixture could be used for the localization of disease-susceptibility genes by a test for the allelic associations with disease that are generated by admixture. Admixture of populations often leads to extended linkage disequilibrium (LD); the term “mapping by admixture linkage disequilibrium” (MALD) was introduced by Stephens et al.,10 and later, McKeigue11 further outlined the theory of this approach. MALD, or admixture mapping, has been applied in African Americans and has achieved respectable successes recently.12–16 In comparison with alternative mapping methods that rely on testing for allelic association, admixture mapping is an economical and theoretically powerful approach. The primary attractions of this approach are: it can use an affected-only design, and it requires far fewer genetic markers than do direct association studies.17–19 Where admixed populations exist, a feasible and efficient application of admixture mapping depends on the availability of genome-wide panels of ancestry-informative markers (AIMs) for an inference of the ancestry of the chromosomal regions of admixed individuals. Such panels are already available for disease-gene discovery in African Americans,20–23 Hispanics/Latinos,21,24,25 and Mexican Americans (the largest subgroup of Hispanics/Latinos).20,26 However, no such a map is yet available for Uyghur populations, although admixture mapping should hold equal promise for Uyghurs because of their mixture of ancestries from different continents and their large population size.
In this study, as a follow-up and extension of our previous study,4 we conducted a genome-wide analysis of admixture for two Uyghur population samples (HGDP-UG and PanAsia-UG), collected from northern and southern regions of Xinjiang, respectively. We first investigated their genetic structure and their genetic relationship with EAS and EUR populations and estimated their respective admixture times. We then screened AIMs using SNP data from several large-scale genomic projects, including the International HapMap Project,27–29 the Human Genome Diversity Project (HGDP),30 and the PanAsia SNP Project.31 All of the AIMs in this panel were validated in six EAS population samples and nine EUR population samples and showed little variation of allele frequency within both EAS and EUR population samples. Finally, we evaluated the effectiveness of this admixture map for the localization of disease genes in Uyghur populations.
Subjects and Methods
Populations and Samples
Overall, 26 Uyghur samples (PanAsia-UG) were collected at Hetian, in southern Xinjiang. They are part of 40 Uyghur samples used in our previous study.4 Another 10 Uyghur samples (HGDP-UG), part of the HGDP panel, were originally collected from Yili, at the northern part of Xinjiang. Besides the 10 HGDP-UG samples, 10 Mongola samples, 10 northern Han Chinese (Han-NChina) samples, 35 Han Chinese samples (Han), 29 Japanese samples (Japanese), and 8 European population samples (29 French, 24 Basque, 28 Sardinian, 14 Italian, 8 Tuscan, 16 Orcadian, 17 Adygei, and 25 Russian) were selected from the HGDP-CEPH panel.30 Genotype data of 60 CEU (Utah residents with ancestry from northern and western Europe), 45 CHB (Han Chinese individuals in Beijing), and 45 JPT (Japanese individuals in Tokyo) were obtained from the database of the International HapMap Project.27–29
Data Sets
Genotype data of 26 PanAsia-UG samples generated with the Affymetrix Genechip Human Mapping 50K Xba array were obtained from the Pan-Asia Project.31 A set of 58,960 SNPs was genotyped with the Affymetrix XBA50K chip in 26 PanAsia-UG samples, according to the manufacturer's protocols. Genotype data of 60 CEU, 45 CHB, and 45 JPT samples were obtained from the International HapMap Project27–29 (HapMap publicly released #23a, 2008-04-01). Genotype data of the other samples generated with the Illumina HumanHap650K Beadchip were obtained from the database of the Human Genome Diversity Project-Centre d'Etude du Polymorphisme Humain (HGDP-CEPH) sample of populations.30 The detailed information about data filtration and data quality control was described elsewhere.31
Combined Data Set for Population-Structure Analysis
We integrated three data sets (HapMap data, PanAsia 50K data, and HGDP-CEPH 650K data) according to SNP ID (rs number). This effort yielded 19,934 SNPs genotyped in all 17 population samples. By a comparison of the duplicated genotypes of five Melanesian samples in both data sets, a total of 80 genotypes were found to be inconsistent in two datasets, indicating that the genotyping concordance between Affymetrix and Illumina technologies is greater than 99.9%. The physical positions of SNPs were based on the Homo sapiens Genome Build 36. The average spacing between adjacent markers is 137.7 kb, with a minimum of 17 bp and a maximum of 29.6 Mb; the median between-marker distance (BMD) is 65.4 kb.
Given the large number of markers in our dataset, genetic analyses can be performed at the level of the individual, making no presumption of group membership. We applied a model-based clustering algorithm, implemented by the computer program STRUCTURE,32,33 to infer the genetic ancestry of individuals. Our approach is based solely on genotype without the incorporation of any information on sampling location or population affiliation of each individual. STRUCTURE analysis was performed without any prior population assignment and was performed ten times for each number of population groups (K), with 20,000 replicates and 30,000 burn-in cycles under the admixture model. The log likelihood of each analysis at varying Ks is also estimated in the STRUCTURE analysis (Figure S1, available online) and, as expected, favored two population groups in both PanAsia-UG samples and HGDP-UG samples.
FST Calculation, Molecular Phylogeny, and Principal-Component Analysis
FST is a measurement of genetic differentiation between populations. We calculated FST for all pairs of populations by using the population allele frequencies across all 19,934 autosomal SNPs. The algorithm considers the variation of sample size, following Weir and Hill.34 We performed principal-component analysis at the individual level; eigenvectors of each individual were calculated using EIGENSOFT.35 To construct the phylogenetic tree, we used both the maximum-likelihood method, implemented in the CONTML program of the PHYLIP36 package, and the Neighbor-Joining method,37 implemented in MEGA.38
Genetic Map of SNPs
Although available analyses suggest that there are differences in meiotic recombination frequency across different genomic intervals in different human populations,39 a previous study showed that similar power was achieved for different genetic maps.22 In the current study, we use the Rutgers combined linkage-physical map40 to locate markers on the genetic map. The Rutgers map combines genotyping data from both deCODE and CEPH families and incorporates the latest human genome assembly Build 36. The positions of SNPs on the Rutgers map were determined by a web-based linkage-mapping server that carried out a smoothing calculation to estimate genetic-map positions, including those markers which were not mapped directly.
Selection of AIMs from HapMap Data
For each SNP, we calculated FST, which adjusts for unequal sample size, following Weir and Hill.34 We identified 271,907 SNPs with an FST > 0.25 from genotypes of 60 unrelated CEU subjects (parents) and 90 unrelated East-Asian subjects (JPT and CHB HapMap data sets) in an initial examination of the combined phase I29 and phase II27 HapMap results (about 3.9 million genotypes are available). High FST values favor selection of markers that are closer to fixation in one or both parental populations.26 Those SNPs that showed deviation from Hardy-Weinberg equilibrium within population were excluded with the use of Fisher's exact test (p < 0.001), in which p was estimated with Arlequin 3.01,41 with 100,000 permutations.
Validation of AIMs in HGDP-CEPH Data
In the initial set of 271,907 AIMs, 63,544 SNPs in the HGDP-CEPH panel were genotyped and thus could be validated in more EAS and EUR populations, and another 405 SNPs with FST > 0.8 between EAS and EUR were also kept for further filtration. Small within-group variations (FST < 0.05) of AIMs were validated in six EAS populations and nine EUR populations; a summary of SNP-screen results is shown in Table S1 and Table S2. AIMs were further screened by the choosing of a maximum of two SNPs in 0.1 cM windows, with a minimum distance of 0.01 cM between SNPs. Additional SNPs were added in regions with lower informativeness, and SNPs were thinned in regions of high informativeness.
AIMs for Studying PanAsia-UG Samples Only
We screened another set of AIMs for assessment of admixture of PanAsia-UG samples. The screening procedure was similar to that aforementioned, except that the intermarker distance between AIMs could not be satisfied because only 58,960 SNPs were genotyped in PanAsia-UG samples.
Simulation Studies and Assessment of Genome-wide Information Content of EAS/EUR AIMs
We used Shannon entropy-related statistics (ri) which is implemented in ANCESTRYMAP42 as a measure of the map informativeness of each AIM, and we used the average of ri across loci (ravg) as a measure of the map power for all AIMs. A rough interpretation of ravg is that 1/ravg times as many samples must be genotyped, relative to a study with perfect information about local ancestry (ravg = 1), for comparable power to be achieved.25
We simulated northern and southern Uyghur samples, using EAS and EUR ancestry proportions estimated in the present study. Genotypes were sampled from the 60 EUR (CEU) samples and the 90 EAS (JPT and CHB) samples used to build our admixture map. For simulated samples of northern and southern Uyghurs, genotypes of each locus were generated on the basis of allele frequencies in HGDP-UG and PanAsia-UG, respectively. Chromosomal segments were created under the assumption of 110 generations for northern Uyghur populations and 130 generations for southern Uyghur populations since admixture, and they were assigned ancestries by the use of admixture proportions estimated in HGDP-UG and PanAsia-UG, respectively.
Disease cases were simulated as by Price et al.25 We assumed an increased disease risk of 1.5 for each chromosome with EAS ancestry at the disease locus, thus raising the proportion of EAS ancestry at that locus and at chromosomal segments containing it.25 Simulations were run with ANCESTRYMAP software,42 which produces a local LOD (log10 odds) score and a genome-wide LOD score on the basis of a locus-genome statistic that compares ancestry of cases at a candidate locus with genome-wide ancestry of cases. We studied only case-control design, with 1000 cases and 1000 controls simulated for each study. To check whether there were false-positive results reported, in control-only runs, 2000 controls were randomly sampled as 1000 pseudocases and 1000 controls.
Calculation of the Number of Samples Needed for Detection of an Admixture Association
To calculate the power of an admixture scan for a population distribution of admixture, we calculate the number of disease samples needed for an expected LOD (log10 odds) score ≥ 5 using the formula described elsewhere.25 The distribution of EAS ancestry was set as [0.42, 0.63] for northern Uyghurs and [0.40, 0.53] for southern Uyghurs, respectively, according to the estimation used in this study. For each population, this quantity is computed under the ideal assumption of perfect information about ancestry, as a function of the relative disease risk conferred by each copy of a particular ancestry at the disease locus. For real disease scans involving a map with imperfect information, the number of samples required to achieve significance needs to be scaled by relative informativeness at the locus. To convert from this number to the actual number of samples required for detection of a disease locus with use of the map, it is necessary to multiply the number by the reciprocal of ravg as estimated above.
Results
Genetic Diversity and Relationship of Populations
Heterozygosity measures the genetic diversity within each population, and both expected heterozygosity (He) and observed heterozygosity (Ho) were calculated for each population with the use of genotypes of 19,934 SNPs (Table 1). The two Uyghur populations (PanAsia-UG and HGDP-UG) show higher Ho than do the other EAS and EUR populations, and PanAsia-UG also shows much higher He than do the other populations, indicating that the Uyghur population has a higher proportion of common alleles (high MAF SNPs), which is expected in admixture populations. Genetic difference between populations was estimated by pairwise FST. Notably, a) FST between the two Uyghur populations (0.0009) is much lower than that of most of the other population pairs and only slightly higher than that between Japanese populations and between Han Chinese populations (Table S3); b) FST values between PanAsia-UG and EAS populations are higher than those between HGDP-UG and EAS populations (Figure 1; paired t test, one-tailed p = 1.16 × 10−6); and c) FST values between PanAsia-UG and EUR populations are lower than those between HGDP-UG and EUR populations (Figure 1; paired t test, one-tailed p = 8.24 × 10−9). The latter two observations suggest that the PanAsia-UG population has a closer genetic relationship with the EUR population, whereas the HGDP-UG population has a closer genetic relationship with the EAS population. Both the maximum-likelihood (Figure 2A) tree and the Neighbor-Joining tree (Figure 2B), including Uyghur populations and their putative ancestral populations EAS and EUR, support the aforementioned relationship.
Table 1.
Observed Heterozygosity and Expected Heterozygosity within Population
| Sample ID | Observed Heterozygosity | Expected Heterozygosity | ||
|---|---|---|---|---|
| JPT | 0.285 | ± 0.184 | 0.282 | ± 0.175 |
| Japanese | 0.282 | ± 0.186 | 0.281 | ± 0.175 |
| Mongola | 0.294 | ± 0.214 | 0.280 | ± 0.177 |
| CHB | 0.287 | ± 0.182 | 0.284 | ± 0.174 |
| Han-NChina | 0.290 | ± 0.216 | 0.274 | ± 0.180 |
| Han | 0.286 | ± 0.186 | 0.282 | ± 0.175 |
| PanAsia-UG | 0.323 | ± 0.165 | 0.319 | ± 0.148 |
| HGDP-UG | 0.323 | ± 0.201 | 0.306 | ± 0.160 |
| Basque | 0.314 | ± 0.171 | 0.309 | ± 0.154 |
| Sardinian | 0.312 | ± 0.169 | 0.309 | ± 0.154 |
| Italian | 0.317 | ± 0.188 | 0.307 | ± 0.157 |
| Tuscan | 0.319 | ± 0.212 | 0.301 | ± 0.164 |
| French | 0.320 | ± 0.164 | 0.315 | ± 0.149 |
| Orcadian | 0.318 | ± 0.183 | 0.308 | ± 0.156 |
| CEU | 0.320 | ± 0.152 | 0.318 | ± 0.144 |
| Adygei | 0.321 | ± 0.178 | 0.312 | ± 0.153 |
| Russian | 0.320 | ± 0.168 | 0.314 | ± 0.150 |
Figure 1.
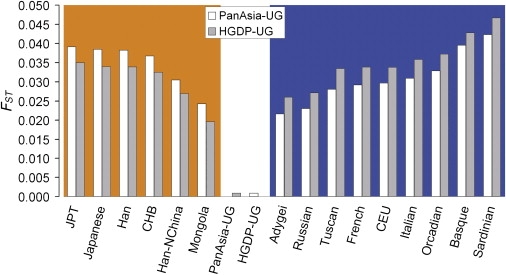
Pairwise FST between the Uyghur Population and Other Populations
Estimations of FST between Uyghur and East-Asian populations are shaded by orange color and sorted in descending order, and those between Uyghur and European populations are shaded by blue color and sorted in ascending order.
Figure 2.
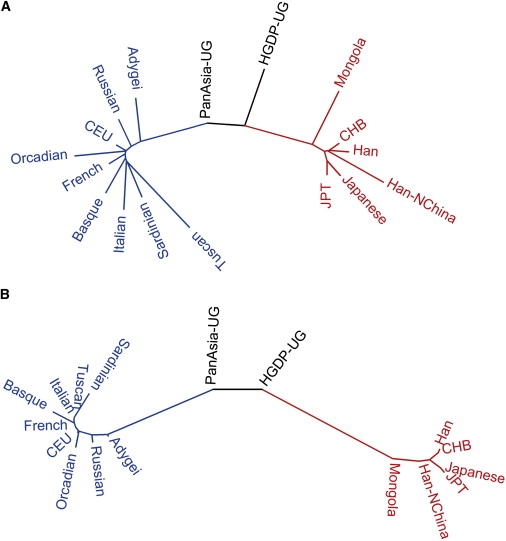
Cluster Relationship of Populations
(A) Maximum-likelihood tree of 17 populations.
(B) Neighbor-Joining tree of 17 populations based on pair-wise FST. Branches are colored according to ethnic groups; blue is used for European populations, red for East-Asian populations, and black for Uyghur populations.
Estimation of Individual Admixture and Population Structure
Using 19,934 SNPs genotyped in all 428 individuals representing 17 populations, we ran STRUCTURE from K = 1 to K = 6, with ten repeats for each K. According to the distribution of Ln(Pr), as shown in Figure S1, the most probable and appropriate number of clusters in our data set should be two, corresponding to EAS and EUR. Figure 3A shows summary plot of individual admixture proportions based on the highest-probability run of ten STRUCTURE runs. The results show that individuals from the same population often share membership coefficients in the inferred cluster, with the exception that one Japanese outlier shows obvious admixture. Mongola, Adygei, and Russian individuals show some degree of admixture as well.
Figure 3.
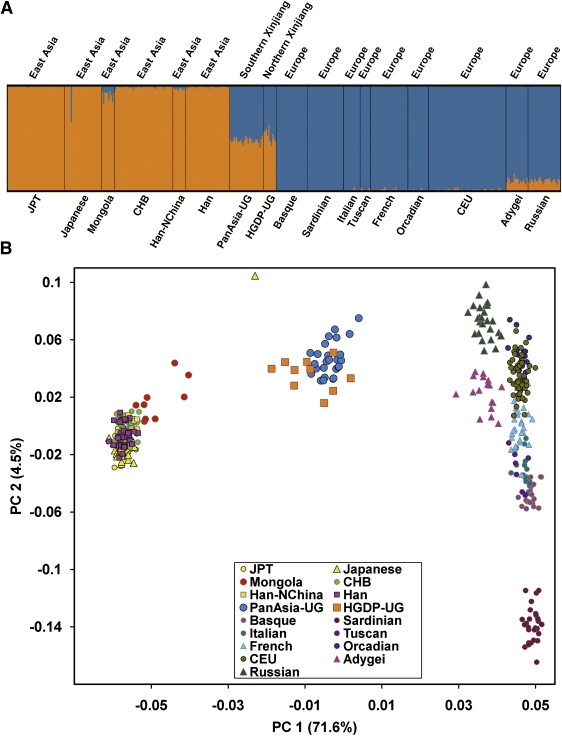
STRUCTURE Analysis and Principle-Component Analysis for 428 Individuals Representing All 17 Populations
(A) Summary plot of individual admixture proportions. The results of individual admixture proportions estimated from 19,934 SNPs. Each individual is represented by a single vertical line broken into two colored segments, with lengths proportional to each of the two inferred clusters; orange indicates East-Asian ancestry proportion and blue indicates European ancestry proportion. The predefined populations IDs are presented at the bottom of the plot, and the geographic region of each population is shown on the top of the plot.
(B) Analysis of the first two principal components. Population IDs of individuals are indicated with colors as shown in the legend.
The two Uyghur population samples (PanAsia-UG and HGDP-UG) display strong admixture of both EUR and EAS. The EUR contributions to PanAsia-UG samples and HGDP-UG samples are 51.9% and 47.0%, respectively, on average. Notably, the admixture proportions among PanAsia-UG individuals are quite similar, with the lowest from EUR contribution as 47.1% and the highest as 59.1%, and the standard deviation is only 2.9%. This deviation is much smaller than the estimation in an African-American population sample43 and is consistent with our previous results based on the data of one chromosome.4 For HGDP-UG samples, the EUR ancestry contribution ranges from 37.2% to 57.1%, and the standard deviation is only 6.2%. The much smaller deviation in Uyghurs suggest a much earlier admixture event for the Uyghur population as compared with the African-American population.43
The mean values of admixture proportions between the two Uyghur population samples are different (Student's t test, one-tailed p = 0.018), with HGDP-UG (53.0%) having more EAS ancestry than PanAsia-UG (48.1%). However, at the individual level, such difference is not large enough to assign individuals into two distinct populations, as shown in Figure S2. There are five HGDP-UG individuals (50%) with EAS ancestry smaller than the largest EAS ancestry of the PanAsia-UG individuals, and there are three HGDP-UG individuals with EAS ancestries even smaller than that of the average of PanAsia-UG individual.
Principal-Component Analysis of Individuals
Principal-component analysis (PCA) provides a useful approach for revealing relationships among individuals. Eigenvectors of each individual were calculated with EIGENSOFT.35 Table S4 shows the eigenvalues, the Tracy-Widom (TW) statistics,35 and the TW significance for the first 11 PCs. The first two PCs are displayed in Figure 3B, and they show extremely strong significance (p values = 0). Together, they explain 76.1% of the primary variation represented by the first ten PCs. The first PC explains 71.6% of the primary variation and shows a separation of the EAS and EUR individuals, with Uyghur (PanAsia-UG and HGDP-UG) individuals lying between the two groups, which is also expected given that Uyghur is the admixture of these two populations.
EAS and EUR AIMs for Admixture Mapping in Uyghurs
The final set of AIMs contains 8150 SNPs distributed on 23 chromosomes, with 7999 SNPs validated in six EAS populations and nine EUR populations (Table S5). The other 151 SNPs were also retained because of their very large FST (> 0.8) between JPT-CHB and CEU and both alleles fixed in parental populations or one allele fixed in one parental population. A scatterplot of frequencies of 8150 AIMs in EUR and EAS populations is displayed in Figure 4. A complete list of markers and the detailed information of 8150 AIMs were provided in Table S7. We also provide five other measurements of ancestry information, allele-frequency difference (δ),44 Wahlund's f,45 Rosenberg's In,46 Fisher's information content (FIC),47 and Shannon information content (SIC),23 for each AIM (see Table S7). For FIC and SIC calculations, admixture proportion from parental populations was assumed as 0.5:0.5 according to the estimation in this study. The final length of the chromosomal region that was covered by AIMs was 4024 cM (Table S5); the average intermarker distance was 0.50 cM, with a median of 0.33 cM. The distribution of intermarker distance is shown in Figure S3: 66.3% of adjacent marker pairs had intermarker distance < 0.5 cM, 89.3% had distance < 1 cM, and 98% had distance < 2 cM. We screened another set of AIMs for the assessment of admixture of PanAsia-UG samples. Altogether, 2750 AIMs were screened, and the marker information is shown in Table S6. A complete list of markers and the detailed information of 2750 AIMs are also available in Table S8.
Figure 4.
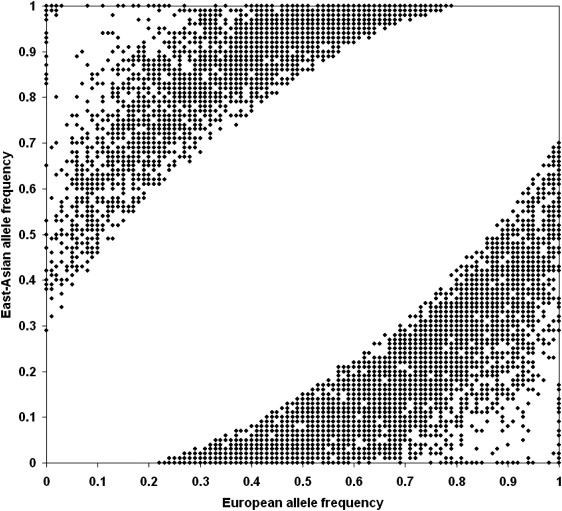
European and East-Asian Allele Frequencies for the 8150 AIMs
Population and Individual Admixture Assessment Based on AIMs
The STRUCTURE results from 19,934 random markers showed that both PanAsia-UG and HGDP-UG were admixed populations with contributions from both EUR and EAS. To ensure that the screened AIMs produced no bias in estimation of admixture proportion, we further performed STRUCTURE analysis with the same parameters for AIMs and estimated the admixture proportion of PanAsia-UG and HGDP-UG. Figure 5B shows the admixture proportions estimated from 7999 AIMs for each individual. PanAsia-UG samples have an average of 51.5% of admixture from EUR populations and 48.5% from EAS populations. HGDP-UG samples have 47.0% of admixture from EUR and 53.0% of admixture from EAS. The proportion of EAS ancestry in PanAsia-UG individuals ranges from 40.9% to 52.9%, and the proportion of EAS ancestry in HGDP-UG individuals ranges from 42.9% to 62.8%. For both PanAsia-UG and HGDP-UG individuals, the mean EAS contribution to autosomal chromosomes was similar, with the use of AIMs and 19,934 random markers (Figure 5A), as indicated by large t test p values (0.232 and 0.975 for PanAsia-UG and HGDP-UG, respectively). The results estimated from each chromosome in HGDP-UG and PanAsia-UG populations are shown in Table 2. Variation of the estimations among chromosomes was mainly due to differences between numbers of markers, whereas the larger variation in HGDP-UG was probably due to its smaller sample size.
Figure 5.
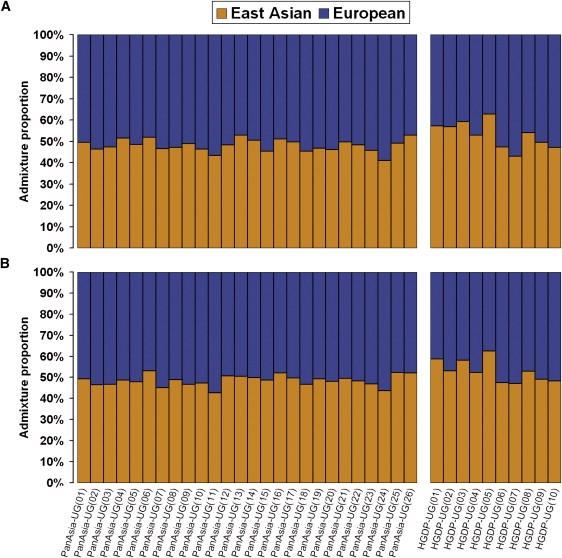
Summary Plot of Individual Admixture Proportions
Each individual is represented by a single vertical line broken into two colored segments, with lengths proportional to each of the two inferred clusters; orange indicates East-Asian ancestry proportion, and blue indicates European ancestry proportion. The ordinate indicates the proportion unit. The individual IDs are presented at the bottom of the plot.
(A) The results of individual admixture proportions estimated from 19,934 SNPs.
(B) The results of individual admixture proportions estimated from AIMs.
Table 2.
Inferred East-Asian Ancestry in Two Uyghur Population Samples
| HGDP-UG (n = 10) |
PanAsia-UG (n = 26) |
|||||||
|---|---|---|---|---|---|---|---|---|
| AIMs Used |
East-Asian Ancestry |
AIMs Used |
East-Asian Ancestry |
|||||
| Chr. | No. | Size (cM) | Mean ± SD (%) | No. | Size (cM) | Mean ± SD (%) | ||
| 1 | 592 | 300.1 | 56.4 | ± 6.5 | 238 | 278.2 | 49.3 | ± 9.2 |
| 2 | 650 | 273.9 | 56.2 | ± 5.8 | 268 | 254.3 | 49.7 | ± 4.9 |
| 3 | 557 | 244.8 | 52.0 | ± 10.0 | 212 | 240.8 | 47.1 | ± 8.3 |
| 4 | 516 | 231.7 | 55.5 | ± 6.2 | 183 | 215.8 | 46.1 | ± 7.2 |
| 5 | 510 | 217.1 | 48.1 | ± 10.2 | 205 | 208.8 | 46.9 | ± 8.2 |
| 6 | 438 | 222.2 | 49.7 | ± 9.0 | 192 | 213.7 | 48.5 | ± 9.6 |
| 7 | 482 | 197.6 | 54.8 | ± 9.1 | 150 | 192.7 | 47.6 | ± 9.9 |
| 8 | 369 | 181.3 | 53.9 | ± 9.5 | 135 | 173.9 | 47.4 | ± 10.3 |
| 9 | 369 | 198.4 | 55.1 | ± 9.4 | 117 | 177.7 | 47.7 | ± 8.7 |
| 10 | 384 | 183.2 | 49.5 | ± 9.6 | 133 | 175.4 | 52.7 | ± 10.7 |
| 11 | 350 | 187.4 | 51.9 | ± 6.5 | 96 | 173.4 | 48.4 | ± 11.6 |
| 12 | 351 | 184.0 | 50.1 | ± 7.1 | 122 | 180.9 | 54.9 | ± 11.4 |
| 13 | 238 | 138.7 | 58.7 | ± 11.9 | 99 | 137.3 | 47.2 | ± 9.2 |
| 14 | 230 | 129.9 | 58.7 | ± 9.0 | 77 | 116.5 | 49.3 | ± 8.9 |
| 15 | 252 | 142.5 | 59.1 | ± 14.4 | 87 | 142.2 | 50.6 | ± 12.3 |
| 16 | 233 | 134.8 | 58.1 | ± 9.9 | 67 | 113.6 | 44.8 | ± 11.5 |
| 17 | 238 | 159.4 | 57.4 | ± 6.0 | 53 | 148.8 | 48.5 | ± 10.8 |
| 18 | 214 | 131.1 | 50.4 | ± 15.2 | 86 | 125.6 | 44.5 | ± 14.8 |
| 19 | 176 | 117.4 | 53.2 | ± 11.1 | 22 | 104.2 | 36.9 | ± 19.1 |
| 20 | 217 | 108.6 | 56.1 | ± 10.0 | 53 | 103.5 | 49.9 | ± 12.5 |
| 21 | 135 | 67.9 | 54.0 | ± 12.5 | 45 | 59.8 | 47.6 | ± 15.8 |
| 22 | 164 | 84.5 | 52.5 | ± 7.2 | 15 | 52.9 | 43.5 | ± 32.1 |
| Total | 7665 | 3836.5 | 53.0 | ± 6.2 | 2655 | 3590.0 | 48.1 | ± 2.9 |
SD denotes the standard deviation of average East-Asian ancestry of individuals.
Empirical Estimation of Admixture Time of Uyghurs
The admixture times of Uyghur populations were inferred from the posterior of recombination parameters estimated by STRUCTURE analysis and ADMIXMAP analysis. These parameters can be converted to the “effective number of generations back to unadmixed ancestors,” because under a model with a single pulse of admixture, this is equivalent to the number of generations since the admixture event.32 The STRUCTURE analysis was performed under the admixture model, with 20,000 replicates and 30,000 burn-in cycles. The posterior distribution of recombination parameter r (breakpoints per cM) is shown in Figure 6A. Both the mean and the median of r for HGDP-UG samples are 1.10, and the 90% confidence interval (CI) is [1.04, 1.16]. For PanAsia-UG samples, the mean and median are 1.29, and the 90% CI is [1.22, 1.36]. Under a model with a single pulse of admixture (or a hybrid isolation [HI] model),48 the admixture event is estimated to have happened about 110 (104–116) generations ago, or 2200 (2080–2320) years ago, for HGDP-UG and 129 (122–136) generations ago, or 2580 (2440–2720) years ago, for PanAsia-UG, with the assumption of 20 years per generation. For ADMIXMAP, the analysis was performed under the random-mating model, with 22,000 replicates and 2000 burn-in cycles. Posterior distribution of the recombination parameter τ (sum-of-intensities per cM) is shown in Figure 6B. Both the mean and that median of τ for HGDP-UG are 0.89, and the 90% CI is [0.84, 0.97], and for PanAsia-UG, the mean is 1.05, the median is 1.03, and the 90% CI is [0.94, 1.20]. Under the assumption of an HI model and a generation time of 20 years, the admixture event happened about 1780 (1680–1940) years ago for HGDP-UG and about 2100 (1880–2400) years ago for PanAsia-UG. The difference between the STRUCTURE and ADMIXMAP estimates is probably due to the difference between these algorithms; the similar discrepancy was also observed in a previous study.26
Figure 6.
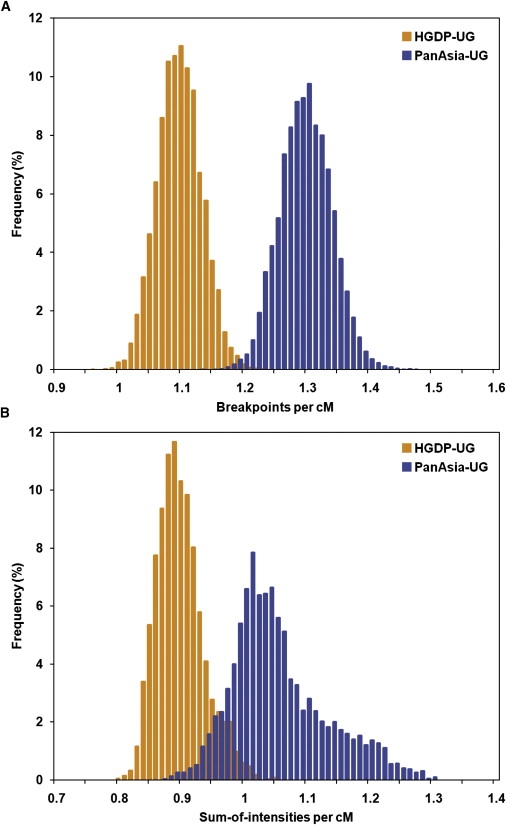
Posterior Distribution of the Recombination Parameters for HGDP-UG and PANASIA-UG
Results estimated from two population samples are indicated with colors as shown in the legend.
(A) Posterior distribution of the recombination parameter r (breakpoints per cM) estimated by STRUCTURE analysis. Both the mean and median of r for HGDP-UG are 1.10, and the 90% CI is [1.04, 1.16], and for PanAsia-UG the mean and median are 1.29, and the 90% CI is [1.22, 1.36].
(B) Posterior distribution of the recombination parameter τ (sum of intensities per cM) estimated by ADMIXMAP analysis. Both the mean and median of τ for HGDP-UG are 0.89, and the 90% CI is [0.84, 0.97]; for PanAsia-UG, the mean is 1.05, the median is 1.03, and the 90% CI is [0.94, 1.20].
Informativeness of the Uyghur Admixture Map
To evaluate the informativeness of the admixture map for inferring the ancestry of chromosomal segments in Uyghur populations, we calculated a percentage of maximum-informativeness statistics (ravg) (see Subjects and Methods). We modeled the ancestral populations with 60 EUR (CEU) and 90 EAS (JPT and CHB) samples (see Subjects and Methods). For northern Uyghurs, the complete panel of 8150 AIMs extracted > 50% of the admixture information for > 99% of the genome and > 70% of the admixture information for > 90% of the genome. For southern Uyghurs, the mapping power was lower due to fewer AIMs; the panel of 2750 AIMs extracted > 50% of the admixture information for > 67% of the genome and > 60% of the admixture information for > 51% of the genome. For each population, the informativeness at each locus in the genome is displayed in Figure 7. Previous studies have suggested that LD in the parental populations might cause false-positive results in admixture mapping.19,49 For the complete AIM panel (8150 SNPs), LD is still present in both parental populations (EAS and EUR). Thus, we examined the subset of AIMs with marker pairs in strong LD removed; the results are shown in Table 3 and the following section. Map informativeness decreased with the decreasing of marker number. For example, in northern Uyghurs, we selected four subsets from 8150 AIMs (Table 3), the subset panel of 4039 AIMs extracted an average of 69% of the maximum admixture information, the subset of 2714 AIMs extracted 61% of the maximum, and the subset of 1396 AIMs extracted 45% of the maximum; the increase of power of the subset of 1255 AIMs could be due to the high informative of single markers, which was also true for the subset of 1021 AIMs (selected from 2750 AIMs) in southern Uyghurs.
Figure 7.
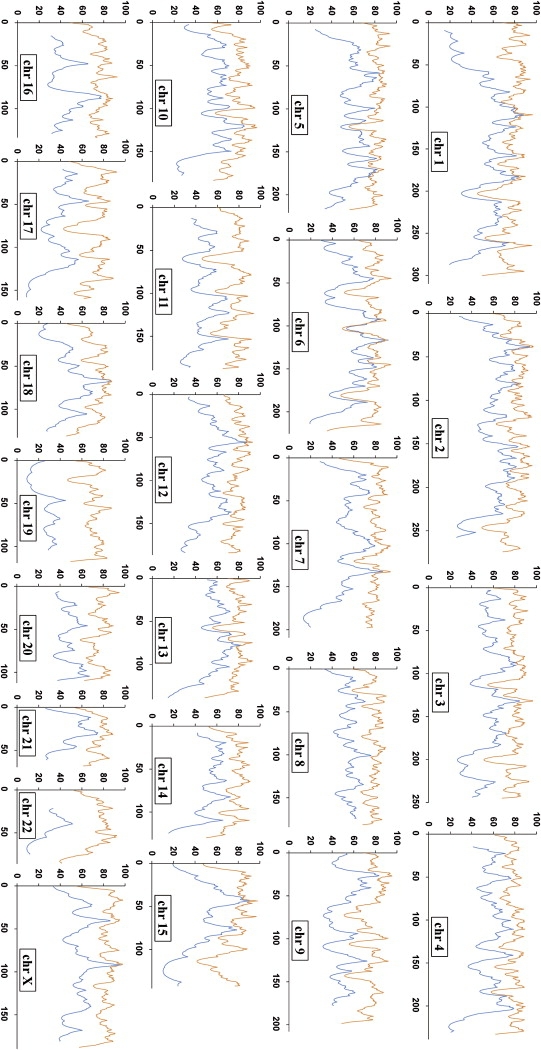
Distribution of Admixture-Mapping Information for Each Chromosome
The admixture-mapping information (ordinate) is shown for each position on the Rutger map (abscissa). The information was determined via the ANCESTRYMAP analysis of genotyping results, with the use of 7999 SNP AIMs for Uyghur (in orange) and 2750 AIMs for PANASIA-UG (in blue).
Table 3.
Results of Simulated Diseases Studies in Two Uyghur Populations
| Uyghur Population | No. of Marker | Mapping Power (%) | No. of Cases Neededa | LD in Parental Populationb | False-Positive Resultsc | Control-Only LOD |
Case-Control LOD |
||
|---|---|---|---|---|---|---|---|---|---|
| Local | Global | Local | Global | ||||||
| Northern | 7,999 | 78 | 451 | ++ | − | 0.3 | −2.9 | 10.8 | 7.6 |
| Northern | 4,039 | 69 | 509 | + | − | −1.3 | −3.3 | 5.4 | 2.4 |
| Northern | 2,714 | 61 | 576 | − | − | −0.1 | −2.4 | 7.3 | 4.6 |
| Northern | 1,396 | 45 | 781 | − | − | 0.1 | −2.2 | 5.2 | 2.6 |
| Northern | 1,255 | 54 | 651 | − | − | 0.4 | −1.8 | 11.9 | 9.3 |
| Southern | 2,750 | 54 | 577 | + | − | 0.7 | −1.4 | 7.8 | 4.9 |
| Southern | 1,804 | 43 | 724 | − | − | −0.8 | −3.1 | 8.3 | 5.5 |
| Southern | 1,021 | 46 | 677 | − | − | 0.4 | −1.5 | 13.2 | 10.3 |
Assumed increased disease risk of 1.5 with EAS ancestry at the disease locus.
LD presents in EAS and EUR for a few adjacent marker pairs, ++ denotes strong LD (r2 >0.5), + denotes moderate LD (r2 >0.2), and - denotes weak or no LD (r2 <0.2).
False-positive results are indicated by genome-wide LOD score in control-only study, + denotes false-positive results (global LOD >0) present, and - denotes no false-positive results (global LOD <0).. Local and global LOD scores were averaged across ten control-only simulations and ten case-control simulations.
Performance of the Admixture Map in Disease Studies
To examine the effects of LD in parental populations and to evaluate the performance of our admixture map in actual disease studies, we simulated disease cases using the panel of complete AIMs and its subsets with different densities (Table 3). For each AIM panel, ten disease loci were chosen at which the mapping information (ravg) most closely matched its genome-wide average (Table 3). False-positive results were examined by control-only simulations, with 1000 pseudocase samples and 1000 control samples drawn from simulated controls. In all cases, i.e., for the complete AIM panel and its subsets, ANCESTRYMAP reported no false-positive results, as indicated by genome-wide LOD scores (< 0) and maximum local LOD scores (< 1) in control-only simulations (Table 3). In contrast, ANCESTRYMAP reported positive results in case-control simulations, as indicated by genome-wide LOD scores (> 2) and maximum local LOD scores (> 5) (Table 3). These results are encouraging: they implied that our maps could identify the disease loci correctly, and the influence of residual LD in parental populations was limited, at least in simulation studies.
Expected Power of Admixture Mapping in Uyghurs
The expected power of admixture mapping in Uyghurs could be indicated by an estimation of the number of cases that would be needed to detect an admixture association. In the initial estimation, we assumed perfect information about ancestry at each locus in the genome, and we used the distribution of ancestries of individual samples, which was estimated in this study (see Subjects and Methods). For this analysis, northern Uyghurs (HGDP-UG) and southern Uyghurs (PanAsia-UG) were considered separately. The results were displayed in Figure 8. The southern Uyghur population showed a little higher statistical power per sample for admixture mapping (fewest samples needed) than did the northern Uyghur population, because of the relatively larger proportions of both EAS (0.40∼0.53) and EUR (0.47∼0.60) ancestry in the southern Uyghur population than that in the northern Uyghur population (0.42∼0.63 for EAS; 0.37∼0.58 for EUR). Considering the information lost in real mapping, for detection of a locus with genome-wide average mapping information (Table 3) in which EAS ancestry confers, on average, a 1.5-fold increased risk for disease, the number of cases needed [450 ∼ 800] and are displayed in Table 3. With the moderate density of the AIM panel, for example, 2714 AIMs for the northern Uyghur population and 2750 AIMs for the southern Uyghur population, the number of cases needed is about 600 (Table 3).
Figure 8.
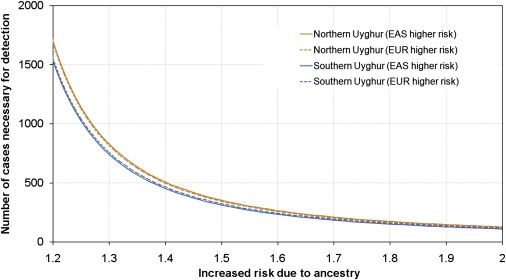
Sample Size Needed for Detection of a Disease Locus with the use of Admixture Mapping in Uyghur Populations
“Northern Uyghurs” refers to the populations represented by HGDP-UG, “Southern Uyghurs” refers to the populations represented by PanAsia-UG. For each population, this quantity is computed under the ideal assumption of perfect information about ancestry, as a function of the relative disease risk conferred by each copy of a particular ancestry at the disease locus.
Discussion
In this study, we analyzed genome-wide admixture for two Uyghur populations sampled from northern and southern Xinjiang. Various analyses of genetic structure in the Uyghur samples consistently revealed that the Uyghur populations in Xinjiang are typical admixtures with both EAS and EUR genetic components, suggesting that they were products of the admixture of EAS and EUR ancestries. The estimated admixture proportions from genome-wide data are 53:47 (EAS:EUR) in the northern Uyghur population and 48:52 (EAS:EUR) in the southern Uyghur population. This estimation is consistent with that of a previous study based on a single chromosome,4 although the two are not exactly identical. Our previous estimation was 40:60 (EAS:EUR), which was based on 83 AIMs selected from chromosome 21 data in 40 southern Uyghur samples. The difference could be due to both the difference of sample size and the difference of markers, but the latter plays a relatively more important role, which could be seen from the variation of estimations among chromosomes, as shown in Table 2. We suggest the current estimation for southern Uyghurs is more precise and, thus, is closer to the true admixture proportion. Another Uyghur population sample (HGDP-UG) used in this study was originally collected from Yili, which is located in northern Xinjiang. It was believed that more interaction occurred recently between Han Chinese and Uyghurs in northern Xinjiang. The estimation of EUR ancestry contribution to northern Uyghur populations from previous studies varied from 30%5 or 36.3%6 to 50%7 or even 55%.8 However, previous studies suffered from the small number of markers5–7 or, even, the single locus that they used.8 Our estimation of 53%, based on genome-wide markers within the middle values of previous estimates, could be attributed to the use of a much larger number of markers and, therefore, could be closer to the truth. Our estimation also confirmed that there is a greater contribution of EAS ancestry than of EUR ancestry (47%) in northern Uyghurs. However, the current estimation could suffer from the small sample size (n = 10). At the individual level, the distribution of admixture proportions among Uyghur individuals is quite similar, and the standard deviation is only 6.2% for northern Uyghur samples and even less (2.9%) for southern Uyghur samples. This result suggests a much earlier history of admixture for the Uyghur population compared to that of more recently admixed populations, such as African Americans, of which individual admixture proportions vary much more (SD = 19.7%).43
We further constructed what is, to our knowledge, the first admixture map that constitutes a practical resource for admixture mapping in Uyghurs. Although the Uyghur population is one presenting a typical admixture of eastern and western anthropological traits, its potential utility in admixture mapping has been largely ignored because of its uncharacterized and suspected earlier history of admixture. Typical admixture populations used for admixture mapping often involve those formed by recent admixture between groups originating on different continents as a result of European maritime expansion during the past few hundred years.18 On the basis of recombination information, which can be extracted from the current data, we estimated that the time since admixture of the two Uyghur population samples was more than 100 generations for both population samples. This number is substantially greater than that of African Americans, to which admixture mapping has been applied and achieved many successes. This much earlier admixture history leads to a requirement for a much larger set of AIMs for saturation of the recombination intervals and extraction of admixture-mapping information. Considering the admixture-mapping power, however, the near-equal admixture of parental populations (50:50 EAS:EUR) compensates to some degree for the lower information content of AIMs, because the mapping power will decrease in populations when there is a much larger contribution from only one parental population and will increase when the admixture of parental populations is nearly equal.11,42 Therefore, the admixture mapping could also be powerful in Uyghurs. Furthermore, the benefit of the earlier history of admixture in the HGDP-UG population is that we can also expect to have sharper mapping resolution, resulting in a reduced effort in the subsequent fine-mapping study.
To our knowledge, our map constitutes the first practical resource for admixture mapping in Uyghurs, but it is far from being perfect. There are still many gaps due to the consideration of validation in both EAS and EUR populations (small variation within groups), and there are a few AIMs that do not show a large difference in allele frequency between EAS and EUR (FST < 0.30) and thus provide limited map information. Furthermore, residual LD is still present in both parental populations (EAS and EUR), and this could cause false-positive results. However, much higher density of markers is needed for the Uyghur population for saturation of the recombination intervals due to its weak LD,4 which inevitably results in the presence of residual LD in parental populations. On the one hand, we demonstrated in simulation studies that the influence of the presence of residual LD in parental populations was limited (Table 3). On the other hand, concerning potentially possible false-positive results due to LD, a possible supplemental strategy is this: at the genotyping stage, dense AIMs (even those with strong LD in EAS and EUR) could be genotyped in samples. At the analysis stage, markers could be analyzed separately, with a set of markers being analyzed at each time so that no LD confounds the single analysis. Finally, the results of the separate analyses could be ultimately combined. The cost of this strategy, which avoids LD and, thus, false-positive results, is that the map power will decrease in each subset of markers and a larger sample size will be needed (as shown in Table 3). New methods based on the Markov-HMM (MHMM) algorithm have recently been developed and account for LD in parental populations.50,51 However, the power and, hence, efficiency of the use of whole-genome SNP-association test panels in admixture mapping is not yet clear.22 Another important question is whether admixture mapping will be a useful methodology in the age of dense genome-wide scans with hundreds of thousands of markers.25 The first advantage of admixture mapping is the potentially much lower genotyping cost as compared with the cost of a dense whole-genome scan. Apart from the lower genotyping workload, there are other advantages in the use of a panel of markers that are preselected to be informative: markers for which allele frequencies vary within continental groups can be excluded, the marker spacing can be large enough to ensure no allelic association within subpopulations, and the computational burden is reduced.18 In addition, the coarse granularity of the admixture signal reduces the number of hypotheses tested (or, in Bayesian terms, increases the prior probability of each causal hypothesis) compared to the hundreds of thousands of hypotheses tested in dense genome-wide scans.25 Furthermore, the use of a locus-genome statistic that considers only local ancestry estimates of disease cases, with no noise introduced from controls, leads to an improvement in power by a factor of 2.25,42
The most obvious applications of admixture mapping are those to diseases in which risk varies between ethnic groups. There are relatively few diseases for which epidemiological criteria (based on migrant studies and the relationship of risk to individual admixture proportions) support genetic explanations for ethnic variation in risk.18 A few common diseases or traits are known differ between East-Asian and European populations, such as cardiovascular disease,52–56 hypertension,57,58 obesity and type II diabetes,54,57,59–62 longevity,63–65 etc. Female breast cancer is seen more commonly in North America and Europe and is relatively uncommon in Asia and Latin America, with a 3-fold difference between high and low risk areas.66 Primary liver cancer (i.e., cancer originating in the liver, rather than spreading to the liver from other sites) is striking in its extremely low incidence in Western countries, Latin America, and India and its high incidence in east and southeast Asia.66 The number of diseases showing a difference in risk between EAS and EUR populations could potentially be much greater than that known at the present time, due to lack of thorough investigations. However, admixture mapping is not necessarily limited to these diseases.18 The ability to detect a locus by admixture mapping depends not on the number of disease alleles at the locus but only on whether the pool of disease alleles at the locus is distributed differentially between the ancestral subpopulations. It is possible that such loci exist even where no overall ethnic variation in disease risk is detectable;17 for instance, when two or more loci have effects in opposite directions.18
Finally, we should point out that our empirical assessment of Uyghur populations could be not comprehensive because of the few populations and small sample sizes used. We especially emphasize that many differences that we observed between southern and northern Uyghur populations, including those in admixture level, admixture time, and mapping power, could result from the small sample size (especially northern Uyghurs) used in this study. Additional studies are needed for clarification of those differences. In addition, the existence of more than two ancestral origins (K = 3) makes the potential use of AIMs for admixture mapping more complicated. We constructed the admixture map assuming two ancestral origins (K = 2) based on STRUCTURE posterior probability, which does not definitively exclude the possibility of the third ancestral origin. To examine the possibility that more ancestral origins of Uyghurs could be found, in the case that more reference populations became available, we performed STRUCTURE analysis for recently published 650K SNP data30 and 377 STR data67 in the HGDP-CEPH panel. From the results of both SNP and STR data (Figure S4), we found only a very small proportion of the third component (apart from the EAS and EUR components) in the Uygur population. This component is shared by Central-Asian populations, which are, per se, a mixture of EAS and EUR (seen from small Ks); therefore, the third component observed in Uygur could also derive from EUR or EAS ancestry. However, the few populations and small sample size in this study could cause uncertainties in some estimations; our results can be improved further by genotyping of higher-density markers and inclusion of more Uyghur populations, with a larger sample size.
Supplemental Data
Supplemental data include four figures and eight tables and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
ADMIXMAP program, http://homepages.ed.ac.uk/pmckeigu/admixmap/index.html
ANCESTRYMAP program, http://genepath.med.harvard.edu/∼reich/Software.htm
Arlequin ver 3.11 program, http://cmpg.unibe.ch/software/arlequin3/
EIGENSOFT program, http://genepath.med.harvard.edu/∼reich/Software.htm
The HGDP-CEPH Diversity Panel Database, http://www.cephb.fr/hgdp-cephdb/
International HapMap Project, http://www.hapmap.org/downloads/index.html.en
PanAsia Project, http://pasnp.org/index.php
SNP Genetic Mapping, http://actin.ucd.ie/cgi-bin/rs2cm.cgi
STRUCTURE 2.2.2 program, http://pritch.bsd.uchicago.edu/structure.html
Acknowledgments
We are grateful to J. Tan, F. Zhang, and Z. Xu for their help in collecting Uyghur samples. We would like to express our appreciation to the PASNPI Consortium for its allowing us to use the genotype data. We thank N. Patternson, D. Reich, and A. Tandon for their helpful comments on ANCESTRYMAP analysis. This work was supported by grants from the Chinese High-Tech Program (863) (2002BA711A10), the Basic Research Program of China (973) (2004CB518605, 2002CB512900), the National Natural Science Foundation of China (NSFC30571060), and the Shanghai Science and Technology Committee (04DZ14003). This work was also supported by the Shanghai Leading Academic Discipline Project (No. B111).
References
- 1.Ai Q., Xiao H., Zhao J., Xu Y., Shi F. A survey on physical characteristics of Uigur Nationality. ACTA Anthropologica Sinica. 1993;12:357–365. [Google Scholar]
- 2.Yao Y.G., Kong Q.P., Wang C.Y., Zhu C.L., Zhang Y.P. Different matrilineal contributions to genetic structure of ethnic groups in the silk road region in china. Mol. Biol. Evol. 2004;21:2265–2280. doi: 10.1093/molbev/msh238. [DOI] [PubMed] [Google Scholar]
- 3.Wells R.S., Yuldasheva N., Ruzibakiev R., Underhill P.A., Evseeva I., Blue-Smith J., Jin L., Su B., Pitchappan R., Shanmugalakshmi S. The Eurasian heartland: a continental perspective on Y-chromosome diversity. Proc. Natl. Acad. Sci. USA. 2001;98:10244–10249. doi: 10.1073/pnas.171305098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu S., Huang W., Qian J., Jin L. Analysis of genomic admixture in Uyghur and its implication in mapping strategy. Am. J. Hum. Genet. 2008;82:883–894. doi: 10.1016/j.ajhg.2008.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Du R. Human Population Genetics Studies in China. Bulletin of Biology. 1997;32:9–12. [Google Scholar]
- 6.Xiao F.X., Yang J.F., Cassiman J.J., Decorte R. Diversity at eight polymorphic Alu insertion loci in Chinese populations shows evidence for European admixture in an ethnic minority population from northwest China. Hum. Biol. 2002;74:555–568. doi: 10.1353/hub.2002.0050. [DOI] [PubMed] [Google Scholar]
- 7.Zhao T.M., Lee T.D. Gm and Km allotypes in 74 Chinese populations: a hypothesis of the origin of the Chinese nation. Hum. Genet. 1989;83:101–110. doi: 10.1007/BF00286699. [DOI] [PubMed] [Google Scholar]
- 8.Comas D., Calafell F., Mateu E., Perez-Lezaun A., Bosch E., Martinez-Arias R., Clarimon J., Facchini F., Fiori G., Luiselli D. Trading genes along the silk road: mtDNA sequences and the origin of central Asian populations. Am. J. Hum. Genet. 1998;63:1824–1838. doi: 10.1086/302133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chakraborty R., Weiss K.M. Admixture as a tool for finding linked genes and detecting that difference from allelic association between loci. Proc. Natl. Acad. Sci. USA. 1988;85:9119–9123. doi: 10.1073/pnas.85.23.9119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stephens J.C., Briscoe D., O'Brien S.J. Mapping by admixture linkage disequilibrium in human populations: limits and guidelines. Am. J. Hum. Genet. 1994;55:809–824. [PMC free article] [PubMed] [Google Scholar]
- 11.McKeigue P.M. Mapping genes that underlie ethnic differences in disease risk: methods for detecting linkage in admixed populations, by conditioning on parental admixture. Am. J. Hum. Genet. 1998;63:241–251. doi: 10.1086/301908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Darvasi A., Shifman S. The beauty of admixture. Nat. Genet. 2005;37:118–119. doi: 10.1038/ng0205-118. [DOI] [PubMed] [Google Scholar]
- 13.Oksenberg J.R., Barcellos L.F., Cree B.A., Baranzini S.E., Bugawan T.L., Khan O., Lincoln R.R., Swerdlin A., Mignot E., Lin L. Mapping multiple sclerosis susceptibility to the HLA-DR locus in African Americans. Am. J. Hum. Genet. 2004;74:160–167. doi: 10.1086/380997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reich D., Patterson N., De Jager P.L., McDonald G.J., Waliszewska A., Tandon A., Lincoln R.R., DeLoa C., Fruhan S.A., Cabre P. A whole-genome admixture scan finds a candidate locus for multiple sclerosis susceptibility. Nat. Genet. 2005;37:1113–1118. doi: 10.1038/ng1646. [DOI] [PubMed] [Google Scholar]
- 15.Reich D., Patterson N., Ramesh V., De Jager P.L., McDonald G.J., Tandon A., Choy E., Hu D., Tamraz B., Pawlikowska L. Admixture mapping of an allele affecting interleukin 6 soluble receptor and interleukin 6 levels. Am. J. Hum. Genet. 2007;80:716–726. doi: 10.1086/513206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu X., Luke A., Cooper R.S., Quertermous T., Hanis C., Mosley T., Gu C.C., Tang H., Rao D.C., Risch N. Admixture mapping for hypertension loci with genome-scan markers. Nat. Genet. 2005;37:177–181. doi: 10.1038/ng1510. [DOI] [PubMed] [Google Scholar]
- 17.Hoggart C.J., Shriver M.D., Kittles R.A., Clayton D.G., McKeigue P.M. Design and analysis of admixture mapping studies. Am. J. Hum. Genet. 2004;74:965–978. doi: 10.1086/420855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKeigue P.M. Prospects for admixture mapping of complex traits. Am. J. Hum. Genet. 2005;76:1–7. doi: 10.1086/426949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith M.W., O'Brien S.J. Mapping by admixture linkage disequilibrium: advances, limitations and guidelines. Nat. Rev. Genet. 2005;6:623–632. doi: 10.1038/nrg1657. [DOI] [PubMed] [Google Scholar]
- 20.Collins-Schramm H.E., Phillips C.M., Operario D.J., Lee J.S., Weber J.L., Hanson R.L., Knowler W.C., Cooper R., Li H., Seldin M.F. Ethnic-difference markers for use in mapping by admixture linkage disequilibrium. Am. J. Hum. Genet. 2002;70:737–750. doi: 10.1086/339368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith M.W., Lautenberger J.A., Shin H.D., Chretien J.P., Shrestha S., Gilbert D.A., O'Brien S.J. Markers for mapping by admixture linkage disequilibrium in African American and Hispanic populations. Am. J. Hum. Genet. 2001;69:1080–1094. doi: 10.1086/323922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tian C., Hinds D.A., Shigeta R., Kittles R., Ballinger D.G., Seldin M.F. A genomewide single-nucleotide-polymorphism panel with high ancestry information for African American admixture mapping. Am. J. Hum. Genet. 2006;79:640–649. doi: 10.1086/507954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith M.W., Patterson N., Lautenberger J.A., Truelove A.L., McDonald G.J., Waliszewska A., Kessing B.D., Malasky M.J., Scafe C., Le E. A high-density admixture map for disease gene discovery in african americans. Am. J. Hum. Genet. 2004;74:1001–1013. doi: 10.1086/420856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mao X., Bigham A.W., Mei R., Gutierrez G., Weiss K.M., Brutsaert T.D., Leon-Velarde F., Moore L.G., Vargas E., McKeigue P.M. A genomewide admixture mapping panel for Hispanic/Latino populations. Am. J. Hum. Genet. 2007;80:1171–1178. doi: 10.1086/518564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Price A.L., Patterson N., Yu F., Cox D.R., Waliszewska A., McDonald G.J., Tandon A., Schirmer C., Neubauer J., Bedoya G. A genomewide admixture map for Latino populations. Am. J. Hum. Genet. 2007;80:1024–1036. doi: 10.1086/518313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tian C., Hinds D.A., Shigeta R., Adler S.G., Lee A., Pahl M.V., Silva G., Belmont J.W., Hanson R.L., Knowler W.C. A genomewide single-nucleotide-polymorphism panel for Mexican American admixture mapping. Am. J. Hum. Genet. 2007;80:1014–1023. doi: 10.1086/513522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frazer K.A., Ballinger D.G., Cox D.R., Hinds D.A., Stuve L.L., Gibbs R.A., Belmont J.W., Boudreau A., Hardenbol P., Leal S.M. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.The International HapMap Consortium The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 29.The International HapMap Consortium A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li J.Z., Absher D.M., Tang H., Southwick A.M., Casto A.M., Ramachandran S., Cann H.M., Barsh G.S., Feldman M., Cavalli-Sforza L.L. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- 31.The HUGO Pan-Asian SNP Consortium Mapping Human Genetic History in Asia. Science. 2008 [Google Scholar]
- 32.Falush D., Stephens M., Pritchard J.K. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pritchard J.K., Stephens M., Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weir B.S., Hill W.G. Estimating F-statistics. Annu. Rev. Genet. 2002;36:721–750. doi: 10.1146/annurev.genet.36.050802.093940. [DOI] [PubMed] [Google Scholar]
- 35.Patterson N., Price A.L., Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Felsenstein J. PHYLIP–Phylogeny Inference Package (Version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- 37.Saitou N., Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 38.Kumar S., Tamura K., Nei M. MEGA3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief. Bioinform. 2004;5:150–163. doi: 10.1093/bib/5.2.150. [DOI] [PubMed] [Google Scholar]
- 39.Evans D.M., Cardon L.R. A comparison of linkage disequilibrium patterns and estimated population recombination rates across multiple populations. Am. J. Hum. Genet. 2005;76:681–687. doi: 10.1086/429274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kong X., Murphy K., Raj T., He C., White P.S., Matise T.C. A combined linkage-physical map of the human genome. Am. J. Hum. Genet. 2004;75:1143–1148. doi: 10.1086/426405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schneider S., Roessli D., Excoffier L. Genetics and Biometry Lab, Dept of Anthropology University of Geneva; Geneva, Switzerland: 2000. Arlequin: A Software for Population Genetics Data Analysis. User Manual ver 2.0. [Google Scholar]
- 42.Patterson N., Hattangadi N., Lane B., Lohmueller K.E., Hafler D.A., Oksenberg J.R., Hauser S.L., Smith M.W., O'Brien S.J., Altshuler D. Methods for high-density admixture mapping of disease genes. Am. J. Hum. Genet. 2004;74:979–1000. doi: 10.1086/420871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu S., Huang W., Wang H., He Y., Wang Y., Wang Y., Qian J., Xiong M., Jin L. Dissecting linkage disequilibrium in african-american genomes: roles of markers and individuals. Mol. Biol. Evol. 2007;24:2049–2058. doi: 10.1093/molbev/msm135. [DOI] [PubMed] [Google Scholar]
- 44.Shriver M.D., Smith M.W., Jin L., Marcini A., Akey J.M., Deka R., Ferrell R.E. Ethnic-affiliation estimation by use of population-specific DNA markers. Am. J. Hum. Genet. 1997;60:957–964. [PMC free article] [PubMed] [Google Scholar]
- 45.Wahlund S. Zusammensetzung von Populationen und Korrelationserscheinungen von Standpunkt der Vererbungslehre aus betrachtet. Hereditas. 1928;11:65–106. [Google Scholar]
- 46.Rosenberg N.A., Li L.M., Ward R., Pritchard J.K. Informativeness of genetic markers for inference of ancestry. Am. J. Hum. Genet. 2003;73:1402–1422. doi: 10.1086/380416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pfaff C.L., Barnholtz-Sloan J., Wagner J.K., Long J.C. Information on ancestry from genetic markers. Genet. Epidemiol. 2004;26:305–315. doi: 10.1002/gepi.10319. [DOI] [PubMed] [Google Scholar]
- 48.Long J.C. The genetic structure of admixed populations. Genetics. 1991;127:417–428. doi: 10.1093/genetics/127.2.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reich D., Patterson N. Will admixture mapping work to find disease genes? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005;360:1605–1607. doi: 10.1098/rstb.2005.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tang H., Coram M., Wang P., Zhu X., Risch N. Reconstructing genetic ancestry blocks in admixed individuals. Am. J. Hum. Genet. 2006;79:1–12. doi: 10.1086/504302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sundquist A., Fratkin E., Do C.B., Batzoglou S. Effect of genetic divergence in identifying ancestral origin using HAPAA. Genome Res. 2008;18:676–682. doi: 10.1101/gr.072850.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chaturvedi N., Coady E., Mayet J., Wright A.R., Shore A.C., Byrd S., Mc G.T.S.A., Kooner J.S., Schalkwijk C.G., Hughes A.D. Indian Asian men have less peripheral arterial disease than European men for equivalent levels of coronary disease. Atherosclerosis. 2007;193:204–212. doi: 10.1016/j.atherosclerosis.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 53.Tillin T., Dhutia H., Chambers J., Malik I., Coady E., Mayet J., Wright A.R., Kooner J., Shore A., Thom S. South Asian men have different patterns of coronary artery disease when compared with European men. Int J Cardiol. 2007 doi: 10.1016/j.ijcard.2007.07.129. in press. Published online November 19 2007. [DOI] [PubMed] [Google Scholar]
- 54.Barnett A.H., Dixon A.N., Bellary S., Hanif M.W., O'Hare J.P., Raymond N.T., Kumar S. Type 2 diabetes and cardiovascular risk in the UK south Asian community. Diabetologia. 2006;49:2234–2246. doi: 10.1007/s00125-006-0325-1. [DOI] [PubMed] [Google Scholar]
- 55.Budoff M.J., Nasir K., Mao S., Tseng P.H., Chau A., Liu S.T., Flores F., Blumenthal R.S. Ethnic differences of the presence and severity of coronary atherosclerosis. Atherosclerosis. 2006;187:343–350. doi: 10.1016/j.atherosclerosis.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 56.Whincup P.H., Gilg J.A., Papacosta O., Seymour C., Miller G.J., Alberti K.G., Cook D.G. Early evidence of ethnic differences in cardiovascular risk: cross sectional comparison of British South Asian and white children. BMJ. 2002;324:635. doi: 10.1136/bmj.324.7338.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baskar V., Kamalakannan D., Holland M.R., Singh B.M. Does ethnic origin have an independent impact on hypertension and diabetic complications? Diabetes Obes. Metab. 2006;8:214–219. doi: 10.1111/j.1463-1326.2005.00485.x. [DOI] [PubMed] [Google Scholar]
- 58.Brown D.E., Sievert L.L., Aki S.L., Mills P.S., Etrata M.B., Paopao R.N., James G.D. Effects of age, ethnicity and menopause on ambulatory blood pressure: Japanese-American and Caucasian school teachers in Hawaii. Am. J. Hum. Biol. 2001;13:486–493. doi: 10.1002/ajhb.1080. [DOI] [PubMed] [Google Scholar]
- 59.Abate N., Chandalia M. Ethnicity and type 2 diabetes: focus on Asian Indians. J. Diabetes Complications. 2001;15:320–327. doi: 10.1016/s1056-8727(01)00161-1. [DOI] [PubMed] [Google Scholar]
- 60.Abate N., Chandalia M. Ethnicity, type 2 diabetes & migrant Asian Indians. Indian J. Med. Res. 2007;125:251–258. [PubMed] [Google Scholar]
- 61.Chambers J.C., Kooner J.S. Diabetes, insulin resistance and vascular disease among Indian Asians and Europeans. Semin. Vasc. Med. 2002;2:199–214. doi: 10.1055/s-2002-32043. [DOI] [PubMed] [Google Scholar]
- 62.Zimmet P. Type 2 (non-insulin-dependent) diabetes–an epidemiological overview. Diabetologia. 1982;22:399–411. doi: 10.1007/BF00282581. [DOI] [PubMed] [Google Scholar]
- 63.Mayila·Wufuer, Zhou W., Gu M., Fang M., Cheng Z., Qiu C. Association of polymorphisms of HLA-DRB gene and ACE gene with natural longevity in the Xin Jiang Uigur People. Basic & Clinical Medicine. 2005;25:492–497. [Google Scholar]
- 64.Jiang W.X., Qiu C.C., Cheng Z.H., Niu W.Q. Comparative study of APOB gene 3′ VNTR polymorphisms between natural longevity and controls in Uighur nationality. Chinese Journal of Medical Genetics. 2006;23:523–527. [PubMed] [Google Scholar]
- 65.Ling S., Quan X., Zuheng C., Changchun Q. The Correlation of the Polymorphisms of CETP Gene with the Natural Longevity in Uygur Population. Journal of Medical Research. 2007;36:27–30. [Google Scholar]
- 66.The National Cancer Institute of Canada (2005) International variation in cancer incidence, 1993–1997.
- 67.Rosenberg N.A., Pritchard J.K., Weber J.L., Cann H.M., Kidd K.K., Zhivotovsky L.A., Feldman M.W. Genetic structure of human populations. Science. 2002;298:2381–2385. doi: 10.1126/science.1078311. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.