

Abstract
Although kidney stone formation due to hypercalcemic states is rare, it is important for urologists to understand the pathophysiology of these conditions, methods of diagnosis, and treatments. This should foster a quicker diagnosis and institution of appropriate therapy. The latter typically leads to the attenuation of kidney stone activity. Moreover, these patients have a systemic disease, and therapy has other health benefits.
Key words: Nephrolithiasis, Hypercalcemia, Calcium homeostasis, Vitamin D, Calcitriol, Parathyroid hormone, Hyperparathyroidism, Sarcoidosis
The majority of patients with nephrolithiasis have calcium-containing stones.1 Therefore, processes that influence the delivery of calcium to the kidney may impact stone formation. A significant number of such stone formers have increased urinary calcium excretion. However, a minority has a systemic disease causing chronic hypercalcemia and promotion of stone formation.2 This article will focus on basic mechanisms of calcium balance, the pathophysiology of hypercalcemic states associated with kidney stone formation, and methods of detecting and correcting these disorders.
Sites of Calcium Homeostasis Control
Calcium homeostasis is normally under very tight control. There are 3 main sites of control: the gastrointestinal tract, bone, and kidney.
Under normal conditions, approximately 30% to 40% of dietary calcium is absorbed in the gastrointestinal tract. Animal experiments have demonstrated that the majority, nearly 90%, is absorbed in the small intestine, whereas the remainder of uptake occurs in the colon.3 The stomach is not considered to play a significant role in calcium absorption.4 Studies using radiolabeled calcium in rats have shown that 65% to 88% of daily calcium intake is absorbed in the ileum, 4% to 17% in the jejunum, and 7% to 8% in the duodenum.5,6 A similar pattern has been reported in dogs.7,8 Intestinal calcium perfusion studies in humans have shown the jejunum to have the highest rate of calcium absorption per unit length.9 Despite the high rate of absorption in the jejunum, the primary determinant of total intestinal absorption seems to be transit time in each segment, with the longest transit time occurring in the ileum.3 As a result, total human intestinal calcium absorption is highest in the ileum, with segmental absorption following the pattern seen in the previously mentioned animal studies.10,11
For calcium to be absorbed by the gut, it must be ionized and in solution.12 Mechanical and chemical digestion combined with the acidic environment of the stomach causes calcium to dissociate from non-ionic components of food. The acidity also brings ionized calcium into solution.12,13 Despite these processes, only a small portion of total calcium is soluble in the luminal space, and the solubility decreases as chyme traverses the intestine and becomes less acidic.3
Intestinal calcium absorption occurs by 3 primary routes. An active transcellular route is primarily localized in the upper duodenum.12,14 This transport system consists of 3 sequences. First, calcium moves from the lumen down an electrochemical gradient to enter the intestinal cell through non-voltage-gated epithelial calcium channels located in the brush border membrane. Calcium then passes through the cytosol to the basolateral membrane, a process that is facilitated by calbindin 9, a cytosolic protein that acts as an intracellular calcium “ferry” or “chaperone.” This trans-cytosolic calcium movement is faster than that occurring with passive diffusion. Finally, calcium is pumped out of the cell into the lamina propria and up an electrochemical gradient by an adenosine triphosphateactivated calcium pump.15 This mechanism can be completely saturated in the presence of high levels of intestinal calcium. Calcitrol [1,25(OH)2D3] serves to stimulate the synthesis of epithelial calcium channels, calbindin 9, and the calcium adenosine triphosphatase pump.15 Thus, calcitriol acts to upregulate calcium absorption by the transcellular route.
The second mechanism of calcium absorption is nonsaturable paracellular transport, which is also influenced by calcitriol.13 In this mechanism, calcium travels out of the lumen, through the paracellular tight junctions between adjacent enterocytes, and into the lamina propria. This mechanism requires a calcium concentration in the lumen of 2 to 6 mmol/L to overcome the electrochemical gradient.15 Thus, at luminal calcium concentrations of less than 2 mmol/L, passive paracellular absorption plays a minimal role. At luminal calcium concentrations greater than 2 to 6 mmol/L, the rate of paracellular absorption is directly proportional to luminal concentration. Calcitriol acts to change the assembly and permeability of tight junctions in a way that leads to a net increase in conductance of calcium in the presence of a favorable gradient.16
The third mechanism is vesicular transport, which is also calcitriol dependent and occurs primarily in the duodenum. The relative contribution of this pathway to calcium absorption under normal physiologic conditions is not yet clearly defined.13,15,17
The concentration of soluble, ionized calcium in the intestinal lumen influences the mode of calcium absorption. When the calcium concentration is low the majority is absorbed through the active transcellular route. When the calcium concentration is high, the paracellular route predominates.12 The active transport system is more efficient than paracellular transport. Thus, the efficiency with which dietary calcium is absorbed varies with its concentration; there is a higher percentage of dietary calcium absorption when total calcium intake is low than when it is high.1
Once calcium has entered the extracellular space, it circulates throughout the body. Bone acts as a reservoir that holds 99% of the body’s total calcium at any given time, but this reservoir is not static. Osteoblastic activity and osteoclastic activity occur simultaneously, such that calcium from bone is being constantly exchanged with extracellular calcium in a process of continual bone remodeling.18 Every day approximately 300 mg of calcium is exchanged between the extracellular pool and the bone storage pool.19
Excess calcium is excreted by both the gut and the kidney. Most of the calcium excreted in feces is accounted for by sources of dietary calcium that could not be completely absorbed, but some calcium in the gut comes from endogenous sources. In the proximal gut, endogenous calcium comes primarily from gastric juices, whereas most endogenous calcium in the distal gut comes from mucus and colonic mucosal cells.20 Endogenous calcium that enters the gut undergoes absorption similar to calcium from exogenous sources, but the relatively low efficiency of calcium absorption leads most endogenous calcium that enters the gut to be excreted.20 In healthy adults, daily endogenous fecal calcium loss is approximately the same as urinary excretion of calcium.21,22
Extracellular calcium exists in 3 fractions. Approximately 50% of extracellular calcium is ionized and is free to bind receptors and exert biologic activity, 40% is bound to plasma proteins, and 10% is complexed to anions such as citrate, phosphate, sulfate, bicarbonate, and lactate.23 Albumin accounts for 60% of plasma proteins and is the primary protein that binds calcium, at a ratio of 0.8 mg/dL calcium for every 1 g/dL of albumin at a normal pH.23 The remainder of the bound calcium is bound to serum globulin proteins.
The majority of calcium delivered to the kidney is reabsorbed. Calcium that is ionized or complexed to anions is ultrafiltered at the glomerulus, whereas calcium that is bound to proteins is not typically filtered by the kidney.23 Normally, 98% to 99% of the filtered calcium is reabsorbed: 50% to 60% is reabsorbed in the proximal convoluted tubule in a passive, paracellular fashion, 15% in the thick ascending limb by passive paracellular and active mechanisms, and 10% to 15% in the distal convoluted tubule mostly by an active, transcellular process.19
Modulators of Calcium Homeostasis
Vitamin D is an important component of calcium homeostasis. It is derived from the diet or generated by ultraviolet radiation to the skin, which photo-isomerizes precursors.24 Vitamin D is activated in a 2-step process. The liver hydroxylates vitamin D at the 25 carbon through the cytochrome P450 system in a constitutive manner, yielding 25(OH)D3.24 The latter is converted to calcitriol in the proximal tubule of the kidney in a tightly controlled step by the enzyme 1-alpha hydroxylase.13,24 Calcitriol binds to a high-affinity receptor, the vitamin D receptor (VDR), which is transported to the nucleus, where it regulates messenger ribonucleic acid (mRNA) transcription.24 The influences of calcitriol on gut calcium transport have been reviewed above.
Calcitriol also has important actions at the skeletal level.25 Providing sufficient calcium for bone matrix protein is one of its important roles. It also influences the content of bone matrix. For example, calcitriol suppresses the production of type I collagen and induces osteocalcin synthesis, 2 of the major bone matrix proteins.13 Calcitriol also stimulates osteoclast differentiation.13
Calcitriol also modulates parathyroid gland function. When the VDR is activated by calcitriol, it inhibits the synthesis of parathyroid hormone (PTH).24 Calcitriol also modulates expression of VDR and calcium sensing receptor (CaSR) in the parathyroid glands. Increased levels of calcitriol enhance VDR expression, though this mechanism may represent indirect control through increased serum calcium.24 Calcitriol stimulates the expression of CaSR in the parathyroid glands.24 The net effect of calcitriol on the parathyroid tissue is to reduce PTH secretion and parathyroid cell differentiation.24
PTH is another important component of calcium balance. Parathyroid chief cells synthesize, store, and secrete PTH in response to serum calcium levels, and PTH serves to provide the minute-to-minute control of serum calcium levels.13 PTH is synthesized as a pro-hormone and cleaved to a metabolically active form by the kidney and liver. Control of PTH secretion occurs in 3 time frames. Short-term control is modulated by the CaSR. In a low serum calcium state, the absence of CaSR stimulation leads to upregulation of PTH secretion from vesicular stores.13 Medium-term control is by increased production of PTH mRNA. Long-term control is by cellular replication. When chronically stimulated, as in the presence of chronically low serum calcium, parathyroid cells will replicate and the parathyroid gland can become hyperplastic, leading to an increased ability to secrete PTH.13
The action of PTH is mediated by its complexation with a cell-surface G protein receptor. The binding of PTH with this receptor stimulates a number of secondary messengers that potentiate the actions of PTH on its endorgan targets. Its influences at these sites are described next.
In the kidney, PTH exerts its primary function on the distal nephron, where it upregulates transcellular calcium reabsorption by increasing the transmembrane voltage gradient, thus hyperpolarizing the cell.13 PTH also reduces phosphate reabsorption in the proximal and distal tubules, primarily by reducing the expression of sodium phosphate co-transporter proteins necessary for transcellular reabsorption of potassium. PTH further acts on the kidney by inducing transcription of the 1-alpha hydroxylase gene, leading to increased production of calcitriol. Thus, the net effect of PTH on the kidney is to increase extracellular calcium and decrease extracellular phosphate levels.13
Interestingly, PTH acts to stimulate both bone reabsorption and bone deposition. Although the net effect of PTH is to stimulate bone reabsorption, the local effects vary widely.13 When PTH receptors on preosteoblasts are activated, this inhibits their maturation into osteoblasts. Stimulation of PTH receptors on osteoblasts decreases production of bone matrix proteins.13 Although osteoclasts do not have PTH receptors, cell signaling from PTH-activated osteoblasts promotes osteoclastic activity, resulting in bone resorption.
Parathyroid hormone-related protein (PTHrP) also binds to PTH receptor and potentiates the same actions as PTH. PTHrP seems to primarily act in an autocrine or paracrine fashion and is only found in systemic circulation in disease states such as malignancy.
Calcitonin serves to rapidly decrease the serum calcium level when given exogenously through reduction in tubular resorption of calcium and impairment of osteoclastic bone resorption.13 Calcitonin seems to work through a G protein receptor in the same family as the PTH receptor but is not known to play a role in the day-to-day control of calcium homeostasis.13
Homeostasis
A complex interplay between PTH and calcitriol and the mineral ions maintains calcium levels within a narrow range (Figure 1). When serum calcium is low, PTH is secreted, leading to an increase in bone resorption and renal conservation of calcium. Calcitriol is generated, leading to increased intestinal absorption of dietary calcium.13 If serum calcium is high, PTH is suppressed, calcitriol is suppressed, intestinal absorption of calcium decreases, and the kidney excretes more calcium.13
Figure 1.
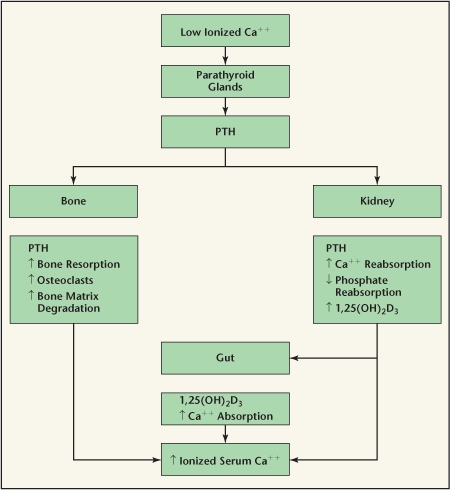
Calcium homeostasis. PTH, parathyroid hormone.
Failures of Calcium Homeostasis
Calcium regulation can go awry at many different steps in the control mechanisms, but only rarely does this lead to nephrolithiasis. Diseases that result in hypercalcemia and hypercalciuria may be associated with stone formation (Table 1). The 2 most prevalent are hyperparathyroidism and sarcoidosis.
Table 1.
Hypercalcemic States That Lead to Nephrolithiasis
| Hyperparathyroidism |
| Sarcoidosis |
| Other granulomatous disease |
| Prolonged immobilization |
| Milk-alkali syndrome |
| Hypervitaminosis D |
| Malignancy (rarely causes nephrolithiasis) |
Hyperparathyroidism
Primary hyperparathyroidism is due to a sporadic solitary parathyroid adenoma 80% to 85% of the time.26,27 Parathyroid carcinoma accounts for 1% to 4% of primary hyperparathyroidism, whereas parathyroid hyperplasia accounts for up to 15%.26,28,29 Rarely, patients with primary hyperparathyroidism may have multiple endocrine neoplasia syndromes type I or IIa (MEN I or MEN IIa). Hyperparathyroidism occurs in approximately 95% of patients with MEN I. Hyperparathyroidism is due to hyperplasia of all 4 parathyroid glands in this condition. MEN I usually manifests in the second or third decade of life.13 In contrast, 5% to 20% of patients with MEN IIa will have hyperparathyroidism, and the disease typically presents later in life.13 Similar to MEN I, it is usually the result of hyperplasia.26 Subtotal parathyroidectomy is performed in patients with MEN I and MEN II. The recurrence rate in such patients is higher than in those with hyperparathyroidism due to a solitary adenoma.13
In hyperparathyroidism, the serum PTH level is inappropriately increased, leading to increases in bone resorption, reabsorption of calcium in the nephron, and intestinal absorption. The net effect is a rise in the serum calcium concentration.13 This increases the filtered load of calcium in the glomerulus, which may override the impact of the increased absorption in the nephron, leading to hypercalciuria. The hypercalcemia of hyperparathyroidism typically is not to the degree reached in other hypercalcemic states.
As screening and diagnostic tools for hyperparathyroidism have improved, patients with primary hyperparathyroidism are now frequently diagnosed while still asymptomatic.30 As a result, the incidence of urolithiasis in primary hyperparathyroidism has decreased from nearly 80% in early series to 7% to 20% in recent series.31,32 Broadus and colleagues33 reported that those who develop stones have higher urinary calcium excretion than those who do not. There are patients with primary hyperparathyroidism and stones who have normal calcium excretion, suggesting that other stone risk factors may be playing a role.34–36 Patients with primary hyperparathyroidism and stones typically form calcium phosphate, calcium oxalate, or mixed calcium stones.1,37
Less than 1% of stone formers in the community setting have primary hyperparathyroidism.38 A higher prevalence is encountered in referral centers (1.65%–13.3%).39–42
Although only a small percentage of patients with stones have primary hyperparathyroidism, screening for hyperparathyroidism is still recommended. Screening with serum calcium testing is inexpensive, and when the diagnosis is established the disease can be surgically cured and stone activity significantly reduced.30 Mollerup and colleagues30 examined 107 patients with renal calculi and hyperparathyroidism at presentation. After surgical correction of patients’ hyperparathyroidism, the stone recurrence rate was 30% at 5 years, significantly lower than the rate before operation and comparable to the rate of recurrence in idiopathic stone formers.30 Deaconson and associates43 reported similar reductions in stone events after parathyroidectomy. Contrary to this evidence, Vestergaard and Mosekilde44 reported that stone event rates were higher in patients with primary hyperparathyroidism who underwent surgical correction than in those treated conservatively (odds ratio, 2.49; 95% confidence interval, 1.93–3.23). The investigators postulate that the increase in stone events may be due to selection bias when surgery is indicated for individuals more seriously affected by the disease. They also propose that some of this increased incidence may be from renal damage done by previous stone events.
If serum calcium is elevated, intact PTH and phosphorous are measured. In hyperparathyroidism, the intact PTH level is usually high-normal or elevated and phosphorous depressed. Ionized calcium is measured when serum albumin is abnormal because this will enhance detection.45
Although screening for hyperparathyroidism is straightforward, the hypercalcemia associated with hyperparathyroidism may be intermittent, and some patients may not be hypercalcemic at all, with serum calcium in the high-normal range. As a result, it is necessary to measure serum calcium levels over time to fully evaluate for hyperparathyroidism in symptomatic patients.27 Alternatively, one may consider obtaining an intact PTH level for patients with a high-normal serum calcium level and symptoms attributable to hyperparathyroidism, such as recurrent stone formation, especially if forming calcium phosphate stones.46,47 The administration of a short course of thiazide diuretic or indapamide therapy will sometimes unmask the diagnosis in equivocal cases, the patients in such cases becoming hypercalcemic on therapy.
PTH acts to decrease calcium binding to albumin and globulin proteins. As a result, patients with early hyperparathyroidism can have elevated ionized calcium levels and the associated sequela of hypercalcemia but have normal total serum calcium levels. This may be the underlying mechanism for normocalcemic hyperparathyroidism.39–41,48,49
(99m)Tc-sestamibi scans have been used successfully to identify a number of metabolically active tissues, including parathyroid adenomas. They are mainly used when the diagnosis of primary hyperparathyroidism is highly suspected on the basis of serum calcium and PTH testing. In this setting this study may identify a parathyroid adenoma and facilitate minimally invasive parathyroidectomy.50,51 It may also help in the detection of ectopic parathyroid adenomas and in the evaluation of patients suspected of having normocalcemic hyperparathyroidism.48,52 One must be aware of the sensitivity and specificity of this test when making treatment decisions. (99m)Tc-sestamibi scans have a sensitivity of 88.44% for solitary parathyroid adenomas, but the sensitivity for other parathyroid pathology is significantly worse: 44.46% for multigland hyperplasia, 29.95% for double adenomas, and 33% for carcinoma.52,53 False-positive results on sestamibi scans are also possible, particularly in the setting of thyroid pathology or reactive lymph nodes.
Sarcoidosis
Sarcoidosis is a granulomatous disease that can lead to hypercalcemia. Hypercalcemia has been reported to occur in 2% to 63% of patients with sarcoidosis, with most estimates falling around 10%.54 Hypercalciuria is approximately 3 times more common than hypercalcemia in sarcoidosis, occurring in up to 50% of patients.55 Harrell and Fisher56 reported a relationship between elevated serum vitamin D and sarcoidosis in 1939. Subsequent investigations have demonstrated that hypercalcemia in sarcoidosis is caused by extrarenal production of calcitriol by 1-alpha hydroxylase present in the macrophages of the granulomas.54 Calcitriol is postulated to act as an immune regulator in a fashion similar to other cytokines, but its exact mechanism in the granulomas has not been elucidated.54
Hypercalcemia due to sarcoidosis can be treated effectively with corticosteroid administration. In adults, the administration of 20 to 40 mg of prednisone daily leads to reduction in serum calcium and calcitriol and resolution of hypercalciuria, typically in 5 to 7 days.54
Other Granulomatous Diseases
Other granulomatous diseases have been associated with hypercalcemia, including leprosy,57 coccidioidomycosis,58 tuberculosis,40 plasma cell granuloma,59 Wegener’s granulomatosis,60 and silicosis.61 In all of these diseases except for tuberculosis, the mechanism of hypercalcemia seems to be increased production of calcitriol in granulomas. In tuberculosis calcitriol may play a role, but there may be other mechanisms because serum calcitriol levels in such cases are not always elevated.62
Malignancy
Malignancy may cause hypercalcemia due to many mechanisms. The most prominent involves the production of PTHrP. PTHrP production is normally regulated by the CaSR on the cell surface. Certain tumors, such as breast tumor cells, have an abnormal regulation in which CaSR stimulates production of PTHrP instead of inhibiting it.63 As a result, PTHrP levels greatly exceed the normal concentrations that are just sufficient to act in an autocrine or paracrine fashion. As serum levels rise, PTHrP begins to act similarly to PTH, resulting in hypercalcemia. However, this infrequently results in stone formation. Perhaps this is owing to shorter periods of hypercalcemia due to cancer mortality or modulation of other stone risk parameters.64,65
Familial Hypocalciuria Hypercalcemia
Familial hypocalciuria hypercalcemia is an autosomal dominant mutation of parathyroid CaSR.66 Patients with this disorder have moderately elevated serum calcium levels and varied symptoms, such as fatigue, headache, or weakness. As the name implies, this disorder does not lead to hypercalciuria and therefore does not lead to urolithiasis.66
Milk-Alkali Syndrome
The occurrence of milk-alkali syndrome as a cause for hypercalcemia dramatically decreased with the introduction of H2 blockers and proton pump inhibitors for treatment of peptic ulcer disease. Milk-alkali syndrome leads to the classic triad of hypercalcemia, metabolic alkalosis, and renal failure.67 It is caused by ingestion of calcium and alkali substances together in susceptible individuals. New evidence indicates that incidence of this disorder may again be on the rise because calcium supplementation is now emphasized for prevention of osteoporosis and because calcium carbonate is being used to reduce secondary hyperparathyroidism in chronic renal failure.68–70 The syndrome has been associated with urolithiasis, and the diagnosis should be considered when other causes of hypercalcemia have been ruled out.37 Removal of the sources of alkali and calcium leads to rapid improvement of the hypercalcemia and the metabolic alkalosis. Renal function typically improves with treatment, but may not return to baseline.68
Hypervitaminosis D
Hypervitaminosis D has been reported to cause urolithiasis, but vitamin D intoxication is now rarely encountered in clinical practice.37,71 Occasionally mild forms of vitamin D intoxication occur as the result of treatment of hypoparathyroidism with large doses of vitamin D and oral calcium, but even this does not usually lead to hypercalciuria sufficient to cause nephrolithiasis.37
Prolonged Immobilization
Total body immobilization as is found in orthopedic disorders and paralysis leads to hypercalciuria, hyperphosphaturia, and in some patients, hypercalcemia. This may also occur during space travel, a low-gravity environment in which mobility may be limited. It seems that the hormonal control of bone is not sufficient to maintain its crystalline microstructure. Rather, mechanical stress serves to alter signals in bone that combine with the previously discussed hormonal regulation to lay down and maintain mechanically strong bones.72–74 Decreased muscle activity and decreased weight bearing in immobilized patients lead to changes in calcium homeostasis, resulting in the resorption of skeletal bone and eventuating in the aforementioned metabolic changes.37,75–77 In the setting of immobility, hypercalcemia is usually only found in children or young adults with recent fractures, but on occasion may be found in older adults with underlying disorders of calcium metabolism.37 It may lead to kidney stone formation, with affected patients usually forming calcium phosphate stones. Remobilization of the patient typically leads to resolution of these metabolic changes.37,75,76 Similar metabolic changes can occur during space travel owing to the microgravity environment.78
Conclusion
Although kidney stone formation due to hypercalcemic states is rare, it is important for urologists to understand the pathophysiology of these conditions, methods of diagnosis, and treatments. This should foster a quicker diagnosis and institution of appropriate therapy. The latter typically leads to the attenuation of kidney stone activity. Moreover, these patients have a systemic disease, and therapy has other health benefits.
Main Points.
The majority of patients with nephrolithiasis have calcium-containing stones. A significant number of such stone formers have increased urinary calcium excretion, but a minority has a systemic disease causing chronic hypercalcemia and promotion of stone formation.
Calcium homeostasis is normally under very tight control. A complex interplay between parathyroid hormone and calcitriol and the mineral ions maintains calcium levels within a narrow range. Calcium regulation can go awry at many different steps in the control mechanisms, but only rarely does this lead to nephrolithiasis.
Of the diseases/conditions that result in hypercalcemia and hypercalciuria and that may be associated with stone formation, the 2 most prevalent are hyperparathyroidism and sarcoidosis. Others include other granulomatous diseases (eg, leprosy, coccidioidomycosis, tuberculosis, plasma cell granuloma, Wegener’s granulomatosis, and silicosis), prolonged immobilization, milkalkali syndrome, hypervitaminosis D, and (rarely) malignancy.
Although only a small percentage of patients with stones have primary hyperparathyroidism, screening for hyperparathyroidism is still recommended. Screening with serum calcium testing is inexpensive, and when the diagnosis is established the disease can be surgically cured and stone activity significantly reduced.
Hypercalcemia due to sarcoidosis can be treated effectively with corticosteroid administration. In adults, the administration of 20 to 40 mg of prednisone daily leads to reduction in serum calcium and calcitriol and resolution of hypercalciuria, typically in 5 to 7 days.
References
- 1.Campbell MF, Wein AJ, Kavoussi LR, editors. Campbell-Walsh Urology. Philadelphia: Saunders Elsevier; 2007. [Google Scholar]
- 2.Bushinsky DA, Parker WR, Asplin JR. Calcium phosphate supersaturation regulates stone formation in genetic hypercalciuric stone-forming rats. Kidney Int. 2000;57:550–560. doi: 10.1046/j.1523-1755.2000.00875.x. [DOI] [PubMed] [Google Scholar]
- 3.Duflos C, Bellaton C, Pansu D, Bronner F. Calcium solubility, intestinal sojourn time and paracellular permeability codetermine passive calcium absorption in rats. J Nutr. 1995;125:2348–2355. doi: 10.1093/jn/125.9.2348. [DOI] [PubMed] [Google Scholar]
- 4.Hoenderop JG, Nilius B, Bindels RJ. Calcium absorption across epithelia. Physiol Rev. 2005;85:373–422. doi: 10.1152/physrev.00003.2004. [DOI] [PubMed] [Google Scholar]
- 5.Marcus CS, Wasserman RH. Ca and Mg levels in gastrointestinal mucosa of fed, fasted, and lactose-treated rats. J Appl Physiol. 1966;21:1063–1067. doi: 10.1152/jappl.1966.21.3.1063. [DOI] [PubMed] [Google Scholar]
- 6.Cramer CF, Copp DH. Progress and rate of absorption of radiostrontium through intestinal tracts of rats. Proc Soc Exp Biol Med. 1959;102:514–517. doi: 10.3181/00379727-102-25301. [DOI] [PubMed] [Google Scholar]
- 7.Cramer CF. In vivo intestinal transport of calcium and water from solutions recycled through healed cut loops in dogs. J Nutr. 1964;84:118–124. doi: 10.1093/jn/84.2.118. [DOI] [PubMed] [Google Scholar]
- 8.Cramer CF. Sites of calcium absorption and the calcium concentration of gut contents in the dog. Can J Physiol Pharmacol. 1965;43:75–78. doi: 10.1139/y65-009. [DOI] [PubMed] [Google Scholar]
- 9.Krejs GJ, Nicar MJ, Zerwekh JE, et al. Effect of 1,25-dihydroxyvitamin D3 on calcium and magnesium absorption in the healthy human jejunum and ileum. Am J Med. 1983;75:973–976. doi: 10.1016/0002-9343(83)90877-x. [DOI] [PubMed] [Google Scholar]
- 10.Weiser MM, Bloor JH, Dasmahapatra A. Intestinal calcium absorption and vitamin D metabolism. J Clin Gastroenterol. 1982;4:75–86. doi: 10.1097/00004836-198202000-00014. [DOI] [PubMed] [Google Scholar]
- 11.Ireland P, Fordtran JS. Effect of dietary calcium and age on jejunal calcium absorption in humans studied by intestinal perfusion. J Clin Invest. 1973;52:2672–2681. doi: 10.1172/JCI107461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bronner F, Pansu D. Nutritional aspects of calcium absorption. J Nutr. 1999;129:9–12. doi: 10.1093/jn/129.1.9. [DOI] [PubMed] [Google Scholar]
- 13.Larsen PR. Williams Textbook of Endocrinology. 10th ed. Philadelphia: Saunders; 2003. [Google Scholar]
- 14.Bronner F. Mechanisms of intestinal calcium absorption. J Cell Biochem. 2003;88:387–393. doi: 10.1002/jcb.10330. [DOI] [PubMed] [Google Scholar]
- 15.Wasserman RH, Chandler JS, Meyer SA, et al. Intestinal calcium transport and calcium extrusion processes at the basolateral membrane. J Nutr. 1992;122(3 suppl):662–671. doi: 10.1093/jn/122.suppl_3.662. [DOI] [PubMed] [Google Scholar]
- 16.Chirayath MV, Gajdzik L, Hulla W, et al. Vitamin D increases tight-junction conductance and paracellular Ca2+ transport in Caco-2 cell cultures. Am J Physiol. 1998;274(2 pt 1):G389–G396. doi: 10.1152/ajpgi.1998.274.2.G389. [DOI] [PubMed] [Google Scholar]
- 17.Nemere I. Vesicular calcium transport in chick intestine. J Nutr. 1992;122(3 suppl):657–661. doi: 10.1093/jn/122.suppl_3.657. [DOI] [PubMed] [Google Scholar]
- 18.Hadjidakis DJ, Androulakis II. Bone remodeling. Ann N Y Acad Sci. 2006;1092:385–396. doi: 10.1196/annals.1365.035. [DOI] [PubMed] [Google Scholar]
- 19.Brenner BM, Rector FC. Brenner & Rector’s The Kidney. Philadelphia: Saunders; 2004. [Google Scholar]
- 20.Davies KM, Rafferty K, Heaney RP. Determinants of endogenous calcium entry into the gut. Am J Clin Nutr. 2004;80:919–923. doi: 10.1093/ajcn/80.4.919. [DOI] [PubMed] [Google Scholar]
- 21.Heaney RP, Abrams SA. Improved estimation of the calcium content of total digestive secretions. J Clin Endocrinol Metab. 2004;89:1193–1195. doi: 10.1210/jc.2003-031660. [DOI] [PubMed] [Google Scholar]
- 22.Heaney RP, Recker RR. Determinants of endogenous fecal calcium in healthy women. J Bone Miner Res. 1994;9:1621–1627. doi: 10.1002/jbmr.5650091016. [DOI] [PubMed] [Google Scholar]
- 23.Bushinsky DA, Monk RD. Electrolyte quintet: calcium. Lancet. 1998;352:306–311. doi: 10.1016/s0140-6736(97)12331-5. [DOI] [PubMed] [Google Scholar]
- 24.Dusso AS, Brown AJ, Slatopolsky E. Vitamin D. Am J Physiol Renal Physiol. 2005;289:F8–F28. doi: 10.1152/ajprenal.00336.2004. [DOI] [PubMed] [Google Scholar]
- 25.Hendy GN, Hruska KA, Mathew S, Goltzman D. New insights into mineral and skeletal regulation by active forms of vitamin D. Kidney Int. 2006;69:218–223. doi: 10.1038/sj.ki.5000091. [DOI] [PubMed] [Google Scholar]
- 26.Johnson SJ, Sheffield EA, McNicol AM. Best practice no 183. Examination of parathyroid gland specimens. J Clin Pathol. 2005;58:338–342. doi: 10.1136/jcp.2002.002550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Siperstein AE, Shen W, Chan AK, et al. Normocalcemic hyperparathyroidism. Biochemical and symptom profiles before and after surgery. Arch Surg. 1992;127:1156–1157. doi: 10.1001/archsurg.1992.01420100015003. discussion 1161–1163. [DOI] [PubMed] [Google Scholar]
- 28.Rodgers SE, Perrier ND. Parathyroid carcinoma. Curr Opin Oncol. 2006;18:16–22. doi: 10.1097/01.cco.0000198019.53606.2b. [DOI] [PubMed] [Google Scholar]
- 29.Shane E. Clinical review 122: parathyroid carcinoma. J Clin Endocrinol Metab. 2001;86:485–493. doi: 10.1210/jcem.86.2.7207. [DOI] [PubMed] [Google Scholar]
- 30.Mollerup CL, Vestergaard P, Frøkjaer VG, et al. Risk of renal stone events in primary hyperparathyroidism before and after parathyroid surgery: controlled retrospective follow up study. BMJ. 2002;325:807. doi: 10.1136/bmj.325.7368.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klugman V, Favus MJ. Diagnosis and treatment of calcium kidney stones. Adv Endocrinol Metab. 1995;6:117–142. [PubMed] [Google Scholar]
- 32.Silverberg SJ, Shane E, Jacobs TP, et al. Nephrolithiasis and bone involvement in primary hyperparathyroidism. Am J Med. 1990;89:327–334. doi: 10.1016/0002-9343(90)90346-f. [DOI] [PubMed] [Google Scholar]
- 33.AE Broadus, RL Horst, R Lang, et al. The importance of circulating 1,25-dihydroxyvitamin D in the pathogenesis of hypercalciuria and renalstone formation in primary hyperparathyroidism. N Engl J Med. 1980;302:421–426. doi: 10.1056/NEJM198002213020801. [DOI] [PubMed] [Google Scholar]
- 34.Khanam A, Rahman MA. Parathyroid hormone in urinary stone patients. Mol Cell Biochem. 1993;121:1–4. doi: 10.1007/BF00928693. [DOI] [PubMed] [Google Scholar]
- 35.D’Angelo A, Lodetti MG, Giannini S, et al. Hyperparathyroidism: cause or consequence of recurrent calcium nephrolithiasis? Miner Electrolyte Metab. 1992;18:359–364. [PubMed] [Google Scholar]
- 36.Odvina CV, Sakhaee K, Heller HJ, et al. Biochemical characterization of primary hyperparathyroidism with and without kidney stones. Urol Res. 2007;35:123–128. doi: 10.1007/s00240-007-0096-2. [DOI] [PubMed] [Google Scholar]
- 37.Thomas WC., Jr. Urinary calculi in hypercalcemic states. Endocrinol Metab Clin North Am. 1990;19:839–849. [PubMed] [Google Scholar]
- 38.Derrick FC., Jr. Renal calculi in association with hyperparathyroidism: a changing entity. J Urol. 1982;127:226. doi: 10.1016/s0022-5347(17)53709-x. [DOI] [PubMed] [Google Scholar]
- 39.Sedlack JD, Kenkel J, Czarapata BJ, et al. Primary hyperparathyroidism in patients with renal stones. Surg Gynecol Obstet. 1990;171:206–208. [PubMed] [Google Scholar]
- 40.Fuss M, Pepersack T, Gillet C, et al. Calcium and vitamin D metabolism in granulomatous diseases. Clin Rheumatol. 1992;11:28–36. doi: 10.1007/BF02207080. [DOI] [PubMed] [Google Scholar]
- 41.Dimkovic NB, Wallele AA, Oreopoulos DG. Renal stone disease, elevated iPTH level and normocalcemia. Int Urol Nephrol. 2002;34:135–141. doi: 10.1023/a:1021331728317. [DOI] [PubMed] [Google Scholar]
- 42.Lavan JN, Neale FC, Posen S. Urinary calculi. Clinical, biochemical and radiological studies in 619 patients. Med J Aust. 1971;2:1049–1061. doi: 10.5694/j.1326-5377.1971.tb92706.x. [DOI] [PubMed] [Google Scholar]
- 43.Deaconson TF, Wilson SD, Lemann J., Jr. The effect of parathyroidectomy on the recurrence of nephrolithiasis. Surgery. 1987;102:910–913. [PubMed] [Google Scholar]
- 44.Vestergaard P, Mosekilde L. Cohort study on effects of parathyroid surgery on multiple outcomes in primary hyperparathyroidism. BMJ. 2003;327:530–534. doi: 10.1136/bmj.327.7414.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Monchik JM. Normocalcemic hyperparathyroidism. Surgery. 1995;118:917–923. doi: 10.1016/s0039-6060(05)80094-7. [DOI] [PubMed] [Google Scholar]
- 46.Levy FL, Adams-Huet B, Pak CY. Ambulatory evaluation of nephrolithiasis: an update of a 1980 protocol. Am J Med. 1995;98:50–59. doi: 10.1016/S0002-9343(99)80080-1. [DOI] [PubMed] [Google Scholar]
- 47.Preminger GM. The metabolic evaluation of patients with recurrent nephrolithiasis: a review of comprehensive and simplified approaches. J Urol. 1989;141(3 pt 2):760–763. doi: 10.1016/s0022-5347(17)41004-4. [DOI] [PubMed] [Google Scholar]
- 48.Lowe H, McMahon DJ, Rubin MR, et al. Normocalcemic primary hyperparathyroidism: further characterization of a new clinical phenotype. J Clin Endocrinol Metab. 2007;92:3001–3005. doi: 10.1210/jc.2006-2802. [DOI] [PubMed] [Google Scholar]
- 49.Ladenson JH, Lewis JW, McDonald JM, et al. Relationship of free and total calcium in hypercalcemic conditions. J Clin Endocrinol Metab. 1979;48:393–397. doi: 10.1210/jcem-48-3-393. [DOI] [PubMed] [Google Scholar]
- 50.AP Dackiw, JJ Sussman, HA Fritsche, Jr, et al. Relative contributions of technetium Tc 99m sestamibi scintigraphy, intraoperative gamma probe detection, and the rapid parathyroid hormone assay to the surgical management of hyperparathyroidism. Arch Surg. 2000;135:550–555. doi: 10.1001/archsurg.135.5.550. discussion 555–557. [DOI] [PubMed] [Google Scholar]
- 51.Rubello D, Mariani G, Al-Nahhas A, et al. Minimally invasive (99m)Tc-sestamibi radioguided surgery of parathyroid adenomas. Panminerva Med. 2005;47:99–107. [PubMed] [Google Scholar]
- 52.Kettle AG, O’Doherty MJ. Parathyroid imaging: how good is it and how should it be done? Semin Nucl Med. 2006;36:206–211. doi: 10.1053/j.semnuclmed.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 53.Palestro CJ, Tomas MB, Tronco GG. Radionuclide imaging of the parathyroid glands. Semin Nucl Med. 2005;35:266–276. doi: 10.1053/j.semnuclmed.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 54.Sharma OP. Vitamin D, calcium, and sarcoidosis. Chest. 1996;109:535–539. doi: 10.1378/chest.109.2.535. [DOI] [PubMed] [Google Scholar]
- 55.Zeimer HJ, Greenaway TM, Slavin J, et al. Parathyroid-hormone-related protein in sarcoidosis. Am J Pathol. 1998;152:17–21. [PMC free article] [PubMed] [Google Scholar]
- 56.Harrell GT, Fisher S. Blood chemical changes in Boeck’s sarcoid with particular reference to protein, calcium and phosphatase values. J Clin Invest. 1939;18:687–693. doi: 10.1172/JCI101084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Couri CE, Foss NT, Dos Santos CS, de Paula FJ. Hypercalcemia secondary to leprosy. Am J Med Sci. 2004;328:357–359. doi: 10.1016/s0002-9629(15)33948-3. [DOI] [PubMed] [Google Scholar]
- 58.Parker MS, Dokoh S, Woolfenden JM, Buchsbaum HW. Hypercalcemia in coccidioidomycosis. Am J Med. 1984;76:341–344. doi: 10.1016/0002-9343(84)90799-x. [DOI] [PubMed] [Google Scholar]
- 59.Helikson MA, Havey AD, Zerwekh JE, et al. Plasma-cell granuloma producing calcitriol and hypercalcemia. Ann Intern Med. 1986;105:379–381. doi: 10.7326/0003-4819-105-3-379. [DOI] [PubMed] [Google Scholar]
- 60.Shaker JL, Redlin KC, Warren GV, Findling JW. Case report: hypercalcemia with inappropriate 1,25-dihydroxyvitamin D in Wegener’s granulomatosis. Am J Med Sci. 1994;308:115–118. doi: 10.1097/00000441-199408000-00011. [DOI] [PubMed] [Google Scholar]
- 61.Kozeny GA, Barbato AL, Bansal VK, et al. Hypercalcemia associated with silicone-induced granulomas. N Engl J Med. 1984;311:1103–1105. doi: 10.1056/NEJM198410253111707. [DOI] [PubMed] [Google Scholar]
- 62.Jacobs TP, Bilezikian JP. Clinical review: rare causes of hypercalcemia. J Clin Endocrinol Metab. 2005;90:6316–6322. doi: 10.1210/jc.2005-0675. [DOI] [PubMed] [Google Scholar]
- 63.Chattopadhyay N. Effects of calcium-sensing receptor on the secretion of parathyroid hormonerelated peptide and its impact on humoral hypercalcemia of malignancy. Am J Physiol Endocrinol Metab. 2006;290:E761–E770. doi: 10.1152/ajpendo.00350.2005. [DOI] [PubMed] [Google Scholar]
- 64.Coe FL, Parks JH. Nephrolithiasis: Pathogenesis and Treatment. 2nd ed. Chicago: Year Book Medical Publishers; 1988. [Google Scholar]
- 65.Rodman JS, Mahler RJ. Kidney stones as a manifestation of hypercalcemic disorders. Hyperparathyroidism and sarcoidosis. Urol Clin North Am. 2000;27:275–285. doi: 10.1016/s0094-0143(05)70257-3. viii. [DOI] [PubMed] [Google Scholar]
- 66.D’souza-Li L. The calcium-sensing receptor and related diseases. Arq Bras Endocrinol Metabol. 2006;50:628–639. doi: 10.1590/s0004-27302006000400008. [DOI] [PubMed] [Google Scholar]
- 67.Orwoll ES. The milk-alkali syndrome: current concepts. Ann Intern Med. 1982;97:242–248. doi: 10.7326/0003-4819-97-2-242. [DOI] [PubMed] [Google Scholar]
- 68.Picolos MK, Lavis VR, Orlander PR. Milk-alkali syndrome is a major cause of hypercalcaemia among non-end-stage renal disease (non-ESRD) inpatients. Clin Endocrinol (Oxf) 2005;63:566–576. doi: 10.1111/j.1365-2265.2005.02383.x. [DOI] [PubMed] [Google Scholar]
- 69.Picolos MK, Orlander PR. Calcium carbonate toxicity: the updated milk-alkali syndrome; report of 3 cases and review of the literature. Endocr Pract. 2005;11:272–280. doi: 10.4158/EP.11.4.272. [DOI] [PubMed] [Google Scholar]
- 70.Beall DP, Scofield RH. Milk-alkali syndrome associated with calcium carbonate consumption. Report of 7 patients with parathyroid hormone levels and an estimate of prevalence among patients hospitalized with hypercalcemia. Medicine (Baltimore) 1995;74:89–96. doi: 10.1097/00005792-199503000-00004. [DOI] [PubMed] [Google Scholar]
- 71.Freedman P. Renal colic and persistent hypercalcuria following self-administration of vitamin D. Lancet. 1957;272:668–669. doi: 10.1016/s0140-6736(57)91125-x. [DOI] [PubMed] [Google Scholar]
- 72.Rabin R, Gordon SL, Lymn RW, et al. Effects of spaceflight on the musculoskeletal system: NIH and NASA future directions. FASEB J. 1993;7:396–398. doi: 10.1096/fasebj.7.5.8462780. [DOI] [PubMed] [Google Scholar]
- 73.Money KE. Biological effects of space travel. Can Aeronaut Space J. 1981;27:195–201. [PubMed] [Google Scholar]
- 74.Schneider VS, McDonald J. Skeletal calcium homeostasis and countermeasures to prevent disuse osteoporosis. Calcif Tissue Int. 1984;36(suppl 1):S151–S154. doi: 10.1007/BF02406149. [DOI] [PubMed] [Google Scholar]
- 75.Hwang TI, Hill K, Schneider V, Pak CY. Effect of prolonged bedrest on the propensity for renal stone formation. J Clin Endocrinol Metab. 1988;66:109–112. doi: 10.1210/jcem-66-1-109. [DOI] [PubMed] [Google Scholar]
- 76.Deitrick JE, Whedon GD, Shorr E. Effects of immobilization upon various metabolic and physiologic functions of normal men. Am J Med. 1948;4:12. doi: 10.1016/0002-9343(48)90370-2. [DOI] [PubMed] [Google Scholar]
- 77.Jeantet A, Giachino G, Rossi P, et al. Immobilization: a cause of resorptive hypercalciuria. Contrib Nephrol. 1984;37:31–35. doi: 10.1159/000408545. [DOI] [PubMed] [Google Scholar]
- 78.Zerwekh JE. Nutrition and renal stone disease in space. Nutrition. 2002;18:857–863. doi: 10.1016/s0899-9007(02)00911-5. [DOI] [PubMed] [Google Scholar]