

Abstract
The granulocyte colony-stimulating factor receptor (G-CSFR) is a critical regulator of granulopoiesis, but the mechanisms controlling its surface expression are poorly understood. Recent studies using transfected cell lines have suggested the activated G-CSFR is routed to the lysosome and not the proteasome. Here, we examined the role of the ubiquitin/proteasome system in regulating G-CSFR surface expression in both ts20 cells that have a temperature-sensitive E1 ubiquitin-activating enzyme and in primary human neutrophils. We show that the G-CSFR is constitutively ubiquitinated, which increases following ligand binding. In the absence of a functional E1 enzyme, ligand-induced internalization of the receptor is inhibited. Pre-treatment of ts20 transfectants with either chloroquine or MG132 inhibited ligand-induced G-CSFR degradation, suggesting a role for both lysosomes and proteasomes in regulating G-CSFR surface expression in this cell line. In neutrophils, inhibition of the proteasome but not the lysosome was found to inhibit internalization/degradation of the activated G-CSFR. Collectively, these data demonstrate the requirement for a functional ubiquitin/proteasome system in G-CSFR internalization and degradation. Our results suggest a prominent role for the proteasome in physiologic modulation of the G-CSFR, and provide further evidence for the importance of the ubiquitin/proteasome system in the initiation of negative signaling by cytokine receptors.
Keywords: G-CSFR, ubiquitination, proteasome, internalization, neutrophil
1. Introduction
A remarkable feature of granulopoiesis is the regulated production and release of neutrophils (PMN) to maintain homeostatic levels in the circulation. The granulocyte colony-stimulating factor receptor (G-CSFR) is a critical regulator of this process, and transduces signals for myeloid cell survival, proliferation, and neutrophilic maturation and also for stem cell mobilization [1–8]. The G-CSFR is a transmembrane protein that belongs to the cytokine receptor family. G-CSF binding induces a conformational change in the receptor that activates Jak kinases and downstream signaling cascades that include signal transducer and activators of transcription (Stat) kinases, Ras/mitogen-associated protein (Ras/MAP) kinase, and phosphatidylinositol-3 (PI3) kinase/Akt pathways [9,10]. Simultaneous with intracellular activation, mechanisms that down-regulate the G-CSFR in target cells are rapidly turned on to attenuate receptor signaling and protect cells from over-stimulation. These mechanisms include the recruitment of SHIP and SHP-1 to the G-CSFR complex and the production of SOCS proteins [11–13].
Despite the central role the G-CSFR plays in regulating granulopoiesis, the mechanisms that modulate its surface expression remain poorly understood. For the receptor tyrosine kinase (RTK) family of growth factor receptors that includes the epidermal growth factor receptor (EGFR) and platelet-derived growth factor receptor (PDGFR), ligand binding has been shown to trigger receptor endocytosis, internalization, and subsequent intracellular degradation [14–18]. Tyrosine- or dileucine-containing internalization motifs in the cytoplasmic tails of these receptors have been identified that mediate ligand-induced endocytosis through interactions with adaptor proteins and clathrin. Conjugation of ubiquitin, a 76 amino acid polypeptide, to the cytoplasmic domains of these receptors has also been shown to contribute to receptor endocytosis [19]. Enzymatic attachment of ubiquitin to lysine residues present in the receptors was shown to provide a recognition tag for their delivery to either the lysosome or proteasome. In the acidic environment of the lysosome, bound ligands dissociate from their respective receptors, while in the proteasome, the ubiquitinated receptors are degraded.
For the Type I cytokine receptors, endocytosis and degradation has been best characterized for the growth hormone receptor (GH-R), interleukin-2 receptor (IL-2R), and the erythropoietin receptor (EpoR) [20–25]. These receptors all undergo ligand-induced ubiquitination. Both the GHR and IL-2R are routed to the lysosome for degradation, while the EpoR is degraded by both proteasomes and lysosomes. Endocytosis and lysosomal degradation of the GHR was shown to require an active ubiquitin/proteasome system, while the role of the ubiquitin/proteasome system in internalization and lysosomal routing of the IL-2R remains controversial. Recent studies of the murine G-CSFR indicate it is also ubiquitnated and sorted to the lysosome [26,27].
In the current study, we have examined the role of the ubiquitin/proteasome system in G-CSFR internalization and intracellular routing using transfected cell lines and for the first time primary human neutrophils. We show that the G-CSFR is ubiquitinated via ligand-dependent and independent mechanisms and that disruption of the ubiquitin machinery inhibits endocytosis of the G-CSFR. Pre-treatment of both transfected cells and neutrophils with proteasome inhibitors was also found to inhibit ligand-induced degradation of the activated G-CSFR. These data indicate that ubiquitination of the G-CSFR itself is a key event in the initiation of negative signaling by the G-CSFR, and suggest an important role for the proteasome in modulating G-CSFR surface expression.
2. Materials and methods
2.1 Reagents and cell culture
The ts20 Chinese hamster cell line was derived from the Chinese hamster lung cell line V-79 [28] and was generously provided by Dr. Ger Strous (University Medical Center Utrecht, The Netherlands). This cell line has a temperature-sensitive E1 ubiquitin-activating enzyme. In ts20 cells, the E1 enzyme is required for a functional ubiquitin/proteasome system and is active at the permissive temperature of 30°C but inactive at 42°C. ts20 cells were cultured in Eagle’s minimal essential medium (MEM-α) supplemented with 10% fetal bovine serum (FBS), penicillin/streptomycin, and 4.5 gm/L glucose. Recombinant human G-CSF was a generous gift from Amgen (Thousand Oaks, CA). Media and cell culture reagents were purchased from GIBCO/Invitrogen (Carlsbad, CA).
2.2 Plasmid construction and transfection
The entire open reading frame of the wild-type (WT) G-CSFR was amplified using Advantage-HF2 DNA polymerase (Clonetech, Palo Alto, CA) and cloned into the pcDNA3.1D-TOPO mammalian expression vector (Invitrogen). PCR reactions were performed with an initial 3 min denaturation at 94°C, followed by 25 cycles of denaturation at 94°C for 30 sec, annealing at 63°C for 30 sec, elongation at 68°C for 90 sec, and a final 3 min elongation at 68°C. The forward primer used for amplification contained a Kozak consensus sequence [29] (5′-CCATGGCAAGGCTGGGAAACTGC-3′) and the reverse primer (5′-GAAGCTCCCCAGCGCCTCCATC-3′) lacked a stop codon to allow read through to the V5 and His6 epitope tags contained within the vector sequence. The WT G-CSFR was epitope-tagged with V5 and His6 (pcDNA3.1D/WTv5h6). We have previously described the generation of the Δ716 G-CSFR form [30]. For generation of the Myc-tagged Δ716 G-CSFR construct (pcDNA3.1D/Δ716+myc), the same forward primer but a different reverse primer (5′-TCACAGATCCTCTTCTGAGATGAGTTTTTGTTCTAGGCCACAAGGGCCAC-3′) were used in PCR reactions. For these reactions, the first five cycles were carried out under the identical conditions used for amplification of the WT G-CSFR, but for the last twenty cycles denaturation was done at 94°C for 30 sec, which was then followed by a combined annealing/elongation step at 68°C for 90 sec. PCR reactions with this primer pair amplify the entire open reading frame of the Δ716 G-CSFR fused to a Myc-tag followed by a stop codon.
The tagged G-CSFR constructs were subcloned into the mammalian expression vector pcDNA6 (Invitrogen) from pcDNA3.1D, and positive clones were selected in blasticidin. The fidelity of the entire open reading frame of each expression vector was confirmed by automated DNA sequencing. Transfections of ts20 cells were performed in 60 mm dishes using 1 mg of DNA and Effectene (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Cells (5×105/mL) were either harvested at 24–36 hours after transfection and transiently transfected cells were used in some experiments, or the cells were transferred to media containing G418 (75 mg/mL) to isolate positive clones stably expressing the various receptor forms. Single clones were isolated by limiting dilution and were screened for G-CSFR expression by immunoblot analysis and flow cytometry. For some experiments, ts20 cells were transiently co-transfected with either the V5-tagged WT G-CSFR cDNA or myc-tagged Δ716 G-CSFR cDNA and a hemagluttinin (HA)-tagged ubiquitin cDNA using Effectene. The HA-tagged ubiquitin cDNA was generously provided by Dr. Dirk Bohmann (University of Rochester, Rochester, NY).
2.3 Flow cytometry and cell sorting
Cells were collected in cell dissociation solution (Sigma, St. Louis, MO), incubated on ice for 1 h with 50 μL of a phycoerythrin (PE)-conjugated monoclonal anti-G-CSFR antibody (LMM- 741, BD PharMingen, San Diego, CA) in 0.5 mL staining buffer (phosphate buffered saline (PBS), 2% FBS, and 0.1% sodium azide), washed in staining buffer, resuspended in 3 mL of MEMα, and counted. PE-positive cells were isolated using a BD FACSVantage, and the sorted cells (1–2×106) were collected into polystyrene tubes containing MEMα supplemented with 30% FBS. Unstained and phycoerythrin isotype (Caltag, Invitrogen) stained cells served as controls. The sorted cells were spun down, transferred to MEMα media, and incubated at 30°C with 5% CO2 in a humidified incubator.
2.4 Immunoprecipitations and immunoblot analysis
Cells (2×107/mL) were lysed in ice-cold lysis buffer (1.5% triton X-100, 0.1% SDS, 0.1 mM sodium deoxycholate, 500 mM NaCl, 5 mM EDTA, 10 mM N-ethylmaleimide, 25 mM HEPES, pH 7.8) containing a cocktail of protease inhibitors (Roche, Indianapolis, IN), incubated on ice for 20 min, and centrifuged at 4°C at 10 000 g. The supernatants were collected from each sample and protein concentrations determined using the BCA reagent (Pierce, Rockford, IL). A total of 150 mg of protein from each sample in equivalent final volumes was mixed 1:1 with IP buffer (10% glycerol, 100 mM KCl, 5 mM MgCl2, 50 mM Tris, pH 8), and incubated with monoclonal antibodies (2 mg) recognizing either the V5 or Myc epitope (Invitrogen) and 40 mL of protein G-agarose (Invitrogen) overnight at 4°C. Immunoprecipitates were collected by centrifugation at 2 000 g at 4°C, washed 3x with a 1:1 mix of lysis and IP buffers, and resuspended in LDS sample buffer. The samples were heated to 70°C for 10 min, resolved on 4–12% Bis-Tris acrylamide gels using a MOPS running buffer (Invitrogen), and transferred to nitrocellulose. For analysis of whole cell lysates, 50 μg of protein from each sample was resolved by SDS-PAGE and subjected to immunoblot analysis. Immunoreactive bands were visualized using the enhanced chemiluminescence (ECL) reagent (Amersham, Piscataway, NJ). For analysis of G-CSFR ubiquitination in cells transiently co-expressing HA-tagged ubiquitin and either the WT G-GSFR or the Δ716 G-CSFR, cells were transferred 24 h after co-transfections to serum-free MEMα media containing 20 mM HEPES, penicillin/streptomycin, 4.5 gm/L glucose, and 1% BSA, and incubated with 100 ng/mL G-CSF at 37°C for varying times. Cells were lysed as described above, immunoprecipitated with protein A or G agarose and anti-V5 or anti-Myc monoclonal antibody (Invitrogen) for the WT G-CSFR and Δ716 G-CSFR, respectively, or immunoprecipitated with an anti-HA antibody (Santa Cruz, Santa Cruz, CA). Proteins were transferred to nitrocellulose and immunoblotted with a biotin-conjugated antibody to the G-CSFR (BD PharMingen), antibodies to hemagluttinin, V5 or Myc, or anti-ubiquitin antibodies raised against the FK1 and FK2 clones (Affiniti Research, Exeter, UK). Immune complexes were detected by ECL. For densitometric analysis, ImageJ software made available through the National Institutes of Health was used.
2.5 Biotinylation of surface proteins
Cells (5×105) expressing the WT G-CSFR cultured in 60 mm dishes as described above and grown to 80% confluency were washed three times in PBS (pH 8), and surface proteins labeled with biotin using the membrane impermeable agent sulfo-NHS-LC-Biotin (Pierce). To each dish, 900 μL PBS (pH 8) and 100 μL sulfo-NHS-LC-biotin (5 mg/mL) were added, and the cells incubated on an orbital shaker at room temperature for 30 min. Reactions were terminated by the addition of 500 μL 50 mM Tris/HCl in PBS (pH 8), and the cells washed twice in the same buffer. To examine the effects of lysosome and proteasome inhibitors on G-CSFR internalization and degrdation, cells were pretreated with 25 μM lactacystin (Calbiochem, San Diego, CA) alone, 200μM chloroquine (Sigma) alone, 20 μM MG132 (Sigma) alone, or lactacystin plus chloroquine for 1hr at 37ºC. The cells were then stimulated with G-CSF (100 ng/mL) for varying times at 37ºC, lysed, immunoprecipitated with anti-V5 antibody, and immunoblotted with streptavidin-HRP (Pierce).
2.6 Immunofluorescence Microscopy
Stably transfected ts20 cells (7.5×104) were grown overnight on glass cover-slips at 30°C in a humidified incubator with 5% CO2. The adherent cells were washed with cold PBS, then with cold serum-free MEMα containing 20 mM HEPES, penicillin/streptomycin, 4.5 gm/L glucose, and 1% BSA, and incubated for 1 h at 30°C (permissive temperature) or 42°C (non-permissive temperature) in the absence of G-CSF. Cells were then stimulated with G-CSF (100 ng/mL) for 1 h at 4°C, washed with serum-free media, and incubated at 30°C or 42°C for 0, 30, and 60 min. At the specified times, the cells were washed with PBS, fixed in 3% paraformaldehyde for 1 h, and incubated with PBS containing 0.1% sodium azide and 5% BSA for 30 min at room temperature (RT). The cells were incubated overnight with a biotin-conjugated mouse anti-human G-CSFR (CD114) antibody (BD PharMingen) at 4°C, washed with PBS containing 0.1% sodium azide, and incubated for 1 h with Streptavidin-Cy5 (Caltag) as the secondary antibody. Nuclei were stained with Hoechst stain (Molecular Probes, Invitrogen) for 30 min at RT. Cover slips containing the adherent cells were mounted on glass slides using Pro-Long Antifade mounting media (Molecular Probes) and examined under a Zeiss LSM 510 multiphoton confocal immunofluorescence microscope (Zeiss, Oberkochen, Germany) equipped with a c-Apochromat 63×/1.2 corr objective. Zeiss LSM5 Image software was used for image processing.
2.7 Statistical Analysis
Individual ts20 cells expressing the WT G-CSFR detected by Cy5 fluorescence were analyzed from multiple confocal images (10–15 cells). The fluorescence intensity of an individual cell in a single slice was measured in three different channels through the entire width of the cell. The side-to-side diameter and fluorescence intensity of the outer 0.35 μm of each cell analyzed were quantified using LSM5 software. Data collected from multiple cells for each set of experimental conditions were used to determine the average fluorescence intensity and the standard deviation calculated. The Student’s t test was used to calculate the two-tailed p value for WT G-CSFR expression at 30°C and 42°C.
2.8 Neutrophil isolation and analysis
Neutrophils were isolated from the whole blood of healthy donors using Ficoll-Paque PLUS (Amersham Biosciences, Pittsburgh, PA), 3% Dextran sulfate, and ACK lysis buffer (Sigma), then washed twice in a PBS/0.2% BSA solution. Aliquots of 1×106 cells were resuspended in PBS/BSA. G-CSF (100 ng/mL) was added to each sample and allowed to bind to the cells for 1 h at 4ºC, after which the cells were incubated at 37ºC for varying times. The cells were washed twice with the PBS/BSA containing 0.1% sodium azide, then incubated with phycoerythrin (PE)-conjugated antibody to the G-CSFR (BD PharMingen) for 45 min at 4ºC. The cells were fixed in 1% paraformaldehyde and analyzed using a BD FACS Calibur cytometer.
For inhibitor studies, neutrophils (1×106/sample) were isolated as described above and pre-treated for 1 h at 37ºC with varying concentrations of chloroquine (10–500 μM), lactacystin (2.5–100 μM), or MG132 (5–200 μM). For all subsequent experiments, the concentration used for each inhibitor was: chloroquine at 10 μM, lactacystin at 100, and MG132 at 5 μM. Following pre-treatment with inhibitors, the cells were then stimulated with G-CSF (100 ng/mL), washed, stained, fixed, and analyzed as described above.
3. Results
3.1 Constitutive and ligand-induced ubiquitination of the G-CSFR
To determine whether the G-CSFR undergoes ubiquitination and the potential role of the carboxy-terminal tail of the receptor in this process, we initially expressed the WT G-CSFR and Δ716 G-CSFR truncation mutant in the ts20 cell line. The Δ716 G-CSFR lacks the C-terminal 98 aa. of the G-CSFR, and is the most frequently detected mutant G-CSFR form in patients with severe congenital neutropenia (SCN) transforming to acute myelogenous leukemia (AML). The ts20 cell line which has a temperature-sensitive E1 ubiquitin-activating enzyme readily permits the identification of ubiquitin-dependent processes by mere temperature shifting. Due to a lack of efficient commercially available antibodies to the G-CSFR for performing immunoprecipitations, the WT G-CSFR was epitope-tagged with a V5-tag fused to its carboxy-terminus and the Δ716 G-CSFR was tagged with a C-terminal Myc-tag. The tagged G-CSFR forms were individually co-transfected along with an HA-tagged ubiquitin into ts20 cells and grown at 30°C. The use of HA-tagged ubiquitin permitted us to readily detect ubiquitinated proteins in transfected cells. Whole cell lysates were prepared from cells 24 h after co-transfections, and immunoprecipitated with anti-HA antibody then immunoblotted with a biotin-conjugated anti-G-CSFR antibody. As shown in Figure 1A, both the WT G-CSFR (left panel) and Δ716 G-CSFR (right panel) were expressed as ubiquitinated proteins that appeared as broad bands. As expected, ubiquitinated G-CSFR forms were undetectable in either untransfected cells (UNT) or cells transfected with the WT or truncated G-CSFR forms without HA-tagged ubiquitin. We next examined the effect of ligand binding (G-CSF, 100 ng/mL) on G-CSFR ubiquitination for varying times after ligand exposure. For these experiments, whole cell lysates were immunoprecipitated with antibody to the relevant epitope-tag then immunoblotted with anti-HA antibody. Notably, a low level of ubiquitination of the WT G-CSFR was detected even prior to exposure of the cells to G-CSF. In response to G-CSF, ubiquitination of the WT G-CSFR was observed to rapidly and transiently increase as indicated by the smeared band pattern above the ~ 120 kDa size of the WT G-CSFR. Maximal ubiquitination of the WT G-CSFR was detected at 10 min (Figure 1B, upper left panels). Like the WT G-CSFR, the truncated receptor was also constitutively ubiquitinated at low levels, as shown in the upper right panel of Figure 1B. However, in contrast to the WT G-CSFR, G-CSF stimulation did not induce increased ubiquitination of the Δ716 G-CSFR over basal levels (Figure 1B, upper right panels). Even at 120 min after G-CSF stimulation, we were unable to detect increased ubiquitination of the Δ716 G-CSFR in five independent experiments (data not shown). To confirm that our inability to detect G-CSF-induced ubiquitination of the truncated G-CSFR was not due to differences in receptor protein loading, the same blots were stripped and reblotted with an antibody to the G-CSFR (Figure 1B, lower panels). Densitometric analysis confirmed increased ubiquitination of the WT but not the Δ716 G-CSFR in response to ligand binding (Figure 1C). Both the WT and Δ716 G-CSFR forms appeared as doublets (Figure 1B, lower panels), with the top bands likely representing glycosylated forms of the receptors. When the same blots were stripped and reblotted with antibody to native ubiquitin, expression of HA-tagged ubiquitin appeared to only modestly increase the overall intensity of ubiquitinated protein bands (data not shown). There was no evidence that the observed ubiquitination of the G-CSFR was due to ubiquitin overexpression.
Fig. 1.

Constitutive and ligand-induced ubiquitination of the WT G-CSFR. (A) Whole cell lysates prepared from untransfected ts20 cells (UNT), ts20 cells transfected with V5-tagged WT G-CSFR (left panel) or Myc-tagged Δ716 G-CSFR (right panel), or co-transfected with either tagged receptor form and HA-tagged ubiquitin (HA) were immunoprecipitated with anti-HA antibody, then blotted with antibody to the G-CSFR. (B) ts20 cells co-expressing V5-tagged WT G-CSFR and HA-ubiquitin (left panels) or Myc-tagged Δ716 G-CSFR and HA-ubiquitin (right panels) were stimulated with G-CSF (100 ng/mL) for the indicated times, lysed, then immunoprecipitated with anti-V5 (left) or anti-Myc (right) antibody and blotted with anti-HA antibody (upper panels) or anti-G-CSFR antibody (lower panels). Untransfected cells (UNT) and cells transfected with HA-ubiquitin only (HA) are shown as controls. (C) To control for receptor protein loading, histograms of the density ratio between bands in the upper and lower panels (B) were generated using ImageJ software.
3.2 Ligand binding induces mono- and polyubiquitination of the G-CSFR
The observation that different ubiquitin chain lengths appear to target proteins to different intracellular compartments prompted us to next examine whether the WT G-CSFR undergoes mono- or polyubiquitination after ligand-induced activation. For these experiments, we used the FK1 and FK2 (P4D1) commercially available antibodies that have been used extensively to analyze ubiquitination of a variety of growth factor receptors, including the EGFR and PDGFR [31]. The FK1 antibody recognizes polyubiquitinated chains in a mixture of ubiquitin monomers and polymers but not single ubiquitin moieties. The FK2 monoclonal antibody recognizes mono-ubiquitinated proteins as efficiently as polyubiquitinated proteins. Whole cell lysates prepared from cells expressing the V5-tagged WT-G-CSFR were stimulated with G-CSF (100 ng/mL) for varying times, immunoprecipitated with anti-V5 antibody, then immunoblotted with either the FK1 or FK2 antibody. As shown in Figure 2A, a faint smeary band that increased in intensity transiently after 5 min of ligand stimulation was detected with the FK1 antibody. When the same blot was stripped and reblotted with the FK2 antibody, more prominent smeary bands that increased in intensity with G-CSF stimulation were detected above the ~ 120 kDa size of the WT G-CSFR (Figure 2B). To control for receptor protein loading, the same blot was reblotted with anti-V5 antibody (Figure 2C). The increased level of ubiquitination observed after ligand binding using the FK2 antibody was not due to differences in receptor protein loading as indicated by densitometric analysis. These results demonstrate that the WT G-CSFR undergoes mono- and polyubiquitination in response to ligand binding.
Fig. 2.

Ligand binding induces mono-/polyubiquitination of the G-CSFR. ts20 cells expressing V5-tagged WT G-CSFR were incubated at 37°C with G-CSF (100 ng/mL) for the indicated times, lysed, then immunoprecipitated with anti-V5 antibody and blotted with the indicated antibodies. The FK1 antibody (A) recognizes polyubiquitinated proteins whereas the FK2 antibody (B) recognizes both poly- and mono-ubiquitinated proteins. (C) The blot was stripped and re-blotted with anti-V5 antibody. Histograms are shown to the right of each blot depicting the density ratio between bands of FK1 and FK2 to anti-V5, respectively, as a control for receptor protein loading.
3.3 Ligand-induced internalization of the G-CSFR is ubiquitin-dependent
To determine the role of ligand-induced ubiquitination on internalization of the G-CSFR, we examined the effect of inhibition of the E1 enzyme on the cellular localization of the G-CSFR in stably transfected ts20 cells. For these experiments, confocal immunofluorescence microscopy was utilized and methods similar to those previously described for the growth hormone receptor (GHR) and the thyrotropin-releasing hormone receptor (TRHR) [17,32]. As shown in Figure 3 (upper panels, A-C), at the permissive temperature of 30°C, the WT G-CSFR was rapidly internalized after ligand-binding as indicated by the vesicular fluorescent intracellular distribution pattern that was apparent within 30 min. Notably, when the temperature was shifted to 42°C to inactivate the E1 enzyme, the WT G-CSFR remained predominantly membrane-bound (upper panel, D, arrows). These results demonstrate that ligand-induced internalization of the G-CSFR is ubiquitin-dependent. In contrast to the WT G-CSFR, in response to ligand binding the truncated Δ716 G-CSFR remained membrane-bound at the permissive temperature (Figure 3, lower panels, arrows). There were no appreciable differences in the distribution pattern of the truncated receptor when the cells were exposed to ligand and incubated at either the permissive or non-permissive temperature (data not shown).
Fig. 3.
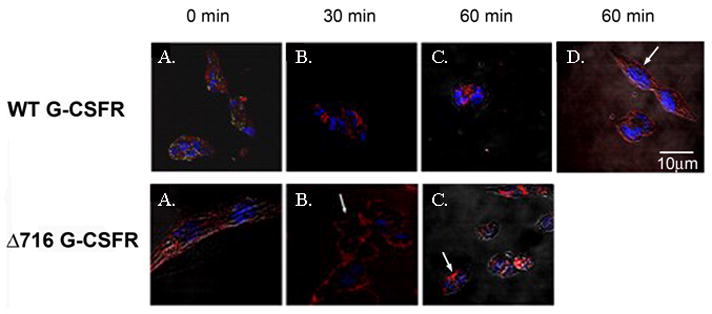
Internalization of the G-CSFR requires an intact ubiquitin-conjugation system. ts20 cells expressing the WT (upper panels) or Δ716 (lower panels) G-CSFR were stimulated with G-CSF (100 ng/mL) for 1h at 4oC then incubated at 30°C for 0 (A), 30 min (B), and 60 min (C). Cells were fixed, sequentially incubated with biotinylated antibody to the G-CSFR, streptavidin-Cy5, and Hoechst nuclear stain, then examined by confocal microscopy. (A-C) Cellular distribution of WT and Δ716 G-CSFR (red) and nuclei (blue). (D) WT G-CSFR-expressing cells incubated at 42°C (non-permissive temperature) for 60 min. Arrows indicate predominant membrane localization of the WT G-CSFR when ubiquitination is inhibited at 42– and of the Δ716 G-CSFR at the permissive temperature.
To quantify differences in cell surface expression that were statistically significant at the permissive versus non-premissive temperatures, confocal immunofluorescence images were analyzed using LSM5 Image software. For these analyses, G-CSFR surface expression was quantified using the average fluorescence intensity from at least 10 individual cells for each set of experimental conditions. Data from a representative experiment are shown in Figure 4. In five independent experiments, a statistically significant decrease in receptor surface expression following ligand binding was detected only in cells expressing the WT G-CSFR and only at the permissive temperature of 30°C (p<0.0001). No statistically significant changes in surface expression of the WT G-CSFR were detected when ubiquitination was inhibited by shifting the cells to 42°C (p = 0.13) even at 60 min following ligand binding (Figure 4B). Additionally, no statistical difference in surface expression of the truncated receptor was detected when the cells were stimulated with G-CSF and incubated at the permissive (p=0.66) temperature.
Fig. 4.

Inhibition of the E1 ubiquitin-activating enzyme blocks G-CSFR internalization. G-CSFR surface expression was quantified from multiple confocal images using LSM5 software to measure the Cy5 fluorescence intensity over the outer 0.35 mM of individual ts20 cells transfected with the WT or Δ716 G-CSFR. The cells were stimulated with G-CSF (100 ng/mL) for 1h at 4°C, then incubated at 30°C or shifted to 42°C to inactivate the E1 enzyme. Cells were fixed and sequentially incubated with biotin-conjugated mouse anti-human G-CSFR antibody (CD114, BD PharMingen) and Streptavidin-Cy5 (Caltag) to detect the G-CSFR. (A) Yellow arrow (left panel) depicts the region used to measure fluorescence intensity from a representative image of a cell expressing the Δ716 G-CSFR incubated for 30 min at 30°C. Histogram (right panel) shows the fluorescence intensity of Cy5 (red) and DAPI (blue) staining in the region corresponding to the yellow arrow. (B) Bar graphs show the mean Cy5 fluorescence intensity corresponding to G-CSFR surface expression on cells stimulated with G-CSF and incubated for the indicated times at either 30°C or 42°C.
3.4 Both lysosomes and proteasomes degrade the activated G-CSFR in ts20 cells
We next examined the effect of inhibition of the lysosome and/or proteasome on G-CSFR surface expression in ts20 cells expressing the WT G-CSFR. For these experiments, we used the lysosomal inhibitor chloroquine (200 μM), the proteasomal inhibitor lactacystin (25 μM), and MG132 (20 μM), which has been shown to have inhibitory effects on both the lysosome and proteasome [33]. Cells were biotinylated, left untreated or pre-treated with chloroquine alone, lactacystin alone, both chloroquine and lactacystin, or MG132 alone, then stimulated with or without G-CSF (100 ng/mL) at 37°C for different times, and lysed. Whole cell lysates were immunoprecipitated with anti-V5 antibody and analyzed by Western blotting using HRP-labeled streptavadin. As shown in the blots in Figure 5A and the densitometric analyses in Figure 5B, decreased surface expression of the WT G-CSFR was readily apparent within 1 h after the addition of ligand, and by 2 h there was a more than 50% reduction in receptor surface expression. In the absence of ligand, surface expression of the WT G-CSFR remained relatively unchanged at 2 h (Figure 5A, last lane) and also at 4 h (data not shown). Notably, in the presence of chloroquine, membrane expression of the WT G-CSFR remained largely unchanged even at 2 h after the addition of ligand. In contrast, lactacystin alone failed to inhibit ligand-induced internalization and degradation of the receptor. When cells were pre-treated with both chloroquine and lactacystin, or with MG132 alone to inhibit both proteasomes and lysosomes [33], internalization/degradation of the G-CSFR was inhibited. These results demonstrate that in transfected ts20 cells, the WT G-CSFR undergoes ligand-induced internalization and is processed intracellularly via both lysosome- and proteasome-mediated pathways.
Fig. 5.
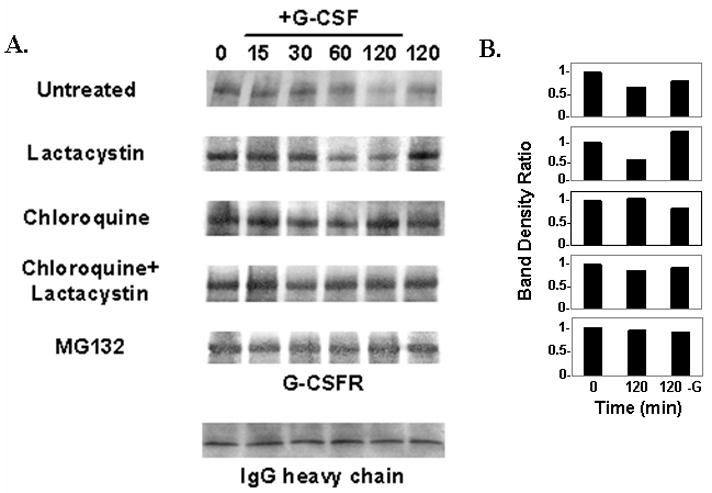
Effect of lysosomal and proteasomal inhibitors on ligand-induced degradation of the G-CSFR. Surface proteins from ts20 cells expressing the WT G-CSFR were labeled by biotinylation. After biotinylation, cells were left untreated, or pre-treated with the following inhibitors: lactacystin (25μM) alone, chloroquine (200μM) alone, MG132 (20μM) alone, or lactacystin plus chloroquine. Cells were then stimulated with or without G-CSF (100ng/ml) at 37ºC for the indicated times, and lysed. (A) Whole cell lysates were immunoprecipitated with anti-V5 antibody and immunoblotted with HRP-labeled streptavidin to detect the biotinylated G-CSFR. Blot with IgG heavy chain (bottom panel) is included to show equal protein loading. (B) Densitometric analysis of bands in (A) for times 0 and 120 min after G-CSF stimulation and at 120 min in the absence of G-CSF.
3.5 Ligand-induced internalization and trafficking of the G-CSFR in neutrophils involves the proteasome
To determine the physiologic relevance of our observations in ts20 cells, we next examined internalization and trafficking of the G-CSFR in neutrophils isolated from healthy donors. We initially examined the effect of G-CSF binding on surface expression of the G-CSFR. Neutrophils were isolated as described, stimulated with G-CSF (100 ng/mL) for varying times at 37ºC, stained with PE-conjugated antibody to the G-CSFR, fixed, then analyzed by flow cytometry. As shown in Figure 6 (upper left histogram), G-CSF receptors internalize rapidly in neutrophils in response to ligand binding as indicated by the shift to the left. The reduction observed in G-CSFR expression at 60 min was similar to the reduction in G-CSFR surface expression observed at 15 min, suggesting that internalization of the G-CSFR in response to ligand binding is complete in neutrophils by 15 min.
Fig. 6.

Effect of lysosome and proteasome inhibitors on G-CSFR internalization in human neutrophils. Human neutrophils were isolated from whole blood using Ficoll-Paque PLUS, 3% Dextran sulfate, and ACK lysis buffer. After isolation, neutrophils were left untreated, or pre-treated with the following inhibitors for 1 h at 37ºC: lactacystin (100 μM) alone, chloroquine (10 μM) alone, or MG132 (5 μM) alone. Neutrophils were then stimulated with or without G-CSF (100 ng/ml) for 1 h at 4ºC, then transferred to 37ºC for the indicated time points. Cells were then stained using PE-conjugated anti-G-CSFR antibody for 45 min at 4ºC, fixed in 1% paraformaldehyde, and analyzed. Unstained controls are indicated in the solid grey shading. A shift to the left is indicative of decreased G-CSFR expression and consistent with G-CSFR internalization/degradation.
We next investigated the effects of lysosome and proteasome inhibitors on G-CSFR internalization in neutrophils. Neutrophils were pre-treated with varying concentrations of chloroquine (10–500 mM), lactacystin (2.5–100 mM), or MG132 (5–200 mM), and G-CSFR surface expression analyzed by flow cytometry. For the histograms shown in Figure 6, a shift to the left would indicate decreased G-CSFR surface expression or receptor internalization, whereas inhibition of receptor internalization/degradation should yield a profile that is shifted to the right of t=60 (with no inhibitors) and approaches t=0. Only lactacystin and MG132 were found to inhibit G-CSFR internalization/degradation, which was dose-dependent. Chloroquine failed to inhibit G-CSFR internalization at all concentrations examined. A representative experiment is shown in Figure 6 using optimal concentrations of each inhibitor. These results were reproducible in two independent experiments, and demonstrate that the activated G-CSFR in human neutrophils is internalized and processed via proteasome-dependent processes.
4. Discussion
Activation of growth factor receptors by their cognate ligands is rapidly followed by desensitization processes that control the amplitude and duration of intracellular signaling. Deregulation of these processes has been shown to be associated with severe human pathologies. Evidence for the importance of downregulation of G-CSFR surface expression comes from studies of patients with SCN transforming to AML in whom acquired point mutations in the G-CSFR gene producing truncated G-CSFR forms have been identified [9,34,35]. Our laboratory and others have previously shown that mutant G-CSFR forms from these patients lack a critical domain in their cytoplasmic tails that mediates receptor internalization and degradation. In response to ligand-binding, surface expression of the mutant G-CSFR forms was found to be increased, which correlated with sustained intracellular activation and enhanced growth and survival responses to G-CSF [10,29,30,36]. Increased expression of the G-CSFR either at the mRNA or protein level has also been reported in patients with AML without antecedent SCN, and in patients with CML and acute and chronic lymphoid leukemias [37–43].
Ligand-induced ubiquitination has been shown to be a crucial initial event in the desensitization of many receptors [44,45]. Ubiquitin-conjugation is an evolutionarily conserved mechanism for the post-translational modification of proteins whereby a ubiquitin polypeptide covalently attaches via its C-terminal glycine as a single moiety (mono-), multiple single moieties (multi-), or a chain of four or more units (poly-) to lysine residues in a target protein [19,46]. Sequential ATP-dependent activation of ubiquitin by an E1 activating enzyme initiates this process followed by transfer to a carrier protein (E2), then linkage of activated ubiquitin to the target protein either directly or in cooperation with an ubiquitin ligase (E3). For most species, only one E1 but multiple E2 and E3 enzymes have been described. For short-lived cytosolic and nuclear proteins as well as misfolded intralumenal and transmembrane proteins, ubiquitination generally serves as a recognition tag for destruction by the proteasome. Attachment of multiple ubiquitin molecules generally results in the delivery of target proteins to the proteasome for degradation, while attachment of a single ubiquitin molecule to a single lysine residue or to multiple different lysines in a target protein typically leads to delivery to the lysosome.
Several members of the Type I cytokine receptor family have been shown to undergo ligand-induced ubiquitination and internalization [17,20–25]. Perhaps the best characterized of these is the GHR. Using the ts20 cell line with a temperature-sensitive E1 ubiquitin-activating enzyme, Strous et al. showed that the GHR is ubiquitinated in response to ligand binding and demonstrated a requirement for the ubiquitin cellular machinery in internalization of the GHR.
In the current study, we have also utilized the ts20 cell line to examine the role of ubiquitination in regulation of membrane expression of the G-CSFR. Like the GHR, constitutive ubiquitination of the G-CSFR was observed. In response to ligand binding, an increase in ubiquitination of the WT G-CSFR was also observed. Increased ubiquitination was detected as early as 10 min after ligand binding, a time frame in which we have previously shown that the G-CSFR is internalized using 125I-labeled G-CSF [30]. Thus, ligand-induced ubiquitination of the G-CSFR appears to be an early event that correlates with ligand-induced internalization. At the permissive temperature of 30°C, the transfected WT G-CSFR was found to be rapidly internalized. A vesicular intracellular distribution pattern was readily apparent within 30 min after ligand binding in cells expressing the full-length G-CSFR. In contrast, when the temperature was shifted to 42°C to inactivate the E1 enzyme, the WT G-CSFR remained predominantly at the cell surface. These results indicate that an intact ubiquitin-conjugation system is required for ligand-induced internalization of the G-CSFR.
We next investigated whether the aberrant internalization of the Δ716 G-CSFR truncation mutant that we previously described [30] was due to defective ubiquitination. Like the WT G-CSFR, the Δ716 G-CSFR was also found to be constitutively ubiquitinated. In contrast to the WT G-CSFR, however, there was no evidence for ligand-induced ubiquitination of the Δ716 G-CSFR in five independent experiments.
Confocal microscopy of ts20 transfectants confirmed that the WT G-CSFR in response to ligand binding remained predominantly at the cell surface at the non-permissive temperature of 42°C. In addition, the Δ716 G-CSFR was also found predominantly at the cell surface following ligand binding at the non-permissive temperature but also at the permissive temperature. These results suggest that defective ligand-induced ubiquitination of the Δ716 G-CSFR underlies its aberrant internalization.
The ubiquitin-conjugation system could modulate G-CSFR endocytosis by either of two general mechanisms: directly, by ubiquitinating the G-CSFR itself; or indirectly, by ubiquitinating an accessory protein required for G-CSFR internalization. Although the cytoplasmic domain of the G-CSFR lacks a classical ubiquitin-dependent endocytosis motif, it has five lysine residues at positions 632, 672, 680, 681, and 762 that could serve as potential attachment sites for ubiquitin. Irandoust et al. previously reported that mutation of all five G-CSFR cytoplasmic lysines to arginine had no effect on ligand-induced internaliaztion, arguing against a direct mechanism [26]. This observation along with our own findings here suggest the G-CSFR like the GHR [27] undergoes ubiquitination dependent ligand-induced internalization through an indirect mechanism. Such a mechanism is consistent with the involvement of an accessory protein(s) that interacts with the G-CSFR at a site distal to residue 716.
The low level of constitutive ubiquitination observed with both the WT and truncated receptor forms may serve to regulate steady-state G-CSFR levels by promoting constitutive receptor endocytosis, and would necessarily involve one or more of the four proximal cytoplasmic lysines that are retained in the Δ716 mutant as suggested by Irandoust [26]. Notably, using transfected 32Dcl10 cells, Aarts et al. [48] reported that the WD40 and SOCS box protein-2 (Wsb-2) binds to the C-terminal 10–20 residues of the G-CSFR and that fusion of Wsb-2 to a G-CSFR mutant terminating at residue 769 reduced its increased expression to the level of the WT G-CSFR. The WT G-CSFR was ubiquitinated but not the truncated Δ769 G-CSFR fused to Wsb-2, leading the authors to also suggest a role for ubiquitination in regulation of the steady-state distribution of the G-CSFR.
In this paper, we also show that the WT receptor but not the Δ716 truncation mutant undergoes ligand-induced mono-/polyubiquitination, as indicated by reactivity of the ubiquitinated receptor species with the FK2 antibody. Our results differ from a recent study by Erkeland et al. [27], who reported a lack of reactivity of the ubiquitinated murine G-CSFR with the FK2 antibody. The reasons for the discrepancy are not readily apparent but could relate to the different cell lines used by these investigators or inherent differences in ligand-induced modifications of murine versus human G-CSFR.
Our current observation that the Δ716 mutant is not properly ubiquitinated would explain our earlier observation that the Δ716 G-CSFR undergoes aberrant ligand-induced degradation [30], and suggests the activated truncated receptor fails to acquire the necessary ubiquitin recognition tag for proper intracellular routing. Our finding that chloroquine inhibits ligand-induced degradation of the WR G-CSFR supports a role for ubiquitination in routing the G-CSFR in transfected ts20 cells through the endocytic pathway for delivery to lysosomes, and is consistent with the report by Erkeland et al. [27] However, our observations with MG132 and the FK2 antibody also support a role for proteasomes in addition to lysosomes in degradation of the activated G-CSFR in ts20 cells. Thus, the region that is deleted in the Δ716 mutant appears to mediate both ubiquitination-dependent internalization of the activated G-CSFR and intracellular sorting of the ubiquitinated G-CSFR species. The juxtamembrane lysine at position 632 was shown by Irandoust et al. [26] to contribute to lysosomal routing of the G-CSFR in transfected cells that was dependant on SOCS3, which binds to tyrosine 729 that is deleted in the Δ716 G-CSFR.
Since model systems that employ overexpression of transfected receptors are prone to artifacts due to the presence of large intracellular pools of the transfected receptors and limiting quantities of endogenously expressed proteins required for receptor trafficking, they may not appropriately reflect true in vivo events. This is a concern with with previously published trafficking studies of the G-CSFR which have all utilized transfected cell lines and with. To address the physiologic relevance of our observations, we examined G-CSFR trafficking in neutrophils obtained from healthy human donors. Our studies are the first to date to utilize primary human cells expressing endogenous G-CSFR. We show the G-CSFR is rapidly internalizalized within 15 min in response to ligand binding in neutrophils. Pre-treatment of neutrophils with the proteasome inhibitors lactacystin and MG132 significantly inhibited downregulation of the G-CSFR in response to ligand binding. In contast, pre-treatment of neutrophils with the lysosome inhibitor, chloroquine, failed to inhibit G-CSFR internalization and degradation following G-CSF binding.
Our findings extend earlier reports regarding G-CSFR internalization and sorting. We show that an intact ubiquitin-conjugation system is required for both internalization and degradation of the G-CSFR. We also show for the first time using primary human cells that the proteasome plays an important role in physiologic regulation of the activated G-CSFR. Our findings support a mechanism whereby ligand binding leads to increased ubiquitination and internalization of the G-CSFR and subsequent routing of the endocytosed receptor to the ptroteasome where it is degraded. Collectively, the data best fit a model in which the C-terminal region of the G-CSFR that is deleted in the Δ716 mutant recruits an associating protein with ubiquitin ligase activity in response to ligand-binding that mediates the ubiquitination and subsequent internalization and sorting of the G-CSFR. Our findings underscore the importance of the use of physiologically relevant model systems for receptor trafficking studies, and add the G-CSFR to the growing list of growth factor receptors whose expression is modulated by the ubiquitin/proteasome system.
Acknowledgments
This work was supported by grants from the National Institute of Diabetes and Digestive and Kidney Diseases (DK68639), and the National Cancer Institute (CA75226, CA82859, and CA16058).
References
- 1.Demetri GD, Griffin JD. Granulocyte colony-stimulating factor and its receptor. Blood. 1991;78:2791–2808. [PubMed] [Google Scholar]
- 2.Williams GJ, Smith CA, Spooncer E, Dexter TM, Taylor DR. Haematopoietic colony stimulating factors promote cell survival by supporting apoptosis. Nature. 1990;343:76–79. doi: 10.1038/343076a0. [DOI] [PubMed] [Google Scholar]
- 3.Begley CG, Metcalf D, Nicola NA. Binding characteristics and proliferative action of purified granulocyte-colony stimulating factor (G-CSF) on normal and leukemic human promyelocytes. Exp Hematol. 1998;16:71–79. [PubMed] [Google Scholar]
- 4.Avalos BR, Gasson JC, Hedvat C, et al. Human granulocyte colony-stimulating factor: Biologic activities and receptor characterization on hematopoietic cells and small lung cancer cell lines. Blood. 1990;75:851–857. [PubMed] [Google Scholar]
- 5.Hammond WP, Chatta GS, Andrews RG, Dale DC. Abnormal responsiveness of granulocyte committed progenitor cells in cyclic neutropenia. Blood. 1992;79:2536–2539. [PubMed] [Google Scholar]
- 6.Lieschke GJ, Grail D, Hodgson G, et al. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency and impaired neutrophil mobilization. Blood. 1994;84:1737–1746. [PubMed] [Google Scholar]
- 7.Schmitz S, Franke H, Loeffler M, Wichmann HE, Diehl V. Reduced variance of bone-marrow transit time of granulopoiesis – a possible pathomechanism of human cyclic neutropenia. Cell Prolif. 1994;27:655–667. [Google Scholar]
- 8.Schmitz S, Franke H, Brusis J, Wichmann HE. Quantification of the cell kinetic effects of G-CSF using a model of human granulopoiesis. Exp Hematol. 1993;21:755–760. [PubMed] [Google Scholar]
- 9.Avalos BR. Molecular analysis of the granulocyte colony-stimulating factor receptor. Blood. 1996;88:761–777. [PubMed] [Google Scholar]
- 10.Hunter MG, Avalos BR. Granulocyte colony-stimulating factor receptor mutations in severe congenital neutropenia transforming to acute myelogenous leukemia confer resistance to apoptosis and enhance cell survival. Blood. 2000;95:2132–2137. [PubMed] [Google Scholar]
- 11.Hunter MG, Jacob A, O’donnell LC, Agler A, Druhan LJ, Coggeshall KM, Avalos BR. Loss of SHIP and CIS recruitment to the granulocyte colony-stimulating factor receptor contribute to hyperproliferative responses in severe congenital neutropenia/acute myelogenous leukemia. J Immunol. 2004;173:5036–5045. doi: 10.4049/jimmunol.173.8.5036. [DOI] [PubMed] [Google Scholar]
- 12.Dong F, Qiu Y, Yi T, Touw IP, Larner AC. The carboxyl terminus of the granulocyte colony-stimulating factor receptor, truncated in patients with severe congenital neutropenia/acute myeloid leukemia, is required for SH2-containing phosphatase-1 suppression of Stat activation. J Immunol. 2001;167:6447–6452. doi: 10.4049/jimmunol.167.11.6447. [DOI] [PubMed] [Google Scholar]
- 13.Hermans MH, van de Geijn GJ, Antonissen C, Gits J, van Leeuwen D, Ward AC, Touw IP. Signaling mechanisms coupled to tyrosines in the granulocyte colony-stimulating factor receptor orchestrate G-CSF-induced expansion of myeloid progenitor cells. Blood. 2003;101:2584–2590. doi: 10.1182/blood-2002-07-2062. [DOI] [PubMed] [Google Scholar]
- 14.Heldin CH. Dimerization of cell surface receptors in signal transduction. Cell. 1995;80:213–223. doi: 10.1016/0092-8674(95)90404-2. [DOI] [PubMed] [Google Scholar]
- 15.Bagley CJ, Woodcock JM, Stomski FC, Lopez AF. The structural and functional basis of cytokine receptor activation: Lessons from the common b subunit of the granulocyte-macrophage colony-stimulating factor, interleukin-3 (IL-3), and IL-5 receptors. Blood. 1997 Mar 1;89(5):1471–1482. [PubMed] [Google Scholar]
- 16.Vieira AV, Lamaze C, Schmid SL. Control of EGF receptor signaling by clathrin-mediated endocytosis. Science. 1996;274:2086–2089. doi: 10.1126/science.274.5295.2086. [DOI] [PubMed] [Google Scholar]
- 17.Strous GJ, van Kerkhof P, Govers R, Ciechanover A, Schwartz AL. The ubiquitin conjugation system is required for ligand-induced endocytosis and degradation of the growth hormone receptor. EMBO J. 1996;15:3806–3812. [PMC free article] [PubMed] [Google Scholar]
- 18.Sorkin A, Eriksson A, Heldin C-H, Westmark B, Claesson-Welsh L. Pool of ligand-bound platelet-derived growth factor b-receptors remain activated and tyrosine phosphorylated after internalization. J Cell Physiol. 1993;156:373–382. doi: 10.1002/jcp.1041560221. [DOI] [PubMed] [Google Scholar]
- 19.Dikic I, Giordano S. Negative receptor signaling. Curr Opin Cell Biol. 2003 Apr;15(2):128–35. doi: 10.1016/s0955-0674(03)00004-8. [DOI] [PubMed] [Google Scholar]
- 20.Lamaze C, Dujeancourt A, Baba T, Lo CG, Benmerah A, Dautry-Varsat A. Interleukin 2 receptors and detergent-resistant membrane domains define a clathrin-independent endocytic pathway. Mol Cell. 2001;7:661–671. doi: 10.1016/s1097-2765(01)00212-x. [DOI] [PubMed] [Google Scholar]
- 21.Hemar A, Subtil A, Lieb M, Morelon E, Hellio R, Dautry-Varsat A. Endocytosis of interleukin 2 receptors in human T lymphocytes: distinct intracellular localization and fate of the receptor alpha, beta, and gamma chains. J Cell Biol. 1995;129:55–64. doi: 10.1083/jcb.129.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rocca A, Lamaze C. Subtil, Dautry-Varsat A. Involvement of the ubiquitin/proteasome system in sorting of the interleukin 2 receptor beta chain to late endocytic compartments. Mol Biol Cell. 2001;12:1293–1301. doi: 10.1091/mbc.12.5.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yen CH, Yang YC, Ruscetti SK, Kirken RA, Dai RM, Li CC. Involvement of the ubiquitin-proteasome pathway in the degradation of nontyrosine kinase-type cytokine receptors of IL-9, IL-2, and erythropoietin. J Immunol. 2000;165:6372–6380. doi: 10.4049/jimmunol.165.11.6372. [DOI] [PubMed] [Google Scholar]
- 24.Yu A, Malek TR. The proteasome regulates receptor-mediated endocytosis of interleukin-2. J Biol Chem. 2001;276:381–385. doi: 10.1074/jbc.M007991200. [DOI] [PubMed] [Google Scholar]
- 25.Walrafen P, Verdier F, Kadri Z, Chretien S, Lacombe C, Mayeux P. Both proteasomes and lysosomes degrade the activated erythropoietin receptor. Blood. 2005;105:600–608. doi: 10.1182/blood-2004-03-1216. [DOI] [PubMed] [Google Scholar]
- 26.Irandoust MI, Aarts LH, Roovers O, Gits J, Erkeland SJ, Touw IP. Suppressor of cytokine signaling 3 controls lysosomal routing of G-CSF receptor. EMBO J. 2007 Apr 4;26(7):1782–1793. doi: 10.1038/sj.emboj.7601640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Erkeland SJ, Aarts LH, Irandoust M. Novel role of WD40 and SOCS box protein-2 in steady-state distribution of granulocyte colony-stimulating factor receptor and G-CSF-controlled proliferation and differentiation signaling. Oncogene. 2007 Mar 29;26(14):1985–1894. doi: 10.1038/sj.onc.1210004. [DOI] [PubMed] [Google Scholar]
- 28.Kulka RG, Raboy B, Schuster R, et al. A Chinese hamster cell cycle mutant arrested at G2 phase has a temperature-sensitive ubiquitin-activating enzyme, E1. J Biol Chem. 1988 Oct 25;263(30):15725–15731. [PubMed] [Google Scholar]
- 29.Ward AC, van Aesch YM, Schelen AM, Touw IP. Defective internalization and sustained activation of truncated granulocyte colony-stimulated factor receptor found in severe congenital neutropenia/acute myeloid leukemia. Blood. 1999;93:447–458. [PubMed] [Google Scholar]
- 30.Hunter MG, Avalos BR. Deletion of a critical internalization domain in the G-CSFR in acute myelogenous leukemia preceded by severe congenital neutropenia. Blood. 1999 Jan 15;93(2):440–446. [PubMed] [Google Scholar]
- 31.Haglund K, Sigismund S, Polo S, Szymkiewicz I, Di Fiore PP, Dikic I. Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat Cell Biol. 2003 May;5(5):461–466. doi: 10.1038/ncb983. [DOI] [PubMed] [Google Scholar]
- 32.Cook LB, Zhu CC, Hinkle PM. Thyrotropin-releasing hormone receptor processing: role of ubiquitination and proteasomal degradation. Mol Endocrinol. 2003 Sep;17(9):1777–1791. doi: 10.1210/me.2003-0073. [DOI] [PubMed] [Google Scholar]
- 33.Longva KE, Blystad FD, Stang E, Larsen AM, Johannessen LE, Madshus IH. Ubiquitination and proteasomal activity is required for transport of the EGF receptor to inner membranes of multivesicular bodies. J Cell Biol. 2002 Mar 4;156(5):843–854. doi: 10.1083/jcb.200106056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dong F, Hoefsloot LH, Schelen AM, et al. Identification of a nonsense mutation in the granulocyte colony-stimulating factor in severe congenital neutropenia. Proc Natl Acad Sci USA. 1994;91:4480–4484. doi: 10.1073/pnas.91.10.4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dong F, Brynes RK, Tidow N, Welte K, Löwenberg B, Touw IP. Mutations in the gene for the granulocyte colony-stimulating-factor receptor in patients with acute myeloid leukemia preceded by severe congenital neutropenia. New Engl J Med. 1995;333:487–493. doi: 10.1056/NEJM199508243330804. [DOI] [PubMed] [Google Scholar]
- 36.Hermans MHA, Antonissen C, Ward AC, Mayhen AEM, Ploemacher RE, Touw IP. Sustained receptor activation and hyperproliferation in response to granulocyte colony-stimulating factor (G-CSF) in mice with a severe congenital neutropenia/acute myeloid leukemia-derived mutation in the G-CSF receptor gene. J Exp Med. 1999 Feb 15;189(4):683–692. doi: 10.1084/jem.189.4.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shimizu K, Kitabayashi I, Kamada N, et al. AML1-MTG8 leukemic protein induces the expression of granulocyte colony-stimulating factor (G-CSF) receptor through the up-regulation of CCAAT/enhancer binding protein epsilon. Blood. 2000 Jul 1;96(1):288–296. [PubMed] [Google Scholar]
- 38.Da Silva N, Meyer-Monard S, Menot M-L, et al. Functional G-CSF pathways in t(8;21) leukemic cells allow for differentiation induction and degradation of AML1-ETO. Hematol J. 2000;1(5):316–328. doi: 10.1038/sj.thj.6200047. [DOI] [PubMed] [Google Scholar]
- 39.Elsasser A, Franzen M, Kohlmann A, et al. The fusion protein AML1-ETO in acute myeloid leukemia with translocation t(8;21) induces c-jun protein expression via the proximal AP-1 site of the c-jun promoter in an indirect, JNK-dependent manner. Oncogene. 2003 Aug 28;22(36):5646–5657. doi: 10.1038/sj.onc.1206673. [DOI] [PubMed] [Google Scholar]
- 40.Inukai T, Sugita K, Iijima K, et al. Leukemic cells with 11q23 translocations express granulocyte colony-stimulating factor (G-CSF) receptor and their proliferation is stimulated with G-CSF. Leukemia. 1998 Mar;12(3):382–389. doi: 10.1038/sj.leu.2400951. [DOI] [PubMed] [Google Scholar]
- 41.Matsushita K, Arima N. Involvement of granulocyte colony-stimulating factor in proliferation of adult T-cell leukemia cells. Leuk Lymphoma. 1998 Oct;31(3–4):295–304. doi: 10.3109/10428199809059222. Review. [DOI] [PubMed] [Google Scholar]
- 42.Corcione A, Tasso P, Pistoia V. The granulocyte colony-stimulating factor (G-CSF)/G-CSF receptor (G-CSFR) system in B-cell chronic lymphocytic leukemia. Leuk Lymphoma. 1997 Oct;27(3–4):239–246. doi: 10.3109/10428199709059680. [DOI] [PubMed] [Google Scholar]
- 43.De Lau WB, Hurenkamp J, Berendes P, Touw IP, Clevers HC, van Dijk MA. The gene encoding the granulocyte colony-stimulating factor receptor is a target for deregulation in pre-B ALL by the t(1;19)-specific oncoprotein E2A-Pbx1. Oncogene. 1998 Jul 30;17(4):503–510. doi: 10.1038/sj.onc.1201967. [DOI] [PubMed] [Google Scholar]
- 44.Shenoy SK, McDonald PH, Kohout TA, Lefkowitz RJ. Regulation of receptor fate by ubiquitination of activated beta 2-adrenergicreceptor and beta-arrestin. Science. 2001 Nov 9;294(5545):1307–1313. doi: 10.1126/science.1063866. [DOI] [PubMed] [Google Scholar]
- 45.Marchese A, Benovic JL. Agonist-promoted ubiquitination of the G protein-coupled receptor CXCR4mediates lysosomal sorting. J Biol Chem. 2001 Dec 7;276(49):45509–45512. doi: 10.1074/jbc.C100527200. [DOI] [PubMed] [Google Scholar]
- 46.Bonifacino JS, Weissman AM. Ubiquitin and the control of protein fate in the secretory and endocytic pathways. Annu Rev Cell Dev Biol. 1998;14:19–57. doi: 10.1146/annurev.cellbio.14.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Govers R, ten Broeke T, van Kerkhof P, Schwartz AL, Strous GJ. Identification of a novel ubiquitin conjugation motif, required for ligand-induced internalization of the growth hormone receptor. EMBO J. 1999 Jan 4;18(1):28–36. doi: 10.1093/emboj/18.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aarts LH, Roovers O, Ward AC, Touw IP. Receptor activation and 2 distinct COOH-terminal motifs control G-CSF receptor distribution and internalization kinetics. Blood. 2004 Jan 15;103(2):571–579. doi: 10.1182/blood-2003-07-2250. [DOI] [PubMed] [Google Scholar]