

Abstract
We have followed individual ribosomes as they translate single messenger RNA hairpins tethered by the ends to optical tweezers. Here we reveal that translation occurs through successive translocation-and-pause cycles. The distribution of pause lengths, with a median of 2.8 s, indicates that at least two rate-determining processes control each pause. Each translocation step measures three bases—one codon—and occurs in less than 0.1 s. Analysis of the times required for translocation reveals, surprisingly, that there are three substeps in each step. Pause lengths, and thus the overall rate of translation, depend on the secondary structure of the mRNA; the applied force destabilizes secondary structure and decreases pause durations, but does not affect translocation times. Translocation and RNA unwinding are strictly coupled ribosomal functions.
Current understanding of the ribosome and the mechanism of translation has been significantly strengthened and expanded by recent advances in crystallography1-6 and cryo-electron microscopy7-10. The ribosome undergoes several dynamical structural changes as it moves relative to the mRNA and transfer RNAs during translation8,11. Kinetic experiments have given a quantitative description of some of these dynamics during the main steps of the elongation cycle of protein synthesis12. During elongation, the secondary structures present in all mRNAs are disrupted to allow movement of the mRNA through the 30S subunit, and the reading of each codon. This task is aided by the mRNA helicase activity of the ribosome that has been localized to the downstream tunnel of the 30S subunit13. Moreover, interactions of mRNA pseudoknots or hairpins with the helicase region of the ribosome can shift the reading frame of the mRNA to the -1 frame, and play an important role in regulating gene expression in retroviruses14-16.
It is extremely difficult to follow the steps of ribosomes during translational elongation using ensemble methods, because the dynamics of individual ribosomes are stochastic17,18 and it is impossible to synchronize their activity. Here, we have used optical tweezers to follow the step-by-step translation of a single hairpin-forming mRNA molecule by a single ribosome. This approach has allowed us to characterize the dynamics of ribosome translation, measuring the time the ribosome spends at each codon, the number of mRNA nucleotides that move through the ribosome in each translocation step, and the time required per step. We have also determined the effects of mRNA structure on step size and rate, and have studied the effects of internal Shine-Dalgarno sequences19 on translation arrest. These experiments provide a dynamic picture of the movement of a messenger RNA through a ribosome.
In these experiments, we used a single mRNA hairpin with a ribosome stalled at the 5′ end by omission of a required aminoacyl-tRNA; the RNA was attached to two micrometre-sized beads by RNA-DNA handles. One of the beads was held on a micropipette and the other in an optical trap used to measure the changes in distance between the beads (in nanometres) and the forces applied to the hairpin (in piconewtons) (Fig. 1A)20-22. Translation is resumed at the single-molecule level by adding a mixture containing the required aminoacyl-tRNAs. During translation, the ribosome opens the hairpin as it moves through the RNA; thus, each base translocated requires the breaking of a base pair, which corresponds to an increase in the end-to-end distance of the mRNA by about 1 nm at the forces involved in these experiments (15-20 pN)23. Translation can thus be followed in real time by monitoring these changes in distance.
Figure 1. Experimental design.

A, The ribosome was stalled at the 5′ side of the mRNA hairpin construct, which was then held between two polystyrene beads coated with anti-digoxigenin antibody (top; approximately 3 μm in diameter) and streptavidin (bottom; approximately 2 μm). The micropipette was substituted by a second laser trap when VE274hp was used for translation. Drawings are schematic and not to scale. B, All the mRNA constructs used in this report share the same sequence and design except for the hairpin (upper case) and flanking regions (lower case). The Shine-Dalgarno sequence (AGGAG) and AUG start codon are highlighted in cyan, the tetraloop of the hairpin in green, Val codon patches in grey and Glu in yellow. The codons chosen to stall the ribosome are boxed. a, S3hp, 60 bp. b, VE60hp, 60 bp. c, VE274hp, 274 bp. The complementary sequence for VE274hp is not shown.
To establish that translation occurred in a single molecule of RNA held in the optical tweezers apparatus, we used a 60-base-pair (bp) hairpin that contains unique codons where the ribosome can be stalled (S3hp; see a in Fig. 1B). The progress of translation was established by determining the size of the residual hairpin, through its mechanical unfolding (details presented in Supplementary Information and Supplementary Fig. 1). To verify that the change in hairpin size was caused by a stalled ribosome at the corresponding position on the hairpin, we flowed a ribosome-releasing mixture containing puromycin (see Methods) into the reaction chamber. After a few minutes, the hairpin fully refolded, indicating that the ribosome had been released. These control experiments established that an RNA duplex can be unwound by the ribosome as translation proceeds, and that a ribosome can be stably stalled on the RNA at a chosen position.
We also translated the mRNA in bulk in the absence of force (using Escherichia coli S100 enzymes, see Supplementary information). The 60-bp S3hp mRNA and its corresponding wild-type ribosomal S3 mRNA were translated completely, but the hairpin RNA was translated approximately 40% slower (see Supplementary Fig. 2). These results show that ribosomes can translate long helices as used in our experiments, even without the aid of force to unfold them.
Following translation in real time
To follow translation by single ribosomes in real time, we designed another 60-bp hairpin construct, VE60hp, whose first 15 codons in the hairpin region encode only valine (Val) and glutamic acid (Glu), preceded by two phenylalanine codons (Phe) to allow ribosome stalling (see b in Fig. 1B). The hairpin with a ribosome stalled at the Phe codon was held at a constant force of 20 pN—a force below that required to unfold the hairpin (approximately 23 pN, see Supplementary Fig. 3), and the extension change was monitored in time. As expected, hairpin unwinding was not observed before a translation mixture was injected into the reaction chamber to resume elongation. The extension and force trajectories versus time obtained in the presence of the translation mixture are shown in Fig. 2a. Strikingly, the extension shows a repeated step-pause-step pattern. These patterns display clear and discrete transitions (indicated by arrows). The steps were quantitatively analysed by calculating the corresponding pairwise distance distributions23, as shown in Fig. 2b. The distance between steps is 2.7 nm, which corresponds to six nucleotides at 20 pN; that is, the breaking of three hairpin base pairs accompanying a translocation step of three bases. Thus, we identified these transitions as ribosomal steps corresponding to the translation of individual codons. Figure 2c shows a step-pause-step trajectory defining the translocation time, the dwell time (the time between successive steps) and the pause time (the time between translocation events) of one such cycle. Direct analysis of the translocation events (number of events, n = 121) during translation of VE60hp gives a step size of 2.94 ± 0.72 bases occurring in 0.078 ± 0.045 s.
Figure 2. Codon-by-codon translation of VE60hp.

a, Extension and force trajectories during translation. The data were collected at 200 Hz (blue traces) and smoothed to 10 Hz (red). Discrete steps are indicated by arrowheads. The 18 nm rip at 163 s corresponds to spontaneous opening of the remaining approximately 18-bp hairpin ahead of the translating ribosome. b, Pairwise distance analysis of the extension trajectory in a from 147 to 157 s after correction for drift. c, The pattern of the translation process illustrated by a close-up view of the 148-150 s region. d, Distribution of translocation times. The translocation times (n = 121) were binned in 0.025 s intervals, and the probability density plotted and fitted to three possible Poisson equations (see Supplementary Information): one step (black line), two steps (blue) and three steps (red), with R2 values of 0.45, 0.87 and 0.95, respectively. The k value from the best fit (red line) is 40 ± 4 s-1. Attempts to fit the distribution to two or three steps with different rate constants gave equal rate constants as the best fit.
Although in our assay we directly observe the opening of hairpin base pairs and the release of the single-strand chains due to the helicase activity of the ribosome, and not its actual translocation, it is noteworthy that the ribosome opens exactly three base pairs, corresponding to the translation of each codon. The most parsimonious interpretation of this observation is that unwinding and translocation are strictly coupled functions and occur simultaneously; that is, within 0.078 s. Accordingly, we refer here to the transitions observed as translocations. These results constitute the first direct real-time observation, to our knowledge, of the physical steps of the ribosome machine moving along the mRNA during translation.
Translation of a 274-bp hairpin
We used a third mRNA construct (VE274hp; see c in Fig. 1C) harbouring a 274-bp hairpin with patches of Val and Glu codons to improve the statistics of translation dynamics. To follow translation on this longer messenger, we used a dual-trap optical tweezer instrument in which the pipette holding the bottom bead shown in Fig. 1A was replaced by a second optical trap. This instrument provided lower drift that improved data collection during the longer times required to translate this mRNA. However, as expected, the longer RNA also introduced more high-frequency noise.
Translation of VE274hp began by stalling the ribosome at the first Glu codon (E20; see c in Fig. 1B) of the hairpin. The RNA was held at an initial force of 17-18 pN before flowing the translation mixture (containing purified Val-tRNAVal and Glu-tRNAGlu) into the chamber to resume elongation. As the reaction proceeded, opening of the hairpin by the translating ribosome resulted in an increase in RNA extension, and a corresponding decrease in force24, because the two laser beams of the dual-trap tweezers were held fixed during the experiments (Supplementary Fig. 4).
As seen in Fig. 3a, translation dynamics is a complex process, which is different for each reaction. The four translation trajectories shown in Fig. 3a were obtained for different ribosomes under the same reaction conditions, but the patterns vary significantly. Apart from runs of active translation, these trajectories also show variable overall translation rates, the existence of long pauses of variable lengths and locations, and positions where translation stopped altogether. Nonetheless, close-up views of active translation trajectories from different reactions (Fig. 3b) show the characteristic translocation-pause-translocation pattern with three-base steps, or multiples of them.
Figure 3. Translational trajectories and arrests for VE274hp.

Translations were followed on dual-trap optical tweezers35. a, Examples of translation trajectories from four different ribosomes. The five horizontal yellow stripes correspond to the Glu patches (see c). b, Close-up views of trajectories. Data were collected at 2 kHz and smoothed to 10 Hz. c, Translation arrests. The positions (relative to the A-site on the ribosome) where translation arrests occur are indicated by short horizontal bars, blue for the wild-type VE274hp and red for the mutant hairpin VE274hp-G2A. The VE274hp-G2A differs from the wild type only in the first internal Shine-Dalgarno-like sequence (boxed by solid green line).
Only a small fraction of ribosomes (n = 4 out of 29) reach the last Glu patch (labelled E5; see, for example, Fig. 3a, blue trace) of the VE274hp. Most trajectories end earlier; we define these early halting events as ‘translation arrests’. Figure 3c summarizes the positions of translation arrests in the VE274hp construct (underlying blue bars). One-third (9 out of 29) of the ribosomes stopped at a region (positions 67-75) that occurs just downstream from a Shine-Dalgarno-like AGGAGG sequence (positions 56-61, Fig. 3c). Moreover, another internal Shine-Dalgarno sequence (positions 167-172) also seems to favour translational arrests. To test the effects of internal Shine-Dalgarno sequences on translation, the first Shine-Dalgarno sequence AG GAG G in VE274hp was changed to AA GAA G; note that the new codons are recognized by the same isotype of tRNAGlu and the amino-acid sequence is maintained. On this mutant hairpin, VE274hp-G2A, the translation arrest downstream of the Shine-Dalgarno sequence (positions 67-75) has now disappeared (underlying red bars, Fig. 3c). These results suggest that translating ribosomes can be stalled by base pairing between the 3′ end of 16S rRNA and internal Shine-Dalgarno sequences in the mRNA, similar to the interaction during the initiation process. A recent report shows that the Shine-Dalgarno interaction increases the rupture force of the ribosome from the bound mRNA by 10 pN25. The arrests observed in the vicinity of the Shine-Dalgarno sequence in our experiments indicate that this extra binding energy, together with the downstream hairpin structure, is sufficient to block the movement of the ribosome.
Pauses and translocations
As shown in Fig. 3a, active translation runs made up of translocation-pause-translocation cycles are eventually interrupted by longer pauses, which can last up to 1-2 min (Supplementary Fig. 5). The long pauses occur less often in the first valine patch (V1) than in succeeding ones (Supplementary Fig. 6a). These long pauses can be identified from the derivatives of the individual translation trajectories (that is, the derivatives of the traces in Fig. 3a; one is shown in Supplementary Fig. 6b). The valleys between peaks on such plots characterize the long pauses, whereas the peaks on the plot correspond to active translation runs. These peaks show a relatively constant rate of 0.45 ± 0.17 codons per second, corresponding to a mean dwell time of 2.2 s (n = 43 ribosomes); these short pauses, we surmise, represent the constitutive (intrinsic) pauses of a translating ribosome.
The kinetics of translation have been studied in bulk12, and in single-molecule fluorescence resonance energy transfer (FRET) experiments17,18; the rates of the rate-determining (slowest) steps are of order 1-10 s-1. In single-molecule experiments, the distribution of lifetimes for a species depends on the number and rates of the molecular events that transform that species into another. If a single stochastic event determines the lifetime, the distribution of lifetimes follows a single exponential decay from the smallest lifetimes to the longest lifetimes. For two or more successive reactions the distribution has a maximum at intermediate values of the lifetimes (see Supplementary Information)26,27.
We analysed the dwell times—the times between successive steps—during translation of the first 15 valines in the VE274hp sequence. We found that these times for an actively translating subset of ribosomes (mean dwell time approximately 2 s) are not exponentially distributed. Instead, the data (n = 180) can be fitted well (R2 = 0.93) to a difference of two exponentials (Fig. 4a). To ensure that our choice of ribosomes had not biased the results, we also analysed different subsets, and all the ribosomes. A difference of two exponentials was needed to fit each distribution, although the values of the rate constants, k, differed for each distribution. These results indicate that two, or more, successive rate-limiting processes control the dwell times between steps.
Figure 4. Dwell times and force effect.
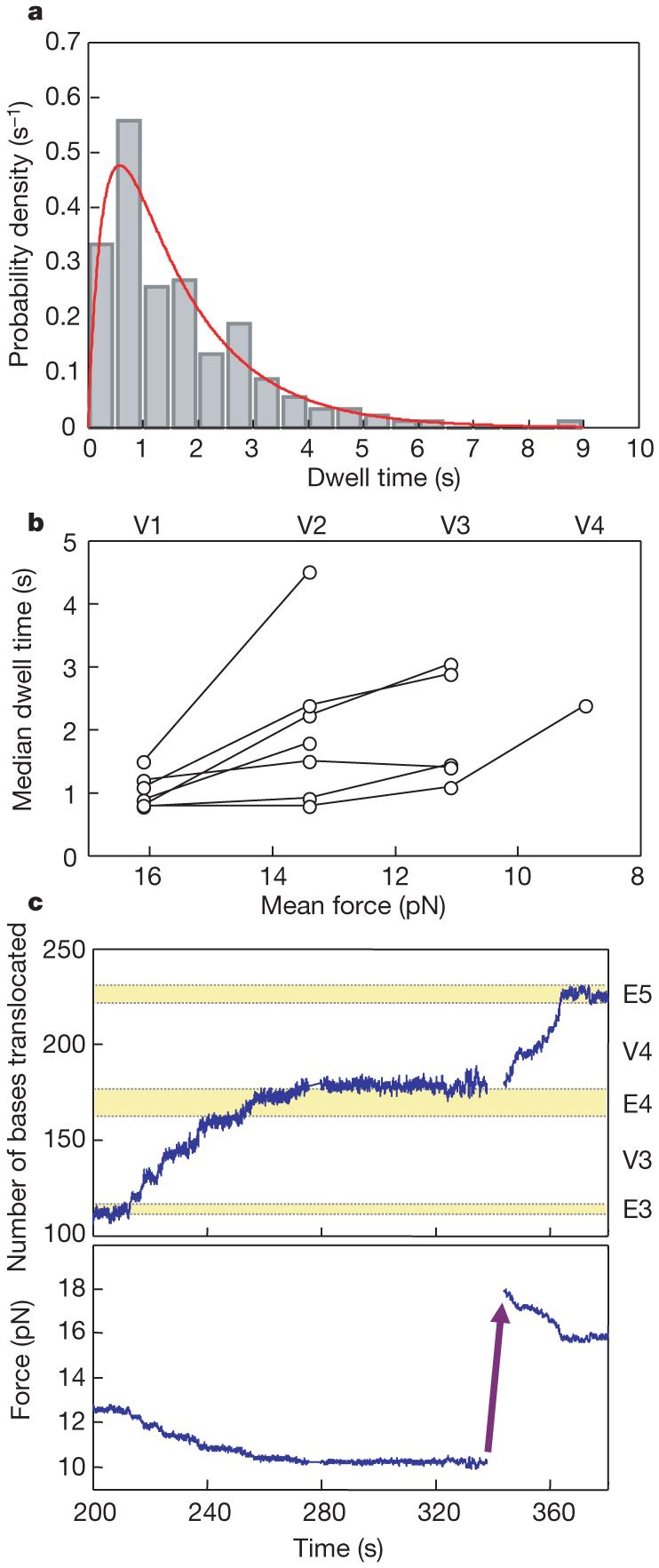
a, Distribution of dwell times from VE274hp. Dwell-time data (n = 180) are pooled from translations of the first Val patch (V1) for 12 ribosomes with mean dwell times less than 2 s. The distribution (0.5 s bins) is well fitted to a mechanism in which two consecutive reactions take place (red line), with k1 = 0.7 ± 0.2 s-1, k2 = 3.4 ± 1.8 s-1 and R2 = 0.93. b, Force effect on dwell times. The median dwell times in each Val patch are plotted as a function of force (mean force in each Val patch, V1-V4). Data points from the same ribosomes are connected by lines. c, Force effect on long pauses. In this example, the translation paused when the force dropped to about 10 pN. After approximately 1 min of pausing, the force was rapidly raised to 18 pN, and the translation resumed immediately.
By contrast, and surprisingly, the distribution of translocation times for VE60hp best fits an equation (see Supplementary Information) with three identical substeps, with a mean lifetime of each of the three substeps of 0.025 ± 0.002 s (k = 40 s-1) (Fig. 2d). This result indicates that multiple processes with similar kinetics underlie the translocation step of three nucleotides. Until now, no technique has allowed characterization of the dynamics of ribosome stepping to this degree of detail. The kinetic processes we identified here could involve, for example, steps in the unlocking rearrangement of the ribosome28; or it is also possible that the ribosome moves over the length of one codon with three consecutive, equally timed, singlebase substeps.
The effect of force on translation rates
Figure 3a shows that translation dynamics (overall rates, see Supplementary Fig. 7, and extents of reactions) are different for each ribosome. However, recurrence of Val patches on VE274hp allowed us to investigate, on the same ribosome, the effect of the force applied to the hairpin. Figure 4b shows that the lengths of pauses increase—translation rates decrease—when the force dropped as translation proceeded from Val segment 1 to 4 (V1-V4). The distributions of dwell times show that decreasing the force (16 to 9 pN) decreases the faster rate constant of the two substeps by a factor of three. At lower forces, the energy barrier impeding strand separation increases, resulting in slower translation rates and longer dwell times. This suggests that a paused ribosome could be rescued by raising the force applied on the ends of the hairpin. This is indeed the case, as shown in Fig. 4c, where raising the force from 10 pN to 18 pN caused translation to immediately resume. Similar force-rescued translations were observed in about one-third of the long-paused ribosomes (14 events in 40 trials). Thus, some of the longer pauses observed in these experiments may be accounted for by the mechanical barriers imposed on the ribosome by the secondary structure of the mRNA.
Surprisingly, the force applied to the hairpin affects only the pause times; there is no detectable change in the translocation times. This observation implies that the rate-limiting step for translocation in our experiments corresponds to an intermediate state of the mRNA-tRNA-ribosome complex whose attainment is prevented, in part, by an activation barrier that is lowered by the force applied to the ends of the RNA. Once the complex has cleared this barrier, helix opening (and translocation) occurs rapidly and independently of force.
Biological implications
We have successfully applied optical tweezers to follow translation by single ribosomes in real time along individual molecules of mRNA. These studies have revealed for the first time that translation occurs not in a continuous manner, but as a series of translocation-pause-translocation events. These intrinsic short pauses, displayed by actively translating ribosomes, have durations that range from a fraction of a second to a few seconds; their mean is 2.2 s. In addition, we observe pauses that are significantly longer, lasting from tens of seconds up to 1-2 min. By contrast, the translocation steps, lasting for less than 0.1 s, were independent of the experimental variables we studied. The ability to measure the time spent at each codon (dwells) and the time required to move to the next codon (translocations) will make it possible to correlate biochemical results obtained in bulk studies with the dynamics of translation at a specific step or substep.
The ubiquitous long pauses observed in these studies may play a role in translation regulation in vivo, and can lead to translational frameshifting29 and to protein misfolding30. A recent cryo-electron microscopy study on frameshifting suggests that mRNA pseudoknots cause conformational changes in the ribosome that create tension on the mRNA, which in turn results in -1 frameshifting16. Pauses caused by the increased barriers as ribosomes move through a hairpin31, as in these experiments, can also lead to frameshifting.
We find that internal Shine-Dalgarno sequences tend to stop translation, presumably by binding to 16S rRNA during long pauses. The ability to measure dwell times at each codon opens the way to understanding the mechanism of translational regulation involving internal Shine-Dalgarno sequences, such as in the expression of the prfB and dnaX genes in E. coli29. The mRNA transcripts of these genes both contain internal Shine-Dalgarno sequences, and can lead to +1 or -1 frameshifts.
The studies on hairpins described here mimic biologically relevant situations of translation. Messenger RNAs are expected to have roughly half of their bases paired32,33, mostly in short hairpins (5-10 bp), but more stable structures, including pseudoknots, are not uncommon. Future efforts should concentrate on single-molecule translation using less structured templates similar to naturally occurring mRNAs, and on measuring the translation signal directly by tethering the ribosome as it moves along the mRNA. By forming tethers34 to the ribosome and the end of the linear mRNA, it will be possible to investigate the motor properties of the ribosome: that is, its stall force and its force-velocity relation. These properties can provide further insight into the mechanism of energy transduction of this essential cellular machine.
METHODS SUMMARY
Preparation of stalling translation mixtures
The stalling translation mixtures were used to stall the ribosome at designated positions on the mRNA. In general, the following components were mixed in 50 μl of Buffer TL: 2 mM ATP, 1 mM GTP, 5 mM PEP (phosphoenol pyruvate), 1 OD total tRNA mixture, 24 μM EF-Tu, 4 μM EF-G, 0.02 mg ml-1 pyruvate kinase (Roche), 1 μl DEAE-purified S-100 enzymes, 40 U RNAguard (RNase inhibitor; Amersham) and amino-acid mixture containing 0.1 mM of each amino acid. Buffer TL contained: 40 mM HEPES-KOH, pH 7.5, 60 mM NH4Cl, 10 mM Mg(OAc)2, 1 mM DTT and 3.6 mM 2-mercaptoethanol. The components of the amino-acid mixture depended on the experiment. For example, to stall the ribosome at the arginine R11 of S3hp, the amino acids used were G, Q, K, V, H, P, N, and I (see a in Fig. 1B). The 50-μl mixture was incubated at 37 °C for 15 min and on ice for 5 min, and then diluted with three volumes of Buffer TL containing 1 mM GTP.
Preparation of initiation and stalled ribosome-mRNA complexes
To make the initiation complex, the mRNA hairpin constructs (0.2 μM) were mixed with 1 mM GTP, 4 μM each of IF1, IF2, IF3, fMet-tRNAfMet, and the 70S ribosome in 10 μl Buffer TL. The mixture was incubated at 37 °C for 15 min. Elongation was performed by mixing 1 μl initiation complex and 9 μl stalling translation mixture and incubating at 37 °C for 2-5 min. The reaction was used to stall the ribosome at the designated position on the mRNA. Finally, the mixture was diluted with 20 volumes of cold Buffer TL to prepare it for tweezer experiments.
Supplementary Material
Acknowledgements
We thank R. Hanna for her early efforts on this project, and S. B. Smith for help with the instrumentation. The work was supported by National Institutes of Health grants (to I.T., C.B. and H.F.N.), and a Grant-in-Aid for Young Scientists (A) from the Japan Society for the Promotion of Science (S.Y.).
APPENDIX
METHODS
General
Configuration of the single-trap tweezers has been described previously36,37. Configuration of the dual-trap tweezers was similar to that described by Moffitt et al.35, except that a 1064-nm laser was used. The E. coli MRE600 ribosomes38, initiation factors (IF1, IF2 and IF3)39, EF-G40, EF-Tu41 and S-100 enzymes42 were purified as per protocols described in the literature. The ribosomes and proteins were made in small aliquots and stored at -80 °C.
Construction of plasmids
The plasmids used in the experiments were originally constructed from pBluescript SK+ (Stratagene). A 769-bp E. coli ribosomal S3 gene43 was inserted between XbaI and NotI sites, and a 619-bp PCR fragment (from pBR322) was inserted into the KpnI site, between the T7 promoter sequence and the S3 gene fragment, to serve as the 5′ handle. To make pS3hp (for S3hp mRNA, see a in Fig. 1B), a 107-bp dsDNA was inserted between NcoI and BsrGI sites (inside the S3 gene), such that the sequence for codons 13-32 of S3 was base-paired to the inserted downstream sequence. pVE60hp (for VE60hp mRNA, see b in Fig. 1B) was derived from pS3hp. A 158-bp dsDNA was inserted to pS3hp between NdeI and BsrGI sites to replace the existing hairpin completely.
pVE274hp (for VE274hp mRNA, see c in Fig. 1B) was derived from pVE60hp and constructed in several steps. Oligonucleotide A (60-base oligonucleotide), pGAAGAAGTG(GTN)14GAAGAGGAG (p = phosphate, N = A, G, C or T; encoding Glu2Val15Glu3), was annealed to an 18-base oligonucleotide connector, CACTTCTTCCTCCTCTTC, which was used to align multiple copies of oligonucleotide A in tandem, followed by ligation. To introduce primer sequences and functional sites, oligonucleotide B (CCATGGAACAAGCTGAGGAATCAAACGTTCTTC) and oligonucleotide C (CGGCCGCCGCCCTAGGGGCTGGCTTTCGCCAGCCCTTTAGTGAGGGTTAATT) were ligated to the 5′ and 3′ sides, respectively. The ligated products were subjected to PCR (primers: TAGGATccatggAACAAGCTGAGGAATCAAACG and AAGGCCtgtacaAATTAACCCTCACTAAAGGGCTGG, NcoI and BsrGI sites are shown in lower case, respectively) and then inserted to pVE60hp between NcoI and BsrGI, replacing the existing hairpin sequence. The cloned sequences were dominated by two copies of oligonucleotide A. The oligonucleotide A regions from two clones were subjected to PCR and ligated to make a construct with essentially four copies of oligonucleotide A (called pRC4). To make a hairpin, the oligonucleotide A region of pRC4 was subjected to PCR (primers: GGACATCCTAATtgtacaAACAAGCTGAGGAATCAAACGTTCTTT and GCTTGTTAggtctcCCTAGGGAAACCTAGGGCGGCGGCCGCTCC, BsrGI and BsaI sites are shown in lower case, respectively; the latter creates a cohesive end compatible to AvrII), and inserted to pRC4 between AvrII and BsrGI, downstream of the oligonucleotide A region. Note that the insertion is in an inverted (tail-to-tail) orientation, such that the insert is complementary to the oligonucleotide A region. RNA was synthesized in vitro with these plasmids with T7 RNA polymerase (Ambion). The synthesized RNA was purified by MEGAclear purification kit (Ambion) and stored at -30 °C.
Aminoacylation of tRNA
Purified tRNAs (tRNAVal, tRNAGlu, tRNAPhe and tRNAfMet) were purchased from Sigma. Total tRNA mixtures were purchased from Sigma and Roche. tRNAs were aminoacylated using DEAE-purified S-100 enzymes38 and extracted with phenol/chloroform. The efficiency of aminoacylation for purified tRNAs was checked by acid gel electrophoresis44. Because of the heterogeneity, the efficiency of aminoacylation for the total tRNA mixtures cannot be determined from the gel.
Preparation of translation mixtures
The translation mixtures prepared here were used to inject into the reaction chamber to resume translation for the stalled ribosome-mRNA complexes. In general, the following components were mixed in a total of 45 μl of Buffer TL: 1 mM GTP, 5 mM PEP, 26.7 μM EF-Tu, and 0.02 mg ml-1 pyruvate kinase. The mixture was incubated at 37 °C for 15 min. Then 5 μl of Val-tRNAVal, Glu-tRNAGlu and Phe-tRNAPhe (each with specified concentrations) were added, incubated at 37 °C for 5 min, and on ice for 5 min. The reaction was diluted with 350 μl (total 400 μl) Buffer TL containing 1 mM GTP, 1 mM ATP, 40 U RNAguard and 1 or 0.1 μM EF-G (final concentrations). Finally, the mixture was filtered (0.22 μm, low protein binding, MILLEX-GV, Durapore membrane, Millipore) and put on ice.
Ribosome-releasing mixture
The ribosome-releasing mixture was used for control experiments to release the ribosome from the stalled ribosome-mRNA complex. The mixture contained the following components in 400 μl Buffer TL: 0.5 mM puromycin, 1 mM GTP, 0.25 μM EF-G, 1 μl DEAE-purified S-100 enzymes, and 40 U RNAguard.
Conversion of translation trajectories
To facilitate data analysis, the extension (end-to-end distance) changes of the VE274hp (or VE274hp-G2A) RNA during translation were converted to the number of bases translocated by the ribosome (base pairs broken), by an empirical method. The force-extension curves before translation were fitted to a worm-like chain (WLC) model24 to obtain the basal WLC curve. A series of curves that mimicked the RNA elasticity corresponding to individual translation steps were calculated. This was done by adding six-nucleotide (three base pairs broken) components each time to the preceding WLC curves, starting from the basal WLC curve. This series of WLC curves was used to calculate the extension change for each step during translation with the consideration of gradual force decreasing. The theoretical relation between the number of bases translocated and extension change was well fitted to a double exponential equation. Finally, the fitted parameters were used to convert the experimentally measured extension change to the number of bases translocated. This empirical method worked well. For example, the force-extension curve after translation in Supplementary Fig. 3a (green trace) shows no apparent rips, suggesting that none or little residual hairpin was left. The converted translation trajectory (the blue trace in Fig. 3a) shows that the ribosome was stalled close to the apical loop of the hairpin, consistent with the experimental data.
Calculation of translation rates
The translation trajectories (number of bases translocated versus time; see Fig. 3a) were analysed by placing a moving window in the region corresponding to a translated Val patch. The window size was adjusted to cover 21 bp (seven codons, about half of a Val patch). The slope from linear regression for the data points in the window was recorded. The window was then moved to scan the whole region in the patch. The translation rate in this Val patch was finally assigned to the maximum slope. The same procedures were repeated for other translated Val patches on the trace.
Analysis of translocation steps
The 200 Hz unsmoothed extension trajectories obtained from the single-trap tweezers were analysed by placing a variable window around a translocation region corresponding to a single step. The window size was varied from 0.025 to 0.50 s, and the linear regression of the 5-100 points in the interval was calculated. The translocation time was assigned to the window size that best fitted a line with the maximum R2 value.
- 36.Smith SB, Cui Y, Bustamante C. Optical-trap force transducer that operates by direct measurement of light momentum. Methods Enzymol. 2003;361:134–162. doi: 10.1016/s0076-6879(03)61009-8. [DOI] [PubMed] [Google Scholar]
- 37.Wen J-D, et al. Force unfolding kinetics of RNA using optical tweezers. I. Effects of experimental variables on measured results. Biophys. J. 2007;92:2996–3009. doi: 10.1529/biophysj.106.094052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moazed D, Noller HF. Interaction of tRNA with 23S rRNA in the ribosomal A, P, and E sites. Cell. 1989;57:585–597. doi: 10.1016/0092-8674(89)90128-1. [DOI] [PubMed] [Google Scholar]
- 39.Lancaster L, Noller HF. Involvement of 16S rRNA nucleotides G1338 and A1339 in discrimination of initiator tRNA. Mol. Cell. 2005;20:623–632. doi: 10.1016/j.molcel.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 40.Wilson KS, Noller HF. Mapping the position of translational elongation factor EF-G in the ribosome by directed hydroxyl radical probing. Cell. 1998;92:131–139. doi: 10.1016/s0092-8674(00)80905-8. [DOI] [PubMed] [Google Scholar]
- 41.Boon K, et al. Isolation and functional analysis of histidine-tagged elongation factor Tu. Eur. J. Biochem. 1992;210:177–183. doi: 10.1111/j.1432-1033.1992.tb17406.x. [DOI] [PubMed] [Google Scholar]
- 42.Traub P, Mizushima S, Lowry CV, Nomura M. In: RNA and Protein Synthesis. Moldave K, editor. Academic Press; New York: 1981. pp. 521–539. [Google Scholar]
- 43.Culver GM, Noller HF. In vitro reconstitution of 30S ribosomal subunits using complete set of recombinant proteins. Methods Enzymol. 2000;318:446–460. doi: 10.1016/s0076-6879(00)18069-3. [DOI] [PubMed] [Google Scholar]
- 44.Varshney U, Lee CP, RajBhandary UL. Direct analysis of aminoacylation levels of tRNAs in vivo. Application to studying recognition of Escherichia coli initiator tRNA mutants by glutaminyl-tRNA synthetase. J. Biol. Chem. 1991;266:24712–24718. [PubMed] [Google Scholar]
References
- 1.Moore PB, Steitz TA. The structural basis of large ribosomal subunit function. Annu. Rev. Biochem. 2003;72:813–850. doi: 10.1146/annurev.biochem.72.110601.135450. [DOI] [PubMed] [Google Scholar]
- 2.Ogle JM, Ramakrishnan V. Structural insights into translational fidelity. Annu. Rev. Biochem. 2005;74:129–177. doi: 10.1146/annurev.biochem.74.061903.155440. [DOI] [PubMed] [Google Scholar]
- 3.Selmer M, et al. Structure of the 70S ribosome complexed with mRNA and tRNA. Science. 2006;313:1935–1942. doi: 10.1126/science.1131127. [DOI] [PubMed] [Google Scholar]
- 4.Yusupov MM, et al. Crystal structure of the ribosome at 5.5 Å resolution. Science. 2001;292:883–896. doi: 10.1126/science.1060089. [DOI] [PubMed] [Google Scholar]
- 5.Schuwirth BS, et al. Structures of the bacterial ribosome at 3.5 Å resolution. Science. 2005;310:827–834. doi: 10.1126/science.1117230. [DOI] [PubMed] [Google Scholar]
- 6.Korostelev A, Trakhanov S, Laurberg M, Noller HF. Crystal structure of a 70S ribosome-tRNA complex reveals functional interactions and rearrangements. Cell. 2006;126:1065–1077. doi: 10.1016/j.cell.2006.08.032. [DOI] [PubMed] [Google Scholar]
- 7.Frank J. Electron microscopy of functional ribosome complexes. Biopolymers. 2003;68:223–233. doi: 10.1002/bip.10210. [DOI] [PubMed] [Google Scholar]
- 8.Frank J, Agrawal RK. A ratchet-like inter-subunit reorganization of the ribosome during translocation. Nature. 2000;406:318–322. doi: 10.1038/35018597. [DOI] [PubMed] [Google Scholar]
- 9.Allen GS, Zavialov A, Gursky R, Ehrenberg M, Frank J. The cryo-EM structure of a translation initiation complex from Escherichia coli. Cell. 2005;121:703–712. doi: 10.1016/j.cell.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 10.Stark H, et al. Ribosome interactions of aminoacyl-tRNA and elongation factor Tu in the codon-recognition complex. Nature Struct. Biol. 2002;9:849–854. doi: 10.1038/nsb859. [DOI] [PubMed] [Google Scholar]
- 11.Horan LH, Noller HF. Intersubunit movement is required for ribosomal translocation. Proc. Natl Acad. Sci. USA. 2007;104:4881–4885. doi: 10.1073/pnas.0700762104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wintermeyer W, et al. Mechanisms of elongation on the ribosome: dynamics of a macromolecular machine. Biochem. Soc. Trans. 2004;32:733–737. doi: 10.1042/BST0320733. [DOI] [PubMed] [Google Scholar]
- 13.Takyar S, Hickerson RP, Noller HF. mRNA helicase activity of the ribosome. Cell. 2005;120:49–58. doi: 10.1016/j.cell.2004.11.042. [DOI] [PubMed] [Google Scholar]
- 14.Alam SL, Atkins JF, Gesteland RF. Programmed ribosomal frameshifting: much ado about knotting! Proc. Natl Acad. Sci. USA. 1999;96:14177–14179. doi: 10.1073/pnas.96.25.14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brierley I, Digard P, Inglis SC. Characterization of an efficient coronavirus ribosomal frameshifting signal: requirement for an RNA pseudoknot. Cell. 1989;57:537–547. doi: 10.1016/0092-8674(89)90124-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Namy O, Moran SJ, Stuart DI, Gilbert RJ, Brierley I. A mechanical explanation of RNA pseudoknot function in programmed ribosomal frameshifting. Nature. 2006;441:244–247. doi: 10.1038/nature04735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blanchard SC, Kim HD, Gonzalez RL, Jr, Puglisi JD, Chu S. tRNA dynamics on the ribosome during translation. Proc. Natl Acad. Sci. USA. 2004;101:12893–12898. doi: 10.1073/pnas.0403884101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blanchard SC, Gonzalez RL, Kim HD, Chu S, Puglisi JD. tRNA selection and kinetic proofreading in translation. Nat. Struct. Mol. Biol. 2004;11:1008–1014. doi: 10.1038/nsmb831. [DOI] [PubMed] [Google Scholar]
- 19.Shine J, Dalgarno L. The 3′-terminal sequence of Escherichia coli 16S ribosomal RNA: complementarity to nonsense triplets and ribosome binding sites. Proc. Natl Acad. Sci. USA. 1974;71:1342–1346. doi: 10.1073/pnas.71.4.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tinoco I, Jr, Li PTX, Bustamante C. Determination of thermodynamics and kinetics of RNA reactions by force. Q. Rev. Biophys. 2006;39:325–360. doi: 10.1017/S0033583506004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liphardt J, Onoa B, Smith SB, Tinoco I, Jr, Bustamante C. Reversible unfolding of single RNA molecules by mechanical force. Science. 2001;292:733–737. doi: 10.1126/science.1058498. [DOI] [PubMed] [Google Scholar]
- 22.Onoa B, et al. Identifying kinetic barriers to mechanical unfolding of the T. thermophila ribozyme. Science. 2003;299:1892–1895. doi: 10.1126/science.1081338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dumont S, et al. RNA translocation and unwinding mechanism of HCV NS3 helicase and its coordination by ATP. Nature. 2006;439:105–108. doi: 10.1038/nature04331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bustamante C, Marko JF, Siggia ED, Smith S. Entropic elasticity of lambda-phage DNA. Science. 1994;265:1599–1600. doi: 10.1126/science.8079175. [DOI] [PubMed] [Google Scholar]
- 25.Uemura S, et al. Peptide bond formation destabilizes Shine-Dalgarno interaction on the ribosome. Nature. 2007;446:454–457. doi: 10.1038/nature05625. [DOI] [PubMed] [Google Scholar]
- 26.Bustamante C, Chemla YR, Forde NR, Izhaky D. Mechanical processes in biochemistry. Annu. Rev. Biochem. 2004;73:705–748. doi: 10.1146/annurev.biochem.72.121801.161542. [DOI] [PubMed] [Google Scholar]
- 27.Tinoco I., Jr. Force as a useful variable in reactions: unfolding RNA. Annu. Rev. Biophys. Biomol. Struct. 2004;33:363–385. doi: 10.1146/annurev.biophys.33.110502.140418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Savelsbergh A, et al. An elongation factor G-induced ribosome rearrangement precedes tRNA-mRNA translocation. Mol. Cell. 2003;11:1517–1523. doi: 10.1016/s1097-2765(03)00230-2. [DOI] [PubMed] [Google Scholar]
- 29.Farabaugh PJ. Programmed translational frameshifting. Microbiol. Rev. 1996;60:103–134. doi: 10.1128/mr.60.1.103-134.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kimchi-Sarfaty C, et al. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;315:525–528. doi: 10.1126/science.1135308. [DOI] [PubMed] [Google Scholar]
- 31.Ivanov IP, Atkins JF. Ribosomal frameshifting in decoding antizyme mRNAs from yeast and protists to humans: close to 300 cases reveal remarkable diversity despite underlying conservation. Nucleic Acids Res. 2007;35:1842–1858. doi: 10.1093/nar/gkm035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Doty P, Boedtker H, Fresco JR, Haselkorn R, Litt M. Secondary structure in ribonucleic acids. Proc. Natl Acad. Sci. USA. 1959;45:482–499. doi: 10.1073/pnas.45.4.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Favre A, Morel C, Scherrer K. The secondary structure and poly(A) content of globin messenger RNA as a pure RNA and in polyribosome-derived ribonucleoprotein complexes. Eur. J. Biochem. 1975;57:147–157. doi: 10.1111/j.1432-1033.1975.tb02285.x. [DOI] [PubMed] [Google Scholar]
- 34.Vanzi F, Takagi Y, Shuman H, Cooperman BS, Goldman YE. Mechanical studies of single ribosome/mRNA complexes. Biophys. J. 2005;89:1909–1919. doi: 10.1529/biophysj.104.056283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moffitt JR, Chemla YR, Izhaky D, Bustamante C. Differential detection of dual traps improves the spatial resolution of optical tweezers. Proc. Natl Acad. Sci. USA. 2006;103:9006–9011. doi: 10.1073/pnas.0603342103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.