

Abstract
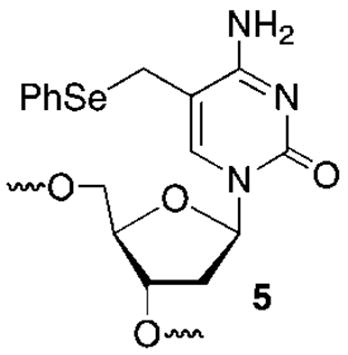
DNA interstrand cross-links have important biological consequences and are useful biotechnology tools. Phenylselenyl substituted derivatives of thymidine (1) and 5-methyl-2′-deoxycytidine (5) produce interstrand cross-links in duplex DNA when oxidized by NaIO4. The mechanism involves a [2,3]-sigmatropic rearrangement of the respective selenoxides to the corresponding methide type intermediates, which ultimately produce the interstrand cross-links. Determination of the rate constants for the selenoxide rearrangements indicates that the rate-determining step for cross-linking is after methide formation. Cross-linking by the thymidine derivative in duplex DNA shows a modest kinetic preference when flanked by pyrimidines as opposed to purines. In contrast, the rate constant for cross-link formation from 5 opposite dG in duplex DNA is strongly dependent upon the flanking sequence and, in general, is at least an order of magnitude slower than that for 1 in an otherwise identical sequence. Introduction of mispairs at the base pairs flanking 5 or substitution of the opposing dG by dI significantly increases the rate constant and yield for cross-linking, indicating that stronger hydrogen bonding between the methide derived from it and dG compared to dA and the respective electrophile derived from 1 limits reaction by increasing the barrier to rotation into the required syn-conformation. Incorporation of 1 or 5 in triplex forming oligonucleotides (TFOs) that utilize Hoogsteen base pairing also yields interstrand cross-links. The dC derivative produces ICLs approximately 10× faster than the thymidine derivative when incorporated at the 5′-termini of the TFOs and higher yields when incorporated at internal sites. The slower, less efficient ICL formation emanating from 1 is attributed to reaction at N1-dA, which requires local melting of the duplex. In contrast, 5 produces cross-links by reacting with N7-dG. The cross-linking reactions of 1 and 5 illustrate the versatility and utility of these molecules as mechanistic probes and tools for biotechnology.
DNA alkylation is a common chemical process that is useful as a chemical tool but can also have biological consequences. For instance, alkylating agents are employed in the laboratory for determining DNA structure. However, alkylated DNA can be genotoxic and/or cytotoxic.1 In addition, a number of molecules, including some antitumor agents, react with DNA by forming covalent bonds to the opposing strands.2 The cross-linked strands are strong blocks to replication and are cytotoxic if unrepaired.3 Modified oligonucleotides containing alkylating agents have also been used to form interstrand cross-links in duplex and triplex DNA.4–6 We recently described a modified thymidine (1) that forms stable base pairs with dA and produces interstrand cross-links (ICLs) (4a, 4b) in duplex DNA under mild oxidative conditions (Scheme 1).7–9 The cross-linking reaction is unusual in that like the antitumor agent N,N′-bis(2-chloroethyl)-nitrosourea (BCNU) the cross-links are produced between opposing nucleotides in the duplex.10 The phenyl selenide (1) has been used to sensitize DNA to the effects of γ-radiolysis and as a tool for detecting single nucleotide polymorphisms.11,12 This efficient cross-linking chemistry has now been extended to produce cross-links between dG and a derivative of 2′-deoxycytidine (5). Mechanistic characterization of the reactivity of 1 and 5 in duplex and triplex DNA is described.
Scheme 1.

Interstrand Cross-Link Formation from 1 under Oxidative Conditions
In general, small molecules and modified oligonucleotides that cross-link DNA are useful tools for mechanistic studies and in biotechnology applications.13–17 Oligonucleotide cross-linking is also potentially useful in therapeutic applications.18–21 A desirable oligonucleotide cross-linking reaction should proceed in high yield, with good selectivity and a high rate constant. The phenyl selenide nucleotide (1) satisfies these requirements. It undergoes transformation into the selenoxide under a variety of conditions, including with oxidants as mild as NaIO4. Cross-linking with the opposing dA in duplex DNA is also induced using reactive oxygen species that are produced in cells, such as H2O2 and 1O2. Cross-links to dA are produced in 40–50% yield with a half-life of ~30 min at room temperature. Examination of molecular models suggested that the analogous phenyl selenide derivative of 5-methyl-2′-deoxycytidine (5) would cross-link an opposing dG when oxidized to the phenyl selenoxide. Similar qualitative examination of 1:dA and 5:dG Hoogsteen base pairs suggested that the modified nucleotides would also be useful for producing interstrand cross-links (ICLs) in triple helical DNA. The investigations described herein reveal that these predictions were borne out, but the chemistry is more complex than models suggest.
Results and Discussion
Synthesis of the Cross-Linking Precursor and Its Incorporation into Oligonucleotides
The synthesis of oligonucleotides containing 1 was reported.7 The monomeric precursor to the methide (8) and respective phosphoramidite (10) for preparing oligonucleotides containing 5 was synthesized in routine fashion from 6 (Scheme 2).9 Following synthesis of the triazole (7), the exocyclic amine was introduced and the acetate protecting groups were cleaved in one pot. A variety of approaches were taken to carry 8 forward. Ultimately, the amine (8) was protected as its acetate without protecting the hydroxyl groups, as it was not advantageous to transiently protect the hydroxyl groups. Good yields of 9 were obtained by carrying out tritylation on the crude acetylated nucleoside. The phosphoramidite was prepared from 9 using standard conditions.
Scheme 2.

Synthesis of Phosphoramidite 10a
a (a) POCl3, triazole, TEA, CH3CN; (b) Concentrated aqueous NH4OH, dioxane; (c) Ac2O, DMF; (d) DMTCl, pyridine; (e) N,N′-diisopropyl 2-cyanoethyl phosphoramidic chloride, N,N′-diisopropylethylamine, CH2Cl2.
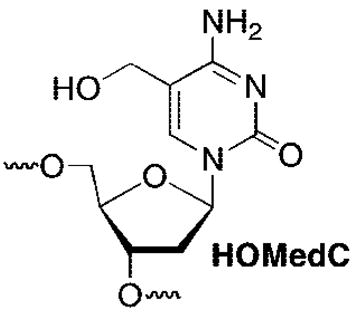
Oligonucleotides containing 5 were prepared using commercially available β-cyanoethyl phosphoramidites containing phenoxyacetyl protecting groups on the exocyclic amines of dA and dG. The exocyclic amine of dC was protected with acetate. Due to the potential oxidation of 5, t-BuOOH was used on the synthesizer in place of I2/pyridine/H2O. In addition, the coupling time for 5 was extended to 5 min. The oligonucleotides containing 5 were deprotected using concentrated aqueous ammonium hydroxide at 25 °C for 3 h. Denaturing polyacrylamide gel electrophoresis was used to separate the oligonucleotides from deletion products. The oligonucleotides were further purified by reversed phase HPLC to separate the respective phenyl selenide containing oligonucleotides from the hydroxy-methyl (e.g., HOMedC) degradation product.
After characterizing the oligonucleotides containing 5 by ESI-MS, the effect of the phenyl selenide on duplex thermal stability was analyzed by measuring the UV-melting temperatures (TM, Table 1). The phenyl selenide reduced the TM 5.3 °C relative to when 5-methyl-2′-deoxycytidine (MedC) was present, which is comparable to the depression observed when 1 was substituted for thymidine opposite dA.7 The effect of a mispair opposite 5 was greater than when 1 was examined, but the reduction was smaller than when dA was placed opposite MedC. Overall, these data suggest that 5 forms a Watson–Crick base pair with dG, albeit one that is less stable than a MedC:dG pair.
Table 1.
Comparison of UV-Melting Temperatures of Duplexes Containing 5 or MedC
| 5′-d(AGA TGG AT X TAG GAC G)
3′-d(TCT ACC TA9N A7Tc CTG C) | |||
|---|---|---|---|
| duplex | X | N | TM (°C) |
| 11a | 5 | G | 58.0 ± 0.6 |
| 11b | 5 | A | 49.2 ± 0.3 |
| 11c | MedC | G | 63.3 ± 0.7 |
| 11d | MedC | A | 49.6 ± 0.8 |
Oxidative Transformation of the Phenyl Selenides into Methide Species
Reaction of 8 with NaIO4 in deuterated phosphate buffer was analyzed using 1H NMR (Figure 1). Analysis 10 min after addition of periodate revealed that starting material (8) was completely consumed (Figure 1A), as evidenced by the absence of diagnostic resonances at δ 6.87 (singlet) and δ 5.88 (triplet) corresponding to the vinyl and C1′-protons of 5, respectively. The resonances corresponding to 8 were replaced by those that were indicative of a diastereomeric mixture of 13 and a second molecule identified as the selenoxide (12), which was also formed as a mixture of diastereomers (Scheme 3). The vinyl protons (~δ 5.62, 5.86) and the C6-proton (~δ 6.03) were used to identify methide 13. Evidence for the selenoxide (12) was gleaned from the appearance of two C1′-protons (~δ 6.14, 6.04) and two C6-vinyl protons (~δ 7.30, 7.21). After incubating the sample at 37 °C for 4 h (Figure 1B), the resonances corresponding to the methide (13) and 12 were replaced by those of the C5-hydroxymethyl product (HOMedC) (Figure 1C). Comparing these data to those obtained upon subjection of the thymidine substrate (1) to the same reaction conditions suggests that the 2′-deoxycytidine selenoxide (12) undergoes the [2,3]-sigmatropic rearrangement more slowly than the respective thymidine analogue (2) for which there was no 1H NMR evidence for the respective selenoxide 10 min after adding the oxidant.8
Figure 1.

1H NMR analysis of oxidative rearrangement of 8 in deuterated phosphate buffer: (A) 10 min after addition of NaIO4; (B) 4 h after addition of NaIO4. (C) Independently synthesized HOMedC.
Scheme 3.

Oxidative Rearrangement of 8
Absorption spectroscopy was used to quantitatively compare the [2,3]-sigmatropic rearrangements of the thymidine (2) and 2′-deoxycytidine selenoxides (12). The disappearance of the selenoxides was followed continuously by taking advantage of the blue-shifted absorption spectra of the rearrangement products (3, 13). The selenoxides followed first-order decay.22 However, 2 ((6.1 ± 0.8) × 10−3 s−1, t1/2 = 1.9 min) rearranged approximately 5 times more rapidly than the 2′-deoxycytidine (12) compound ((1.2 ± 0.1) × 10−3 s−1, t1/2 = 9.6 min).
Interstrand Cross-Linking by 5
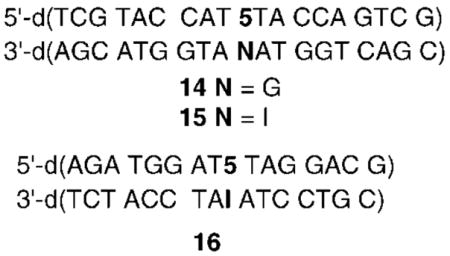
Reaction of 11a or 14 with NaIO4 (5 mM) at 37 °C produced cross-linked product in as high as 47.0 ± 0.5% yield, and the rate of ICL formation at 37 °C followed first-order kinetics (kICL = (1.1 ± 0.2) × 10−4 s−1; t1/2 = 104 min). Cross-linking from 5 is approximately 1 order of magnitude slower than that from the thymidine analogue (1) when it too is flanked by thymidines (kICL = (1.2 ± 0.3) × 10−3 s−1; t1/2 = 9.4 min).12 The molecular weight of the cross-linked product from 11a was determined using ESI-MS.22 This revealed that O2 was not incorporated and that the product resulted from the net loss of benzene selenol from the starting duplex. Insight into the site of electrophilic attack on the opposite strand and the structure of the product was obtained by subjecting duplexes containing 2′-deoxyinosine (dI, 15 and 16) or dA (11b) opposite 5 to the reaction conditions. Although cross-links formed in modest yield when dA (8.2 ± 0.3%) was mispaired opposite 5, the yield of cross-link in a duplex (16) containing dI (52.1 ± 0.5%) was comparable to when dG was present. ESI-MS revealed that the cross-linking product when dI opposed the phenyl selenide (16) also arose from the formal elimination of benzene selenol.22
The nucleotide position(s) at which cross-linking occurred was determined using Hopkins’ hydroxyl radical cleavage method (Figure 2).22,23 As expected, all cross-links in 11a emanated from the position at which 5 is produced (Figure 2A). Analysis of the position of the cross-link in the complementary strand revealed that although the majority of the cross-links involve reaction with the dG opposite 5, 15–20% of the cross-links involve dA9 (Figure 2B). In contrast, labeling the complementary strand of 11a at its 3′-terminus clearly showed that cross-linking did not occur at dA7 (Figure 2C). The lower cross-linking fidelity exhibited by 11a than the thymidine analogue (1)7 was attributed to the reduced nucleophilicity of the opposing nucleotide (dG) and not the lower reactivity of the methide (13) produced from 5. Kinetic experiments and additional product characterization described below attest to this.
Figure 2.
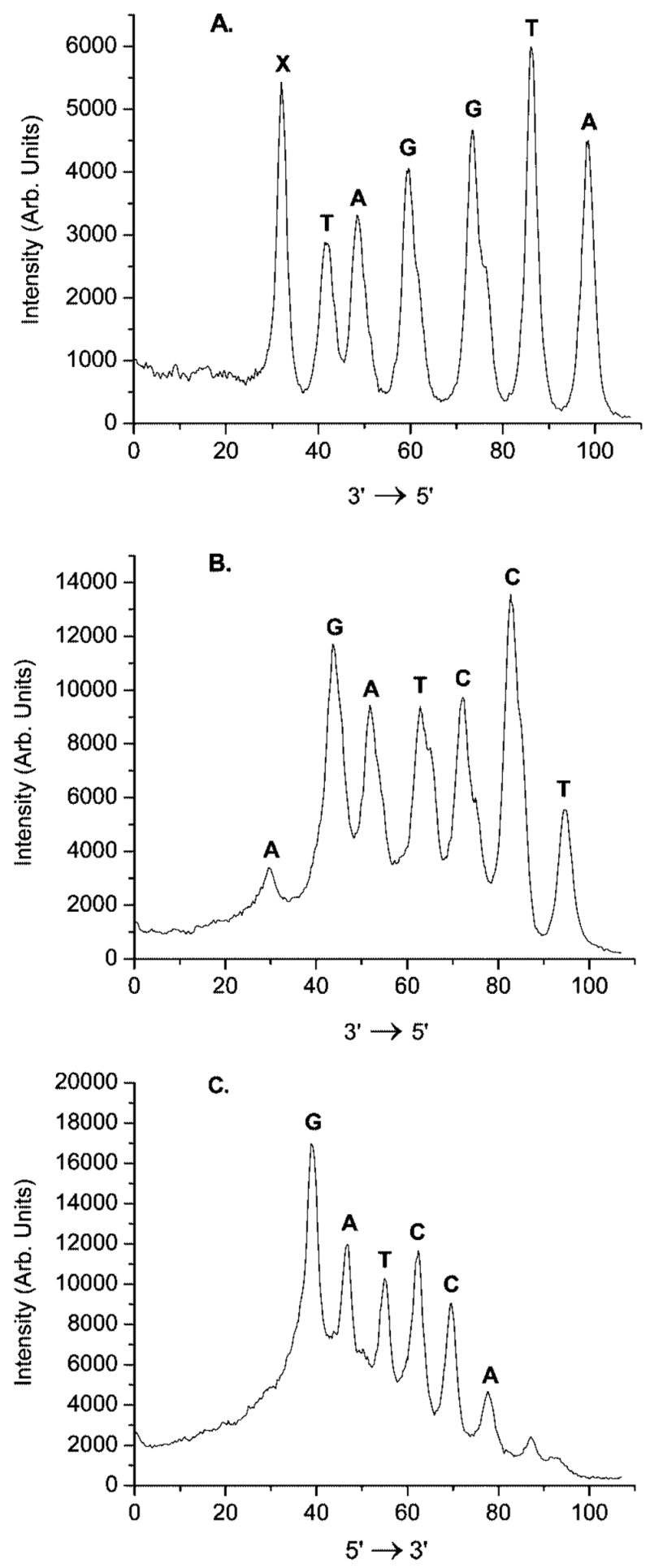
Determination of interstrand cross-linking position via OH• cleavage of NaIO4 treated 11a: (A) 5′-32P-11a, phenyl selenide containing strand labeled; (B) 5′-32P-11a, complementary strand labeled; (C) 3′-32P-11a, complementary strand labeled.
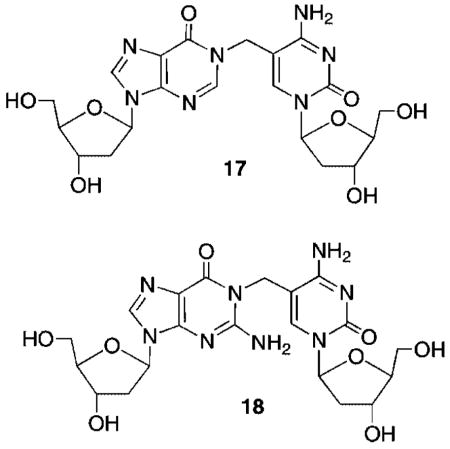
The similarity in cross-linking yields between duplexes containing dG or dI opposite the methide (13) suggested that the electrophile reacted with the N1-position of the purines. This hypothesis was verified upon characterization of the product formed from reaction with dI. Initially, 17 was isolated from a reaction between monomeric 5 and dI. 1H NMR characterization of 17 was consistent with the proposed structure, as evidenced for example by the vinyl proton pattern.22 Formation of 17 in DNA was confirmed by HPLC coinjection, following enzymatic digestion of 16 subjected to NaIO4 treatment. The analogous product (18) from reaction between 13 and dG could not be isolated in pure form in sufficient quantities for NMR structural analysis. However, ESI-MS was consistent with its formation. In contrast to reaction of the thymidine methide (3) with dA, the cross-linked products formed in duplexes in which 13 was opposed by dG or dI were stable upon incubation in the presence of NaN3 (0.3 M) at 37 °C for 5 days. This also is consistent with alkylation at the N1 position of dI or dG by 13, which should give rise to a stable product.24 Reaction at the N2-amino of dG would also give rise to a stable product. Although we cannot rule out reaction at this position, the similarities between reaction with dI and dG suggest that reaction at the N2-amino does not occur.
Flanking Sequence Effects on Cross-Linking in Duplex DNA Containing 5
In contrast to the reactivity of the thymidine analogue (1), we found that the sequence flanking 5 had a significant effect on cross-linking even though NaIO4 induced cross-linking reactions were run at 37 °C instead of 25 °C. Although 5 effectively yielded cross-links when flanked by dT (per above), only 4.0 ± 0.3% ICL was observed when the phenyl selenide was flanked by dA (19). Moreover, no cross-linked product was observed when 5 was surrounded by dC (20) or dG (21) at 37 °C. Because 13 must adopt the syn-conformation in order to react with the opposing strand, local duplex stability was suspected to be the cause of the sequence effect on cross-linking. We postulated that the more stable base pairing with dG than the thymidine analogue opposite dA, as well as flanking sequence effects on duplex stability, affected cross-linking by 5 (and 13). The latter is consistent with the inverse correlation between calculated TM’s (dT < dA < dC < dG)25 for the respective duplexes containing dC instead of 5 and the observed sequence flanking dependence (dT > dA > dC ≈ dG) on cross-linking.
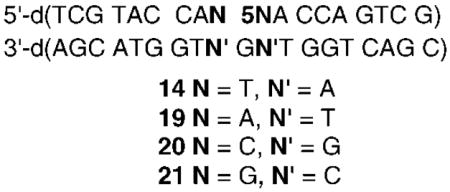
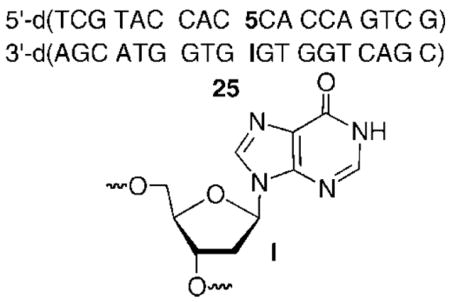
Additional experimental support for the hypothesis that increased base pair stability in the vicinity of 5 adversely affected its ability to react with a nucleotide in the opposing strand was obtained by investigating temperature and sequence effects on cross-linking. Increasing the reaction temperature from 37 to 45 °C increased cross-linking in 19 from 4.0 ± 0.3% to 10.4 ± 0.3% and from <1% to 8.7 ± 0.6% in 20. Moreover, introducing mismatches at the flanking positions resulted in even more significant increases in cross-linking yields at 37 °C (Table 2). The most profound evidence in support of the effect of duplex thermal stability on cross-linking from DNA containing 5 was obtained by substituting dI (15, 25), which forms only two hydrogen bonds with 5 (and 13), for dG opposite the phenyl selenide. High cross-link yields (29.0 ± 1.2%) were observed at 37 °C when dI was flanked by dG (25), whereas a comparable sequence yielded <1% ICL product when 5 was opposite dG (20). In addition, the rate constant for cross-link formation in 25 (kICL = (1.9 ± 0.3) × 10−4 s−1; t1/2 = 61.8 min; 37 °C) was even greater than when 5 was opposite dG and flanked by thymidines (14). However, the rate constant for cross-linking in 25 was ~10-times slower at the same temperature than when the thymidine analogue (1) was flanked by dC (kICL = (1.5 ± 0.2) × 10−3 s−1; 37 °C).12
Table 2.
Effect of Flanking Sequence Mismatches on ICL Formation from 5
| 5′-d(TCG TAC CAA 5AA CCA GTC G)
3′-d(AGC ATG GTN GNT GGT CAG C) | ||
|---|---|---|
| duplex | N | % ICL |
| 19 | T | 4.0 ± 0.3 |
| 22 | G | 18.3 ± 0.4 |
| 23 | C | 38.2 ± 2.6 |
| 24 | A | 49.6 ± 5.6 |
γ-Radiolysis Mediated Cross-Linking
We previously showed that the major pathway for phenyl selenide 1 cross-linking upon γ-radiolysis proceeded through the selenoxide (Scheme 1).11 Consequently, cross-linking due to 137Cs irradiation of duplex DNA containing 5 should parallel the oxidative reaction induced by NaIO4. Indeed, the same local sequence effects on cross-linking by γ-radiolysis were detected as when the substrates were treated with NaIO4. For instance, no cross-linked product was observed when duplexes containing 5 that are flanked by dG or dC were irradiated (data not shown). Furthermore, the rate constants for cross-link formation in 14 (kICL = (1.3 ± 0.1) × 10−4 s−1; t1/2 = 90.9 min; 37 °C) and 25 (kICL = (1.7 ± 0.2) × 10−4 s−1; t1/2 = 69 min; 37 °C) upon exposure to γ-radiolysis were within experimental error of those measured using NaIO4. These experiments not only further verify that cross-linking involves selenoxide formation (12) but also reveal that 5 (unlike 1) will not be useful as a radiosensitizing agent.11
Interstrand Cross-Linking in Triplexes
Inspection of molecular models suggested that incorporating 1 or 5 in a triplex forming oligonucleotide (TFO) that utilizes Hoogsteen base pairing could produce ICLs via the same oxidation/rearrangement mechanism (Scheme 4). The proximity of the electrophile in syn-3 or syn-13 to the nucleophilic N7-position of the purines suggested this position as the likely site of attack when the modified nucleotides adopt the syn-conformation. Hoogsteen triplexes are less stable than Watson–Crick duplexes and are stabilized at slightly acidic pH. Stable triplexes can be formed at physiological pH using synthetically modified nucleotides in the TFO.19–21,26,27 Since our goal was to establish the viability of 1 and 5 as triplex cross-linking agents, for synthetic ease we opted to construct the TFOs using thymidine and 2′-deoxycytidine. This required that we determine that the rearrangement/alkylation chemistry function at pH 5.5 and that triplexes containing the modified pyrimidines be sufficiently stable under these conditions as well.
Scheme 4.

Alkylation in Triplex DNA via Hoogsteen Base Pair Formation
The required cross-linking reaction temperature was determined by measuring the TM’s of triplexes containing 1 and 5 at the 5′-termini or an internal position of a TFO at pH 5.5 (Table 3). In each instance, the triplex containing the modified pyrimidine at an internal position was less stable than that at the 5′-terminus. The lowest TM was 34 °C, leading us to carry out the cross-linking reactions at 25 °C. The compatibility of the ICL process at pH 5.5 (Hepes buffer) was established by examining cross-link formation in duplex DNA. Reducing the pH from 7.2 to 5.5 decreased the ICL yield (25 °C) from 1 in 30 to 19 ± 0.4% from 30.0 ± 1.5%.
Table 3.
UV-Melting Temperatures of Triplexes
| TM(°C) Triplex to Duplex | |
|---|---|
| 5′-d(T CTT C1T TTT TCT CTT T) 5′-d(GTA GAA GAA AAA AGA GAA A)3′-d(CAT CTT CTT TTT TCT CTT T) 26 | 34.0 ± 0.8 |
| 5′-d(1 CTT CTT TTT TCT CTT T) 5′-d(GTA GAA GAA AAA AGA GAA A) 3′-d(CAT CT T CTT TTT TCT CTT T) 27 | 42.0 ± 0.6 |
| 5′-d(T CTT 5TT TTT TCT CTT T) 5′-d(GTA GAA GAA AAA AGA GAA A) 3′-d(CAT CTT CTT TTT TCT CTT T) 28 | 34.7 ± 0.5 |
| 5′-d(5 CTT CTT TTT TCT CTT T) 5′-d(GTG GAA GAA AAA AGA GAA A) 3′-d(CAC CT T CTT TTT TCT CTT T) 29 | 39.0 ± 0.3 |
| 5′-d(AGA TGG AT1 TAG GAC G) 3′-d(TCT ACC TAA ATC CTG C) 30 |
These conditions were utilized for determining the NaIO4 (5 mM) mediated cross-linking of 26–29 at 25 °C (Table 4). No increase in ICL yields was observed when the Mg2+ concentration was increased to 50 mM from 10 mM. ESI-MS analysis of isolated product from reactions of 31 and 29 confirmed the expected coupling between the TFO and the polypurine strand.22 Comparable cross-link yields were obtained when either modified pyrimidine was incorporated at the 5′-termini of the respective TFOs. In addition, the ICL yields in the triplexes were comparable to those measured in the model duplex (30) under the same pH conditions. In contrast, the ICL yield from 1 decreased significantly when it was incorporated at an internal position (26), whereas the reactivity of 13 was unaffected by its position in the oligonucleotide. Although the ICL yields obtained from the TFOs containing 1 or 5 at their 5′-termini were comparable, the rate constant for cross-link formation from 5 in 29 (kICL = (8.6 ± 0.1) × 10−4 s−1; 25 °C) was ~10-fold higher than that from 1 in a comparable triplex (27, kICL = (8.2 ± 0.8) × 10−5 s−1; 25 °C). The relative reactivity of 1 (3) and 5 (13) in triplex DNA is in marked contrast to what was observed in comparable duplexes. The rate constant for cross-linking from 1 situated at the 5′-terminus of the TFO was ~20-fold slower in the triplex than in duplex DNA, whereas the rate constant for ICL formation from 5 increased approximately 8-fold.
Table 4.
Interstrand Cross-Link (ISC) Yields from 1 and 5 in Triple Helical DNA
| % yield ICLa |
|||
|---|---|---|---|
| triplex (phenyl selenide) | NFTb | Δc | Piperidined |
| 26 (1) | 4.0 ± 0.1 | n.d. | 3.6 ± 0.3 |
| 27 (1) | 20.0 ± 0.5 | 2.5 ± 0.6 | 10.0 ± 1.0 |
| 28 (5) | 24.0 ± 0.3 | n.d. | 1.0 ± 0.1 |
| 29 (5) | 25.0 ± 0.5 | 2.2 ± 0.4 | 1.3 ± 0.2 |
| 31 (1) | 13.0 ± 1.0 | 3.0 ± 0.1 | 12.0 ± 0.5 |
| 32 (5) | 11.5 ± 0.5 | 1.0 ± 0.1 | 1.0 ± 0.1 |
% Yields are the average of at least two independent experiments.
NFT = no further treatment after reaction with NaIO4.
90 °C for 20 min at pH 7.2.
1.0 M Piperidine, 90 °C, 20 min.
The differences in reactivity between 1 and 5 in triplex DNA were contrary to expectations based upon the anticipated respective barriers to rotation about their glycosidic bonds. However, the 2′-deoxycytidine derivative should be more electrophilic under the conditions if it is protonated. In addition, reaction at the N7-purine position of dG should be more facile due to its greater nucleophilicity compared that of dA.28 Although these properties help rationalize why 5 is more effective overall at forming ICLs in the triplexes, it does not explain the effect of position in the TFO on the reactivity of the TFOs containing 1.
In order to provide further insight into the reactivity of the TFOs containing 1 and 5, we examined the stability and reactivity of the ICL products. The stability and alkaline lability of DNA alkylation products depend upon the reaction site. The cross-links formed from 1 were almost completely destroyed upon heating at 90 °C (pH 7.2) for 25 min, which is consistent with reaction at N7- or N1- of dA, but ruled out reaction of 3 with the N6-amino group. In addition, reaction at N3-dA was considered unlikely, as that nucleophile is in the minor groove. The cross-linked products derived from 5 were also almost completely decomposed upon heating, which is consistent with reaction occurring at the N7-position of dG as well. In contrast, the cross-links derived from 1 and 5 exhibited different levels of stability to 1 M piperidine treatment (90 °C, 20 min). Mild alkaline hydrolysis of N7-alkylated purines produces formamidopyrimidines, which are labile to the harsher piperidine conditions.29 Hence, if N7-dA and N7-dG were the sites attacked by 3 and 13, respectively, the cross-links should decompose upon treatment with piperidine. Surprisingly, piperidine treatment of the ICLs derived from 1 in 27 produced ~50% cleavage. In addition, a small amount of a product that migrated even faster than γ-32P-ATP in 20% denaturing PAGE was observed.22 This product was also observed when cross-linked 29 was treated with piperidine. In addition, the ICL formed from 29 was almost completely destroyed by piperidine treatment. The formation of a radioactive product that migrated even more rapidly than γ-32P-ATP suggested that 3 and 13 reacted with the 5′-terminal dG in the purine strand of the duplexes (27 and 29). N7-Alkylation of the terminal dG would yield the alkylated formamidopyrimidine under the alkaline conditions. Concomitant cleavage of this lesion would ultimately result in the release of the 5′-terminal 32P-phosphate (Figure 3). Alternatively, deglycosylation of the N7-alkylated product would yield an abasic site directly, which would also release the radiolabeled phosphate upon alkaline hydrolysis.
Figure 3.

Phosphate release upon alkaline treatment following N7-alkylation of the 5′-terminal dG in triple helical DNA. (A) Cartoon illustration of alkylation of the 5′-terminal dG by 13. (B) Phosphate release upon alkaline treatment (* = 32P).
The hypothesis that the methides produced at the 5′-termini of TFOs could react at a distal dG was tested by substituting thymidine for the 5′-terminal dG in triplexes 27 and 29. Indeed, subjection of cross-linked 32 to either heating or piperidine resulted in almost complete destruction of the product. However, free phosphate was not detected following alkaline treatment. These observations suggest that 13 produced in triplex 32 did not react at the 5′-terminal thymidine. The lability of the ICL to alkaline conditions or heat in phosphate buffer is consistent with reaction at N7-of the opposing dG, as predicted by simple modeling of the Hoogsteen pairing (Scheme 4). Incorporation of thymidine at the 5′-terminus of the polypurine-containing strand in 31 also clarified the reactivity of 3 in triple helical DNA. As in the experiments with 26 and 27, heating cross-linked 31 destroyed almost all of the ICL (Table 4). However, reaction with piperidine had essentially no effect on the ICL produced in 31. These data suggest that although alkylation of the opposing dA by 3 produces a product with a labile glycosidic bond, the resistance to cleavage by alkali indicates that N7-dA is not the position attacked because the respective formamidopyrimidine formed under these conditions should be labile to piperidine.29 Of the other nitrogens on dA, only reaction at N1 would produce a product with the observed chemical behavior due to its ability to isomerize to an N6-alkylamino derivative (Scheme 5).7,30,31 Although reaction at N1-dA is unexpected based upon inspection of molecular models, this nitrogen is more nucleophilic than the N7-dA.28 Reaction at N1-dA requires that the Watson–Crick duplex undergo local melting. This conformational restriction helps explain why the rate constant for cross-linking by 3 is so much slower in the triplex than in the duplex and even slower than the reaction of 13. It may also help explain why the ICL yield in 26 is so much lower than that in 27. Placing the phenyl selenide at the 5′-terminus of the TFO enables it to react with the dG of the duplex not involved in triplex formation. In addition, fraying of the TFO in triplex 27 facilitates the required conformational change of the opposing dA. In comparison, the internal methide (3) in 26 may in effect be more rigid, making it more difficult for the dA to undergo the rotation necessary to enable N1 to react with the electrophile.
Scheme 5.

Effects of Alkali Treatment and Heat on N1-dA:3 Cross-Link

Conclusions
Oligonucleotides containing phenyl substituted derivatives of thymidine (1) and 5-methyl-2′-deoxycytidine (5) form stable Watson–Crick duplexes and triplexes via Hoogsteen base pairs. The phenyl selenides undergo rapid oxidation to selenoxides (2, 12), followed by [2,3]-sigmatropic rearrangements to electrophilic methides (3, 13). The rearrangements are facile at 25 °C, with the slower one (12 → 13) occurring with a < 10 min half-life. Adoption of the respective syn-conformations enables 3 and 13 to cross-link with the opposing strand. Interstrand cross-link formation from 5 (13) in duplex DNA is significantly affected by a flanking sequence when it is opposite dG. Varying temperature, introducing mismatches, and most significantly replacing the opposing dG with dI reveal that stronger hydrogen bonding between 5 (13) and dG compared to 1 (3) and dA increases the importance of the flanking sequence (and its commensurate effect on TM) on cross-linking, presumably by increasing the barrier faced by 13 to adopt the syn-conformation. While this limits the utility of 5 as a cross-linking agent in duplex DNA, it opens the possibility of using it in other applications. For instance, the presence of the methide in the major groove potentially provides a tool for probing protein–DNA cross-linking.
The barriers to conformational isomerization do not distinguish the reactivity of 1 and 5 from one another when they are incorporated into triplex forming oligonucleotides (TFOs) that recognize purines via Hoogsteen base pairing. The syn-isomers of the respective methides are well positioned to react at N7 of the opposing purine. The 2′-deoxycytidine derivative (5) produces cross-links via reaction at this position with the opposing dG. In contrast, the greater nucleophilicity of the N1-position of adenine overcomes the requirement to break its hydrogen bonds with the opposing dT to react with 3 in the TFO. Although cross-linking from 5 occurs equally well when it is incorporated at terminal or internal positions of the TFO, the required local melting of the duplex to form interstrand cross-links with 3 limits the yield of this covalent modification when the modified nucleotide is incorporated at an internal position of the TFO. In addition, the triple helix cross-linking reactions were carried out under nonoptimal pH conditions for ICL formation in order to stabilize the triplexes. Based upon our results in duplexes, we expect that higher ICL yields will be obtained using modified TFOs that are compatible with carrying out the experiments at physiological pH. Overall, the phenyl selenide derivatives provide a means for efficient cross-linking under mild reaction conditions. In addition, the requirement of a mild oxidant allows for temporal control of the cross-linking reaction.
Supplementary Material
Complete description of experimental procedures. NMR spectra of synthetic molecules, ESI-MS of cross-linked products, UV–vis spectra describing selenoxide rearrangements, HPLC analysis of enzyme digests, and autoradiograms of cross-linking reactions. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We are grateful for support of this research by the National Institute of General Medical Sciences (GM-054996). This research was supported in part by the Intramural Research Program of the NIH, National Institute on Aging.
References
- 1.Gates KS, Nooner T, Dutta S. Chem Res Toxicol. 2004;17:839–856. doi: 10.1021/tx049965c. [DOI] [PubMed] [Google Scholar]
- 2.Veldhuyzen WF, Parde P, Rokita SE. J Am Chem Soc. 2003;125:14005–14013. doi: 10.1021/ja036943o. [DOI] [PubMed] [Google Scholar]
- 3.Noll DM, Mason TM, Miller PS. Chem Rev. 2006;106:277–301. doi: 10.1021/cr040478b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou Q, Rokita SE. Proc Nat Acad Sci USA. 2003;100:15452–15457. doi: 10.1073/pnas.2533112100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nagatsugi F, Kawasaki T, Usui D, Maeda M, Sasaki S. J Am Chem Soc. 1999;121:6753–6754. [Google Scholar]
- 6.Kawasaki T, Nagatsugi F, Ali MM, Maeda M, Sugiyama K, Hori K, Sasaki S. J Org Chem. 2005;70:14–23. doi: 10.1021/jo048298p. [DOI] [PubMed] [Google Scholar]
- 7.Hong IS, Greenberg MM. J Am Chem Soc. 2005;127:3692–3693. doi: 10.1021/ja042434q. [DOI] [PubMed] [Google Scholar]
- 8.Hong IS, Greenberg MM. J Am Chem Soc. 2005;127:10510–10511. doi: 10.1021/ja053493m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hong IS, Ding H, Greenberg MM. J Am Chem Soc. 2006;128:485–491. doi: 10.1021/ja0563657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fischhaber PL, Gall AS, Duncan JA, Hopkins PB. Cancer Res. 1999;59:4363–4368. [PubMed] [Google Scholar]
- 11.Hong IS, Ding H, Greenberg MM. J Am Chem Soc. 2006;128:2230–2231. doi: 10.1021/ja058290c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peng X, Greenberg MM. Nucleic Acids Res. 2008;36:e31. doi: 10.1093/nar/gkn052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Povsic TJ, Strobel SA, Dervan PB. J Am Chem Soc. 1992;114:5934–41. [Google Scholar]
- 14.Ferentz AE, Keating TA, Verdine GL. J Am Chem Soc. 1993;115:9006–14. [Google Scholar]
- 15.Osborne SE, Cain RJ, Glick GD. J Am Chem Soc. 1997;119:1171–1182. [Google Scholar]
- 16.Coleman RS, Pires RM. Nucleic Acids Res. 1997;25:4771–4777. doi: 10.1093/nar/25.23.4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glick GD. In: Current Protocols in Nucleic Acid Chemistry. Beaucage SL, editor. Vol. 2. John Wiley & Sons, Inc.; New York: 2003. pp. 5.7.1–5.7.13. [Google Scholar]
- 18.Li H, Broughton-Head VJ, Peng G, Powers VEC, Ovens MJ, Fox KR, Brown T. Bioconjugate Chem. 2006;17:1561–1567. doi: 10.1021/bc0601875. [DOI] [PubMed] [Google Scholar]
- 19.Puri N, Majumdar A, Cuenoud B, Natt F, Martin P, Boyd A, Miller PS, Seidman MM. Biochemistry. 2002;41:7716–7726. doi: 10.1021/bi025734y. [DOI] [PubMed] [Google Scholar]
- 20.Puri N, Majumdar A, Cuenoud B, Miller PS, Seidman MM. Biochemistry. 2004;43:1343–1351. doi: 10.1021/bi035808l. [DOI] [PubMed] [Google Scholar]
- 21.Nagatsugi F, Sasaki S, Miller PS, Seidman MM. Nucleic Acids Res. 2003;31:e31. doi: 10.1093/nar/gng031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.See Supporting Information.
- 23.Millard JT, Weidner MF, Kirchner JJ, Ribeiro S, Hopkins PB. Nucleic Acids Res. 1991;19:1885–1892. doi: 10.1093/nar/19.8.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weinert EE, Frankenfield KN, Rokita SE. Chem Res Toxicol. 2005;18:1364–1370. doi: 10.1021/tx0501583. [DOI] [PubMed] [Google Scholar]
- 25. HyTher is available at: http://ozone2.chem.wayne.edu/Hyther/hytherm1main.html.
- 26.Singleton SF, Dervan PB. Biochemistry. 1992;31:10995–1003. doi: 10.1021/bi00160a008. [DOI] [PubMed] [Google Scholar]
- 27.Povsic TJ, Dervan PB. J Am Chem Soc. 1989;111:3059–61. [Google Scholar]
- 28.Bloomfield VA, Crothers DM, Tinoco I., Jr . Nucleic Acids Structures, Properties, and Functions. University Science Books; Sausalito, CA: 2000. [Google Scholar]
- 29.Haraguchi K, Delaney MO, Wiederholt CJ, Sambandam A, Hantosi Z, Greenberg MM. J Am Chem Soc. 2002;124:3263–3269. doi: 10.1021/ja012135q. [DOI] [PubMed] [Google Scholar]
- 30.Jones JW, Robins RK. J Am Chem Soc. 1963;85:193–201. [Google Scholar]
- 31.Chang C-j, DaSilva J, Byrn SR. J Org Chem. 1983;48:5151–5160. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Complete description of experimental procedures. NMR spectra of synthetic molecules, ESI-MS of cross-linked products, UV–vis spectra describing selenoxide rearrangements, HPLC analysis of enzyme digests, and autoradiograms of cross-linking reactions. This material is available free of charge via the Internet at http://pubs.acs.org.