

Summary
ISO 15189 5.5.1 “The laboratory shall use examination procedures, … which meet the needs of the users of laboratory services and are appropriate for the examinations.”
Requirements for analytical quality include:
understanding the analytical goal
seeking an assay that fulfills those goals
establishing your own performance with that assay
setting warning and action limits for your assay
applying quality control tools to every important step.
Introduction
The first characteristic of good analysis is that we know exactly what we are measuring – ‘the measurand’. Ideally we should also know what cross reactants there are in the assay so that these can be taken into account if necessary. The second characteristic is that we know how to express the amount measured in a standardised manner. Ideally this would be expressed in SI units and traceable to a definitive method.
The final characteristic of good quantitative analysis is that we can repeatedly get the same result. It is generally impossible to get exactly the same result every time, but it is very important that we think about how close those repeated measurements should be to each other. This is the quality specification that defines whether our measurement is worthwhile.
Strategies to Define Quality Specifications
I was fortunate enough to attend the 1999 consensus meeting in Stockholm on analytical quality specifications that brought together the World Health Organization (WHO), International Federation of Clinical Chemistry (IFCC), International Union of Pure and Applied Chemistry (IUPAC) and members of the International Standards Organization Technical Committee 212 (ISO TC212). The proceedings of this meeting were published together with a consensus on strategies to set global quality specifications in laboratory medicine.1 The consensus was largely concerned with enumerating the different approaches available and then defining a hierarchy of these approaches from worst to best (Table 1). I will start by addressing the poorest approach which seems paradoxically categorised as ‘state of the art’.
Table 1.
Stockholm consensus of the hierarchy of strategies to set global quality specifications in laboratory medicine.
| Level | Approach | Advantage | Disadvantage |
|---|---|---|---|
| 1 | Clinical Outcome | Based on what will happen to patient | Studies rarely available |
| 2a | Clinical Survey | Based on what doctors will do | Doctor’s action may not affect patient |
| 2b | Biological Variability | Based on improving signal to noise ratio | Variability may differ between patient groups |
| 3 | Expert Opinion | Based on best experience available | May still not know what is achievable |
| 4 | EQA / Proficiency Testing | Based on what is routinely achievable | What is achievable may not be good enough |
| 5 | State of the Art | Based on what others tell us they achieve | May not be routinely achievable or adequate |
State of the Art Approach
This approach requires an assessment of available information from other laboratories where the analytical performance has been measured. This may have been published or provided through other means of personal communication. If this is the best that other laboratories can achieve then we should aim for at least the same standard. The problem with this approach is that we may have been told the best possible performance under optimal conditions rather than typical performance expected with routine patient samples. However, the main problem is that we do not explicitly know if this ‘state of the art’ performance is actually good enough to fulfil the medical need for the test.
External Quality Assurance (EQA) Approach
EQA (or proficiency testing), also provides an assessment of the state of the art, however at least because of its external nature, it tends to be an objective measure. Furthermore if EQA samples are handled as if they were patient specimens (what should happen) the state of the art represents what is usually achieved rather than what may be optimally achieved. However, once again, we still cannot be sure if the typical performance in EQA (e.g. median performance) or the highest level of performance in EQA (e.g. 20th centile) is sufficient to fulfil the medical need for that test.
Expert Opinion Approach
Professional societies often form advisory groups to develop guidelines, including guidelines on what is an acceptable analytical performance. When we consider the qualifications and expertise of such committees, it is surprising to think that this would not be considered the best approach available. The problem here is that even the best experts will often form their opinions around what is achievable rather than what is required. Ideally the experts should state the rationale they used to form their consensus opinion.
Stated another way, how could we know that a higher level of performance is practically required if this is rarely or never achieved? Conversely, and almost just as bad, is that when an extremely high level of performance is achieved by most laboratories, experts are likely to set the standard at this level rather than letting it slip, even if there is little evidence that this has any clinical advantage.
Impact on Patient Classification Clinical Survey Approach
The first way to consider this patient classification approach is by surveying clinicians. We could ask them what observed change in a test result would lead to a change in their diagnosis or patient management. By defining this (consensus) value, we would have defined the analytical specification of our assay because if such an observed change was solely due to our analytical error, the doctor would be treating our results rather than the patient!
Impact on Patient Classification Biological Variability Approach
Another way of ensuring that the result we provide is more patient signal than analytical noise is by knowing what the usual variability in the patient signal is. There is a wealth of literature on the usual day to day biological variability of measurements in healthy patients. We have assumed for many years that this variation in stable health is similar in stable disease. For example a healthy patient may vary their ALT between 10 and 20 IU/L while a relatively stable individual with chronic hepatitis C may vary their level between 100 and 200 IU/L, which is the same relative variability. Recent literature has shown that the variation in disease is often slightly greater than that in health,2 but this may be because of the awkward expectation of such an entity as stable disease. Anyway the point is that we do know the biological variability of almost every analyte in the laboratory, so we should try to ensure that the analytical noise is less than this variation and that the changes in patient results are more likely to be due to patient changes than analytical changes. Cotlove et al. described this principle many years ago as an imprecision requirement where CVA (analytical) < ½ CVI (intraindividual biological variability).3 Fraser has refined this simple approach to include the desirable bias characteristics so that patient groups (e.g. healthy and sick) can be distinguished from each other.4
While application of either the clinician survey or biological variability will ensure that our analytical errors will not lead to a change in the way we classify patients (e.g. healthy/sick or stable/deteriorating/improving), we cannot be sure that either approach will actually lead to an improved clinical outcome.
Clinical Outcome Approach
The highest level of the hierarchy to define analytical quality specifications is based on the analytical performance required to manage an improved clinical outcome. The best example of this is the Diabetes Control and Complications Trial (DCCT) for HbA1c 5 where we know that patients with a HbA1c greater than 8.0% have increased risk of diabetic complications and those with HbA1c below 7.0% do not. Clearly we must be able to distinguish these patients, otherwise their clinical outcome could be affected. Our analytical imprecision (and bias) should never allow HbA1c results of 7.0% and 8.0% to be interchanged.
Unfortunately these sorts of clinical outcome studies are very few and we have to rely on the next level down. Clinical surveys are also fairly rare but, as already stated, biological variability figures are known for almost all common clinical laboratory analytes. Biological variability has therefore become the highest level and most easily defined approach available to the laboratory.
Table 2 lists desirable imprecision goals defined by biological variability together with what is achieved by the 20th, 50th and 90th centile of laboratories in Australasia. We can see that most laboratories achieve desirable imprecision for most analytes.
Table 2.
Performance of laboratories in a 2007 Australasian EQA program compared to desirable imprecision goals based on biological intraindividual variability.6 EQA data reproduced with permission from the RCPA Chemical Pathology QAP.
| Analyte | Biological Intraindividual Variability (CVI) | Desirable Imprecision (½CVI) | 2007 EQA Performance (centiles)
|
||
|---|---|---|---|---|---|
| 20th | 50th | 90th | |||
|
| |||||
| Imprecision (CVA) | |||||
| Iron | 26.5% | 13.3% | 2.0% | 2.9% | 6.7% |
| ALT | 24.3% | 12.2% | 2.2% | 2.9% | 4.9% |
| Lactate | 27.2% | 13.6% | 2.6% | 3.6% | 8.4% |
| Bilirubin | 25.6% | 12.8% | 2.5% | 3.4% | 6.0% |
| Lipase | 23.1% | 11.6% | 2.7% | 4.1% | 10.9% |
| Creatine Kinase | 22.8% | 11.4% | 3.2% | 4.3% | 10.7% |
| Triglyceride | 21.0% | 10.5% | 3.2% | 4.0% | 6.7% |
| GGT | 13.8% | 6.9% | 2.0% | 2.8% | 4.9% |
| AST | 11.9% | 6.0% | 2.1% | 2.8% | 5.3% |
| Amylase | 9.5% | 4.8% | 1.9% | 2.6% | (7.5%) |
| Phosphate | 8.5% | 4.3% | 1.8% | 2.4% | (4.3%) |
| Cholesterol | 6.0% | 3.0% | 1.7% | 2.3% | (4.0%) |
| LDH | 6.6% | 3.3% | 1.9% | 2.6% | (4.4%) |
| Potassium | 4.8% | 2.4% | 1.6% | 2.0% | (2.9%) |
| Glucose | 6.5% | 3.3% | 2.1% | 2.8% | (4.4%) |
| Ferritin | 14.9% | 7.5% | 4.6% | 6.7% | (13.3%) |
| HDL Cholesterol | 7.1% | 3.6% | 2.3% | 3.3% | (6.0%) |
| ALP | 6.4% | 3.2% | 3.0% | (4.4%) | (7.3%) |
| Creatinine | 4.3% | 2.2% | (2.5%) | (3.4%) | (5.5%) |
| Magnesium | 3.6% | 1.8% | (2.3%) | (3.0%) | (4.9%) |
| Protein | 2.7% | 1.4% | (1.7%) | (2.3%) | (3.7%) |
| Albumin | 3.1% | 1.6% | (2.1%) | (2.8%) | (4.9%) |
| Transferrin | 3.0% | 1.5% | (2.1%) | (2.8%) | (5.0%) |
| Chloride | 1.2% | 0.6% | (1.0%) | (1.3%) | (2.5%) |
| Calcium | 1.9% | 1.0% | (1.7%) | (2.2%) | (3.6%) |
| Bicarbonate | 4.0% | 2.0% | (3.4%) | (4.8%) | (8.0%) |
| Sodium | 0.7% | 0.4% | (0.9%) | (1.1%) | (2.0%) |
EQA results in brackets indicate where that percentage of laboratories failed to achieve the desirable imprecision goal according to biological variability.
Establishing your Quality Specifications
Using the hierarchies listed in Table 1, laboratories should define the level of performance required for each particular analyte. When choosing an assay, or an analytical system, they should focus on those analytes that are difficult to measure (e.g. sodium and calcium), rather than wasting effort on analytes that are done very well by most methods (e.g. total bilirubin and triglycerides). It is easy to forget in our costconscious laboratories there are simple ways of improving imprecision such as by running duplicates
Establishing your Analytical Quality Performance
The underlying principle of ‘measurement uncertainty’ is that a laboratory should know how precisely they can measure any particular analyte. There are three important reasons for this:
So that they can be sure that their performance fulfills their a priori quality specification as discussed above.
So that this information is available to the laboratory user (i.e. clinician) as they may need to take the analytical ‘noise’ into account when managing their patient on the basis of any particular laboratory result.
So that they can monitor their analytical performance on a day to day basis to ensure that they are maintaining their stated performance. Furthermore on this point, so that they can quickly understand which factors are most likely to have caused an analytical failure.
There are a few ways of obtaining the analytical performance expectation for your analytical method:
Manufacturers’ kit inserts may contain a reasonable estimate of the expected performance of their method. However, apart from salesmanship, there are many reasons why the stated performance may not be reliable. For example, was the performance measured in a laboratory as busy as yours with the same quality of staff? Furthermore are your instruments maintained and operated exactly like those used elsewhere? The manufacturers’ kit inserts should only be used as a rough guide.
EQA programs often list the various methods in the program and their observed performance. This is a little more reliable but the two main flaws are the few values that some EQA programs use to calculate imprecision and the unsupported expectation that your laboratory will be like the typical laboratory in the program.
Commercial QC material is often supplied not only with the expected mean values obtained by different methods for that lot of material, but also the expected variability observed for each of those methods. Unfortunately, the ‘expected imprecision’ figures for QC material are often very wide. There are a number of possible reasons for this, apart from avoiding a frustrated customer whose ‘QC results are always out with this new material’. The more valid reasons are that they may have been derived over several instruments or in several laboratories with widely varying operating conditions.
Doing it for yourself is, as usual, the safest method. It does take time and effort to repeat the analyses over and over again in your own laboratory but there are many easily available guidelines for this. An important point to remember is the more often you repeat, the better your estimate will be. Table 3 shows the increase in your estimate of measurement uncertainty due to not having enough data points. You can see that there are large gains in going up to 30 repetitions but little to gain by going to 50. Interestingly, what is not included in the table is that insufficient data points will generally cause you to underestimate your CVA because you haven’t given yourself the opportunity to pick up the more outlying variations.
Table 3.
Table showing the expected increase in measurement uncertainty through not having enough repeat data points.
| Repeats N | Percent Increase in Measurement Uncertainty |
|---|---|
| 2 | 76% |
| 3 | 52% |
| 4 | 42% |
| 5 | 36% |
| 10 | 24% |
| 20 | 16% |
| 30 | 13% |
| 50 | 10% |
Referring to Figure 1, it is generally considered ‘acceptable’ to be performing above the minimum standard but below the optimal standard. It is ‘desirable’ to be performing as close to the optimal standard as you can. It is similarly ‘undesirable’ to be performing close to the minimum standard as a further small deterioration in performance will lead to an ‘unacceptable’ performance.
Figure 1.

Graphical representation of quality standards. Warnings and failures of analytical performance.
Unacceptable performance may be below the minimum standard for imprecision that your review of quality specifications indicated was required for patient care, and therefore may result in adverse patient outcomes.
Alternatively, the minimum standard may be the analytical performance you have stated to your laboratory users that you will maintain (your measurement uncertainty for that analyte). Allowing analytical performance outside your measurement uncertainty breaks your agreement with laboratory users and may lead to an incorrect expectation that could also affect patient care.
Setting the Minimum Standard for Analysis
The traditional performance of analytical internal quality control uses the principle of warning and action. If a discrepant QC result is unlikely to be due to chance alone (e.g. outside the 95% confidence intervals of ±2 CVAs), then this should be a warning to investigate. You may be too close to the minimum standard. However, if the result is almost impossibly due to chance alone (e.g. outside the 99% confidence interval of ±3 CVAs), then action should be taken to hold back results as you already suspect you are below your minimum standard.
The 2 CVA or 3 CVA approach to the minimum standards commonly used in a Levey Jennings approach to QC is more closely associated, according to the hierarchy of strategies (Table 1) with levels 4 and 5, i.e. ‘what is achievable’ rather than what is required.
An alternative approach may be to apply level 3 or an expert defined limit. Many examples exist including the ±20% minimum standard for TSH measurement (especially applied to the lower limits of quantitation), the ±10% minimum standards for troponin measurement (also especially applied to the lower limits of 99th centile of upper reference limit). Other examples include the National Cholesterol Education Program (NCEP) imprecision goals for cholesterol (3.0%) and triglyceride (5.0%), or the American Diabetes Association (ADA) imprecision goal for glucose (5.0%).
Higher levels of definition are available at level 2, and Fraser has defined the ‘minimum standard’ for imprecision based on biological variability as ¾ of the biological CVI.6 It is not surprising that the TSH experts say a maximum imprecision of ±20% is required while the intraindividual biological variability for TSH is almost the same (CVI = 19.7%). Figure 2 also shows the inter-relationship of the biological variability approach with observed CVA traditionally applied to the total error limits of the Levey Jennings graph.
Figure 2.
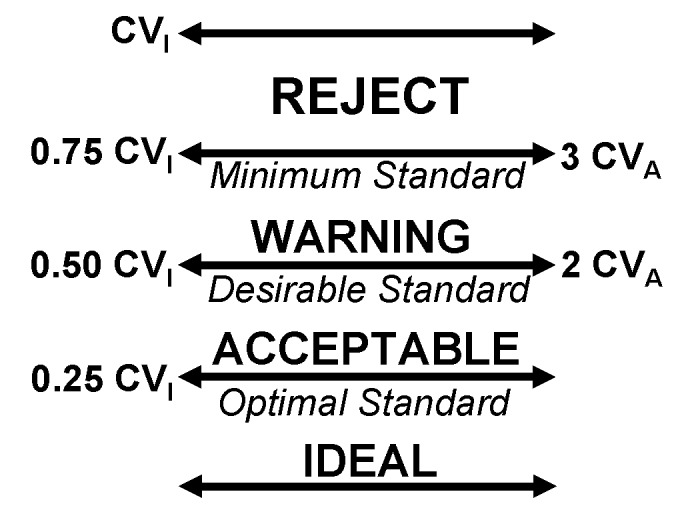
The inter-relationship between optimal / desirable / minimum standards based on biological variability and total error limits traditionally applied to Levey Jennings graphs.
A special case deserves specific mention. When analytical imprecision is far better than biological requirements, on the one hand there may be a larger degree of safety that the failing method imprecision is unlikely to lead to patient mismanagement. However on the other hand, a 3 CVA failure is still a warning that the method is clearly not behaving as it should and therefore there should be little confidence that the next patient analysis will perform predictably.
Definition of minimum standards could also be based on equivalent or higher levels in the hierarchy including the surveyed opinion of clinicians or clinical outcome studies. We have already mentioned the example of distinguishing the DCCT trial’s HbA1c values of 7.0% and 8.0%. For a laboratory to be able to confidently separate these two values requires a CVA that is 2.8 times smaller (i.e. √2 * 1.96SD = 2.78) or between 2 and 3%.
Quality Control Procedures
Quality control procedures in the laboratory help to maintain the expected quality of analysis. Although usually considered to be totally concerned with running internal QC material, they are much more and can be classified into three categories; preanalytical (input monitoring), analytical (process monitoring) and postanalytical (product monitoring).7
Preanalytical Quality Control Procedures
We know that to cook a good meal you usually need good ingredients. Controlling the ‘inputs’ to analysis similarly helps to guarantee a good result. These ingredients include not only good reagents (in date, stored appropriately and correctly reconstituted), but also such obvious things as similarly reliable calibrators and water purity. Furthermore the inputs include properly maintained and checked equipment and properly trained staff. These are vitally important issues that laboratory inspectors expect evidence of. Finally the sample is probably the most important analytical input and ensuring that a correctly identified sample is properly transported, thoroughly centrifuged, appropriately stored and also checked for significant contamination (e.g. haemolysis) prior to analysis, are all vital steps in assuring the quality of results of analysis.
Analytical Control Procedures
Most analysis that we perform today is highly automated with complex analysers containing hundreds of moving parts as well as complex microprocessor controls. Unfortunately, despite the ever increasing cost of these analysers we cannot be confident that they will always work as expected. We therefore need to occasionally check that a sample with an expected result has given an appropriate response (± measurement uncertainty) while we are releasing patient results. This sample with an expected result may be from a previous run, a patient pool that we have prepared or a synthetic patient pool in the form of a commercial quality control serum. Duplicate measurements can also be used to see if the method imprecision is failing but will not necessarily pick up bias drifts.
Postanalytical Control Procedures
Even once the result has been produced with high quality inputs and analytical quality control procedures that are ‘incheck’, there are still some tricks that we have up our sleeve to try to ensure this result makes sense. If it is a high result, are all of the results high on this run (the patient average has shifted upwards)? Was the patient’s previous result also high or have they had an unexpectedly large change (‘delta’)? Finally do they have other results or clinical notes that support the likelihood that these results are real? If these observations are incompatible, it is better to check the analysis than risk releasing this inconsistency for the clinician to cope with.
Conclusion
Analytical performance should ideally be designed to match the clinical application of the test rather than settling for what is generally available. Laboratories should be able to confirm their measurement uncertainty and have this information available to laboratory users. Quality control tools include preanalytical (input control) and postanalytical (product control) tools, which together with traditional methods of analytical quality process control help to ensure reliable results performing according to expectations. These tools may either warn that the minimum acceptable standard is being approached or may demand the action that the results are not released until confidence in analysis can be restored. The more familiar laboratory staff are with the major contributions to their assay’s measurement uncertainty, the less commonly unacceptable results will be missed and the more quickly assay performance will be addressed.
Footnotes
Competing Interests: None declared.
References
- 1.Kenny D, Fraser CG, Hyltoft Petersen P, Kallner A. Consensus Agreement: Conference on strategies to set global quality specifications in laboratory medicine. Scand J Clin Lab Invest. 1999;59:585. [PubMed] [Google Scholar]
- 2.Ricos C, Iglesias N, Garcia-Lario JV, Simon M, Cava F, Hernandez A, et al. Within-subject biological variation in disease: collated data and clinical consequences. Ann Clin Biochem. 2007;44:343–52. doi: 10.1258/000456307780945633. [DOI] [PubMed] [Google Scholar]
- 3.Cotlove E, Harris EK, Williams GZ. Biological and analytical components of variation in long-term studies of serum constituents in normal subjects. 3. Physiological and medical implications. Clin Chem. 1970;16:1028–32. [PubMed] [Google Scholar]
- 4.Fraser CG, Petersen PH, Ricos C, Haeckel R. Proposed quality specifications for the imprecision and inaccuracy of analytical systems for clinical chemistry. Eur J Clin Chem Clin Biochem. 1992;30:311–7. [PubMed] [Google Scholar]
- 5.The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes. The Diabetes Control and Complications Trial Research Group. N Engl J Med. 1993;329:977–86. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 6.Fraser CG. Biological Variability: From Principles to Practice. Washington, DC, USA: AACC Press; 2001. [Google Scholar]
- 7.Hinckley CM. Defining the best quality control systems by design and inspection. Clin Chem. 1997;43:873–9. [PubMed] [Google Scholar]