

Abstract
Childhood absence epilepsy (CAE) is an idiopathic generalised epilepsy characterised by absence seizures manifested by transitory loss of awareness with 2.5-4Hz spike-wave complexes on ictal EEG. A genetic component to aetiology is established but the mechanism of inheritance and the genes involved are not fully defined. Available evidence suggests that genes encoding brain expressed voltage-gated calcium channels, including CACNG3 on chromosome 16p12-p13.1, may represent susceptibility loci for CAE. The aim of this work was to further evaluate CACNG3 as a susceptibility locus by linkage and association analysis. Assuming locus heterogeneity, a significant HLOD score (HLOD=3.54, α=0.62) was obtained for markers encompassing CACNG3 in 65 nuclear families with a proband with CAE. The maximum NPL score was 2.87 (p<0.002). Re-sequencing of the coding exons in 59 patients did not identify any putative causal variants. A linkage disequilibrium (LD) map of CACNG3 was constructed using 23 single nucleotide polymorphisms (SNPs). Transmission disequilibrium was sought using individual SNPs and SNP-based haplotypes with the pedigree disequilibrium test in 217 CAE trios and the 65 nuclear pedigrees.
Evidence for transmission disequilibrium (p≤0.01) was found for SNPs within a ∼35kb region of high LD encompassing the 5′UTR, exon 1 and part of intron 1 of CACNG3. Re-sequencing of this interval was undertaken in 24 affected individuals. Seventy-two variants were identified: 45 upstream; two 5′UTR; and 25 intronic SNPs. No coding sequence variants were identified, although four variants are predicted to affect exonic splicing.
This evidence supports CACNG3 as a susceptibility locus in a subset of CAE patients.
Keywords: Absence epilepsy, linkage, association, CACNG3, genetics, splice variants
Introduction
The absence epilepsies are a group of idiopathic generalised epilepsies (IGEs) which vary in their age of onset, seizure frequency and pattern of evolution. The typical absence seizure is manifested as a transitory loss of awareness with 2.5–4 Hz spike-wave complexes on ictal EEG. The International League Against Epilepsy (ILAE) classification recognises a number of distinct absence epilepsy syndromes including childhood absence epilepsy (CAE), juvenile absence epilepsy (JAE), epilepsy with myoclonic absences, eyelid myoclonus with absences and juvenile myoclonic epilepsy (JME)1. However, it is uncertain whether they represent a ‘biological continuum’ or distinct entities. There is some evidence that CAE and JAE share a close genetic relationship, allowing them to be considered as one phenotype in genetic studies2,3.
Twin studies demonstrate that the IGEs have a significant heritability4, with regards to both occurrence and type of seizure and syndrome5. The molecular genetic basis of CAE in humans is presently unknown, but studies on the mechanism by which spike-wave seizures are generated, isolation of genes causing spike-wave seizures in rodents and initial linkage and association studies in humans have allowed candidate genes and chromosomal regions to be identified.
Four mouse models of spike-wave epilepsy are caused by mutations in genes for different subunits of voltage-gated calcium channels (VGCCs): tottering tg, Cacna1a6; lethargic lh, Cacnb47; ducky du, Cacna2d28; and stargazer stg, CACNG29. There is some evidence that the gamma subunits may function as transmembrane receptor regulatory proteins (TARPs)10, involved in trafficking11.
Genome-wide linkage analysis of IGE-multiplex families has demonstrated evidence for susceptibility loci on chromosomes 3q26, 14q23 and 2q3612. Furthermore, loci for three similar forms of absence epilepsy have been identified on chromosomes 8q24 (ECA1), 5q31.1 (ECA2) and 3q26 (ECA3)13,14,15,16. An association in humans has been documented between polymorphisms in CACNA1A (chromosome 19p13.2-p13.1) and IGE including CAE17. Finally, 12 missense mutations in CACNA1H (chromosome 16p13.3) have been found in 14 sporadic Chinese Han patients with CAE but not in any of 230 unrelated controls18. However, Heron et al. screened exons 9 to 11 of CACNA1H (in which 75% of the missense mutations were found) in 192 patients with IGE or generalised epilepsy with febrile seizures plus but did not find any of those identified by Chen et al.19. Furthermore, we did not find any of the 12 missense mutations in our resource of CAE families and trios; nor did we find any evidence for linkage to the CACNA1H locus20.
Previous analysis of 33 nuclear families with CAE under the assumption of heterogeneity produced evidence supportive of linkage to the CACNG3 locus on 16p12-p13.121, with an HLOD score of 0.55 (α=0.35) and an NPL score of 1.21. Although the HLOD score did not reach statistical significance, this may reflect the lack of power in the family resource and locus heterogeneity. This gene was prioritised for further analysis because it had the second most positive HLOD score, albeit not statistically significant, and because it is a compelling candidate on biological grounds. The GABA cluster on chromosome 15q, which had the most positive HLOD score, has also been investigated further but the results are not presented here. Those genes with HLOD scores of zero and without any supportive evidence from NPL analysis were not pursued in the larger resource.
Available evidence therefore suggests that genes encoding brain expressed voltage-gated calcium channels including CACNG3 may contribute to the aetiology of CAE. The aim of this work was to test this hypothesis by linkage analysis using microsatellite loci spanning CACNG3 in a resource of 65 nuclear families each with a proband with CAE, expanded from the original resource of 33 families and by association analysis using 23 SNPs distributed across CACNG3 in the nuclear families and 217 parent-affected child trios.
Materials and Methods
Subjects and Samples
The 217 trios (affected child and both parents) and 65 nuclear pedigrees (with a total of 145 AE cases, including 25 of the pedigrees originally used by Robinson et al.), were all of Caucasian origin and ascertained from European populations including the UK, France, Germany, Austria, the Netherlands, Denmark, Sweden, Finland and Italy. Clinical data on subjects categorised as affected are provided in the supplementary data. Appropriate informed consent was obtained from all participants. Diagnostic criteria based on the ILAE classification of absence epilepsies were applied as described in the supplementary data1.
Genomic DNA was extracted from whole blood or cheek swab samples according to standard protocols.
Linkage Analysis
Linkage analysis was performed using three fluorescently-labelled microsatellite markers: D16S420, situated 5′ of the gene; URB036 in the gene; a novel marker, UCL1032122 situated 3′ of the gene (chromosome 16: 24416648-24416860). They span a genetic distance of 0.83cM and a physical distance of 273Kb (Figure 1). This is a smaller distance than originally tested for linkage by Robinson et al. because we are specifically targeting CACNG3 whereas markers to include CACNA1H as well were previously used.
Figure 1.

Genotyping was performed on the ABI 373 Sequence Analyser using the Genescan® and Genotyper® software. All pedigrees were checked for Mendelian inheritance using the PedCheck program23. Any pedigrees which failed this test were re-genotyped. Multipoint linkage analysis was performed using GeneHunter 2.124. Parametric analysis was performed under the assumption of autosomal dominant inheritance with a penetrance of 50%. A disease allele frequency of 0.01 and a phenocopy rate of 0.0001 were assumed. These values are compatible with the observed population prevalence and sibling recurrence risk ratio attributable to the locus, based on the original calculations of Risch25. HLOD scores as well as an estimate of α, which represents the proportion of pedigrees consistent with linkage at a specific locus, were calculated. The nonparametric linkage (NPL) statistic, along with the corresponding degree of significance, was also calculated by GeneHunter. The NPL statistic assesses the degree of allele-sharing in affected individuals only and is valuable as it is a “model-free” form of analysis thus bypassing the inherent problems of a parametric analysis such as misspecification of parameters.
Re-sequencing of Coding Exons
Bi-directional re-sequencing of the coding exons (chromosome 16: 24174862-24175823; 24265106-24266056; 24273263-24274226; and 24279814-24281595) was undertaken in 59 cases taken from the 65 nuclear pedigrees. This re-sequencing was performed in conjunction with ABC at Imperial College, London, using standard Sanger dideoxy protocols.
SNP Analysis
Genomic DNA was typed for 23 SNPs by KBiosciences using both the Amplifluor™ and Taqman™ chemistries. SNPs were chosen at ∼10kb intervals encompassing the putative promoter region through to the 3′ UTR (Figure 2). The SNPs have been numbered from one to 23 for ease of reference. One of these is a novel SNP identified via previous sequencing of a subset of the nuclear pedigrees; the remaining 22 can be found on the NCBI SNP database (Table 1).
Figure 2.
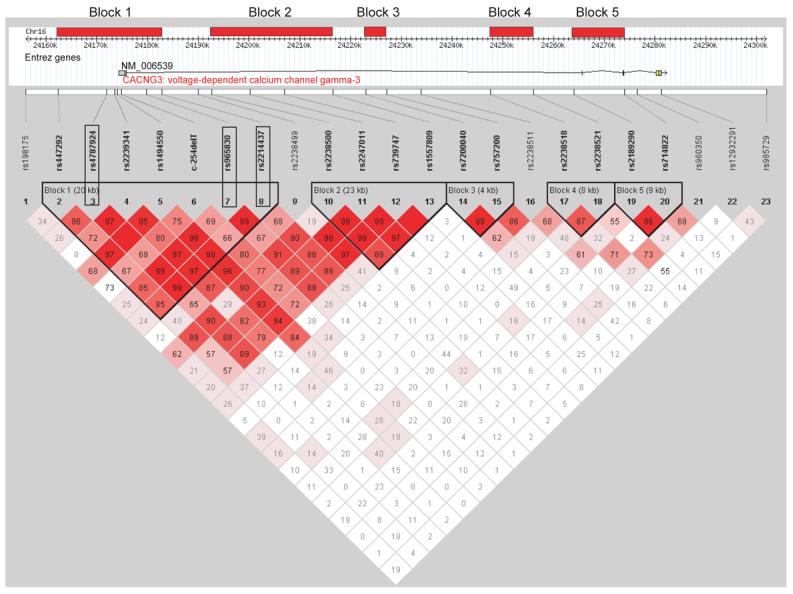
Table 1.
Details of SNPs used for association analysis.
|
MINOR ALLELE FREQUENCY (allele 2 unless otherwise indicated) |
|||||
|---|---|---|---|---|---|
|
SNP NUMBER |
refSNP ID |
based on dbSNP data, Caucasians only |
in unrelated cases |
Allele1 | Allele2 |
| SNP1 | rs198175 | 0.24 (allele1) | 0.28 | G | A |
| SNP2 | rs447292 | 0.37 | 0.37 | G | A |
| SNP3 | rs4787924 | 0.50 0.51(allele1) |
0.51 | A | G |
| SNP4 | rs2239341 | 0.24 | 0.23 | A | C |
| SNP5 | rs1494550 | 0.32 | 0.28 | T | C |
| SNP6 | c-597delT | unavailable | 0.06 | T | delT |
| SNP7 | rs965830 | 0.51(allele1) | 0.52 | T | G |
| SNP8 | rs2214437 | 0.47 | 0.48 | T | A |
| SNP9 | rs2238499 | 0.32 | 0.28 | G | A |
| SNP10 | rs2238500 | 0.47(allele1) | 0.45 | G | A |
| SNP11 | rs2247011 | 0.28 | 0.32 | A | G |
| SNP12 | rs739747 | 0.27(allele1) | 0.30 | A | T |
| SNP13 | rs1557809 | 0.23 | 0.16 | T | C |
| SNP14 | rs7200040 | 0.36(allele1) | 0.34 | A | G |
| SNP15 | rs757200 | 0.32 | 0.26 | C | T |
| SNP16 | rs2238511 | 0.26 | 0.24 | T | C |
| SNP17 | rs2238518 | 0.43 | 0.48 | T | C |
| SNP18 | rs2238521 | 0.18 | 0.09 | G | A |
| SNP19 | rs2189290 | 0.36 | 0.34 | T | C |
| SNP20 | rs714822 | 0.16 | 0.13 | C | G |
| SNP21 | rs960350 | 0.51 | 0.43 | T | C |
| SNP22 | rs12932291 | 0.20 | 0.21 | C | G |
| SNP23 | rs985729 | 0.50(allele1) | 0.46 | T | C |
These SNPs were typed in the entire resource and the genotypes were used to construct LD blocks with Haploview 3.226. Blocks were defined as a solid-spine of LD, i.e. the first and last marker in a block are in strong LD with all intermediate markers (one slight mismatch is allowed by the programme), but these intermediate markers are not necessarily in LD with one another. A minimum D′ of 0.7 was used as the cut-off point for strong LD. The program's standard colour scheme was employed, with pairwise D′ values less than 1 shown and the degree of pink/red shading representing a pairwise LOD ≥ 2. GeneHunter was used to construct haplotypes based on the largest blocks identified. Intrafamilial association analysis was performed on individual SNPs using the PDT27. The PDT produces two measures of association, the PDT-AVE and the PDT-SUM. The former gives all families equal weight in the analysis whereas the latter gives more weight to more informative families28. Association analysis was also performed on the SNP haplotypes. Each haplotype was assigned a single number so that the analysis could be performed essentially as though each haplotype was a single locus with multiple alleles. This is necessary because the PDT cannot simultaneously analyse multiple loci.
The block structure of the CACNG3 locus was also determined using the HapMap genotyped SNPs (see supplementary data).
Re-sequencing and Variant Analysis
Bi-directional direct re-sequencing of ∼35kb of genomic DNA (chromosome 16, 24155960-24190949; accession number GI 51511732, NCBI Nucleotide Database) encompassing SNPs 1-9 from 24 affecteds was performed. Cases were chosen from families compatible with linkage to the CACNG3 locus and included individuals whose haplotypes demonstrated the most significant disease association. This re-sequencing work was performed by Polymorphic DNA Technologies Inc., using standard Sanger dideoxy sequencing protocols. The potential functional affect of all identified variants was assessed by searching for predicted regulatory motifs contained within the TransFac and Biobase databases via the Softberry NSITE portal. This website also contains the FPROM program which predicts the position of potential promoters and enhancers. GeneSplicer30 was used to predict whether any variant might affect the splicing of the gene by identifying exon-intron boundaries and scoring them. ESEfinder31 was used to predict the presence of any exonic splicing enhancers in exon 1. This program identifies putative binding sites for 4 SR-rich proteins thought to be involved in the control of splicing. Prediction is based on a scoring system developed from weighted matrices for each motif consensus sequence; when a certain threshold score is achieved, the motif is recognised. The default values suggested by the program authors were used throughout. Standard BLAST analyses were performed to check for sequence conservation between species.
Results
Linkage Analysis
The maximum HLOD score was 3.54 (α=0.62) located 0.15cM upstream of the distal marker, UCL10321. The non-parametric analysis is also statistically significant: maximum NPL statistic of 2.87 (p<0.002) occurring at UCL10321 (Figure 1).
Re-sequencing of Coding Exons
Bi-directional re-sequencing of the coding exons and surrounding intron-exon boundaries in 59 cases identified 34 variants: four were upstream of CACNG3; six in the 5′UTR; five in intron 1; five in intron 2; nine in intron 3; one synonymous SNP in exon 4 (A2121G, Pro307Pro); two in the 3′UTR; and two downstream of CACNG3.
LD Block Structure
Analysis of LD based on the whole resource identified 5 LD ‘blocks’ (Figure 2). The LD block structure predicted by the HapMap project genotyped SNPs (based on CEPH Caucasian data only), identified 11 blocks of LD across the same region (see supplementary data).
SNP-based Association Analysis
Three SNPs showed significant transmission disequilibrium (p≤0.01) with at least one of the test statistics: SNP3; SNP7; and SNP8 (Table 2). SNP3 is located approximately 2kb upstream of CACNG3 whilst SNPs 7 and 8 are all located in intron 1. All three SNPs are in the first block of LD (Figure 2).
Table 2.
SNPs showing statistically significant disease association (p≤0.01) in at least one PDT test statistic in the entire resource. Full details can be found in the supplementary information.
| SUM PDT | AVE PDT | ||||||
|---|---|---|---|---|---|---|---|
| SNP | Allele | Transmitted |
Not transmitted |
Z (1df) | p-value | Z (1df) | p-value |
| 3 | 2(G) | 300 | 254 | 2.65 | 0.008 | 2.26 | 0.024 |
| 7 | 2(G) | 265 | 221 | 2.64 | 0.008 | 2.32 | 0.020 |
| 8 | 2(A) | 286 | 235 | 2.95 | 0.003 | 2.63 | 0.009 |
Haplotype-based Association Analysis
Block-based haplotype association analysis was performed on the entire data set using the PDT. No single complete haplotype within a block was sufficiently common to allow demonstration of disease association on the global level; however, if a ‘sliding window’ approach was used on each block, associated haplotypes were identified. Using this approach there are 13 haplotypes in Block 1, composed of combinations of SNPs 2-8, which demonstrate overtransmission and disease association (p≤0.05; Table 3). The individual haplotypes which are overtransmitted within each window together form a larger haplotype composed of the alleles 2211122. This haplotype has a frequency of 26.4% in our parental population.
Table 3.
SNP-based sliding-window analysis of Block 1 showing windows which demonstrated significant (p<0.05) global transmission disequilibrium in the entire resource when analysed using the PDT. Only haplotypes showing significant (p<0.05) overtransmission are shown. Full details can be found in the supplementary information.
| SNP |
Frequency in parents (%) |
Transmitted |
Not transmitted |
SUM PDT | GLOBAL | AVE PDT | GLOBAL | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 3 | 4 | 5 | 6 | 7 | 8 | Z (1df) |
p- value |
χ2(df) |
p- value |
Z (1df) |
p- value |
χ2(df) |
p- value |
|||
| 2 | 2 | 31.5 | 207 | 163 | 2.29 | 0.022 | 10.79(3) | 0.013 | 2.60 | 0.009 | 9.33(3) | 0.025 | |||||
| 2 | 2 | 1 | 30.2 | 183 | 143 | 2.16 | 0.031 | 17.41(7) | 0.015 | 2.50 | 0.013 | 14.76(7) | 0.039 | ||||
| 2 | 1 | 44.1 | 262 | 217 | 2.67 | 0.008 | 10.41(3) | 0.015 | 2.43 | 0.016 | 7.93(3) | 0.048 | |||||
| 2 | 1 | 1 | 40.0 | 243 | 195 | 2.84 | 0.005 | 13.88(6) | 0.031 | 2.62 | 0.009 | 10.74(6) | 0.097 | ||||
| 2 | 1 | 1 | 1 | 34.1 | 191 | 141 | 3.20 | 0.001 | 21.57(11) | 0.028 | 3.16 | 0.002 | 21.15(11) | 0.032 | |||
| 2 | 1 | 1 | 1 | 2 | 33.6 | 193 | 131 | 2.98 | 0.003 | 26.61(16) | 0.046 | 3.29 | 0.001 | 28.34(16) | 0.029 | ||
| 1 | 1 | 48.4 | 277 | 231 | 2.69 | 0.007 | 10.98(3) | 0.012 | 2.37 | 0.018 | 8.06(3) | 0.045 | |||||
| 1 | 1 | 1 | 42.2 | 222 | 173 | 3.04 | 0.002 | 16.31(6) | 0.012 | 2.93 | 0.003 | 15.15(6) | 0.019 | ||||
| 1 | 1 | 2 | 36.8 | 210 | 158 | 3.25 | 0.001 | 22.83(7) | 0.002 | 3.18 | 0.002 | 22.78(7) | 0.002 | ||||
| 1 | 1 | 2 | 2 | 36.3 | 206 | 151 | 3.47 | 0.001 | 29.75(12) | 0.003 | 3.48 | 0.001 | 29.81(12) | 0.003 | |||
| 1 | 2 | 40.2 | 225 | 174 | 3.20 | 0.001 | 16.74(3) | 0.001 | 3.10 | 0.002 | 16.53(3) | 0.001 | |||||
| 1 | 2 | 2 | 39.4 | 222 | 162 | 3.92 | 0.000 | 29.95(6) | 0.000 | 3.64 | 0.000 | 27.32(6) | 0.000 | ||||
| 2 | 2 | 45.8 | 277 | 221 | 3.21 | 0.001 | 17.60(3) | 0.001 | 3.09 | 0.002 | 16.17(3) | 0.001 | |||||
The sliding window approach also produces some significant results in Block 2 which runs from SNPs 10-13, although these data are not as significant as for Block 1 (see supplementary data for details).
Variant Detection
Intra-familial association analysis suggested that any functional variant underlying the observed transmission disequilibrium was most likely to be found between SNPs 1 and 9. Consequently, re-sequencing of the ∼35kb of genomic sequence in this region was undertaken. Of the 48 chromosomes from 24 affecteds that were sequenced, 19 were of the most common haplotype, 2211122, which also shows the greatest evidence for disease association. The remaining 25 chromosomes which were sequenced were composed of a variety of different haplotypes. A total of 72 sequence variants were identified, including the 9 previously typed (Figure 3; full details can be found in the supplementary information). Forty-five of these are within 20kb upstream of the gene, two in the 5′UTR and the remaining 25 are in intron 1.
Figure 3.

An initial assessment of which of the identified variants were most likely to be causal was based on whether the minor allele frequency (MAF) was different in the 24 sequenced cases from that quoted on the NCBI database (if that information was available). Any variants in which this did seem to be the case were typed in our entire resource so that intrafamilial association analysis could be performed. Three variants (rs392728, rs11860647 and rs8048987) were genotyped across the resource for this reason. However, intra-familial association analysis with the PDT did not provide any evidence for preferential transmission of either allele (data not shown).
Bioinformatics tools were also used to ascertain which of these 72 variants might be functional. Those considered to be most likely to have a functional effect are summarised in Table 4 (see supplementary information for full details). Of these, rs2021512 and rs1494550 are conserved at the nucleotide level in the chimpanzee (see supplementary data). rs11646957 has been typed in our resource of pedigrees and trios and intrafamilial association analysis performed. The results were not significant (data not shown). Intrafamilial association analysis had already been performed on rs1494550 and n20 as they are SNPs numbers 5 and 6 of the original 23 which were used. Neither demonstrated any disease association in these analyses (see supplementary information). However, rs2021512 did demonstrate significant transmission disequilibrium (SUM PDT χ2(1)=7.91, p=0.005; AVE PDT χ2(1)=4.90, p=0.027), with the reference allele being overtransmitted to cases (457 transmitted:422 not transmitted).
Table 4.
Details of those variants most likely to have a functional effect based on the bioinformatics analyses performed.
| Site Disrupted/Created | ||||
|---|---|---|---|---|
| Variant | ESE Abolished | Novel Acceptor Site Created | ||
| Start position | Sequencea | End position | ||
| rs1164957 | 2 SF2/ASF motifs | |||
| rs1494550 (SNP 5) | 24160423 | AG | 24160424 | |
| n20 (SNP 6) | 24174105 | CT | 24174106 | |
| rs2021512 | 24174976 | AGTTT | 24174980 | |
The position of the indicated variant is shown in bold. This is not shown for SNP6 because the nucleotide is deleted. The splice-site dinucleotide is underlined.
Discussion
Optimal strategies for the genetic analysis of ‘complex’ traits such as childhood absence epilepsy have been much discussed in the recent literature and the potential problems are well recognised. These include uncertainties surrounding the delineation of a categorical phenotype and the unpredictable relationship between phenotype and genotype exemplified by current ignorance of the genetic architecture of most ‘complex’ traits, and the recognition that even epilepsy phenotypes demonstrating simple Mendelian inheritance display immense heterogeneity and variable expressivity. All these uncertainties preclude confident predictions about the likely power and validity of any strategy selected, but these data arise from approaches which have some reasonable degree of support and expectation of success given certain assumptions.
Childhood absence epilepsy is a fairly well defined and homogeneous phenotype with an electrophysiological hallmark and clear evidence of a genetic aetiology. Although there is some debate as to whether a narrower phenotype definition should be adopted – in part to allow clearer prognostic predictions – the clinical criteria adopted here provide the reasonable expectation that the patients ascertained represent a homogenous clinical phenotype. It is known that a variety of IGE phenotypes may cluster in families with a proband with absence epilepsy, but analysis reveals an increased clustering of CAE and JAE2,3 suggesting that they may share susceptibility loci. For this reason the minority of pedigrees in which first degree relatives of a proband with CAE had a diagnosis of JAE were included and such individuals were categorised as affected.
A further advantage of this phenotype for genetic analysis lies in the existing level of understanding of the molecular neurophysiological basis of the ‘spike-wave’ seizures which are their hallmark1. A substantial body of evidence implicates voltage-gated calcium channel genes in the aetiology of spike-wave seizures in rodents and absence seizures in humans. In particular, the stargazer phenotype arises from mutations in Cacng2, one of a family of so-called gamma-subunit genes which have been further defined as a family of transmembrane AMPA receptor regulatory proteins (TARPS)10 that mediate surface expression of AMPA receptors. Preliminary analysis in a limited family resource provided support for CACNG3 as a CAE susceptibility locus. It is noteworthy that the expression pattern of γ3 is specific to the cortex and hippocampus with low levels in the cerebellum, consistent with a role in epileptogenesis. A candidate gene approach therefore seems justified.
The relevant merits of linkage or association in the analysis of a complex trait depend on the genetic architecture of the trait which cannot of course be known in advance. Linkage can detect a locus of moderate effect in a set of small nuclear pedigrees or sib-pairs provided the proportion of linked families is adequate. It is robust to any allelic heterogeneity. Association has more power to detect loci of small effect, but does of course depend on allelic homogeneity and allele frequencies conferring adequate power. Both parametric and non-parametric linkage analysis provided significant evidence for linkage, indicating CACNG3 is a susceptibility loci for CAE in a subset of the 65 nuclear pedigrees analysed. Sequencing of coding regions did not however identify any plausible causal sequence variants.
The role of CACNG3 was therefore further analysed by intra-familial association analysis using the indirect approach based on genotyping of a set of common single nucleotide polymorphisms spanning the CACNG3 gene. The pattern of LD across CACNG3 was established and confirmed by HapMap data. A number of assumptions underlie this strategy and restrict its power to identify causal sequence variants. It is assumed that the actual SNPs typed are not causal but are in sufficiently tight LD with causal SNPs of adequately matching allele frequency to permit their detection. Power diminishes rapidly if these conditions are not fulfilled32. It is also assumed that any causal SNPs will be within the associated interval, although in practice the interval is difficult to define33.
Three SNPs in the first ‘block’ of LD showed significant transmission disequilibrium (SNPs 3, 7 and 8) using the pedigree disequilibrium test in the entire patient resource. Using a ‘sliding window’ approach 13 haplotypes comprising SNPs 2-8 within this ‘block’ demonstrated transmission disequilibrium. Together these form an extended haplotype composed of the alleles 2211122. Only two of these SNPs (SNPs 5 and 6) could be implicated on functional grounds. In each case the minor allele is predicted to lead to creation of a splice acceptor site. However, neither SNP showed significant transmission disequilibrium either alone or in combination. A correction for multiple testing was not applied to these analyses because although methods for calculating the effective number of independent tests have been developed34, their validity in the presence of haplotype block structure has been questioned35 and these methods are not yet established tools. Thus the association evidence described must be considered as tentative but requiring independent replication.
These observations suggested that causal variants underlying the observed transmission disequilibrium might lie within the genomic region between SNP1 and SNP9 and were most likely to be found on chromosomes of the haplotype 2211122. Re-sequencing of 35kb of genomic sequence in 48 chromosomes identified a total of 72 sequence variants. Evaluation of these variants encompassed the use of bioinformatics tools to determine any potential functionality and genotyping in the entire resource to investigate disease association. Four SNPs were predicted to have some functional effect by disrupting exonic splicing enhancer binding motifs or creating novel acceptor sites. One of these, rs2021512, demonstrated significant transmission disequilibrium with the reference sequence allele (G) being overtransmitted in preference to the variant allele (A). This suggests that the variant form is protective. Analysis indicated that the variant allele could potentially create an acceptor splice site although it is unclear how this might affect the function of the protein as rs2021512 is non-genic and approximately 14kb upstream of CACNG3. However, it is possible that this SNP has a subtle regulatory effect which was not identified with the bioinformatics used. Indeed, a paper earlier this year demonstrated that a non-genic variant can have a gain-of-function effect on another gene by creating a new transcriptional promoter36. This is not necessarily what is occurring in this situation but it is clear that variants some distance from a gene can still exert a powerful effect on them. Furthermore, it is still possible that rs2021512 is not a causal variant but is in LD with an unidentified causal variant.
It is possible that the linkage observed is spurious and CACNG3 is not a susceptibility locus for the CAE trait. A false positive result is of course feasible even with the fairly stringent threshold for significance utilised. The transmission disequilibrium observed could be a false positive result, although not due to population substructure. Alternatively the observed association is real, but driven by causal variants outside the sequenced region or too infrequent and heterogeneous to be detected in the limited number of chromosomes sequenced. It has been demonstrated that long range LD can exist generating ‘genetically indistinguishable SNPs’ (giSNPs) which are many kilobases apart37. The power to detect a homogeneous causal variant with a population frequency of 5% is approximately 92% when 48 chromosomes are sequenced but of course a heterogeneous collection of low frequency variants might go undetected. Finally it is possible that the observed SNPs demonstrating transmission disequilibrium have functional consequences which are not apparent.
In conclusion, these observations provide genetic evidence that CACNG3 is a susceptibility locus for childhood absence epilepsy. Common variants showing transmission disequilibrium have been identified. Definitive evidence to confirm or exclude this locus will require re-sequencing across an extended genomic region encompassing CACNG3 in a larger number of patients. Replication studies in similar resources of CAE patients would demonstrate whether rs2021512 is associated in other patient groups, and functional work to establish what the exact biological mechanism could be is needed.
Supplementary Material
Acknowledgements
This work was supported by the MRC (UK), Wellcome Trust, Action Medical Research and Epilepsy Research Foundation. We are very grateful to the families for participating in this study and to all our 142 collaborating clinicians, including Dr Lina Nashef. We would like to thank Généthon for their assistance in collecting the French samples and Richard Sharp for his technical help. Austrian financial support came from the Austrian Research Foundation (awarded to Harald Aschauer, MD), grant number P10460-MED. Thomas Sander, MD, was awarded a grant by the Deutsche Forschungsgemeinschaft (Sa434/3-1), the German National Genome Research Network (01GS0479). Dutch financial support came from the Netherlands Organisation for Health, Research and Development (ZonMW, 940-33-030) and the Dutch National Epilepsy Fund – ‘The power of the small’ (NEF – ‘De macht van het kleine’). Danish support (Mogens Friis, MD, and Marianne Kjeldsen, MD) came from the NINDS grant (NS-31564).
Footnotes
Web Resources
- ■ Advanced Biotechnology Centre, Imperial College, http://bm-abc01.cx.med.ic.ac.uk/
- ■ KBioscience, http://www.kbioscience.co.uk
- ■ NCBI Nucleotide Database, http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=nuccore
- ■ Polymorphic DNA Technologies, http://www.polymorphicdna.com
- ■ Softberry NSITE Portal, http://www.softberry.com
- ■ GeneSplicer, http://www.tigr.org/tdb/GeneSplicer/gene_spl.html
- ■ ESEFinder, http://rulai.cshl.edu/tools/ESE/
Reference List
- 1.Engel J., Jr. A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: report of the ILAE Task Force on Classification and Terminology. Epilepsia. 2001;42:796–803. doi: 10.1046/j.1528-1157.2001.10401.x. [DOI] [PubMed] [Google Scholar]
- 2.Berkovic SF, Andermann F, Andermann E, Gloor P. Concepts of absence epilepsies: discrete syndromes or biological continuum? Neurology. 1987;37:993–1000. doi: 10.1212/wnl.37.6.993. [DOI] [PubMed] [Google Scholar]
- 3.Marini C, Scheffer IE, Crossland KM, et al. Genetic architecture of idiopathic generalized epilepsy: clinical genetic analysis of 55 multiplex families. Epilepsia. 2004;45:467–478. doi: 10.1111/j.0013-9580.2004.46803.x. [DOI] [PubMed] [Google Scholar]
- 4.Berkovic SF, Howell RA, Hay DA, Hopper JL. Epilepsies in twins: genetics of the major epilepsy syndromes. Ann Neurol. 1998;43:435–445. doi: 10.1002/ana.410430405. [DOI] [PubMed] [Google Scholar]
- 5.Kjeldsen MJ, Corey LA, Christensen K, Friis ML. Epileptic seizures and syndromes in twins: the importance of genetic factors. Epilepsy Res. 2003;55:137–146. doi: 10.1016/s0920-1211(03)00117-7. [DOI] [PubMed] [Google Scholar]
- 6.Fletcher CF, Lutz CM, O'Sullivan TN, et al. Absence epilepsy in tottering mutant mice is associated with calcium channel defects. Cell. 1996;87:607–617. doi: 10.1016/s0092-8674(00)81381-1. [DOI] [PubMed] [Google Scholar]
- 7.Burgess DL, Jones JM, Meisler MH, Noebels JL. Mutation of the Ca2+ channel beta subunit gene Cchb4 is associated with ataxia and seizures in the lethargic (lh) mouse. Cell. 1997;88:385–392. doi: 10.1016/s0092-8674(00)81877-2. [DOI] [PubMed] [Google Scholar]
- 8.Barclay J, Balaguero N, Mione M, et al. Ducky mouse phenotype of epilepsy and ataxia is associated with mutations in the Cacna2d2 gene and decreased calcium channel current in cerebellar Purkinje cells. J Neurosci. 2001;21:6095–6104. doi: 10.1523/JNEUROSCI.21-16-06095.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Letts VA, Felix R, Biddlecome GH, et al. The mouse stargazer gene encodes a neuronal Ca2+-channel gamma subunit. Nat Genet. 1998;19:340–347. doi: 10.1038/1228. [DOI] [PubMed] [Google Scholar]
- 10.Tomita S, Chen L, Kawasaki Y, et al. Functional studies and distribution define a family of transmembrane AMPA receptor regulatory proteins. J Cell Biol. 2003;161:805–816. doi: 10.1083/jcb.200212116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen L, Chetkovich DM, Petralia RS, et al. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 2000;408:936–943. doi: 10.1038/35050030. [DOI] [PubMed] [Google Scholar]
- 12.Sander T, Schulz H, Saar K, et al. Genome search for susceptibility loci of common idiopathic generalised epilepsies. Hum Mol Genet. 2000;9:1465–1472. doi: 10.1093/hmg/9.10.1465. [DOI] [PubMed] [Google Scholar]
- 13.Fong GC, Shah PU, Gee MN, et al. Childhood absence epilepsy with tonic-clonic seizures and electroencephalogram 3-4-Hz spike and multispike-slow wave complexes: linkage to chromosome 8q24. Am J Hum Genet. 1998;63:1117–1129. doi: 10.1086/302066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haug K, Warnstedt M, Alekov AK, et al. Mutations in CLCN2 encoding a voltage-gated chloride channel are associated with idiopathic generalized epilepsies. Nat Genet. 2003;33:527–532. doi: 10.1038/ng1121. [DOI] [PubMed] [Google Scholar]
- 15.Sugimoto Y, Morita R, Amano K, et al. Childhood absence epilepsy in 8q24: refinement of candidate region and construction of physical map. Genomics. 2000;68:264–272. doi: 10.1006/geno.2000.6296. [DOI] [PubMed] [Google Scholar]
- 16.Wallace RH, Marini C, Petrou S, et al. Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat Genet. 2001;28:49–52. doi: 10.1038/ng0501-49. [DOI] [PubMed] [Google Scholar]
- 17.Chioza B, Wilkie H, Nashef L, et al. Association between the alpha(1a) calcium channel gene CACNA1A and idiopathic generalized epilepsy. Neurology. 2001;56:1245–1246. doi: 10.1212/wnl.56.9.1245. [DOI] [PubMed] [Google Scholar]
- 18.Chen Y, Lu J, Pan H, et al. Association between genetic variation of CACNA1H and childhood absence epilepsy. Ann Neurol. 2003;54:239–243. doi: 10.1002/ana.10607. [DOI] [PubMed] [Google Scholar]
- 19.Heron SE, Phillips HA, Mulley JC, et al. Genetic variation of CACNA1H in idiopathic generalized epilepsy. Ann Neurol. 2004;55:595–596. doi: 10.1002/ana.20028. [DOI] [PubMed] [Google Scholar]
- 20.Chioza B, Everett K, Aschauer H, et al. Evaluation of CACNA1H in European patients with childhood absence epilepsy. Epilepsy Res. 2006;69:177–181. doi: 10.1016/j.eplepsyres.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 21.Robinson R, Taske N, Sander T, et al. Linkage analysis between childhood absence epilepsy and genes encoding GABAA and GABAB receptors, voltage-dependent calcium channels, and the ECA1 region on chromosome 8q. Epilepsy Res. 2002;48:169–179. doi: 10.1016/s0920-1211(01)00335-7. [DOI] [PubMed] [Google Scholar]
- 22.Jeganathan D, Chodhari R, Meeks M, et al. Loci for primary ciliary dyskinesia map to chromosome 16p12.1-12.2 and 15q13.1-15.1 in Faroe Islands and Israeli Druze genetic isolates. J Med Genet. 2004;41:233–240. doi: 10.1136/jmg.2003.014084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O'Connell JR, Weeks DE. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES. Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet. 1996;58:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 25.Risch N. Linkage strategies for genetically complex traits. I. Multilocus models. Am J Hum Genet. 1990;46:222–228. [PMC free article] [PubMed] [Google Scholar]
- 26.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 27.Martin ER, Monks SA, Warren LL, Kaplan NL. A test for linkage and association in general pedigrees: the pedigree disequilibrium test. Am J Hum Genet. 2000;67:146–154. doi: 10.1086/302957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin ER, Bass MP, Kaplan NL. Correcting for a potential bias in the pedigree disequilibrium test. Am J Hum Genet. 2001;68:1065–1067. doi: 10.1086/319525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chapman JM, Cooper JD, Todd JA, Clayton DG. Detecting disease associations due to linkage disequilibrium using haplotype tags: a class of tests and the determinants of statistical power. Hum Hered. 2003;56:18–31. doi: 10.1159/000073729. [DOI] [PubMed] [Google Scholar]
- 30.Pertea M, Lin X, Salzberg SL. GeneSplicer: a new computational method for splice site prediction. Nucleic Acids Res. 2001;29:1185–1190. doi: 10.1093/nar/29.5.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31:3568–3571. doi: 10.1093/nar/gkg616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Bakker PI, Yelensky R, Pe'er I, Gabriel SB, Daly MJ, Altshuler D. Efficiency and power in genetic association studies. Nat Genet. 2005;37:1217–1223. doi: 10.1038/ng1669. [DOI] [PubMed] [Google Scholar]
- 33.Soranzo N, Cavalleri GL, Weale ME, et al. Identifying candidate causal variants responsible for altered activity of the ABCB1 multidrug resistance gene. Genome Res. 2004;14:1333–1344. doi: 10.1101/gr.1965304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nyholt DR. A simple correction for multiple testing for single-nucleotide polymorphisms in linkage disequilibrium with each other. Am J Hum Genet. 2004;74:765–769. doi: 10.1086/383251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salyakina D, Seaman SR, Browning BL, Dudbridge F, Muller-Myhsok B. Evaluation of Nyholt's procedure for multiple testing correction. Hum Hered. 2005;60:19–25. doi: 10.1159/000087540. [DOI] [PubMed] [Google Scholar]
- 36.De Gobbi M, Viprakasit V, Hughes JR, et al. A regulatory SNP causes a human genetic disease by creating a new transcriptional promoter. Science. 2006;312:1215–1217. doi: 10.1126/science.1126431. [DOI] [PubMed] [Google Scholar]
- 37.Lawrence R, Evans DM, Morris AP, et al. Genetically indistinguishable SNPs and their influence on inferring the location of disease-associated variants. Genome Res. 2005;15:1503–1510. doi: 10.1101/gr.4217605. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.