

Abstract
Progress in management of critically ill neurological patients has led to improved survival rates. However, severe residual neurological impairment, such as persistent coma, occurs in some survivors. This raises concerns about whether it is ethically appropriate to apply aggressive care routinely, which is also associated with burdensome long-term management costs. Adapting the management approach based on long-term neurological prognosis represents a major challenge to intensive care. Magnetic resonance imaging (MRI) can show brain lesions that are not visible by computed tomography, including early cytotoxic oedema after ischaemic stroke, diffuse axonal injury after traumatic brain injury and cortical laminar necrosis after cardiac arrest. Thus, MRI increases the accuracy of neurological diagnosis in critically ill patients. In addition, there is some evidence that MRI may have potential in terms of predicting outcome. Following a brief description of the sequences used, this review focuses on the prognostic value of MRI in patients with traumatic brain injury, anoxic/hypoxic encephalopathy and stroke. Finally, the roles played by the main anatomical structures involved in arousal and awareness are discussed and avenues for future research suggested.
Introduction
Severe brain impairment, most notably persistent coma, may follow traumatic brain injury (TBI), anoxic/hypoxic encephalopathy, or stroke. Although progress in the management of critically ill neurological patients has led to improved survival rates [1], some survivors remain in a persistent vegetative or minimally conscious state. Up to 14% of patients with TBI remain in a persistent vegetative state after 1 year [2-4], and their medical cost has been estimated at US$1 to 7 billion per year in the USA [5]. The possibility that aggressive medical management may lead to survival with severe brain impairment raises ethical issues. Adapting the level of medical care to long-term neurological prognosis is a major challenge for neurological intensive care. The first step in meeting this challenge is validation of tools that accurately predict long-term neurological outcome after severe cerebral insult.
Magnetic resonance imaging (MRI) is more sensitive than computed tomography at detecting stroke in the early phase, subtle abnormalities related to anoxic/hypoxic encephalopathy, and diffuse axonal injury (DAI) in patients with TBI. MRI provides valuable diagnostic information, although it is cumbersome to perform in the acute phase in comatose patients who are undergoing mechanical ventilation. Several MRI sequences and techniques have been used to explore the structures, metabolism and functions of the brain. The data supplied by these methods could be used to predict long-term neurological outcome.
In this review we briefly describe the MRI sequences and techniques used in critically ill neurological patients, and then we discuss their prognostic value in comatose patients with TBI, anoxic/hypoxic encephalopathy, or stroke. Finally, we discuss the prognostic influences of the main anatomical structures that are involved in arousal and awareness, and we suggest avenues for future research.
Magnetic resonance imaging sequences and techniques
Conventional magnetic resonance imaging
Conventional MRI relies chiefly on four sequences [6]. Fluid-attenuated inversion recovery (FLAIR) is the primary sequence used in neuroradiology (Figure 1). It detects brain contusion, brain oedema and subarachnoid or intraventricular haemorrhage, as well as the resulting ventricular dilatation or herniation. The T2*-weighted sequence is more sensitive to intraparenchymal blood than is FLAIR. This sequence can also reveal haemorrhagic DAI [7,8]. The T2-weighted sequence completes the FLAIR sequence and provides greater detail on brainstem and central grey matter. Finally, diffusion weighted imaging (DWI) is sensitive to random movement of water molecules. This sequence shows cerebral oedema and distinguishes cytotoxic from vasogenic oedema. It is used chiefly in patients with ischaemic stroke.
Figure 1.
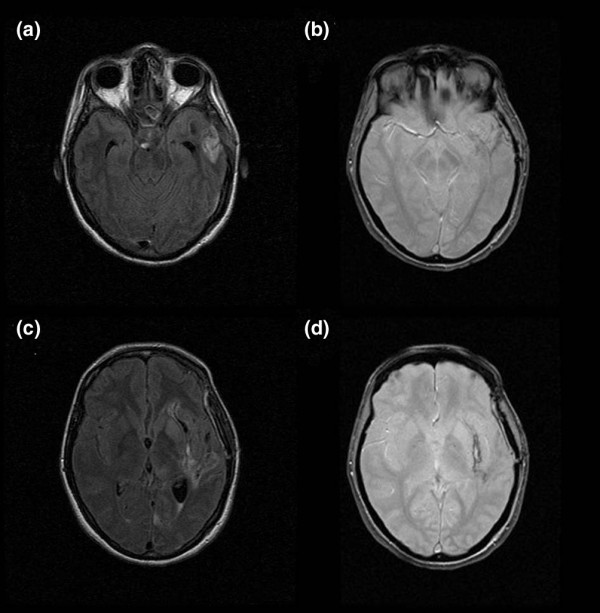
FLAIR and T2* sequences in a patient with an arteriovenous malformation. (a) Axial fluid-attenuated inversion recovery (FLAIR) sequence showing hypersignal in the left temporal lobe. (b) Axial T2* sequence showing mild hyposignal in the same area suggestive of bleeding. (c) Different section of the axial FLAIR sequence showing hypersignal surrounded by hyposignal. Bleeding cannot be confirmed. (d) Axial T2* sequence clearly showing hyposignal lateral to the left putamen. The patient has bleeding from the arteriovenous malformation.
Conventional MRI provides an initial evaluation of brain lesions. However, when it is used alone it fails to predict outcome accurately.
Magnetic resonance spectroscopy
This sequence is a noninvasive technique for assessing brain metabolism in vivo. Proton-magnetic resonance spectroscopy (MRS) is most commonly used. Four main markers are studied: the peak of N-acetyl-aspartate (NAA), an amino acid present in neurones, which reflects the status of neuronal tissue; creatine, found in glia and neurones, which serves as a point of reference because its level is believed to be stable; choline, a constitutive component of cell membranes, which reflects glial proliferation or membrane breakdown [9]; and lactate, a marker of anaerobic metabolism and therefore of ischaemia [10]. As shown in Figure 2, three main pons monovoxel profiles may be observed in patients with TBI.
Figure 2.

Magnetic resonance spectroscopy profile of the pons after traumatic brain injury. (a) Normal profile. The peak of N-acetyl-aspartate (NAA) is higher than the peaks of choline (Cho) and creatine (Cr). (b) Neuronal loss profile. The NAA peak is decreased, nearly to the level of the Cr peak. The NAA/Cr ratio is lower than in panel a. (c) Gliosis profile: increased Cho peak with no change in the Cr or NAA peak. Adapted from [17].
Diffusion tensor magnetic resonance imaging
Diffusion tensor imaging (DTI), derived from DWI, measures the degree and direction of water diffusion (anisotropy). Water diffusion anisotropy reflects the integrity of white matter tracts. Pathophysiological mechanisms that can alter water diffusion anisotropy include DAI, effects of intracranial hypertension and disconnection of white matter tracts.
Magnetization transfer imaging
This sequence is based on the principle that structure-bound protons undergo T1 relaxation coupling with protons in the aqueous phase. Saturated protons in macromolecules exchange longitudinal magnetization with protons in the aqueous phase, leading to a reduction in signal intensity. Magnetization transfer imaging has been found to be sensitive for detecting white matter lesions in several neurological conditions [11,12].
Functional magnetic resonance imaging
Functional MRI may reveal foci of cerebral dysfunction in regions that look structurally intact on conventional MRI. Imaging is based on changes in the oxidative state of haemoglobin, which reflects regional brain activation. Functional MRI remains difficult to perform in critically ill unstable patients and, consequently, few teams have acquired the equipment and experience necessary to apply this technique [13]. The few available studies conducted in comatose patients with TBI showed a correlation between prefrontal/cingulated cortical activation disturbation and cognitive impairments [14,15]. However, functional MRI was performed in these studies at a distance from the injury.
Magnetic resonance imaging findings in specific critical neurological conditions
Traumatic brain injury
Conventional magnetic resonance imaging
MRI was first used to investigate patients with TBI in a 1986 study of 50 patients [16]. The three main findings, which have since been confirmed, were as follows: MRI identified lesions more frequently than did computed tomography; brain lesions were common after TBI; and although patients who regained consciousness rapidly had no lesions in fundamental deep brain structures, some of them had severe cortical lesions. Several descriptions of MRI lesions in TBI patients have been reported since that initial study was published (Table 1) [17-21], although few of them focused on the prognostic value of MRI [17-20]. Conventional MRI findings that strongly predicted outcome included DAI, total lesion burden and DAI in the brainstem.
Table 1.
Conventional magnetic resonance in traumatic brain injury
| Authors (ref.) | |||||||||
| Kampfl, 1998 [19] | Firsching, 1998 [18] | Pierallini, 2000 [30] | Yanagawa, 2000 [28] | Paterakis, 2000 [27] | Firsching, 2001 [29] | Firsching, 2002 [95] | Wedekind, 2002 [31] | Carpentier, 2006 [17] | |
| Study design | Case-control | Prospective | Prospective | Prospective | Prospective | Prospective | Prospective | Retrospective | Prospective |
| Sequences | T1, T2 | T1, T2 | T1, T2, FLAIR | T2, T2* | T1, T2 | T1, T2 | T1, T2 | T1, T2, T2* | MRS, T2, T2* |
| Inclusion criteria | VS between 6 and 8 weeks | Admission in coma (duration >24 hours) | GCS score <8, coma >1 week, post-traumatic amnesia >4 weeks | Alive after 1 week | Discrepancy between CT scan and neurological status | Admission in coma (duration >24 hours) | GCS score <8 | GCS score <8 | Severe TBI |
| Number of patients | 80 | 61 | 37 | 34 | 33 | 102 | 100 | 40a | 40 |
| Delay to MRI | 6 to 8 weeks | <7 days | 60 to 90 days | <3 weeks | <48 hours | <8 days | <7 days | 1 to 39 days | 17.5 ± 6.4 |
| Outcome Variable of Interest | GOS score (2 versus 3–5) at 2, 3, 6, 9 and 12 months | Mortality | Clinical assessment at 3, 6 and 12 months | GOS score at 3 months | GOS score (2–3 versus 4–5) at 6 months | Mortality and outcome at 3 months to 3 yearsb | Mortality at 6 months | GOS score, DRS >6 months (mean delay: 11.3 months) | GOS score (1–2 versus 4–5) and DRS at 18 months |
| Main results | Independent factor of poor outcome on multivariate analysis. Corpus callosum: OR 213.8 (95% CI 14.2 to 3213.3). Brainstem lesions OR 6.9 (95% CI 1.1 to 42.9) | Brainstem lesions: mortality rate of 44%. Bilateral brainstem lesions: mortality rate of 100% | Volume of FLAIR corpus callosum lesions correlated with first clinical evaluation. Volume of FLAIR frontal lobe lesion correlated with clinical outcome at 1 year | Number of T2 lesions correlated with GOS score. Number of T2* lesions correlated with GOS score | DAI stages correlated with outcome. No patient with good outcome had haemorrhagic DAI | Bilateral pons lesions: mortality rate of 100%. Outcome correlated with presence/absence and unilateral/bilateral brainstem lesions | Bilateral upper pontine lesion predicts mortality | More lesions of corpus callosum, basal ganglia and (para-)hippo-campal lesions in patients with brainstem lesions | Total burden of FLAIR and T2* lesions correlated with DRS and GOS score |
aTwenty patients with brainstem lesions were matched to 20 patients without brainstem lesions. bAt last examination. CI, confidence interval; DAI, diffuse axonal injury; DRS, disability rating scale; FLAIR, fluid-attenuated inversion recovery; GCS, Glasgow Coma Scale; GOS, Glasgow Outcome Scale; MRI, magnetic resonance imaging; MRS, magnetic resonance spectroscopy; NA, not applicable; OR, odds ratio; T2*, T2* weighted sequence; TBI, traumatic brain injury; VS, vegetative state.
DAI is the most common primary lesion in TBI patients [22,23] and may be the most common cause of poor outcome [22-24]. DAI may be ischaemic or haemorrhagic [7,8]. Ischaemic DAI is seen as a hypersignal on DWI or FLAIR, with no abnormality on the T2* sequence [25]. The hypersignal with DWI disappears within about 2 weeks. Conversely, haemorrhagic DAI appears as a hyposignal on the T2* sequence, with normal DWI findings. It has been proposed [22] that DAI location could be classified into the following stages: stage 1, frontal and temporal white matter; stage 2, lobar white matter and posterior part of corpus callosum; and stage 3, dorsolateral midbrain and pons. With outcomes defined as Glasgow Outcome Scale [26] scores of 2 to 3 versus 4 to 5, none of the 33 patients with good outcome in another study [27] had haemorrhagic DAI (Table 1). DAI appears to be a major determinant of poor outcomes, although its use as an outcome predictor in the individual patient remains difficult. Whether the correlation between DAI and outcome is due to the total lesion burden or to DAI location remains debated.
In several prospective studies, lesion burden was associated with outcome irrespective of DAI location (Table 1) [17,19,28]. Among 40 prospectively enrolled patients with severe TBI, lesions by FLAIR and T2*-weighted sequences increased progressively with GOS score groups 1 to 2, 3, and 4 to 5 [17]. Similar results were obtained in a study comparing 42 patients with persistent vegetative state with 38 patients who recovered consciousness [19].
A number of studies have focused on the value of DAI location in predicting outcome [19,29-31]. Brainstem lesions in the pons and mesencephalon appear to be the most potent markers of poor prognosis, most notably when they are bilateral and symmetrical [18,19,29,31]. In a prospective study conducted in 61 patients (Table 1) who were studied within 7 days of TBI [18], all patients with bilateral pontine lesions died as compared with 9% of patients with no brainstem lesions. These results were confirmed by the same group in a prospective study of 102 comatose patients [29] using the following four-stage grading system: grade I, lesions of the hemispheres only; grade II, unilateral lesions of the brainstem at any level with or without supratentorial lesions; grade III, bilateral lesions of the mesencephalon with or without supratentorial lesions; and grade IV, bilateral lesions of the pons with or without any of the lesions of lesser grades. Mortality increased gradually from 14% with grade I lesions to 100% with grade IV lesions. These findings were corroborated by two independent studies [19,31] (Table 1). We recently confirmed the prognostic value of brainstem lesions in the upper pons and lower midbrain in a study of 73 patients [32]. Bilateral pontine lesions carry a high mortality rate and predict poor neurological outcomes.
Three studies showed that corpus callosum lesions were associated with poor outcomes [19,30,31] (Table 1). However, these lesions may merely represent markers for severe initial injury. In addition to lesion burden, both total lesion volume and frontal lobe lesion volume on FLAIR images correlated significantly with clinical outcomes [30]. Nevertheless, evaluating DAI lesion volume is difficult (most notably when the lesions are small), time consuming, cumbersome and subject to inter-rater variability.
The presence of severe DAI and a heavy lesion burden are associated with permanent neurological impairment. However, these factors are difficult to use in the individual patient, especially to distinguish GOS score 2 from GOS score 3. In TBI patients, brainstem lesions are easily identified by MRI. In our experience, they are associated with poor outcomes, most notably when they are posterior and bilateral. Posterior brainstem lesions in the periaqueductal grey matter are probably more relevant than anterior brainstem lesions as predictors of poor outcomes in patients with brainstem stroke [21] or TBI [19]. In clinical practice, treatment limitation may deserve consideration in patients who have large bilateral lesions in the posterior part of the pons after TBI.
Magnetic resonance spectroscopy
Several MRS studies have been conducted in TBI patients (Table 2). Some of them were purely descriptive [33], others assessed only the neuropsychological outcomes [34,35], and yet others focused on global outcome as evaluated using the GOS or Disability Rating Scale [17,36-42].
Table 2.
Outcome of traumatic brain injury by magnetic resonance spectroscopy
| Authors (ref.) | |||||||||
| Choe, 1995 [43] | Ricci, 1997 [39] | Ross, 1998 [40] | Friedman, 1999 [36] | Garnett, 2000 [37] | Sinson, 2001 [41] | Uzan, 2003 [42] | Carpentier, 2006 [17] | Marino, 2006 [38] | |
| Study design | Case-control | Prospective | Prospective | Case-control | Prospective | Prospective | Case-control | Prospective | Case-control |
| Delay | 2 weeks to 11 months | 1 to 90 months | 1 to 74 days | 45 ± 21 days/6 months | 12 days (3–35)/6.2 months (2.9–50.6) | 41 days (median) | 6 to 8 months | 17.5 ± 6.4 days | 48 to 72 hours |
| Number of patients | 10 TBI patients versus 10 control individuals | 14 VS TBI patients | 25 TBI patients (12 children) | 14 TBI patients versus 14 control individuals | 26 patients. Early study: 21. Late study: 15. Both: 10 | 30 TBI patients | 14 VS TBI patients versus 5 control individuals | 40 TBI patients | 10 TBI patients versus 10 control individuals |
| Grey matter voxel location | NA | NA | Occipitoparietal | Occipitoparietal | Frontal | NA | Thalamus | NA | Mesial cortex |
| White matter voxel location | Frontoparietal | Frontal | Occipitoparietal | Occipitoparietal | Frontal | Splenium of corpus callosum | NA | Pons | Corpus callosum, mostly white matter |
| Outcome variable of interest | GOS score after MRI | GOS score (1–2 versus 3–5) at follow upa | ROS at discharge and follow upb | GOS score and neuropsychological performance | GOS score, DRS at 6 months | GOS score at 3 months (1–4 versus 5) | Aware versus not aware at >6 months | GOS score (1–2 versus 4–5), DRS at 18 months | GOS score at 3 months |
| Main results | NAA/Cr ratio lower in TBI patients. NAA/Cr ratio correlated with GOS score | NAA/Cr ratio and NAA/Cho ratio lower, Cho/Cr ratio elevated, and NAA/Cho lower in GOS score 1–2 versus GOS 3–5 | NAA levels diminished. NAA/Cr ratio correlated with outcome | NAA levels in white matter lower in TBI patients. Early NAA levels in grey matter correlated with GOS | NAA/Cr ratio lower in TBI patients. Cho/Cr elevated in TBI patients. NAA/Cr ratio correlated with GOS score and DRS | NAA/Cr ratio lower. NAA/Cr correlated with GOS score | NAA/Cr ratio lower in VS. NAA/Cr ratio lower in patients remained in VS compared with patients who regained awareness | NAA/Cr ratio correlated to GOS score and DRS. No correlation between NAA/Cr ratio and lesions burden on FLAIR or T2* | NAA/Cr and NAA/all metabolites ratios lower. La/Cr and La/all metabolites ratios increased in TBI |
aNo further information. bUp to 2 years, except for four out of 25 patients. Cho, choline; Cr, creatinine; DRS, disability rating scale; FLAIR, fluid-attenuated inversion recovery; GOS, Glasgow Outcome Scale; La, lactate; MRI, magnetic resonance imaging; NA, not applicable; NAA, N-acetyl-aspartate; ROS, Rancho Los Amigos Medical Centre Outcome Score; T2*, T2* weighted sequence; TBI, traumatic brain injury; VS, vegetative state.
Compared with control individuals, TBI patients exhibited decreased NAA levels, decreased NAA/creatine ratios and increased choline levels (Table 2) in all brain regions evaluated [35-39,41,42]. Increased lactate levels were seldom found in TBI patients, contrary to patients with other brain injuries [38]. The NAA/creatine ratio appeared to be the best outcome predictor. Low NAA/creatine values correlated with poor outcomes when they were located in the frontal [37,39], frontoparietal [43], or occipitoparietal lobes [36,40]; the splenium of the corpus callosum [41]; the thalami [42]; the pons [17]; or a voxel including the corpus callosum, the white matter, and part of the hemispheric cortex [38].
These studies are heterogeneous (Table 2) in terms of patient selection, time from TBI to MRS, voxel location, method of outcome assessment and timing of outcome assessment. For instance, among studies of patients with TBI, one included only patients in a vegetative state [42], another included patients with severe TBI [17] and a third excluded patients with early initial coma [36]. These differences in patient selection may be associated with differences in severity of brain oedema and in associated hypoxia and herniation, thereby introducing bias into the interpretation of the results. MRS findings vary greatly according to time since TBI. Four phases may be distinguished: an acute phase, which lasts 24 hours after TBI; an early subacute phase, which spans from the days 1 to 13; a late subacute phase, from days 14 to 20; and a chronic phase, which starts on day 21. Only two studies included patients at the acute phase [38,40], and only one of these included all patients before 72 hours [38]. Two studies were conducted from the early subacute phase to the first month [17,37] and one began inclusion in the late subacute phase but included patients up to 11 months after TBI [43]. Four studies focused on the chronic phase; in two of these studies, patients were included 3 weeks to 6 months after TBI [36,39] and in the other two studies they were included 2 months to 8 months after TBI [39,42].
Although NAA/creatine ratios were similar across studies, the results should be interpreted with caution because experimental in vitro and in vivo data suggest differences in the underlying pathophysiological mechanisms and in the time course of the lesions [44-46]. To interpret these results reliably, information on NAA values over time are needed. Experiments conducted in vitro [44] and in vivo [45,46] show an early NAA decrease starting within a few minutes after TBI and reaching the trough value within 48 hours. This finding explains why spectroscopic disturbances may require 48 hours for visualization [47]. NAA levels remain stable within the first month after TBI, supporting the validity of MRS assessment during the second or third week [48,49]. Later on, between 6 weeks and 1 year after TBI, NAA levels may decrease [9,37]. Partial recovery of NAA levels has been suggested and may indicate recovery of mitochondrial function [41].
Another important factor that varied across studies was MRS voxel location (Table 2). Voxels were located in the hemisphere (the occipitoparietal, frontoparietal, or frontal lobes), corpus callosum, thalamus, or brainstem (the pons). Because whole brain analysis is time consuming, voxels are typically restricted to the areas most affected by DAI, namely the lobar white matter, corpus callosum and upper brainstem [50]. Estimation of NAA in the whole brain may improve the prognostic value of MRS [41]. A good compromise may be a voxel encompassing the corpus callosum, white matter and part of the hemispheric cortex [38].
Studies also differed in their definitions of poor and good GOS outcome groups: comparisons involved GOS score 1 to 2 versus GOS score 3 to 5 [39], GOS score 1 to 4 versus GOS score 5 [41], or GOS score 1 to 2 versus GOS score 4 to 5 [17]. Finally, the time from TBI to outcome assessment varied from 3 to 18 months (Table 2), further complicating comparisons because neurological status may improve for up to 1 year after TBI.
Although MRS has superseded conventional MRI, the combination of these two techniques may be useful [17]. Variations in the NAA/creatine ratio over time have not been studied in a large TBI patient population. The above-mentioned variability in NAA levels constitutes the main limitation of this technique. To overcome this limitation, repeated studies at intervals of 1 to 2 weeks are probably needed. In our experience, variations in the NAA/creatine ratio are minimal in many patients. We agree with Sinson and coworkers [41] that whole brain NAA estimation might improve the prognostic value of MRS. Absence of dysfunction by MRS is a valuable finding; in a patient with normal results by both conventional MRI and MRS, a poor outcome is unlikely. However, we have seen a few patients with normal conventional MRI and MRS findings who had poor outcomes, probably related to white matter damage detected as DTI abnormalities.
Diffusion tensor magnetic resonance imaging
Initial reports of DTI in TBI patients suggest that this technique may demonstrate alterations in white matter connections that are missed by conventional MRI [51]. DTI provides information on the physiological status of fibre bundles, thus complementing the metabolic and biochemical information supplied by MRS. At present, little is known about the prognostic value of DTI in patients with TBI. DTI findings correlated with clinical status in patients with multiple sclerosis or neurodegenerative disease [52,53]. In a mouse model of TBI, DTI parameters were significantly reduced in the injured brain, whereas conventional MRI showed no significant changes [54]. Furthermore, changes in relative anisotropy correlated significantly with the density of stained axons on histological sections.
In a study comparing 20 TBI patients and 15 healthy control individuals, fractional anisotropy was reduced in the internal capsule and splenium of the corpus callosum and correlated with Glasgow Coma Scale score and Rankin score at discharge in the TBI patients [55]. Similar findings have been reported in children [56]. Anecdotal case reports of DTI abnormalities in TBI patients have been reported [57,58]. In two patients who recovered partially after 6 years and 19 years, respectively, in a minimally conscious state, DTI disclosed increased anisotropy within the midline cerebellar white matter over an 18-month period [59]. This anisotropy increase correlated with an increase in resting metabolism, measured using positron emission tomography, which suggests that axonal regrowth might underlie increases in anisotropy. Larger studies of DTI variations over time are needed. In our institution, comatose patients have been included in a prospective DTI study for the past 3 years. Patients with major connectivity abnormalities in both hemispheres and the brainstem were at increased risk for poor outcomes. A large multicentre prospective study is ongoing in France to assess the usefulness of combining DTI with MRS.
Magnetization transfer imaging
Magnetization transfer imaging is sensitive for detecting white matter lesions in patients with multiple sclerosis, progressive multifocal leukoencephalopathy, or wallerian degeneration [11,12]. Preliminary results in TBI are promising [60,61]. The magnetization transfer ratio was decreased in TBI patients [60,61]. Out of 28 TBI patients, eight had abnormal magnetization transfer ratios, and all eight had persistent neurological deficits [62]. In another study, however, no correlation was found between GOS score and abnormal magnetization transfer ratio [41].
Anoxic/hypoxic encephalopathy
Anoxic/hypoxic encephalopathy is a devastating condition; its development after prolonged cerebral hypoxia is often difficult to predict on clinical grounds. No controlled studies of routine MRI in large numbers of cardiac arrest patients have been reported. Anecdotal case reports and small series are available [63-67]. As with TBI, MRI findings in hypoxic/anoxic encephalopathy go through four phases [66]: an acute phase, which lasts 24 hours after anoxia or hypoxia; an early subacute phase, from days 1 to 13; a late subacute phase, from days 14 to 20; and a chronic phase, starting on day 21. MRI findings in patients with hypoxic brain damage are complex but distinctive. Brain swelling, cortical laminar necrosis, hypersignal of basal ganglia, delayed white matter degeneration and atrophy occur in succession, as shown in Table 3[63,66,67]. During the acute and early subacute phases, DWI and T2-weighted sequence show hypersignals in the cortex, thalamus and basal ganglia. DWI may be more sensitive for detecting mild hypoxic/anoxic injury within the first few hours, and the hypersignal may occur first in the cerebral cortex and later in the basal ganglia. During the late subacute phase the hypersignals previously seen by DWI tend to fade, and diffuse white matter abnormalities denoting delayed anoxic leukoencephalopathy may develop [68]. During the chronic phase diffuse atrophy and dilatation of the ventricles are visible, whereas DWI is normal.
Table 3.
Chronological magnetic resonance imaging findings in anoxic/hypoxic encephalopathy
| Acute phase (<24 hours) | Early subacute phase (24 hours to day 13) | Late subacute phase (days 14 to 20) | Chronic phase (>21 days) | |
| Characteristics | Brain swelling | Brain swelling | Absence of brain swelling | Diffuse atrophy and dilatation of the ventricles |
| DWI | Hypersignals in the cortex, in the thalamus and in the basal ganglia | Hypersignals in the cortex, in the thalamus and in the basal ganglia | Progressive disappearance of hypersignals found previously | Normal |
| T2 | Hypersignals in the cortex, in the thalamus and in the basal ganglia | Hypersignals in the cortex, in the thalamus and in the basal ganglia. Possible subcortical hyposignals | Hypersignals of the cortex, the thalamus, the basal ganglia and the pons | Normal or possible hypersignals of the cortex, the thalamus, the basal ganglia and the pons |
| T1 | No abnormalities | No abnormalities | Possible spontaneous subcortical and basal ganglia hypersignals | Can be normal |
| T1 with gadolinium enhancement | No abnormalities | Possible subcortical enhancement suggestive of cortical laminar necrosis | Possible subcortical enhancement suggestive of cortical laminar necrosis | No abnormalities |
| Comments | DWI seems more sensitive to mild hypoxic/anoxic injury in the first hours, and the hypersignal in cerebral cortex seems more precocious than in the basal ganglia | Hypersignals on both DWI and T2 become more intense, particularly in the thalamus and the basal ganglia | In some cases, appearance of diffuse white matter, abnormalities of delayed anoxic leukoencephalopathy on both DWI and T2 | In some cases, hypersignals of the cortex and hyposignals in the subcortical zone on both T2 and T1, suggestive of cortical laminar necrosis |
DWI, diffusion weighted imaging; T1, T1 weighted sequence; T2, T2 weighted sequence. Adapted from [66,67].
The three main series published to date included ten [66], eight [67] and six [63] patients. Although the small numbers of patients is a limitation, the succession of four phases was confirmed in several case reports and supported by findings of histological and animal studies [9,12,16,67], indicating far greater vulnerability of grey matter to hypoxia as compared with white matter. This difference in vulnerability may explain why some brain regions are more susceptible than others to diffuse insults such as hypoxia or anoxia [2,11,29,66].
A few studies recorded both MRI findings and long-term outcomes in patients with hypoxic/anoxic encephalopathy [64,67,69]. Diffuse cortical abnormalities by DWI in the acute or early subacute phase appear to be of unfavourable prognostic significance. Of six patients with hypoxic encephalopathy investigated by sequential MRI, the only patient who recovered a GOS score greater than 3 had hypersignals in watershed zones in the parieto-occipito-temporal cortex without cortical hypersignal by DWI. In a study of 10 patients who had suffered a cardiac arrest, FLAIR and DWI showed that eight patients had diffuse abnormalities in the cerebellum, thalamus, frontal and parietal cortices, and hippocampus [69]. None of the patients with cortical structure abnormalities recovered beyond a severely disabled state. In another prospective study, the prognostic value of DWI was evaluated in 12 patients within 36 hours after global cerebral hypoxia [64]. DWI findings correlated with clinical outcomes after 6 months. The three patients with short resuscitation times had a good recovery and normal DWI findings. Of the remaining nine patients, all had DWI abnormalities and developed a vegetative state. Thus, diffuse cortical hypersignals by DWI appear to predict a poor outcome. Conversely, several reports describe delayed anoxic encephalopathy with a good final outcome and resolution of MRI abnormalities. Therefore, finding diffuse hypersignals in the white matter by either DWI or T2/FLAIR weighted sequences should not lead to treatment limitation decisions.
In general, whether MRI findings can be used to guide treatment limitation decisions remains unclear. In our unit, treatment limitation is considered in patients with diffuse cortical hypersignals by DWI or cortical laminar necrosis images after prolonged cardiac arrest, provided the MRI findings are consonant with the clinical examination or electrophysiological data. In contrast, a patient with normal MRI findings after anoxia should probably be re-evaluated 1 or 2 weeks later by clinical examination, electrophysiological testing and MRI.
Few data are available on MRS findings after anoxia [70,71]. No studies were specifically designed to assess the prognostic value of DTI in patients with anoxic/hypoxic encephalopathy. The unique ability of DTI to distinguish between white matter and grey matter, allowing separate quantitative assessment of these two tissues, should be of particular interest in anoxic/hypoxic encephalopathy.
Severe hypoglycaemia has been likened to hypoxic encephalopathy. Imaging study data in patients with hypoglycaemic coma are scant [63,72,73]. Interestingly, DWI abnormalities can mimic stroke in patients with hypoglycaemic coma [74,75]. Rapid improvements in DWI and MRI abnormalities after glucose infusion were recently reported [76].
Ischaemic stroke
Ischaemic stroke causes coma in two main settings, namely malignant stroke and basilar artery occlusion. We focus on these two situations, and we do not discuss the prognostic value of MRI after stroke without coma.
In a study of 37 patients with acute middle cerebral artery infarction, early quantitative DWI findings predicted progression to malignant stroke, which occurred in 11 patients [77]. Factors that predicted malignant stroke were as follows: size of the region with apparent diffusion coefficient (ADC) < 80% greater than 82 ml; ADC in the core of the stroke < 300 mm2/s; and relative ADC within the ADC < 80% of the lesion under 0.62. Another study evaluated 28 patients, of whom 11 experienced malignant stroke [78]. The best predictor of malignant stroke within 14 hours of stroke onset was infarct volume by DWI greater than 145 cm3, which was 100% sensitive and 94% specific. Regarding brainstem stroke, a retrospective study of 47 patients showed that coma, which was a feature in nine patients, was associated with lesions in the posterior pons and lower midbrain [21]. The patients who died had all bilateral brainstem lesions in this area. None of the patients with bilateral lesions survived. Although the number of patients was small in the study, the results are consonant with clinical experience that brainstem stroke with coma and large brainstem lesions has a poor outcome and that some patients who are initially comatose with limited anterior brainstem infarction eventually experience good outcomes.
DTI has been used to assess outcomes after stroke [79], although we are not aware of studies of MRS or DTI to predict outcomes after malignant or brainstem stroke. In a study of 12 patients with subcortical infarcts involving the posterior limb of the internal capsule, a decrease in fractional anisotropy was detected by DTI, indicating secondary degeneration of the fibre tract proximal and distal to the primary ischaemic lesion [80]. Fibre tract degeneration occurred gradually, which might have hampered functional recovery. In patients with brainstem stroke or malignant stroke, DTI may be of considerable value for assessing fibre tract degeneration, thus predicting chances of recovery.
Ascending reticular activating system and prognosis of brain injuries
Several brain areas involved in the prognosis of TBI or stroke play a role in consciousness [17,19,21,81]. Figure 3 shows the anatomical regions involved in arousal and consciousness. Brainstem lesions have been shown to influence the prognosis of patients with coma after TBI or stroke [17,19,21,81]. Bilateral brainstem lesions were associated with poorer outcomes [21,81], and the target area appeared to be the posterior pons and lower midbrain, where the ascending reticular activating system (ARAS) nuclei are located. An MRI study of 88 patients in a vegetative state after TBI confirmed the prognostic importance of lesions in this area [19]. The ARAS projects in part to the basal fore-brain through the hypothalamus by its ventral pathway, as shown in Figure 3. Several pathological studies showed a high rate of basal forebrain lesions in humans who died after head injuries [82], and we found that hypothalamic and basal forebrain lesions were associated with poor outcomes in TBI patients [32]. Histological evidence of neuronal damage in the nucleus basalis of Meynert (the main nucleus of the basal forebrain) was found in most of the patients who died after head injury [82]. The ARAS projects to the reticular thalamic nuclei through its dorsal pathway (Figure 3). Focal damage to the thalami was documented in pathological studies of patients in vegetative state [83,84]. All three pathways lead to cortical arousal. Widespread cortical damage (as described in anoxic/hypoxic encephalopathy [83,85]) and widespread white matter damage (as described in TBI patients [86]) may result in inability to arouse cortical areas (vegetative state). Clinical findings in patients with TBI suggest that impairment in consciousness may correlate with depth of the deepest lesion [20,87]. Although lesions to the ARAS or its projections may correlate with severity of the initial injury or the existence of herniation, another possibility is that they directly contribute to the prognosis. Studies involving multimodal investigations would provide valuable insight in this area [88].
Figure 3.
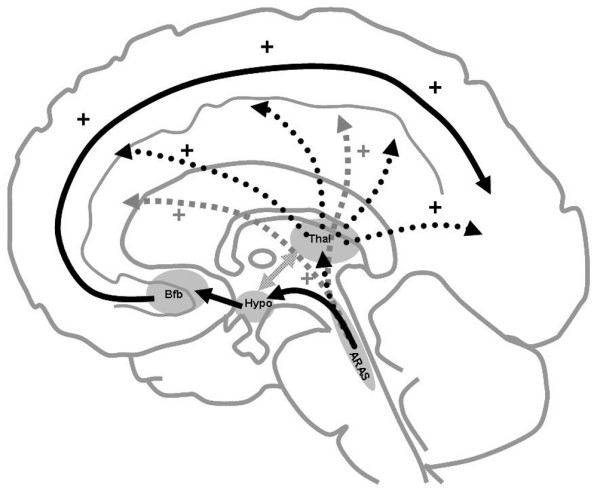
Anatomical substratum of arousal and awareness. Consciousness involves two main components: arousal and awareness of oneself and of the environment. Awareness is dependent on the integrity of specific anatomical regions [89]. The ascending reticular activating system (ARAS), the primary arousal structure, is located in the upper pons and lower midbrain in the posterior part of the upper two-thirds of the brainstem [90,91]. A ventral pathway (black solid arrows) projects to the hypothalamus (hypo) and basal forebrain (Bfb); a dorsal pathway (black dashed arrows) projects to the reticular nuclei of the thalamus (thal); and a third pathway (light grey arrows) projects directly into the cortical regions [90]. From the basal forebrain, two main bundles project diffusely to several cortical areas [92]. The reticular nuclei of the thalamus connect to other nuclei in the thalamus. They are involved in a thalamocortical circuit [93] that controls cortical activity. Some regions of the cerebral cortex may also make specific contributions to consciousness [94].
Avenues for research
Data from patients with TBI, stroke, or anoxic encephalopathy suggest that specific MRI findings may hold promise for outcome prediction. Large studies are not yet available, even in patients with TBI. Given the major ethical, human and economic issues involved, there is an urgent need for large prospective multicentre studies. Only small numbers of patients eligible for such studies are admitted to medical or surgical intensive care units, and few neurosurgical or neurological intensive care units exist; therefore, a multicentre design is essential to ensure recruitment of a sufficiently large population. In our institution, which is a neurosurgical intensive care unit in a tertiary hospital, multimodal prospective imaging by conventional MRI, MRS and DTI is performed routinely in all patients who are still comatose after 2 weeks. A multicentre study funded by the French Ministry of Health is under way.
Conclusion
Patients with severe brain injury, most notably those who remain comatose, generate huge health care costs. Adapting the level of medical care to the neurological outcome is a major challenge currently faced by neurological intensive care. Meeting this challenge will require the development of tools that reliably predict long-term neurological outcomes.
Most MRI studies to date were conducted in patients with TBI. By conventional imaging, presence of bilateral lesions in the dorsolateral upper brainstem appears to be the factor of greatest adverse prognostic significance. With MRS, low NAA/creatine ratio in the hemispheres and in the pons predicts a poor outcome. In anoxic/hypoxic encephalopathy, the factor of greatest adverse significance appears to be the presence of diffuse cortical abnormalities by DWI. However, data are scarcer than in the field of TBI. Finally, regarding brainstem stroke, posterior lesions appear to be associated with poor outcome.
The prognostic value of imaging studies could be improved by combining several techniques and sequences, for instance by combining several MRI sequences or by combining MRI with electrophysiological studies or clinical data. Complete destruction of arousal structures is consistently associated with poor outcome. Multimodal MRI is a promising technique that can be expected to provide accurate prediction of neurological outcome in the near future.
Abbreviations
ADC = apparent diffusion coefficient; ARAS = ascending reticular activating system; DAI = diffuse axonal injury; DTI = diffusion tensor imaging; DWI = diffusion weighted imaging; FLAIR = fluid-attenuated inversion recovery; GOS = Glasgow Outcome Scale; MRI = magnetic resonance imaging; MRS = magnetic resonance spectroscopy; NAA = N-acetyl-aspartate; TBI = traumatic brain injury.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Nicolas Weiss, Email: nic.weiss@wanadoo.fr.
Damien Galanaud, Email: galanaud@dat.org.
Alexandre Carpentier, Email: alexandre.carpentier@lrb.aphp.fr.
Lionel Naccache, Email: lionel.naccache@wanadoo.fr.
Louis Puybasset, Email: louis.puybasset@psl.aphp.fr.
References
- Oddo M, Schaller MD, Feihl F, Ribordy V, Liaudet L. From evidence to clinical practice: effective implementation of therapeutic hypothermia to improve patient outcome after cardiac arrest. Crit Care Med. 2006;34:1865–1873. doi: 10.1097/01.CCM.0000221922.08878.49. [DOI] [PubMed] [Google Scholar]
- Celesia GG. Persistent vegetative state. Neurology. 1993;43:1457–1458. doi: 10.1212/wnl.43.8.1457. [DOI] [PubMed] [Google Scholar]
- Jennett B. Thirty years of the vegetative state: clinical, ethical and legal problems. Prog Brain Res. 2005;150:537–543. doi: 10.1016/S0079-6123(05)50037-2. [DOI] [PubMed] [Google Scholar]
- Payne K, Taylor RM, Stocking C, Sachs GA. Physicians' attitudes about the care of patients in the persistent vegetative state: a national survey. Ann Intern Med. 1996;125:104–110. doi: 10.7326/0003-4819-125-2-199607150-00004. [DOI] [PubMed] [Google Scholar]
- Anderson CV, Wood DM, Bigler ED, Blatter DD. Lesion volume, injury severity, and thalamic integrity following head injury. J Neurotrauma. 1996;13:35–40. doi: 10.1089/neu.1996.13.35. [DOI] [PubMed] [Google Scholar]
- Brandstack N, Kurki T, Tenovuo O, Isoniemi H. MR imaging of head trauma: visibility of contusions and other intraparenchymal injuries in early and late stage. Brain Inj. 2006;20:409–416. doi: 10.1080/02699050500487951. [DOI] [PubMed] [Google Scholar]
- Gerber DJ, Weintraub AH, Cusick CP, Ricci PE, Whiteneck GG. Magnetic resonance imaging of traumatic brain injury: relationship of T2*SE and T2GE to clinical severity and outcome. Brain Inj. 2004;18:1083–1097. doi: 10.1080/02699050410001672341. [DOI] [PubMed] [Google Scholar]
- Scheid R, Preul C, Gruber O, Wiggins C, von Cramon DY. Diffuse axonal injury associated with chronic traumatic brain injury: evidence from T2*-weighted gradient-echo imaging at 3 T. AJNR Am J Neuroradiol. 2003;24:1049–1056. [PMC free article] [PubMed] [Google Scholar]
- Brooks WM, Friedman SD, Gasparovic C. Magnetic resonance spectroscopy in traumatic brain injury. J Head Trauma Rehabil. 2001;16:149–164. doi: 10.1097/00001199-200104000-00005. [DOI] [PubMed] [Google Scholar]
- Garnett MR, Cadoux-Hudson TA, Styles P. How useful is magnetic resonance imaging in predicting severity and outcome in traumatic brain injury? Curr Opin Neurol. 2001;14:753–757. doi: 10.1097/00019052-200112000-00012. [DOI] [PubMed] [Google Scholar]
- Filippi M, Rocca MA. Magnetization transfer magnetic resonance imaging in the assessment of neurological diseases. J Neuroimaging. 2004;14:303–313. doi: 10.1177/1051228404265708. [DOI] [PubMed] [Google Scholar]
- Horsfield Ma. Magnetization transfer imaging in multiple sclerosis. J Neuroimaging. 2005;(Suppl):58S–67S. doi: 10.1177/1051228405282242. [DOI] [PubMed] [Google Scholar]
- Pickard JD, Hutchinson PJ, Coles JP, Steiner LA, Johnston AJ, Fryer TD, Coleman MR, Smielewski P, Chatfield DA, Aigbirhio F, et al. Imaging of cerebral blood flow and metabolism in brain injury in the ICU. Acta Neurochir Suppl. 2005;95:459–464. doi: 10.1007/3-211-32318-x_94. [DOI] [PubMed] [Google Scholar]
- Azouvi P. Neuroimaging correlates of cognitive and functional outcome after traumatic brain injury. Curr Opin Neurol. 2000;13:665–669. doi: 10.1097/00019052-200012000-00009. [DOI] [PubMed] [Google Scholar]
- Fontaine A, Azouvi P, Remy P, Bussel B, Samson Y. Functional anatomy of neuropsychological deficits after severe traumatic brain injury. Neurology. 1999;53:1963–1968. doi: 10.1212/wnl.53.9.1963. [DOI] [PubMed] [Google Scholar]
- Jenkins A, Teasdale G, Hadley MD, Macpherson P, Rowan JO. Brain lesions detected by magnetic resonance imaging in mild and severe head injuries. Lancet. 1986;2:445–446. doi: 10.1016/S0140-6736(86)92145-8. [DOI] [PubMed] [Google Scholar]
- Carpentier A, Galanaud D, Puybasset L, Muller JC, Lescot T, Boch AL, Riedl V, Cornu P, Coriat P, Dormont D, et al. Early morphologic and spectroscopic magnetic resonance in severe traumatic brain injuries can detect 'invisible brain stem damage' and predict 'vegetative states'. J Neurotrauma. 2006;23:674–685. doi: 10.1089/neu.2006.23.674. [DOI] [PubMed] [Google Scholar]
- Firsching R, Woischneck D, Diedrich M, Klein S, Ruckert A, Wittig H, Dohring W. Early magnetic resonance imaging of brainstem lesions after severe head injury. J Neurosurg. 1998;89:707–712. doi: 10.3171/jns.1998.89.5.0707. [DOI] [PubMed] [Google Scholar]
- Kampfl A, Schmutzhard E, Franz G, Pfausler B, Haring HP, Ulmer H, Felber S, Golaszewski S, Aichner F. Prediction of recovery from post-traumatic vegetative state with cerebral magnetic-resonance imaging. Lancet. 1998;351:1763–1767. doi: 10.1016/S0140-6736(97)10301-4. [DOI] [PubMed] [Google Scholar]
- Levin HS, Mendelsohn D, Lilly MA, Yeakley J, Song J, Scheibel RS, Harward H, Fletcher JM, Kufera JA, Davidson KC, Bruce D. Magnetic resonance imaging in relation to functional outcome of pediatric closed head injury: a test of the Ommaya-Gennarelli model. Neurosurgery. 1997;40:432–440. doi: 10.1097/00006123-199703000-00002. discussion 440–441. [DOI] [PubMed] [Google Scholar]
- Parvizi J, Damasio AR. Neuroanatomical correlates of brainstem coma. Brain. 2003;126:1524–1536. doi: 10.1093/brain/awg166. [DOI] [PubMed] [Google Scholar]
- Gentry LR. Imaging of closed head injury. Radiology. 1994;191:1–17. doi: 10.1148/radiology.191.1.8134551. [DOI] [PubMed] [Google Scholar]
- Parizel PM, Ozsarlak , Van Goethem JW, van den Hauwe L, Dillen C, Verlooy J, Cosyns P, De Schepper AM. Imaging findings in diffuse axonal injury after closed head trauma. Eur Radiol. 1998;8:960–965. doi: 10.1007/s003300050496. [DOI] [PubMed] [Google Scholar]
- Wilberger JE, Jr, Deeb Z, Rothfus W. Magnetic resonance imaging in cases of severe head injury. Neurosurgery. 1987;20:571–576. doi: 10.1227/00006123-198704000-00011. [DOI] [PubMed] [Google Scholar]
- Huisman TA. Diffusion-weighted imaging: basic concepts and application in cerebral stroke and head trauma. Eur Radiol. 2003;13:2283–2297. doi: 10.1007/s00330-003-1843-6. [DOI] [PubMed] [Google Scholar]
- Jennett B, Bond M. Assessment of outcome after severe brain damage. Lancet. 1975;1:480–484. doi: 10.1016/S0140-6736(75)92830-5. [DOI] [PubMed] [Google Scholar]
- Paterakis K, Karantanas AH, Komnos A, Volikas Z. Outcome of patients with diffuse axonal injury: the significance and prognostic value of MRI in the acute phase. J Trauma. 2000;49:1071–1075. doi: 10.1097/00005373-200012000-00016. [DOI] [PubMed] [Google Scholar]
- Yanagawa Y, Tsushima Y, Tokumaru A, Un-no Y, Sakamoto T, Okada Y, Nawashiro H, Shima K. A quantitative analysis of head injury using T2*-weighted gradient-echo imaging. J Trauma. 2000;49:272–277. doi: 10.1097/00005373-200008000-00013. [DOI] [PubMed] [Google Scholar]
- Firsching R, Woischneck D, Klein S, Reissberg S, Dohring W, Peters B. Classification of severe head injury based on magnetic resonance imaging. Acta Neurochir (Wien) 2001;143:263–271. doi: 10.1007/s007010170106. [DOI] [PubMed] [Google Scholar]
- Pierallini A, Pantano P, Fantozzi LM, Bonamini M, Vichi R, Zylberman R, Pisarri F, Colonnese C, Bozzao L. Correlation between MRI findings and long-term outcome in patients with severe brain trauma. Neuroradiology. 2000;42:860–867. doi: 10.1007/s002340000447. [DOI] [PubMed] [Google Scholar]
- Wedekind C, Hesselmann V, Lippert-Gruner M, Ebel M. Trauma to the pontomesencephalic brainstem: a major clue to the prognosis of severe traumatic brain injury. Br J Neurosurg. 2002;16:256–260. doi: 10.1080/02688690220148842. [DOI] [PubMed] [Google Scholar]
- Weiss N, Galanaud D, Carpentier A, Tezenas de Montcel S, Naccache L, Coriat P, Puybasset L. A combined clinical and MRI approach for outcome assessment of traumatic head injured comatose patients. J Neurol. 2007. [DOI] [PubMed]
- Cecil KM, Hills EC, Sandel ME, Smith DH, McIntosh TK, Mannon LJ, Sinson GP, Bagley LJ, Grossman RI, Lenkinski RE. Proton magnetic resonance spectroscopy for detection of axonal injury in the splenium of the corpus callosum of brain-injured patients. J Neurosurg. 1998;88:795–801. doi: 10.3171/jns.1998.88.5.0795. [DOI] [PubMed] [Google Scholar]
- Brooks WM, Stidley CA, Petropoulos H, Jung RE, Weers DC, Friedman SD, Barlow MA, Sibbitt WL, Jr, Yeo RA. Metabolic and cognitive response to human traumatic brain injury: a quantitative proton magnetic resonance study. J Neurotrauma. 2000;17:629–640. doi: 10.1089/089771500415382. [DOI] [PubMed] [Google Scholar]
- Friedman SD, Brooks WM, Jung RE, Hart BL, Yeo RA. Proton MR spectroscopic findings correspond to neuropsychological function in traumatic brain injury. AJNR Am J Neuroradiol. 1998;19:1879–1885. [PMC free article] [PubMed] [Google Scholar]
- Friedman SD, Brooks WM, Jung RE, Chiulli SJ, Sloan JH, Montoya BT, Hart BL, Yeo RA. Quantitative proton MRS predicts outcome after traumatic brain injury. Neurology. 1999;52:1384–1391. doi: 10.1212/wnl.52.7.1384. [DOI] [PubMed] [Google Scholar]
- Garnett MR, Blamire AM, Corkill RG, Cadoux-Hudson TA, Rajagopalan B, Styles P. Early proton magnetic resonance spectroscopy in normal-appearing brain correlates with outcome in patients following traumatic brain injury. Brain. 2000;123:2046–2054. doi: 10.1093/brain/123.10.2046. [DOI] [PubMed] [Google Scholar]
- Marino S, Zei E, Battaglini M, Vittori C, Buscalferri A, Bramanti P, Federico A, De Stefano N. Acute metabolic brain changes following traumatic brain injury and their relevance to clinical severity and outcome. J Neurol Neurosurg Psychiatry. 2007;78:501–507. doi: 10.1136/jnnp.2006.099796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci R, Barbarella G, Musi P, Boldrini P, Trevisan C, Basaglia N. Localised proton MR spectroscopy of brain metabolism changes in vegetative patients. Neuroradiology. 1997;39:313–319. doi: 10.1007/s002340050415. [DOI] [PubMed] [Google Scholar]
- Ross BD, Ernst T, Kreis R, Haseler LJ, Bayer S, Danielsen E, Bluml S, Shonk T, Mandigo JC, Caton W, et al. 1H MRS in acute traumatic brain injury. J Magn Reson Imaging. 1998;8:829–840. doi: 10.1002/jmri.1880080412. [DOI] [PubMed] [Google Scholar]
- Sinson G, Bagley LJ, Cecil KM, Torchia M, McGowan JC, Lenkinski RE, McIntosh TK, Grossman RI. Magnetization transfer imaging and proton MR spectroscopy in the evaluation of axonal injury: correlation with clinical outcome after traumatic brain injury. AJNR Am J Neuroradiol. 2001;22:143–151. [PMC free article] [PubMed] [Google Scholar]
- Uzan M, Albayram S, Dashti SG, Aydin S, Hanci M, Kuday C. Thalamic proton magnetic resonance spectroscopy in vegetative state induced by traumatic brain injury. J Neurol Neurosurg Psychiatry. 2003;74:33–38. doi: 10.1136/jnnp.74.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe BY, Suh TS, Choi KH, Shinn KS, Park CK, Kang JK. Neuronal dysfunction in patients with closed head injury evaluated by in vivo 1H magnetic resonance spectroscopy. Invest Radiol. 1995;30:502–506. doi: 10.1097/00004424-199508000-00008. [DOI] [PubMed] [Google Scholar]
- Signoretti S, Marmarou A, Tavazzi B, Lazzarino G, Beaumont A, Vagnozzi R. N-Acetylaspartate reduction as a measure of injury severity and mitochondrial dysfunction following diffuse traumatic brain injury. J Neurotrauma. 2001;18:977–991. doi: 10.1089/08977150152693683. [DOI] [PubMed] [Google Scholar]
- Cecil KM, Lenkinski RE, Meaney DF, McIntosh TK, Smith DH. High-field proton magnetic resonance spectroscopy of a swine model for axonal injury. J Neurochem. 1998;70:2038–2044. doi: 10.1046/j.1471-4159.1998.70052038.x. [DOI] [PubMed] [Google Scholar]
- Rubin Y, Cecil K, Wehrli S, McIntosh TK, Lenkinski RE, Smith DH. High-resolution 1H NMR spectroscopy following experimental brain trauma. J Neurotrauma. 1997;14:441–449. doi: 10.1089/neu.1997.14.441. [DOI] [PubMed] [Google Scholar]
- Alessandri B, al-Samsam R, Corwin F, Fatouros P, Young HF, Bullock RM. Acute and late changes in N-acetyl-aspartate following diffuse axonal injury in rats: an MRI spectroscopy and microdialysis study. Neurol Res. 2000;22:705–712. doi: 10.1080/01616412.2000.11740744. [DOI] [PubMed] [Google Scholar]
- Holshouser BA, Tong KA, Ashwal S, Oyoyo U, Ghamsary M, Saunders D, Shutter L. Prospective longitudinal proton magnetic resonance spectroscopic imaging in adult traumatic brain injury. J Magn Reson Imaging. 2006;24:33–40. doi: 10.1002/jmri.20607. [DOI] [PubMed] [Google Scholar]
- Signoretti S, Marmarou A, Fatouros P, Hoyle R, Beaumont A, Sawauchi S, Bullock R, Young H. Application of chemical shift imaging for measurement of NAA in head injured patients. Acta Neurochir Suppl. 2002;81:373–375. doi: 10.1007/978-3-7091-6738-0_94. [DOI] [PubMed] [Google Scholar]
- Adams JH, Graham DI, Murray LS, Scott G. Diffuse axonal injury due to nonmissile head injury in humans: an analysis of 45 cases. Ann Neurol. 1982;12:557–563. doi: 10.1002/ana.410120610. [DOI] [PubMed] [Google Scholar]
- Levin HS. Neuroplasticity following non-penetrating traumatic brain injury. Brain Inj. 2003;17:665–674. doi: 10.1080/0269905031000107151. [DOI] [PubMed] [Google Scholar]
- Catani M. Diffusion tensor magnetic resonance imaging tractography in cognitive disorders. Curr Opin Neurol. 2006;19:599–606. doi: 10.1097/01.wco.0000247610.44106.3f. [DOI] [PubMed] [Google Scholar]
- Reich DS, Smith SA, Jones CK, Zackowski KM, van Zijl PC, Calabresi PA, Mori S. Quantitative characterization of the corticospinal tract at 3T. AJNR Am J Neuroradiol. 2006;27:2168–2178. [PMC free article] [PubMed] [Google Scholar]
- Mac Donald CL, Dikranian K, Song SK, Bayly PV, Holtzman DM, Brody DL. Detection of traumatic axonal injury with diffusion tensor imaging in a mouse model of traumatic brain injury. Exp Neurol. 2007;205:116–131. doi: 10.1016/j.expneurol.2007.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisman TA, Schwamm LH, Schaefer PW, Koroshetz WJ, Shetty-Alva N, Ozsunar Y, Wu O, Sorensen AG. Diffusion tensor imaging as potential biomarker of white matter injury in diffuse axonal injury. AJNR Am J Neuroradiol. 2004;25:370–376. [PMC free article] [PubMed] [Google Scholar]
- Wilde EA, Chu Z, Bigler ED, Hunter JV, Fearing MA, Hanten G, Newsome MR, Scheibel RS, Li X, Levin HS. Diffusion tensor imaging in the corpus callosum in children after moderate to severe traumatic brain injury. J Neurotrauma. 2006;23:1412–1426. doi: 10.1089/neu.2006.23.1412. [DOI] [PubMed] [Google Scholar]
- Ewing-Cobbs L, Hasan KM, Prasad MR, Kramer L, Bachevalier J. Corpus callosum diffusion anisotropy correlates with neuropsychological outcomes in twins disconcordant for traumatic brain injury. AJNR Am J Neuroradiol. 2006;27:879–881. [PMC free article] [PubMed] [Google Scholar]
- Naganawa S, Sato C, Ishihra S, Kumada H, Ishigaki T, Miura S, Watanabe M, Maruyama K, Takizawa O. Serial evaluation of diffusion tensor brain fiber tracking in a patient with severe diffuse axonal injury. AJNR Am J Neuroradiol. 2004;25:1553–1556. [PMC free article] [PubMed] [Google Scholar]
- Voss HU, Uluc AM, Dyke JP, Watts R, Kobylarz EJ, McCandliss BD, Heier LA, Beattie BJ, Hamacher KA, Vallabhajosula S, et al. Possible axonal regrowth in late recovery from the minimally conscious state. J Clin Invest. 2006;116:2005–2011. doi: 10.1172/JCI27021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H, Meaney DF, McGowan JC, Grossman RI, Lenkinski RE, Ross DT, McIntosh TK, Gennarelli TA, Smith DH. Magnetization transfer imaging of diffuse axonal injury following experimental brain injury in the pig: characterization by magnetization transfer ratio with histopathologic correlation. J Comput Assist Tomogr. 1996;20:540–546. doi: 10.1097/00004728-199607000-00007. [DOI] [PubMed] [Google Scholar]
- McGowan JC, McCormack TM, Grossman RI, Mendonca R, Chen XH, Berlin JA, Meaney DF, Xu BN, Cecil KM, McIntosh TK, et al. Diffuse axonal pathology detected with magnetization transfer imaging following brain injury in the pig. Magn Reson Med. 1999;41:727–733. doi: 10.1002/(SICI)1522-2594(199904)41:4<727::AID-MRM11>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Bagley LJ, McGowan JC, Grossman RI, Sinson G, Kotapka M, Lexa FJ, Berlin JA, McIntosh TK. Magnetization transfer imaging of traumatic brain injury. J Magn Reson Imaging. 2000;11:1–8. doi: 10.1002/(SICI)1522-2586(200001)11:1<1::AID-JMRI1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Fujioka M, Okuchi K, Sakaki T, Hiramatsu K, Miyamoto S, Iwasaki S. Specific changes in human brain following reperfusion after cardiac arrest. Stroke. 1994;25:2091–2095. doi: 10.1161/01.str.25.10.2091. [DOI] [PubMed] [Google Scholar]
- Els T, Kassubek J, Kubalek R, Klisch J. Diffusion-weighted MRI during early global cerebral hypoxia: a predictor for clinical outcome? Acta Neurol Scand. 2004;110:361–367. doi: 10.1111/j.1600-0404.2004.00342.x. [DOI] [PubMed] [Google Scholar]
- Torbey MT, Bhardwaj A. MR imaging in comatose survivors of cardiac resuscitation. AJNR Am J Neuroradiol. 2002;23:738. [PMC free article] [PubMed] [Google Scholar]
- Arbelaez A, Castillo M, Mukherji SK. Diffusion-weighted MR imaging of global cerebral anoxia. AJNR Am J Neuroradiol. 1999;20:999–1007. [PMC free article] [PubMed] [Google Scholar]
- Takahashi S, Higano S, Ishii K, Matsumoto K, Sakamoto K, Iwasaki Y, Suzuki M. Hypoxic brain damage: cortical laminar necrosis and delayed changes in white matter at sequential MR imaging. Radiology. 1993;189:449–456. doi: 10.1148/radiology.189.2.8210374. [DOI] [PubMed] [Google Scholar]
- Kim HY, Kim BJ, Moon SY, Kwon JC, Shon YM, Na DG, Lee KH, Na DL. Serial diffusion-weighted MR Imaging in delayed post-anoxic encephalopathy. A case study. J Neuroradiol. 2002;29:211–215. [PubMed] [Google Scholar]
- Wijdicks EF, Campeau NG, Miller GM. MR imaging in comatose survivors of cardiac resuscitation. AJNR Am J Neuroradiol. 2001;22:1561–1565. [PMC free article] [PubMed] [Google Scholar]
- Wartenberg KE, Patsalides A, Yepes MS. Is magnetic resonance spectroscopy superior to conventional diagnostic tools in hypoxic-ischemic encephalopathy? J Neuroimaging. 2004;14:180–186. doi: 10.1177/1051228403259514. [DOI] [PubMed] [Google Scholar]
- Kucharczyk J, Moseley M, Kurhanewicz J, Norman D. MRS of ischemic/hypoxic brain disease. Invest Radiol. 1989;24:951–954. doi: 10.1097/00004424-198912000-00005. [DOI] [PubMed] [Google Scholar]
- Lo L, Tan AC, Umapathi T, Lim CC. Diffusion-weighted MR imaging in early diagnosis and prognosis of hypoglycemia. AJNR Am J Neuroradiol. 2006;27:1222–1224. [PMC free article] [PubMed] [Google Scholar]
- Yanagawa Y, Isoi N, Tokumaru AM, Sakamoto T, Okada Y. Diffusion-weighted MRI predicts prognosis in severe hypoglycemic encephalopathy. J Clin Neurosci. 2006;13:696–699. doi: 10.1016/j.jocn.2005.02.027. [DOI] [PubMed] [Google Scholar]
- Bottcher J, Kunze A, Kurrat C, Schmidt P, Hagemann G, Witte OW, Kaiser WA. Localized reversible reduction of apparent diffusion coefficient in transient hypoglycemia-induced hemiparesis. Stroke. 2005;36:e20–e22. doi: 10.1161/01.STR.0000155733.65215.c2. [DOI] [PubMed] [Google Scholar]
- Cordonnier C, Oppenheim C, Lamy C, Meder JF, Mas JL. Serial diffusion and perfusion-weighted MR in transient hypoglycemia. Neurology. 2005;65:175. doi: 10.1212/01.wnl.0000167128.14769.7b. [DOI] [PubMed] [Google Scholar]
- Maruya J, Endoh H, Watanabe H, Motoyama H, Abe H. Rapid improvement of diffusion-weighted imaging abnormalities after glucose infusion in hypoglycaemic coma. J Neurol Neurosurg Psychiatry. 2007;78:102–103. doi: 10.1136/jnnp.2006.096776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomalla GJ, Kucinski T, Schoder V, Fiehler J, Knab R, Zeumer H, Weiller C, Rother J. Prediction of malignant middle cerebral artery infarction by early perfusion- and diffusion-weighted magnetic resonance imaging. Stroke. 2003;34:1892–1899. doi: 10.1161/01.STR.0000081985.44625.B6. [DOI] [PubMed] [Google Scholar]
- Oppenheim C, Samson Y, Manai R, Lalam T, Vandamme X, Crozier S, Srour A, Cornu P, Dormont D, Rancurel G, et al. Prediction of malignant middle cerebral artery infarction by diffusion-weighted imaging. Stroke. 2000;31:2175–2181. doi: 10.1161/01.str.31.9.2175. [DOI] [PubMed] [Google Scholar]
- Konishi J, Yamada K, Kizu O, Ito H, Sugimura K, Yoshikawa K, Nakagawa M, Nishimura T. MR tractography for the evaluation of functional recovery from lenticulostriate infarcts. Neurology. 2005;64:108–113. doi: 10.1212/01.WNL.0000148477.65273.0C. [DOI] [PubMed] [Google Scholar]
- Liang Z, Zeng J, Liu S, Ling X, Xu A, Yu J, Ling L. A prospective study of secondary degeneration following subcortical infarction using diffusion tensor imaging. J Neurol Neurosurg Psychiatry. 2007;78:581–586. doi: 10.1136/jnnp.2006.099077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firsching R, Woischneck D, Klein S, Ludwig K, Dohring W. Brain stem lesions after head injury. Neurol Res. 2002;24:145–146. doi: 10.1179/016164102101199684. [DOI] [PubMed] [Google Scholar]
- Murdoch I, Nicoll JA, Graham DI, Dewar D. Nucleus basalis of Meynert pathology in the human brain after fatal head injury. J Neurotrauma. 2002;19:279–284. doi: 10.1089/08977150252807018. [DOI] [PubMed] [Google Scholar]
- Kinney HC, Samuels MA. Neuropathology of the persistent vegetative state. A review. J Neuropathol Exp Neurol. 1994;53:548–558. doi: 10.1097/00005072-199411000-00002. [DOI] [PubMed] [Google Scholar]
- Graham DI, Maxwell WL, Adams JH, Jennett B. Novel aspects of the neuropathology of the vegetative state after blunt head injury. Prog Brain Res. 2005;150:445–455. doi: 10.1016/S0079-6123(05)50031-1. [DOI] [PubMed] [Google Scholar]
- Adams JH, Connor RC. The shocked head injury. Lancet. 1966;1:263–264. doi: 10.1016/s0140-6736(66)90080-8. [DOI] [PubMed] [Google Scholar]
- Adams JH, Doyle D, Ford I, Gennarelli TA, Graham DI, McLellan DR. Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology. 1989;15:49–59. doi: 10.1111/j.1365-2559.1989.tb03040.x. [DOI] [PubMed] [Google Scholar]
- Ommaya AK, Gennarelli TA. Cerebral concussion and traumatic unconsciousness. Correlation of experimental and clinical observations of blunt head injuries. Brain. 1974;97:633–654. doi: 10.1093/brain/97.1.633. [DOI] [PubMed] [Google Scholar]
- Laureys S, Giacino JT, Schiff ND, Schabus M, Owen AM. How should functional imaging of patients with disorders of consciousness contribute to their clinical rehabilitation needs? Curr Opin Neurol. 2006;19:520–527. doi: 10.1097/WCO.0b013e3280106ba9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laureys S, Owen AM, Schiff ND. Brain function in coma, vegetative state, and related disorders. Lancet Neurol. 2004;3:537–546. doi: 10.1016/S1474-4422(04)00852-X. [DOI] [PubMed] [Google Scholar]
- Parvizi J, Damasio A. Consciousness and the brainstem. Cognition. 2001;79:135–160. doi: 10.1016/S0010-0277(00)00127-X. [DOI] [PubMed] [Google Scholar]
- Plum F, Posner JB. The Diagnosis of Stupor and Coma. 3. UK: Oxford University Press; 1980. [Google Scholar]
- Selden NR, Gitelman DR, Salamon-Murayama N, Parrish TB, Mesulam MM. Trajectories of cholinergic pathways within the cerebral hemispheres of the human brain. Brain. 1998;121:2249–2257. doi: 10.1093/brain/121.12.2249. [DOI] [PubMed] [Google Scholar]
- Steriade M. Central core modulation of spontaneous oscillations and sensory transmission in thalamocortical systems. Curr Opin Neurobiol. 1993;3:619–625. doi: 10.1016/0959-4388(93)90064-6. [DOI] [PubMed] [Google Scholar]
- Laureys S, Goldman S, Phillips C, Van Bogaert P, Aerts J, Luxen A, Franck G, Maquet P. Impaired effective cortical connectivity in vegetative state: preliminary investigation using PET. Neuroimage. 1999;9:377–382. doi: 10.1006/nimg.1998.0414. [DOI] [PubMed] [Google Scholar]
- Firsching R, Woischneck D, Klein S, Ludwig K, Döhring W. Brain stem lesions after head injury. Neurol Res. 2002;24:145–146. doi: 10.1179/016164102101199684. [DOI] [PubMed] [Google Scholar]