

Abstract
Granulocyte macrophage–colony stimulating factor (GM-CSF) is critically involved in development of organ-related autoimmune inflammatory diseases including experimental allergic encephalitis and collagen-induced arthritis. Roles of GM-CSF in the initiation and in the effector phase of the autoimmune response have been proposed. Our study was designed to investigate the mechanisms of GM-CSF in autoimmunity using a model of autoimmune heart inflammatory disease (myocarditis). The pathological sequel after immunization with heart myosin has been shown previously to depend on IL-1, IL-6, IL-23, and IL-17. We found that innate GM-CSF was critical for IL-6 and IL-23 responses by dendritic cells and generation of pathological Th17 cells in vivo. Moreover, GM-CSF promoted autoimmunity by enhancing IL-6–dependent survival of antigen specific CD4+ T cells. These results suggest a novel role for GM-CSF in promoting generation and maintenance of Th17 cells by regulation of IL-6 and IL-23 in vivo.
Recent studies demonstrated a novel proinflammatory subpopulation of IL-17– producing T helper cells, termed Th17, that are distinct from Th1 and Th2. These Th17 cells have been suggested to mediate inflammation associated with several organ-related autoimmune diseases including experimental allergic encephalitis (EAE) (1–3), collagen-induced arthritis (4), and experimental autoimmune myocarditis (EAM) (5, 6). Evidence supporting a new concept in which IL-23–dependent Th17, and not IL-12–dependent Th1, cells are responsible for autoimmune inflammation came from reports showing that IL-12 (p35)–deficient and IFN-γ–deficient mice developed normal or even exaggerated autoimmune inflammation, whereas IL-23 (p19)–deficient and IL-17–deficient mice were protected. Consequently, there has been a strong interest in defining effector cytokines produced by the pathological Th17 cells and the conditions and factors determining development and activity of Th17 cells. Besides IL-17A and IL-17F, Th17 cells have been reported to coexpress the proinflammatory cytokines IL-6, TNF-α (2, 7), and IL-22 (8). Similar to Th1 and Th2 development, the cytokine environment plays a critical role in positive and negative regulation of Th17 development. TGF-β, IL-6, and IL-1 have been shown to induce IL-17 production in naive CD4+ T cells (9–12), whereas IL-23 turned out to be essential for expansion and survival of Th17 effector memory cells (11). IL-6 plays a key role as it inhibits TGF-β–induced Treg (9, 13) differentiation and induces RORγt (14), a transcription factor specifically required for Th17 development. In contrast, IL-27 (15) and IL-25 (16) have been shown to negatively regulate Th17 development.
Granulocyte macrophage–colony stimulating factor (GM-CSF) is secreted in response to inflammatory stimuli such as LPS, IL-1, and TNF-α by a variety of different cells, including endothelium, fibroblasts, muscle cells, and macrophages, and by activated T cells (for review see reference 17). It was first described as a hematopoietic cytokine promoting proliferation and differentiation of macrophages, granulocytes, and DC from precursors (18–20). However, GM-CSF−/− mice showed normal development of myeloid cells, including macrophages and DC, except a defect of alveolar macrophage function resulting in alveolar proteinosis (21). In contrast, analysis of GM-CSF−/− mice demonstrated an essential role of GM-CSF for development of autoimmune inflammatory diseases such EAE and collagen-induced arthritis (22, 23). In addition, anti–GM-CSF therapy from time of immunization or from first signs of clinical disease completely prevented or ameliorated autoimmune inflammation, respectively (23, 24). It has been proposed that GM-CSF promotes T cell proliferation and pathological Th1 responses indirectly by activation of macrophages, DC, and microglia cells in the initiation or the effector phase and possibly the generation of chemoattractant gradients for recruitment of inflammatory cells to the target organ (23). However, the precise mechanisms have remained elusive.
In this study, we have determined the mechanism of action of GM-CSF in development of EAM, an animal model of human dilated cardiomyopathy, the most common form of cardiomyopathy (∼80–90% of cases) for which mortality rates remain unacceptably high. EAM is induced by immunization of susceptible mice with cardiac myosin or a peptide located in the head portion of myosin heavy chain α (myhcα614-629) emulsified in CFA. Previous reports have demonstrated that the cytokines IL-6, IL-1, IL-23, and IL-17 are crucial for development of EAM (5, 25, 26), whereas IFN-γ is a negative regulator of heart inflammation (27, 28). Thus, the cytokine network responsible for pathological inflammation in the heart (EAM) and brain (EAE) appears to be very similar.
We found that GM-CSF was essential to inhibit apoptosis and promote IL-17 production by autoimmune CD4+ T cells. Both activities are not mutually exclusive and can be mediated by IL-6. In fact, we found that GM-CSF was essential for IL-6 and IL-23 production by DC and/or macrophages in the initiation phase of the autoimmune response
RESULTS
GM-CSF−/− mice are protected from autoimmune myocarditis
To investigate the role of GM-CSF in autoimmune myocarditis, we immunized GM-CSF−/− mice and BALB/c controls with a peptide derived from the myhc (myhcα614-629) in CFA on day 0 and boosted on day 7. By day 21, WT mice developed severe cardiac inflammatory lesions, characterized by infiltrates of lymphocytes, histiocytes, and neutrophils. In contrast, GM-CSF−/− mice showed strongly reduced disease prevalence and histological score because of few inflammatory foci (Fig. 1, A and B). The few lesions found in the hearts of GM-CSF−/− mice had a similar cellular composition as the inflammatory infiltrates of WT hearts (unpublished data).
Figure 1.

Reduced autoimmune myocarditis, T cell proliferation, and antibody responses to heart myosin in GM-CSF–deficient mice. GM-CSF−/− (open circles) and WT (closed circles) mice were immunized at days 0 and 7 with myhcα614-629 peptide in CFA. Hearts were removed at day 21 and fixed in 4% formalin. (A) The severity of inflammatory infiltrates was scored as described in Materials and methods. (B) Representative images of Hematoxylin and Eosin stainings of WT and GM-CSF−/− heart sections. Bars: (left) 2 mm; (middle) 0.5 mm; (right) 0.1 mm. (C) CD4+ T cells were isolated from the spleen 21 d after immunization and in vitro restimulated with BM-DC and titrating amounts of myhcα614-629. Proliferation was assessed by 3H-thymidine incorporation after 3 d of culture. (D) Serum titers of myhcα614-629-specific IgM and IgG at day 21 d after immunization measured by ELISA. Horizontal lines represent the mean titer for each group (D) and the median histological scores of groups (A). Data shown are representative of three to four experiments. *, P < 0.05.
EAM is a CD4 T cell–mediated disease (29). To assess activation and function of myosin-specific CD4+ T cells, we purified splenic CD4+ T cells from immunized mice and stimulated these with myhcα614-629 presented by WT primary DC in vitro. As shown in Fig. 1 C, myhcα614-629-specific CD4+ T cells from WT mice proliferated potently, whereas CD4+ T cells from GM-CSF−/− mice failed to proliferate ex vivo. Moreover, GM-CSF−/− mice mounted strongly reduced CD4 T helper cell–dependent IgG antibody responses, whereas myhcα614-629-specific IgM antibody level was comparable to WT controls (Fig. 1 D). Thus, our results suggest that GM-CSF is critical for development of autoimmune myocarditis by promoting autoimmune CD4+ T cell responses.
GM-CSF enhances secretion of proinflammatory cytokines by DC
DC are the key cells for priming T cells in vivo (30). It is known that different stages of maturation have different effects on immune responses (31). Immature (or semimature) DC have been shown to induce T cell anergy or tolerance (32), whereas combined toll-like receptor (TLR) and CD40 stimulation of mature DC can break tolerance and induce autoimmune disease (33).
Primary DC from spleens of WT and GM-CSF−/− mice showed comparable surface expression of the maturation markers MHC class II, CD40, and CD86 before and after stimulation with CpG or LPS, the ligands for TLR9 and TLR4, respectively (Fig. 2 A). Moreover, production of the proinflammatory cytokines IL-6, IL-12, and TNF-α by DC of WT and GM-CSF−/− mice was comparably increased upon stimulation with CpG (Fig. 2 B) or LPS (not depicted). In contrast, WT DC did not produce detectable GM-CSF (unpublished data). Addition of recombinant GM-CSF significantly augmented production of IL-6, IL-12, and TNF-α, and transcription of IL-23p19 by CpG stimulated primary DC of both WT and GM-CSF−/− mice (Fig. 2, B and C) but had no effect on up-regulation of MHC class II, CD40, and CD86 (Fig. 2 A). These results suggest that GM-CSF produced by non-DC can augment production of proinflammatory cytokines by DC during an immune response but that it is not required for homeostatic DC development or up-regulation of classical DC maturation/activation markers.
Figure 2.

GM-CSF enhances TLR-induced production of proinflammatory cytokines by DC. DC isolated from spleens of naive WT and GM-CSF−/− mice were stimulated with 1 μM CpG or 1 μg/ml LPS in vitro. (A) Surface expression of CD86, CD40, and MHC class II was assessed by flow cytometry after 6 h. Unstimulated cells (shaded curve), stimulated WT (solid line), GM-CSF−/− (dashed line), and GM-CSF−/− DC in the presence of 20 ng/ml rGM-CSF (dotted line). (B) ELISA measurement of IL-6, IL-12(p40/p70), and TNF-α in supernatants of DC stimulated for 24 h with CpG in the presence and absence of 20 ng/ml GM-CSF. (C) IL-23p19 measured by quantitative PCR of DC stimulated for 4 h with 1 μM CpG in the presence and absence of GM-CSF. One representative experiment of at least two is shown. Error bars indicate SD.
Reduced IL-6 production by DC in GM-CSF−/− mice
To assess cytokine production by antigen-presenting cells in myhcα614-629/CFA-immunized WT and GM-CSF−/− mice, we purified a gemisch of cells expressing CD11b and/or CD11c, including macrophages and DC (Mac/DC), from spleens at various time points after immunization and cultured them in the absence or presence of agonist anti-CD40 mAb to mimic stimulation by activated T cells through CD40L. As shown in Fig. 3 A, Mac/DC from WT mice produced IL-6 from day 2 peaking at day 7 after immunization, which was two- to threefold enhanced by CD40 stimulation. In contrast, IL-6 production by Mac/DC from KO was markedly impaired without and with CD40 stimulation. IL-6 expression in vivo determined by real time PCR was ∼10-fold reduced in Mac/DC of GM-CSF−/− mice at day 7 after innoculation (Fig. 3 B). Purification of CD11c+ cells, including CD11c+CD11b− and CD11c+CD11b+ DC, were a source of IL-6, IL-23, and IL-12/23p40 in immunized WT mice. Both IL-6 and IL-23 production was impaired in DC from GM-CSF−/− mice (Fig. 3 C). To exclude impaired maturation of DC and macrophages in GM-CSF−/− mice, we measured expression of typical maturation markers directly ex vivo. We found comparable MHC class II, CD80, CD86, and CD40 on cell surface of CD11c+DC in WT and GM-CSF−/− mice isolated at days 4 (Fig. 3 D) and 7 (not depicted) after immunization. Similarly, macrophages (i.e., CD11b+cells) did not show a difference in these surface molecules, although their expression was expectedly weaker than on DC (unpublished data). Comparable expression of CD40 rules out reduced stimulation of Mac/DC from GM-CSF−/− mice by agonistic CD40 mAb. These data demonstrate that GM-CSF is essential for IL-6 and IL-23 production by DC in vivo in response to immunization with autoantigen in CFA and confirm the data showing that exogenous GM-CSF enhances IL-6 and IL-23 production by CpG-stimulated DC (Fig. 2).
Figure 3.

Reduced production of IL-6 in immunized GM-CSF−/− mice. WT and GM-CSF−/− mice were immunized s.c. with myhcα614-629/CFA as described in the Fig. 1 legend. (A) At the times indicated, CD11b+ and CD11c+ cells were isolated by magnetic sorting and cultured in the presence and absence of anti-CD40 for 20 h. IL-6 concentrations in the supernatant was measured by ELISA. (B) Expression of IL-6 relative to β-actin was assessed by quantitative PCR using cDNA generated from CD11b+ cells. (C) At day 7, CD11c+ cells were isolated by magnetic sorting and cultured in the presence and absence of anti-CD40 for 24 h (4 × 105 DC per well). IL-6, IL-23, and IL-12p40 levels in the supernatant were measured by ELISA. (D) Analysis of draining LN cells by flow cytometry at day 4 after immunization. Histograms show expression of CD80, CD86, CD40, and MHC class II gated on CD11c+ cells. Continuous and dotted lines represent cells of immunized WT and GM-CSF−/− mice, respectively. Shaded curves represent cells of naive mice. (E) BM-derived DC of WT and GM-CSF−/− mice were pulsed with 10 μg/ml myhcα614-629 and stimulated with 1 μg/ml LPS and 5 μg/ml anti-CD40 for 4 h before injection into WT and GM-CSF−/− mice at days 0, 2, and 4. At day 10, hearts were removed and myocarditis scored by histology. Horizontal lines indicate median. Error bars in A indicate SD of triplicate culture wells. Cells were isolated from pooled draining LN of three mice per time point. Error bars in C indicate SD of triplicate cultures of draining LN DC of four to five mice per time point. Data shown are representative of at least two experiments. *, P < 0.05.
Activated DC induce mild myocarditis in GM-CSF−/− mice
We next asked whether LPS/CD40 activation of BM-DC grown in the presence of recombinant GM-CSF could restore disease in GM-CSF−/− mice. To this end, BM-DC of WT and GM-CSF−/− mice were loaded with myhcα614-629 and activated with LPS and anti-CD40 mAb before adoptive transfer into both WT and GM-CSF−/− recipients. As shown in Fig. 3 E, both WT and GM-CSF−/− DC induced comparably strong heart inflammation in WT mice confirming the ability of TLR/CD40-activated DC to induce myocarditis and suggesting that DC of GM-CSF−/− mice have no intrinsic defect in proinflammatory cytokine production when grown in exogenous GM-CSF. Interestingly, injection of GM-CSF–derived BM-DC activated with LPS/CD40 partially restored disease development in GM-CSF−/− mice, although the disease score was still significantly reduced compared with WT recipient mice (Fig. 3 E). These results show that BM-DC grown and activated with a combination of GM-CSF, LPS, and CD40 induce mild myocarditis in GM-CSF−/− mice, probably by secretion of IL-6 and IL-23 as indicated in Figs. 2 (B and C) and 3 (A–C). In addition, the data indicate that such activated DC trigger GM-CSF production by other cells, which are key for development of severe myocarditis.
T cell proliferation and differentiation of Th17 cells, but not Th1 and Th2 cells, is impaired in GM-CSF–deficient CD4 T cells in vitro
The results shown in Fig. 1 C suggest that GM-CSF is required for activation, differentiation, expansion, and/or survival of autoimmune CD4+ T cells. To experimentally address these possibilities, we capitalized from crossing DO11.10 transgenic mice expressing OVA323-339-specific TCR with GM-CSF−/− mice. Naive (i.e., CD62Lhi) DO11.10 transgenic T cells were isolated from GM-CSF−/− and WT mice and cultured with both GM-CSF−/− and WT splenic DC in the presence of titrated concentrations of specific antigen to measure T cell activation, proliferation, and Th1/Th2 polarization at days 3 and 5, respectively. T cell activation, measured by up-regulation of CD25 and down-regulation of CD62L, was unaffected in the absence of GM-CSF (Fig. 4 A). In contrast, T cell proliferation was impaired in cultures containing GM-CSF−/− CD4+ T cells, independent of whether DC were derived from GM-CSF−/− or WT mice, and it was reconstituted by adding recombinant GM-CSF (Fig. 4 B). For unknown reasons, a commercially available GM-CSF–blocking antibody failed to inhibit T cell proliferation in this experimental setup. Th1 and Th2 cell differentiation was induced by culturing cells with high and low dose antigen, respectively, as reported previously (34). A proportion of IFN-γ+ (Th1) and IL-4+ (Th2) cells coproduced GM-CSF in WT CD4+ T cells (unpublished data). However, Th1 and Th2 differentiation remained unaffected in GM-CSF-deficient CD4+ T cells. (Fig. 4 C).
Figure 4.

GM-CSF production by CD4+ T cells promotes proliferation and differentiation of Th17, but not Th1 or Th2, cells in vitro. Naive OVA323-339 TCR transgenic CD4+ T cells purified from spleens of DO11.10/GM-CSF−/− or DO11.10/GM-CSF+/+ mice were cocultured with splenic DC purified from GM-CSF-deficient (KO) or WT mice in the presence of indicated concentrations of ovalbumin protein or OVA323-339 peptide. (A) CD4+ T cell activation measured by CD25 and CD62L surface expression after 3 d of culture. (B) CD4 T cell proliferation measured by pulsing cells with 3H-Thymidine for the last 12 h of a 60-h coculture. (C) Th1/Th2 cell differentiation driven by indicated antigen concentrations was determined by intracellular staining of IFN-γ and IL-4 after 5 d of coculture. (D and E) Cocultures were stimulated by 10 μM OVA323-339 and 1 μM CpG for 4 d. IL-17, IFN-γ, and GM-CSF production by CD4+ T cells was determined by intracellular staining in cultures containing GM-CSF+/+ CD4+ T cells and DC (D) or indicated combinations of WT and KO T cells and DC (E). (F) WT and KO CD4+ T cells and DC were cultured with anti-CD3 in Th17-polarizing conditions including 5 ng/ml TGFβ1, 20 ng/ml IL-6, 20 ng/ml IL-21, 1 μM CpG, and 100 μg/ml curdlan. Cytokine production was determined by intracellular staining at day 4.
Th17 cell differentiation in vitro can be induced by TCR stimulation in the presence of TGF-β together with IL-6 or IL-21 (9, 11). In addition, exposure of DC to microbial products, such as CpG and Curdlan, drives Th17 polarization by stimulation of IL-6 and other inflammatory cytokines (i.e., IL-1 and IL-23), which may reflect the more physiological situation. To assess the role of GM-CSF in Th17 polarization in vitro, we performed four-way cocultures of WT or GM-CSF−/− DC with CD4+ T cells from DO11.10 WT or GM-CSF−/− mice and specific antigen in the presence of CpG. WT CD4+ T cells grown in this condition differentiated into populations secreting GM-CSF, IL-17, and/or IFN-γ (Fig. 4 D). Th17 cell differentiation was impaired in cultures containing GM-CSF–deficient CD4+ T cells with KO or WT DC (Fig. 4 E). Furthermore, DC and CD4+ T cells from WT and KO mice were cultured with anti-CD3 in the presence of cytokines (i.e., IL-6/TGF-β and IL-21/TGF-β) or Pathogen associated molecular patterns (PAMPs; i.e., CpG and Curdlan) known to promote Th17 differentiation. Confirming data obtained in antigen-specific cultures shown in Fig. 4 E, Th17 polarization induced by CpG and Curdlan was dependent on GM-CSF production by CD4+ T cells (Fig. 4 F). In contrast, Th17 development induced by exogenous IL-6/TGF-β or IL-21/TGF-β was comparable in cultures containing GM-CSF–deficient and WT CD4+ T cells (Fig. 4 F), suggesting that exogenous IL-6 (or IL-21) overcomes GM-CSF requirement. Notably, DC/CD4 stimulation cultures grown with exogenous GM-CSF together with TGF-β in the absence of IL-6 did not induce Th17 differentiation (unpublished data), indicating that microbial stimulation of the DC is critical.
Collectively, these data suggest that DC produce proinflammatory cytokines, including IL-6 but not GM-CSF, upon microbial stimulation with PAMPs. This promotes GM-CSF production by CD4+ T cells, which acts back on DC to further enhance IL-6-production resulting in Th17 differentiation and proliferation.
GM-CSF is essential for CD4 T cell survival in vivo
To confirm the importance of GM-CSF in T cell proliferation in vivo, we performed four-way adoptive transfer experiments by injection of GM-CSF–deficient or GM-CSF–competent OVA323-339 specific CD4+ T cells (from DO11.10 mice) into GM-CSF−/− and WT mice. CD4+ T cells were labeled with CFSE prior to injection to monitor cell cycling and in vivo tracking. Mice were immunized 1 d later with OVA323-339/CFA, and DO11.10 T cells were monitored longitudinally in the blood. GM-CSF–competent CD4+ T cells expanded considerably in immunized WT hosts until day 7 before they started to collapse. Expansion and collapse of GM-CSF–deficient CD4+ T cells was unaltered in WT hosts. In contrast, no such expansion was observed in GM-CSF−/− recipients (Fig. 5 A). Consistently, a significantly reduced number of DO11.10 T cells was observed in draining LN of GM-CSF−/− compared with WT hosts at days 3 and 5 after immunization (Fig. 5 B).
Figure 5.

GM-CSF prevents apoptosis of specific T cells. CD4+ T cells were purified from DO11.10/GM-CSF+/+ and DO11.10/GM-CSF−/−mice and labeled with CFSE before intravenous injection into WT or GM-CSF−/− mice (5 × 106 cells per mouse) at day 3. Mice were immunized s.c. with 200 μg OVA323-339 peptide in CFA at day 0. (A) KJ1-26+ CD4 T cell expansion was monitored over 10 d in the blood of mice (n = 4). The frequency of KJ1-26+ CD4+ T cells in control animals immunized with CFA only was <0.5%. (B) Total number of KJ1-26+ cells in draining LN at days 3 and 5 after immunization. Filled columns, DO11.10/GM-CSF+/+CD4+→GM-CSF+/+ mice; open columns, DO11.10/GM-CSF−/− CD4+→GM-CSF−/− mice (n = 3). (C) Proliferation of KJ1-26+ CD4+ T cells is shown by CFSE dilution at days 3 (left) and 5 (right). (D) Annexin V (top) and PI (bottom) staining of KJ1-26+ CD4+ T cells in draining LN at days 3 and day 5. (E) Shown is the number of cell division determined by CFSE dilution and percentage of apoptotic (Annexin V+ PI−) KJ1-26+ cells at day 3 after immunization. (F) WT and GM-CSF−/− mice were injected with GM-CSF−/−/DO11.10 or GM-CSF+/+/DO11.10 CD4+ T cells before immunization with OVA323-339/CFA. Shown is the frequency of apoptotic (Annexin V+ PI−) KJ1-26+ cells at day 3 after immunization. Error bars indicate SD of three mice per group. One representative experiment of two is shown. * , P < 0.05; ** , P < 0.01.
To determine whether GM-CSF promoted cell expansion by enhancing the rate of cell division, we measured CFSE dilution (35). Surprisingly, we found that cell cycling was not dependent on GM-CSF (Fig. 5 C). Therefore, we hypothesized that reduced CD4 T cell numbers in the absence of GM-CSF resulted from enhanced cell death. To test this, we measured apoptosis and cell death by staining transgenic CD4+ T cells from draining LN with Annexin-V and propidium iodide (PI) after OVA/CFA immunization. Indeed, we found threefold- and twofold-increased numbers of Annexin-V transgenic T cells in GM-CSF−/− mice at days 3 and 5, respectively (Fig. 5 D). Similarly, PI-positive cells were increased in GM-CSF−/− mice at these days (Fig. 5 D). By gating on cells that had undergone the same number of cycles as measured by CFSE dilution, we found that the percentage of apoptotic cells (Annexin-V+ PI−) increased with the division number and that this increase was much stronger in GM-CSF−/− mice. After five cell divisions, the number of apoptotic T cells in GM-CSF−/− mice was threefold higher (Fig. 5 E). As expected from the results in the previous paragraph (Fig. 5 A), apoptosis was increased in GM-CSF−/− recipients irrespective of the ability of transferred T cell to produce GM-CSF or not (Fig. 5 F). These results demonstrate that GM-CSF produced by non–T cells sustains survival of antigen-specific T cells in vivo.
GM-CSF sustains IL-6–mediated T cell survival
It has been reported that GM-CSF receptor is not expressed by resting and activated splenic T cells (36). Indeed, using sensitive quantitative RT-PCR, we could not detect expression of GM-CSFR-α and the signal transducing β subunit (AIC2B) by naive or activated CD4+ T cells differentiated in Th1, Th2, or Th17 polarizing conditions (Fig. 6 A). In contrast, primary splenic DC expressed both GM-CSFR subunits. This result suggests that GM-CSF does not act on the expanding T cells directly but mediates T cell survival indirectly, possibly by stimulating production of T cell growth/survival factors in GM-CSF–responsive cells such as DC and macrophages. We showed that exogenous and endogenous GM-CSF enhanced IL-6 production by DC and/or macrophages (Fig. 2 B; and Fig. 3, A and B). Consistently, we found that endogenous GM-CSF was critically required for antigen-dependent IL-6 production in T cell/DC cocultures (Fig. 6 B). Neutralization of endogenously produced IL-6 by mAb reduced proliferation in WT cocultures, although not to values seen in GM-CSF–deficient cultures (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20071119/DC1). These data suggest that GM-CSF regulates CD4 T cell expansion, at least partially, through induction of IL-6. Indeed, supplementation of GM-CSF–deficient cocultures with IL-6 reconstituted T cell proliferation (Fig. 6 C) and inhibited apoptosis (Fig. 6 D), whereas rIL-2 or rIL-1β showed a small or no effect, respectively (Fig. 6 E).
Figure 6.

T cell survival is mediated by DC-secreted factors and IL-6. (A) Expression of GM-CSFR-α, GM-CSFR-β (AIC2B), and IL-2Rβ was determined by quantitative RT-PCR using cDNA of cells indicated. Naive CD62L+ CD4+ T cells (lane 1) and splenic CD11c+ DC (lane 5) were purified by flow cytometry (purity > 98%). Th1 (lane 2), Th2 (lane 3), and Th17 (lane 4) polarized CD4+ T cells. (B and C) DC of GM-CSF−/− or GM-CSF+/+ mice were cultured with purified CD4+ T cells from DO11.10/GM-CSF−/− or DO11.10/GM-CSF+/+ mice, respectively, in the presence of titrating amounts of OVA323-339. (B) IL-6 levels in the culture supernatant at day 3 were determined by ELISA. (C) Proliferation measured by 3H-Thymidine incorporation in the absence and in the presence of 20 ng/ml rIL-6. (D) Splenocytes of DO11.10/GM-CSF+/+ or GM-CSF−/− mice were cultured with 1 μM OVA323-339 in the absence and presence of 20ng/ml rIL-6 for 3 d before staining of cells with KJ1-26+ mAb, Annexin V (AV), and PI and analysis by flow cytometry. Dot plot gated on KJ1-26+ cells shows early apoptotic cells (PI−AV+), late apoptotic (PI+AV+), and dead cells (PI+ AV−). (E) GM-CSF–deficient cultures were supplemented with rIL-1, rIL-2, or rIL-6. Proliferation was measured by 3H-Thymidine incorporation after 3 d of culture. Proliferation index was calculated as described in Materials and methods. (F) CD4+ T cells purified from DO11.10/GM-CSF+/+ or DO11.10/GM-CSF−/− mice were injected i.v. into GM-CSF+/+ or GM-CSF−/− mice, respectively. 2 d later, groups of KO and WT mice were implanted with osmotic minipumps containing hIL-6 or were sham operated. Subsequently, mice were immunized with 200 μg OVA323-339 peptide emulsified in CFA. After 7 d, draining LN and spleen cells were analyzed by flow cytometry. Symbols indicate total number of splenic KJ1-26+ cells of individual mice. Horizontal lines indicate averages of groups. Shown is one representative experiment of three performed. *, < 0.05. Error bars indicate SD.
We next studied whether IL-6 supplementation can restore CD4 T cell expansion in GM-CSF–deficient mice in vivo. IL-6 has a fast clearance kinetic in the blood, and periodic IL-6 injection is therefore not suitable to compensate continuous endogenous release (unpublished data). To ensure a continuous supply, we implanted osmotic minipumps containing IL-6 in mice after injection of naive DO11.10 CD4+ T cells. As shown in Fig. 6 F, the total number of DO11.10 CD4+ T cells was reduced in GM-CSF−/− mice compared with WT mice 7 d after immunization. Delivery of IL-6 to GM-CSF−/− mice increased the expansion of DO11.10 T cells. A similar reconstitution was also seen in the blood and LN (unpublished data). Collectively, these data indicate that GM-CSF prevents apoptosis and maintains CD4+ T cell responses, at least partially, by stimulation of IL-6 production in vivo.
IL-17–producing cells CD4 T cells are reduced in GM-CSF−/− mice
IL-6 and IL-23 are key factors for development, maintenance, and pathogenicity of IL-17–producing CD4+ T (Th17) cells. Considering the defect of GM-CSF−/− CD4+ T cells in producing IL-17 in vitro, we studied Th17 development in GM-CSF−/− mice in vivo. To this end, we transferred DO11.10/GM-CSF−/− and DO11.10/GM-CSF+/+ CD4+ T cells to KO and WT mice, respectively. Immunization with OVA323-339/CFA resulted in a high frequency of IL-17+IFN-γ− (Th17) cells and lower frequencies of IFN-γ+IL-17− (Th1) and IL-17+IFN-γ+ coproducers among OVA-specific CD4+ T cells in GM-CSF–competent mice. Th17 and IL-17+IFN-γ+ CD4 populations were strikingly reduced, whereas frequencies of Th1 cells were unaffected in GM-CSF−/− mice (Fig. 7 B). Thus, GM-CSF was critical for development and survival of IL-17–producing cells upon immunization with antigen in CFA in vivo.
Figure 7.

GM-CSF−/− mice show reduced frequencies of IL-17+IFN-γ− and IL-17+IFN-γ+ CD4+ T cell populations. (A) CD4+ T cells purified from DO11.10/GM-CSF+/+ or DO11.10/GM-CSF−/− mice were adoptively transferred to GM-CSF+/+ or GM-CSF−/− mice, respectively, 2 d prior to immunization with 200 μg OVA323-339 peptide emulsified in CFA. Mice were boosted at day 7 and draining LN were removed at day 14 to determine KJ1-26+ CD4+ T cells producing IL-17 or IFN-γ by flow cytometry. Top shows dot plots of a representative mouse per group. Bottom shows the frequency of cytokine-producing cells in individual mice. (B and C) GM-CSF−/− and GM-CSF+/+ mice were immunized with myhcα614-629/CFA at days 0 and 7. (B) IL-17 and IFN-γ expression was determined by real-time PCR using cDNA from CD4+ T cells purified from draining LN at day 8. Values show means of groups (n = 3) of mice ±SD. (C) At day 14, CD4+ T cells were isolated from LN and restimulated with myhcα614-629 as described in Materials and methods. IFN-γ– and IL-17–producing cells were determined by ELISPOT analysis. Values indicate individual mice and means of groups.
Finally, we determined the role of GM-CSF in development of IL-17–producing CD4+ T cells in autoimmune myocarditis. GM-CSF−/− and control mice were immunized at days 0 and 7 with myhca614-629/CFA. Real time PCR analysis showed reduced il17 but normal ifng gene expression in purified CD4+ T cells from LN of GM-CSF−/− mice at day 8 (Fig. 7 B). At day 14, however, we found strongly reduced numbers of both IL-17- and IFN-γ–producing CD4+ T cells in GM-CSF−/− mice by ELISPOT assay (Fig. 7 C). It is tempting to speculate that a majority of IFN-γ–positive cells coproduce IL-17, as indicated by flowcytometry results of T cell transfer studies (Fig. 7 A). These data indicate that GM-CSF is critical for induction of IL-17 rather than IFN-γ production by CD4+ T cells, but it later becomes essential for survival of both IL-17– and IFN-γ–producing cells in autoimmune disease.
DISCUSSION
In this manuscript, we provide data suggesting that GM-CSF is essential for development of autoimmune myocarditis by promoting IL-6–dependent IL-17 production and survival of autoimmune CD4+ T cells. GM-CSF−/− mice were almost completely protected from myocarditis induced by immunization of heart autoantigen (myhcα614-629), which is in agreement with other publications reporting reduced EAE, arthritis, and gastritis in GM-CSF−/− mice (22–24, 37) and suggests that GM-CSF plays a key role in development of organ-related autoimmunity similar to IL-1, IL-6, and IL-23. Although activated DC are among the main producers of the latter proinflammatory cytokines, DC do not produce GM-CSF. Consistently, injection of TLR- and CD40-activated BM-DC from WT mice, which can restore EAM in IL-1R1−/− mice (26), failed to restore full-blown EAM in GM-CSF−/− mice. Nevertheless, injection of activated DC from WT or GM-CSF−/− mice increased disease severity in GM-CSF−/−, mice although not to levels observed in WT mice. These data suggest that GM-CSF produced by other cells is important for DC activation in addition to TLR and CD40 stimulation. Consistently, we found that stimulation of splenic DC from WT and GM-CSF−/− mice with rGM-CSF substantially increased TLR-induced production of the proinflammatory cytokines IL-6, IL-23, and TNF-α. Moreover, we showed that IL-6 and IL-23 production by DC was strongly impaired in myhcα/CFA-immunized GM-CSF−/− mice. Both IL-6 and IL-23 are essential for the development of EAM (5, 25), EAE (38–41), arthritis (42–44), and colitis (45–48), demonstrating the importance of these two cytokines for development of organ-related autoimmune and inflammatory diseases. Although some of these studies showed reduced CD4 T cell proliferation (25) and IFN-γ production (39, 49) in the absence of IL-6, it is now thought that IL-6 mediates autoimmune disease by promoting development of pathogenic Th17 cells (9, 11). However, in addition to Th17 development, IL-6 has been shown to prevent apoptosis of effector and memory T cells (50). Similarly, we found that reduced expansion of antigen-specific CD4+ T cells in immunized GM-CSF−/− mice was caused by enhanced apoptosis and not a defect in T cell activation or cell division rate.
It has been shown previously that a soluble factor secreted by activated DC was able to restore impaired proliferation of CD4+ T cells from GM-CSF−/− mice in vitro (51). We have identified that this factor is IL-6. Delivery of recombinant IL-6 to GM-CSF−/− mice in osmotic minipumps or to APC-T cell cocultures in vitro restored proliferation by inhibition of apoptosis of antigen-specific GM-CSF−/− CD4+ T cells. It should be noted that we failed to reconstitute autoimmune myocarditis in GM-CSF−/− mice and recovery of Th17 cells was achieved partially with variable results by implantation of IL-6–secreting osmotic minipumps or by infection with recombinant adenovirus expressing IL-6 (mAd5-IL6; reference 52). However, these approaches also failed to reconstitute Th17 development and autoimmune myocarditis in IL-6−/− mice (unpublished data), indicating that local availability, concentration, and/or half-life of exogenous and ectopically produced IL-6 are not sufficient to reconstitute endogenous IL-6 levels required for disease development. In addition, restoration of Th17 cells and autoimmunity in GM-CSF−/− mice may require supplementation of both IL-6 and IL-23, as indicated by our results.
Although we cannot exclude that other antiapoptotic cytokines stimulated by GM-CSF show a similar effect than IL-6 for T cell survival, we have shown that addition of IL-1 or IL-2 could not restore expansion of GM-CSF−/− CD4+ T cells. Although GM-CSF production by activated T cells can contribute to T cell proliferation in vitro, particularly when no other GM-CSF–producing cell is present in the microenvironment (Figs. 4 B and 6 C), our in vivo adoptive transfer experiments with TCR tg CD4+ T cells demonstrate that GM-CSF production by non–T cells is critical for survival of antigen-specific CD4+ T cells (Fig. 5 F). Indeed, we found that a gemisch of Macrophages and DC isolated from WT mice at day 7 after myhcα614-629/CFA immunization produced GM-CSF (unpublished data). Interestingly, although CD11c+ DC isolated from naive WT BALB/c mice and BM-DC did not produce GM-CSF upon TLR stimulation (i.e., CpG, LPS) or in cocultures with CD4 T cells (Figs. 2 and 4), we could measure GM-CSF in supernatants of CD11c+ cells purified from immunized mice, although less than in the gemisch of macrophages and DC (not depicted). It is tempting to speculate that blood circulating inflammatory monocytes (CX3CR1lowGr1+), which differentiate into functional DC in inflamed tissues (53), have the ability to produce GM-CSF.
The absence of GM-CSF receptor on naive and effector CD4 T cell subsets excludes direct regulation of Th17 lineage commitment by GM-CSF. Consistently, addition of GM-CSF to DC/CD4 T cell cocultures did not induce Th17 differentiation in vitro. However, stimulation of DC with microbial ligands or immunization with CFA adjuvants promoted DC IL-6 production (Fig. 2, B and C; and Fig. 3, A–C), resulting in GM-CSF production by activated CD4+ T cells, which was required for Th17 polarization (Fig. 4, E and F). Requirement of GM-CSF for Th17 development was overcome in the presence of TGFβ/IL-6 or TGFβ/IL-21, further indicating that GM-CSF acts indirectly by enhancing production of Th17-polarizing cytokines.
Consistent with a key role of IL-6 and IL-23 in generation and maintenance of Th17 cells, we found that impaired IL-6 and IL-23 production was associated with reduced numbers of Th17 cells in immunized GM-CSF−/− mice in vivo. Interestingly, numbers of IFN-γ–producing cells determined by ELISPOT were also strongly reduced in GM-CSF−/− mice immunized with myhcα614-629/CFA, whereas development of IFN-γ+IL-17− (Th1) cells was unaffected in GM-CSF−/− mice after adoptive transfer of DO11.10 transgenic T cells and immunization with OVA/CFA. In contrast, a population of IFN-γ+IL-17+ double producers was also reduced in this setting. Unfortunately, we could not distinguish IFN-γ+IL-17− and IFN-γ+IL-17+ cells specific for myhcα614-629 by ELISPOT, and it is possible that the majority of IFN-γ–producing cells actually represent IFN-γ+IL-17+ double producers. The dynamics of IL-17 and IFN-γ production during differentiation of distinct subpopulations depends on the number of antigen-specific CD4+ T cells and the strengths of TCR stimulation (unpublished data), which may also explain possible differences in adoptive transfer of OVA323-339-specific CD4+ T cells and development of endogenous CD4+ T cells specific for myhcα614-629.
The balance of Th17 and Treg cells is key for development of autoimmunity. Development of Foxp3+ Treg cells is enhanced at the expense of Th17 cells in IL-6–deficient mice immunized with the encephalitogenic peptide MOG35-55, which prevents development of EAE (54). It is important to note that we did not find significant differences in Foxp3+ Treg cells in immunized GM-CSF−/− mice (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20071119/DC1), which may indicate that IL-6 levels in GM-CSF−/− mice are sufficient to balance development of Treg cells. However, it remains to be shown whether Treg number and function are similar in autoimmune myocarditis and EAE and the different genetic backgrounds (BALB/c vs C57BL/6).
Our results do not exclude a role for GM-CSF in the effector phase of the response. Elegant studies have demonstrated previously that GM-CSF production by encephalitogenic T cells is required for the development of EAE, probably by activation of microglial cells (23, 55, 56). Similarly, GM-CSF produced by myosin-specific effector CD4+ T cells may activate heart-resident macrophages. Indeed, GM-CSF mRNA is up-regulated in the heart of rats with autoimmune myocarditis (57), and we found that a high frequency of CD4+ T cells activated in vitro under Th17-polarizing conditions or CD4+ T cells isolated from the CNS of mice with EAE coexpress GM-CSF and IL-17 (unpublished data), demonstrating that GM-CSF is an effector cytokine of Th17 cells.
In summary, our studies demonstrate a previously unrecognized role of GM-CSF in survival of Th17 cells that mediate autoimmunity. GM-CSF regulates autoantigen-specific CD4+ T cells indirectly through stimulation of IL-6 and IL-23 production by DC and macrophages in the presence of microbial products, as illustrated in the scheme shown in Fig. 8. Our results may have important implications for understanding the mechanisms of cancer and other immunotherapies using GM-CSF as potent adjuvant.
Figure 8.
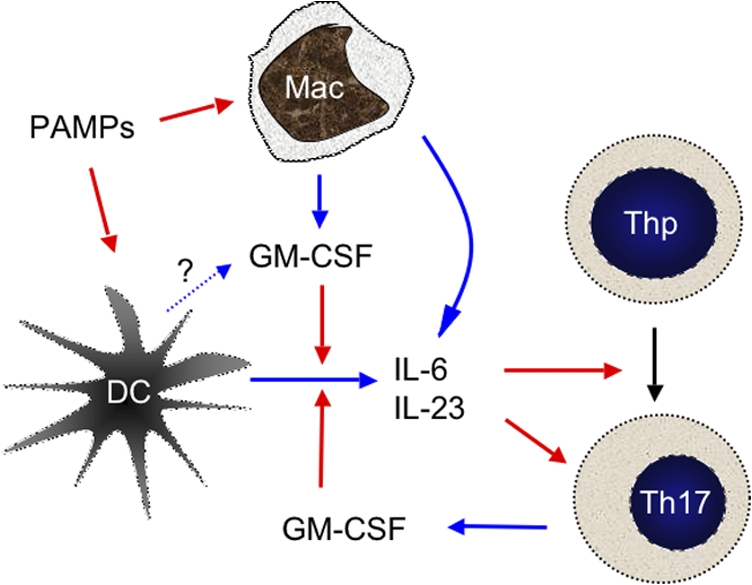
Model for GM-CSF–mediated autoimmunity. PAMPs such as CpG and mycobacterial components in CFA induce DC activation/maturation and production of proinflammatory cytokines, including IL-6, which promote activation of naive CD4 T cells (Thp) and secretion of GM-CSF, IFN-γ, and IL-17. Macrophages (and possibly subpopulations of DC) can produce GM-CSF directly upon encounter of PAMPs. GM-CSF further enhances IL-6 and IL-23 production by DC and macrophages. IL-6 acts at two levels on activated T cells. It enhances survival (both Th1 and Th17) and, together with TGF-β, drives RORγt expression to allow further Th17 polarization. IL-23 can promote Th17 cell maintenance and/or pathogenicity. Blue and red arrows indicate secretion of and stimulation of, respectively.
MATERIALS AND METHODS
Mice.
GM-CSF−/− mice (provided by A. Dunn, Ludwig Institute for Cancer Research, Royal Melbourne Hospital, Victoria, Australia) (21) were backcrossed to BALB/c for seven generations. DO11.10 transgenic GM-CSF−/− mice were obtained by crossing DO11.10 mice (58) with GM-CSF−/− mice. BALB/c WT controls were purchased from Charles River Laboratories. Mice were maintained in isolated ventilated cages in an animal facility free of specific pathogens (BioSupport) and were used for experiments at the age of 8–12 wk. All animal experiments were performed in accordance with Swiss federal legislation and have been approved by the local overseeing body Gesundheitsdirektion Kanton Zürich, Veterinaeramt (permission 148/2005).
Antibodies and cytokines.
Antibodies specific to CD11c, CD86, CD40, CD4, IL-12p40, TNF-α, IFN-γ, IL-17, or Annexin V, conjugated with FITC, PE, or APC were purchased from eBioscience. Anti-KJ1.26 mAb (idiotype-specific DO11.10 TCR) was produced in house and conjugated with biotin. Purification of CD4+ and CD11c+ cells with magnetic beads (MACS; Miltenyi Biotec) was performed according to manufacturer instructions. Intracellular staining for cytokines was performed after fixation with 2% formaldehyde in the presence of 0.5% saponin. Recombinant GM-CSF (PeproTech), IL-1β (provided by G. Spohn, Cytos biotechnology, Schlieren, Switzerland), and hIL-6 (provided by L. Aarden, University of Amsterdam, Amsterdam, Netherlands) were used at concentrations indicated in the text.
Induction of EAM.
Mice were injected s.c. with 100 μg per mouse of the mouse cardiac myhc peptide myhc614-629 Ac-RSLKLMATLFSTYASADR-OH (ANAWA) emulsified in CFA (1 mg/ml H37Ra; DIFCO) on days 0 and 7. At day 0, 400 ng per mouse of Pertussis Toxin (List Biological Laboratories, Inc.) dissolved in PBS containing 1% normal mouse serum was injected i.p. Mice were killed 21 d after the first injection.
Histopathology.
Immunized mice were killed and hearts were removed and fixed in 4% formalin (Sigma-Aldrich). Upon hemotoxylin and eosin staining, myocarditis was histologically scored using grades from 0 to 4 (0, no inflammatory infiltrates; 1, small foci of inflammatory cells between myocytes; 2, larger foci of >100 inflammatory cells; 3, >10% of a cross section involved; 4, >30% of a cross section involved).
ELISA antibody measurement.
ELISA against myhc614-629 was performed as previously described (28). In brief, coating of MaxiSorp microtiter plates (Thermo Fisher Scientific) with 5 μg/ml streptavidin (Invitrogen) was followed by incubation with biotinylated myhc614-629 (ANAWA). Afterward, threefold serial dilutions of serum were incubated for 1 h at room temperature, followed by extensive washing and the addition of Alkaline-phosphatase–labeled anti–mouse IgG (SouthernBiotech). After development with p-nitrophenylphosphate (Sigma-Aldrich), antibody titers were determined at half maximal OD405nm.
DC activation.
Spleens of mice were digested for 1 h with collagenase and purified by MACS using anti-CD11c magnetic beads (Miltenyi Biotec). Afterward, DC were activated with the following different stimuli: 1 μg/ml LPS (Sigma-Aldrich) or titrating amounts of CpG (Microsynth). For surface staining, DC were incubated at 37°C with 5% CO2 for 12 h. Afterward, cells were blocked with anti-CD16/CD32 (eBioscience) and stained for activation markers. Cytokine ELISAs were performed on supernatants from DC culture stimulated for 24 h.
Cytokine production.
IL-6 and IL-12p70 levels were measured by ELISA. Microtiter plates (MaxiSorp) were coated with unlabeled capture anti–IL-6 or anti–IL-12p40 antibodies (eBioscience) at a concentration of 2 μg/ml for 12 h at 4°C. After blocking the plates with 1% BSA and washing, samples were incubated on the plates for 1 h. Detection was performed by incubating the plates with 1 μg/ml biotinylated anti-IL6 or anti-IL12p70 antibodies, respectively (eBioscience), followed by alkaline-phosphatase–labeled streptavidin (SouthernBiotech). Absorption at 405 nm was measured after incubation of the plates with p-nitrophenylphosphate.
In vitro proliferation assay.
Spleens of mice were digested for 1 h with collagenase and purified by MACS using anti-CD11c magnetic beads (Miltenyi Biotec), essentially as previously described (34). CD4+ T cells from DO11.10 tg GM-CSF−/− or GM-SF+/+ mice were isolated by MACS using anti-CD4 magnetic beads. 105 CD4+ T cells from tg mice were cultured with 2 × 104 DC or 105mitomycine C–inactivated splenocytes and titrating concentrations of OVA323-339 (EMC Microcollections) or ovalbumin protein (grade V; Sigma-Aldrich). In experiments indicated, cultures were supplemented with 20 ng/ml IL-1β, IL-2 (1:500 dilution of a supernatant of a hybridoma producing hIL-2), and 20 ng/ml hIL-6. Proliferation was measured by pulsing cultures with 3H-Thymidine (Hartmann Analytic) for the last 12 h of a 3-d culture. Proliferation index was calculated by dividing the cpm at 200 nM OVA323-339 by cpm at baseline (without antigen).
Antigen-specific Th1 and Th2 polarization in vitro.
Spleens of mice were digested for 1 h with collagenase, and DC was isolated by an OptiPrep gradient (Axis-Shield), followed by sorting of CD11c+ cells on a FACS Vantage (BD Biosciences) to a purity >95%. For Th1 and Th2 polarization, naive CD4+ CD62Lhigh T cells from spleens of DO11.10/GM-CSF+/+ or DO11.10/GM-CSF−/− mice were isolated by MACS using anti-CD4 magnetic beads followed by sorting CD62Lhigh cells, using FACS Vantage, to a purity >98%. 104DC and 5 × 104 naive CD4+ T cells were cocultured in the presence of high (1 mM), intermediate (100 μM) or low (10 μM) concentrations of ovalbumin protein (grade V) for 3 d before addition of 104 DC and the same concentrations of antigen for another 2 d. T cell activation was assessed by staining with anti-CD25 mAb and CD62L mAb at day 3 of coculture. Th1/Th2 polarization was analyzed at day 5 by intracellular staining of IL-4 and IFN-γ as previously described (34).
Visualization of CD4 T cell expansion and subset differentiation in vivo.
T cells were isolated from the spleen of DO11.10 GM-CSF+/+ or DO11.10/GM-CSF−/− mice by sorting with anti-CD4 magnetic beads. After labeling of purified CD4+ T cells with CFSE (Invitrogen), 5 × 106 cells per mouse were injected intravenously in WT or GM-CSF−/− mice. One day later, mice were bled and cells stained with KJ1-26 mAb to confirm comparable number of transgenic T cells. Transferred mice were immunized with 200 μg OVA323-339 (ISQAVHAAHAEINEAGR; EMC Microcollection) in CFA (DIFCO). At days 3 and 5 after immunization, proliferation of T cells was analyzed by staining LN cells and FACS analysis.
For induction of Th17 cells in vivo, mice were boosted at day 7. Draining LN were isolated at day 14 and cells were stimulated with 10−7 M PMA and 10−6 M Ionomycin for 5 h. Brefeldin A (Sigma-Aldrich) was added for the last 2 h of activation. Cytokine expression was detected by intracellular staining as described in the previous section.
IL-6 supplementation by minipumps.
T cells were isolated from the spleen of DO11.10, purified, and transferred in GM-CSF+/+ or GM-CSF−/− mice as described in the previous section. After 1–2 d, osmotic minipumps (Alzet 2001; DURECT Corporation) were filled with 26 μg (3 μg/d delivery) rec hIL-6 and s.c. implanted in mice. Shortly after implantation, mice were immunized s.c. with 200 μg OVA323-339 emulsified in CFA.
Quantitative PCR.
Cells used to quantify cytokine receptors were FACS sorted (FACS Aria; Becton Dickinson). All cell populations were >99% pure. After in vitro culture in polarizing conditions for 3 d, cDNA was extracted with Tri-reagent (Molecular Research Center) according to the manufacturer's instructions. Quantitative PCR was performed on an iCycler (Bio-Rad Laboratories) according to the manufacturer's instructions. Oligonucleotides used for PCR amplification of cytokines and receptors were the following: GM-CSFR-α forward, ACGTCATGCACCGTGACC; GM-CSFR-α reverse, CATTGACATCCAGCGTGA; AIC2B forward, ACCAGCAAATGGCACTCAC; AIC2B reverse, GCTGGTGATAGAGGGTCATGT; CD122 forward, GAAGAGGCTCTGAATGTCACAA; CD122 reverse, ACGACCCGAGGATCAGGTT; IL17 forward, GCAAAAGTGAGCTCCAGAAGG; IL17 reverse, AGCTTCCCAGATCACAGAGG; IFN-γ forward, GCTCTGAGACAATGAACGCTAC; and IFN-γ reverse, TTC TAG GCTTTCAATGACTGTGC.
ELISPOT.
Filter plates (96-well; Multi-Screen-IP; Millipore) were coated overnight at 4°C with 2 μg/well of affinity chromatography–purified anti–IFN-γ (mAB AN18,) or with 0.1 μg/well of anti–IL-17 (BD Biosciences) in PBS. Plates were washed twice with PBS and blocked with IMDM/10% FCS for 2 h at RT (200 μl/well). 106/ml BALB/c BM-DC was pulsed with 10 μg myhc614-629/ml for 15 min on ice. DC were adjusted to 0.4 × 106/ml, and 20,000 cells were used as APC. 105 purified CD4+ T cells were incubated 6 h together with peptide-pulsed or unpulsed APCs at 37°C. Setups with 105 T cells only and CD4+ T cells stimulated with 2.5 × 10−7 M PMA and 1.25 μg/ml ionomycin (Sigma-Aldrich) served as controls. Plates were washed five times with PBS, and 2 μg/ml anti–IFN-γ−biotin (mAb XMGD6) or 2 μg/ml anti–IL-17 (BD Biosciences) were added to the respective plate and incubated for 1 h at room temperature. Plates were washed five times with PBS and incubated for 1 h with streptavidin-alkaline-phosphatase (1:1,000 in PBS; Dianova). After five further washing steps, the spots were visualized by using the BCIP/NBT (5-bromo-4-chloro-3-indol-phosphate-toluidine/nitroblue-tetrazoliumchloride) Plus substrate (Mabtech). The enzymatic reaction was stopped by rinsing the wells with water. Plates were counted with an ELISPOT Reader (Autoimmun Diagnostika GmbH). ELISPOT responses of individual mice are expressed as mean number of specific spot-forming cells (experimental sample, negative control with unpulsed APCs) per 100,000 CD4+ T cells from triplicate measurements.
Statistical analysis.
Statistical analysis was performed with the help of Prism (GraphPad Software, Inc.). Two-tailed unpaired Student's t test with a confidence interval of 95% was performed for normal distributed data. Histological scores were analyzed with two-tailed Mann-Whitney test. Results obtained by flow cytometry in Fig. 7 A were evaluated using Kolmogorov-Smirnov statistical analysis for comparisons between frequency distributions. If not mentioned otherwise, error bars indicate SD. P < 0.05 was considered to be significant.
Online supplemental material.
Fig. S1 shows that neutralization of IL-6 by mAb inhibits antigen-specific CD4+ T cell proliferation. Fig. S2 shows that frequencies of Foxp3+ Treg cells are comparable in GM-CSF−/− and GM-CSF+/+ mice. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20071119/DC1.
Supplementary Material
Acknowledgments
This project has been supported by a grant from the Swiss National Foundation (No. 3100A0-100233/1).
The authors have no conflicting financial interest.
Abbreviations used: EAE, experimental allergic encephalitis; EAM, experimental autoimmune myocarditis; GM-CSF, granulocyte macrophage–colony stimulating factor; myhcα, myosin heavy chain α; PAMP, Pathogen associated molecular pattern; PI, propidium iodide; TLR, toll-like receptor.
References
- 1.Komiyama, Y., S. Nakae, T. Matsuki, A. Nambu, H. Ishigame, S. Kakuta, K. Sudo, and Y. Iwakura. 2006. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J. Immunol. 177:566–573. [DOI] [PubMed] [Google Scholar]
- 2.Langrish, C.L., Y. Chen, W.M. Blumenschein, J. Mattson, B. Basham, J.D. Sedgwick, T. McClanahan, R.A. Kastelein, and D.J. Cua. 2005. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 201:233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park, H., Z. Li, X.O. Yang, S.H. Chang, R. Nurieva, Y.H. Wang, Y. Wang, L. Hood, Z. Zhu, Q. Tian, and C. Dong. 2005. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 6:1133–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakae, S., A. Nambu, K. Sudo, and Y. Iwakura. 2003. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J. Immunol. 171:6173–6177. [DOI] [PubMed] [Google Scholar]
- 5.Sonderegger, I., T.A. Rohn, M.O. Kurrer, G. Iezzi, Y. Zou, R.A. Kastelein, M.F. Bachmann, and M. Kopf. 2006. Neutralization of IL-17 by active vaccination inhibits IL-23-dependent autoimmune myocarditis. Eur. J. Immunol. 36:2849–2856. [DOI] [PubMed] [Google Scholar]
- 6.Rangachari, M., N. Mauermann, R.R. Marty, S. Dirnhofer, M.O. Kurrer, V. Komnenovic, J.M. Penninger, and U. Eriksson. 2006. T-bet negatively regulates autoimmune myocarditis by suppressing local production of interleukin 17. J. Exp. Med. 203:2009–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Infante-Duarte, C., H.F. Horton, M.C. Byrne, and T. Kamradt. 2000. Microbial lipopeptides induce the production of IL-17 in Th cells. J. Immunol. 165:6107–6115. [DOI] [PubMed] [Google Scholar]
- 8.Liang, S.C., X.Y. Tan, D.P. Luxenberg, R. Karim, K. Dunussi-Joannopoulos, M. Collins, and L.A. Fouser. 2006. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 203:2271–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bettelli, E., Y. Carrier, W. Gao, T. Korn, T.B. Strom, M. Oukka, H.L. Weiner, and V.K. Kuchroo. 2006. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 441:235–238. [DOI] [PubMed] [Google Scholar]
- 10.Mangan, P.R., L.E. Harrington, D.B. O'Quinn, W.S. Helms, D.C. Bullard, C.O. Elson, R.D. Hatton, S.M. Wahl, T.R. Schoeb, and C.T. Weaver. 2006. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 441:231–234. [DOI] [PubMed] [Google Scholar]
- 11.Veldhoen, M., R.J. Hocking, C.J. Atkins, R.M. Locksley, and B. Stockinger. 2006. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-Producing T Cells. Immunity. 24:179–189. [DOI] [PubMed] [Google Scholar]
- 12.Sutton, C., C. Brereton, B. Keogh, K.H. Mills, and E.C. Lavelle. 2006. A crucial role for interleukin (IL)-1 in the induction of IL-17–producing T cells that mediate autoimmune encephalomyelitis. J. Exp. Med. 203:1685–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pasare, C., and R. Medzhitov. 2003. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 299:1033–1036. [DOI] [PubMed] [Google Scholar]
- 14.Ivanov, I.I., B.S. McKenzie, L. Zhou, C.E. Tadokoro, A. Lepelley, J.J. Lafaille, D.J. Cua, and D.R. Littman. 2006. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 126:1121–1133. [DOI] [PubMed] [Google Scholar]
- 15.Batten, M., J. Li, S. Yi, N.M. Kljavin, D.M. Danilenko, S. Lucas, J. Lee, F.J. de Sauvage, and N. Ghilardi. 2006. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nat. Immunol. 7:929–936. [DOI] [PubMed] [Google Scholar]
- 16.Kleinschek, M.A., A.M. Owyang, B. Joyce-Shaikh, C.L. Langrish, Y. Chen, D.M. Gorman, W.M. Blumenschein, T. McClanahan, F. Brombacher, S.D. Hurst, et al. 2007. IL-25 regulates Th17 function in autoimmune inflammation. J. Exp. Med. 204:161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gasson, J.C. 1991. Molecular physiology of granulocyte-macrophage colony-stimulating factor. Blood. 77:1131–1145. [PubMed] [Google Scholar]
- 18.Hamilton, J.A., and G.P. Anderson. 2004. GM-CSF Biology. Growth Factors. 22:225–231. [DOI] [PubMed] [Google Scholar]
- 19.Inaba, K., M. Inaba, N. Romani, H. Aya, M. Deguchi, S. Ikehara, S. Muramatsu, and R.M. Steinman. 1992. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 176:1693–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inaba, K., R.M. Steinman, M.W. Pack, H. Aya, M. Inaba, T. Sudo, S. Wolpe, and G. Schuler. 1992. Identification of proliferating dendritic cell precursors in mouse blood. J. Exp. Med. 175:1157–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stanley, E., G.J. Lieschke, D. Grail, D. Metcalf, G. Hodgson, J.A. Gall, D.W. Maher, J. Cebon, V. Sinickas, and A.R. Dunn. 1994. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc. Natl. Acad. Sci. USA. 91:5592–5596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Campbell, I.K., M.J. Rich, R.J. Bischof, A.R. Dunn, D. Grail, and J.A. Hamilton. 1998. Protection from collagen-induced arthritis in granulocyte-macrophage colony-stimulating factor-deficient mice. J. Immunol. 161:3639–3644. [PubMed] [Google Scholar]
- 23.McQualter, J.L., R. Darwiche, C. Ewing, M. Onuki, T.W. Kay, J.A. Hamilton, H.H. Reid, and C.C. Bernard. 2001. Granulocyte macrophage colony-stimulating factor: a new putative therapeutic target in multiple sclerosis. J. Exp. Med. 194:873–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cook, A.D., E.L. Braine, I.K. Campbell, M.J. Rich, and J.A. Hamilton. 2001. Blockade of collagen-induced arthritis post-onset by antibody to granulocyte-macrophage colony-stimulating factor (GM-CSF): requirement for GM-CSF in the effector phase of disease. Arthritis Res. 3:293–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eriksson, U., M.O. Kurrer, N. Schmitz, S.C. Marsch, A. Fontana, H.P. Eugster, and M. Kopf. 2003. Interleukin-6-deficient mice resist development of autoimmune myocarditis associated with impaired upregulation of complement C3. Circulation. 107:320–325. [DOI] [PubMed] [Google Scholar]
- 26.Eriksson, U., M.O. Kurrer, I. Sonderegger, G. Iezzi, A. Tafuri, L. Hunziker, S. Suzuki, K. Bachmaier, R.M. Bingisser, J.M. Penninger, and M. Kopf. 2003. Activation of dendritic cells through the interleukin 1 receptor 1 is critical for the induction of autoimmune myocarditis. J. Exp. Med. 197:323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Afanasyeva, M., Y. Wang, Z. Kaya, E.A. Stafford, K.M. Dohmen, A.A. Sadighi Akha, and N.R. Rose. 2001. Interleukin-12 receptor/STAT4 signaling is required for the development of autoimmune myocarditis in mice by an interferon-gamma-independent pathway. Circulation. 104:3145–3151. [DOI] [PubMed] [Google Scholar]
- 28.Eriksson, U., M.O. Kurrer, W. Sebald, F. Brombacher, and M. Kopf. 2001. Dual role of the IL-12/IFN-gamma axis in the development of autoimmune myocarditis: induction by IL-12 and protection by IFN-gamma. J. Immunol. 167:5464–5469. [DOI] [PubMed] [Google Scholar]
- 29.Smith, S.C., and P.M. Allen. 1991. Myosin-induced acute myocarditis is a T cell-mediated disease. J. Immunol. 147:2141–2147. [PubMed] [Google Scholar]
- 30.Banchereau, J., and R.M. Steinman. 1998. Dendritic cells and the control of immunity. Nature. 392:245–252. [DOI] [PubMed] [Google Scholar]
- 31.Mellman, I., and R.M. Steinman. 2001. Dendritic cells: specialized and regulated antigen processing machines. Cell. 106:255–258. [DOI] [PubMed] [Google Scholar]
- 32.Menges, M., S. Rossner, C. Voigtlander, H. Schindler, N.A. Kukutsch, C. Bogdan, K. Erb, G. Schuler, and M.B. Lutz. 2002. Repetitive injections of dendritic cells matured with tumor necrosis factor α induce antigen-specific protection of mice from autoimmunity. J. Exp. Med. 195:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eriksson, U., R. Ricci, L. Hunziker, M.O. Kurrer, G.Y. Oudit, T.H. Watts, I. Sonderegger, K. Bachmaier, M. Kopf, and J.M. Penninger. 2003. Dendritic cell-induced autoimmune heart failure requires cooperation between adaptive and innate immunity. Nat. Med. 9:1484–1490. [DOI] [PubMed] [Google Scholar]
- 34.Ruedl, C., M.F. Bachmann, and M. Kopf. 2000. The antigen dose determines T helper subset development by regulation of CD40 ligand. Eur. J. Immunol. 30:2056–2064. [DOI] [PubMed] [Google Scholar]
- 35.Thompson, B.S., and T.C. Mitchell. 2004. Measurement of daughter cell accumulation during lymphocyte proliferation in vivo. J. Immunol. Methods. 295:79–87. [DOI] [PubMed] [Google Scholar]
- 36.Morrissey, P.J., L. Bressler, L.S. Park, A. Alpert, and S. Gillis. 1987. Granulocyte-macrophage colony-stimulating factor augments the primary antibody response by enhancing the function of antigen-presenting cells. J. Immunol. 139:1113–1119. [PubMed] [Google Scholar]
- 37.Hamilton, J.A. 2002. GM-CSF in inflammation and autoimmunity. Trends Immunol. 23:403–408. [DOI] [PubMed] [Google Scholar]
- 38.Eugster, H.P., K. Frei, M. Kopf, H. Lassmann, and A. Fontana. 1998. IL-6-deficient mice resist myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. Eur. J. Immunol. 28:2178–2187. [DOI] [PubMed] [Google Scholar]
- 39.Samoilova, E.B., J.L. Horton, B. Hilliard, T.S. Liu, and Y. Chen. 1998. IL-6-deficient mice are resistant to experimental autoimmune encephalomyelitis: roles of IL-6 in the activation and differentiation of autoreactive T cells. J. Immunol. 161:6480–6486. [PubMed] [Google Scholar]
- 40.Chen, Y., C.L. Langrish, B. McKenzie, B. Joyce-Shaikh, J.S. Stumhofer, T. McClanahan, W. Blumenschein, T. Churakovsa, J. Low, L. Presta, et al. 2006. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J. Clin. Invest. 116:1317–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cua, D.J., J. Sherlock, Y. Chen, C.A. Murphy, B. Joyce, B. Seymour, L. Lucian, W. To, S. Kwan, T. Churakova, et al. 2003. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 421:744–748. [DOI] [PubMed] [Google Scholar]
- 42.Ohshima, S., Y. Saeki, T. Mima, M. Sasai, K. Nishioka, S. Nomura, M. Kopf, Y. Katada, T. Tanaka, M. Suemura, and T. Kishimoto. 1998. Interleukin 6 plays a key role in the development of antigen-induced arthritis. Proc. Natl. Acad. Sci. USA. 95:8222–8226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sasai, M., Y. Saeki, S. Ohshima, K. Nishioka, T. Mima, T. Tanaka, Y. Katada, K. Yoshizaki, M. Suemura, and T. Kishimoto. 1999. Delayed onset and reduced severity of collagen-induced arthritis in interleukin-6-deficient mice. Arthritis Rheum. 42:1635–1643. [DOI] [PubMed] [Google Scholar]
- 44.Murphy, C.A., C.L. Langrish, Y. Chen, W. Blumenschein, T. McClanahan, R.A. Kastelein, J.D. Sedgwick, and D.J. Cua. 2003. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J. Exp. Med. 198:1951–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Atreya, R., J. Mudter, S. Finotto, J. Mullberg, T. Jostock, S. Wirtz, M. Schutz, B. Bartsch, M. Holtmann, C. Becker, et al. 2000. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in crohn disease and experimental colitis in vivo. Nat. Med. 6:583–588. [DOI] [PubMed] [Google Scholar]
- 46.Yamamoto, M., K. Yoshizaki, T. Kishimoto, and H. Ito. 2000. IL-6 is required for the development of Th1 cell-mediated murine colitis. J. Immunol. 164:4878–4882. [DOI] [PubMed] [Google Scholar]
- 47.Uhlig, H.H., B.S. McKenzie, S. Hue, C. Thompson, B. Joyce-Shaikh, R. Stepankova, N. Robinson, S. Buonocore, H. Tlaskalova-Hogenova, D.J. Cua, and F. Powrie. 2006. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity. 25:309–318. [DOI] [PubMed] [Google Scholar]
- 48.Yen, D., J. Cheung, H. Scheerens, F. Poulet, T. McClanahan, B. McKenzie, M.A. Kleinschek, A. Owyang, J. Mattson, W. Blumenschein, et al. 2006. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J. Clin. Invest. 116:1310–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Okuda, Y., S. Sakoda, H. Fujimura, Y. Saeki, T. Kishimoto, and T. Yanagihara. 1999. IL-6 plays a crucial role in the induction phase of myelin oligodendrocyte glucoprotein 35-55 induced experimental autoimmune encephalomyelitis. J. Neuroimmunol. 101:188–196. [DOI] [PubMed] [Google Scholar]
- 50.Rochman, I., W.E. Paul, and S.Z. Ben-Sasson. 2005. IL-6 increases primed cell expansion and survival. J. Immunol. 174:4761–4767. [DOI] [PubMed] [Google Scholar]
- 51.Wada, H., Y. Noguchi, M.W. Marino, A.R. Dunn, and L.J. Old. 1997. T cell functions in granulocyte/macrophage colony-stimulating factor deficient mice. Proc. Natl. Acad. Sci. USA. 94:12557–12561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Braciak, T.A., S.K. Mittal, F.L. Graham, C.D. Richards, and J. Gauldie. 1993. Construction of recombinant human type 5 adenoviruses expressing rodent IL-6 genes. An approach to investigate in vivo cytokine function. J. Immunol. 151:5145–5153. [PubMed] [Google Scholar]
- 53.Geissmann, F., S. Jung, and D.R. Littman. 2003. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 19:71–82. [DOI] [PubMed] [Google Scholar]
- 54.Korn, T., E. Bettelli, W. Gao, A. Awasthi, A. Jager, T.B. Strom, M. Oukka, and V.K. Kuchroo. 2007. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 448:484–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marusic, S., J.S. Miyashiro, J. Douhan III, R.F. Konz, D. Xuan, J.W. Pelker, V. Ling, J.P. Leonard, and K.A. Jacobs. 2002. Local delivery of granulocyte macrophage colony-stimulating factor by retrovirally transduced antigen-specific T cells leads to severe, chronic experimental autoimmune encephalomyelitis in mice. Neurosci. Lett. 332:185–189. [DOI] [PubMed] [Google Scholar]
- 56.Ponomarev, E.D., L.P. Shriver, K. Maresz, J. Pedras-Vasconcelos, D. Verthelyi, and B.N. Dittel. 2007. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J. Immunol. 178:39–48. [DOI] [PubMed] [Google Scholar]
- 57.Kobayashi, Y., A. Kubo, M. Iwano, Y. Sakaguchi, K. Samejima, Y. Kyoda, K. Yonemasu, and T. Hashimoto. 2002. Levels of MCP-1 and GM-CSF mRNA correlated with inflammatory cytokines mRNA levels in experimental autoimmune myocarditis in rats. Autoimmunity. 35:97–104. [DOI] [PubMed] [Google Scholar]
- 58.Murphy, K.M., A.B. Heimberger, and D.Y. Loh. 1990. Induction by antigen of intrathymic apoptosis of CD4+CD8+TCRlo thymocytes in vivo. Science. 250:1720–1723. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}