

Abstract
Phosphatidylinositol 3-kinases (PI3Ks) play a critical role in regulating B cell receptor– and T cell receptor–mediated signaling. However, their role in natural killer (NK) cell development and functions is not well understood. Using mice expressing p110δD910A, a catalytically inactive p110δ, we show that these mice had reduced NK cellularity, defective Ly49C and Ly49I NK subset maturation, and decreased CD27High NK numbers. p110δ inactivation marginally impaired NK-mediated cytotoxicity against tumor cells in vitro and in vivo. However, NKG2D, Ly49D, and NK1.1 receptor–mediated cytokine and chemokine generation by NK cells was severely affected in these mice. Further, p110δD910A/D910A NK cell–mediated antiviral responses through natural cytotoxicity receptor 1 were reduced. Analysis of signaling events demonstrates that p110δD910A/D910A NK cells had a reduced c-Jun N-terminal kinase 1/2 phosphorylation in response to NKG2D-mediated activation. These results reveal a previously unrecognized role of PI3K-p110δ in NK cell development and effector functions.
NK cells are an important component of innate immunity, capable of mediating cytotoxicity against tumor and virus-infected cells. Effector functions of NK cells are regulated by the coordinated interaction of activating and inhibitory receptors (1). Determining precise signaling events downstream of these receptors is paramount for successful clinical utilization of NK cells. One of the activating receptors, NKG2D, is a lectin type II transmembrane protein expressed on all human and mouse NK cells, and it recognizes MIC-A/B (2) and ULBP-1/2/3 (in humans) (3), and H60 (4, 5), Rae-1α/β/γ/δ/ε (5), and Mult-1 (in mice) (6). Upon activation, NKG2D employs Src family protein tyrosine kinases (PTKs) to initiate two distinct signaling pathways (7–11), leading to effector functions. In the first pathway, activated PTK phosphorylates Tyr-Ile-Asn-Met (YINM) motif-bearing DAP10, which in turn recruits phosphatidylinositol 3-kinase (PI3K) (9). In the second pathway, PTK phosphorylates the immunoreceptor tyrosine-based activation motif (ITAM)–containing KARAP/DAP12, which subsequently triggers Syk and ZAP70 (8–11). Another major activating receptor, Ly49D, which associates with both DAP10 and DAP12 (12, 13), is also a mouse lectin type II transmembrane protein, which interacts with classical MHC class I, H2-Dd (14).
Natural cytotoxicity receptors (NCRs) are immunoglobulin-like transmembrane glycoproteins that recognize unknown ligands on several tumor cells. The NCR family contains three human (NKp46/NCR1, NKp44/NCR2, and NKp30/NCR3) and one mouse (NKp46/NCR1) members (15–18). NKp46 and NKp30 associate with ITAM-bearing CD3ζ (17) and FCRγ (19), respectively, whereas NKp44 recruits DAP12 (20). Although cellular ligands for NCRs have not been found, NCR1 is known to interact with hemagglutinin (HA) of influenza and HA-neuraminidase of Sendai virus (21). NK1.1 (Nkrp1c) is a unique cell marker expressed on NK and NKT cells (22). Although the activating ligands for Nkrp1c have yet to be determined, the inhibitory ligands for its related family members Nkrp1d and Nkrp1f have been defined as the Clr family of C-type lectins (23). NK1.1 physically associates with FcRγ to mediate its signal (24). Several NK inhibitory receptors have been identified, such as KIR, Ly49A, Ly49C, Ly49G2, and Ly49I (25). These inhibitory receptors recognize classical MHC class I molecules. Upon interaction, they recruit phosphatases to the immunoreceptor tyrosine-based inhibitory motif in the cytoplasmic domains (26). Thus, NK cells use a complex set of receptors and signaling pathways to achieve their intended effector functions. Despite recent studies (8–13) that have provided deeper insights regarding the activation pathways, multiple knowledge gaps exist, hindering comprehensive clinical applications of NK cells.
Class I PI3Ks generate secondary lipid messengers that regulate a wide array of intracellular signaling pathways in numerous cell types (27). Several isoforms of regulatory p85 (p85α, p55α, p50α, p85β, and p55γ) and catalytic p110 (p110α, p110β, p110γ, and p110δ) subunits have been described to play distinct functions (27). For example, mice lacking the p85α regulatory or p110δ catalytic subunit show severely impaired B and T cell development and functions (28, 29). Deletion of individual catalytic or regulatory subunits results in altered expression of other subunits (30, 31). Thus, use of gene KO mice precludes proper evaluation of the PI3K isoform-selective functions in lymphocytes. To avoid these inherent complications in using KO mice, we generated mice with a point mutation that completely inactivated the catalytic function of p110δ subunit (further referred to as p110δD910A/D910A mice) (32). This point mutation, Asp910→Ala (D910A), resulted in a complete loss-of-function locus but retained the normal expression levels of p110δ protein. More importantly, this strategy did not result in any compensatory increase of p110α, p110β, and total p85 subunits in thymocytes (32).
In this study, using the p110δD910A/D910A mice, we demonstrate that PI3K-p110δ plays a pivotal role in the development and effector functions of NK cells. p110δD910A/D910A mice had reduced NK cellularity and a selective impairment in the maturation of Ly49C and Ly49I NK subsets. Further, mature NK cell populations, as defined by CD27 and CD11b, were markedly altered in p110δD910A/D910A mice. p110δ inactivation marginally impaired NK cell–mediated cytotoxicity against tumor and virally infected cells. However, NKG2D, Ly49D, and NK1.1 receptor–mediated cytokine and chemokine generation were severely impaired in NK cells from p110δD910A/D910A mice. In addition, cytokine generation by p110δD910A/D910A NK cells in response to virally infected cells was markedly reduced. Biochemical analysis indicates that defects in the activation and phosphorylation of the c-Jun N-terminal kinase (JNK) pathway could be responsible for the impaired cytokine generation in p110δD910A/D910A NK cells. Our results demonstrate that the PI3K-p110δ subunit plays a crucial role in NK cellularity, terminal maturation, and cytokine/chemokine generation.
RESULTS
Unaltered expression of PI3K subunits in p110δD910A/D910A mice
The class I PI3K complex is primarily made up of p85 regulatory and p110 catalytic subunits. Altering expression of one subunit severely affects the expression of the others (31). p110α deletion resulted in significant overexpression of the p85α subunit (33). Similarly, deletion of p85α leads to an increased expression of p85β and a dramatic decrease of p110α, p110β, and p110δ subunits (28, 29). Therefore, we first analyzed the expression of distinct PI3K subunits in IL-2–activated NK cells from p110δD910A/D910A mice that were backcrossed with the C57BL/6 strain for N12 generations (Fig. 1 a). All p110 isoforms were detectable in NK cells derived from both spleen and BM, with the level of the inactive p110δ being similar to that in NK cells from C57BL/6 (WT/WT) mice. No compensatory expressions of p110α, p110β, and p110γ in NK cells from spleen and BM of p110δD910A/D910A mice were found. We also analyzed the presence of total p85 and p85α subunits with pan–anti-p85 and anti-p85α antibodies, respectively. Our results showed no alteration in the quantity of total p85 or p85α subunit in p110δD910A/D910A-derived splenic or BM NK cells (Fig. 1 a). These observations are consistent with our earlier studies, which demonstrated that thymocytes from p110δD910A/D910A mice contain similar expression levels of p110α, p110β, and total p85 subunits compared with those of WT mice (32). Collectively, we conclude that the D910A mutation did not alter the protein expression levels of p110δ itself. Also, there were no alterations in the protein levels of p110α, p110β, and p110γ subunits in the NK cells from p110δD910A/D910A mice. Further, there were no changes in the levels of total p85 or p85α subunits because of the D910A mutation.
Figure 1.

Phenotypic characterization of p110δD910A/D910A mice. (a) Expression levels of PI3K subunits were unaltered after the catalytic inactivation of p110δ. Expression of p110 and total p85 and p85α isoforms was analyzed by Western blotting in IL-2–activated splenic and BM NK cells. (b) Absolute numbers of spleen and BM cells are significantly reduced in p110δD910A/D910A mice. Total cellularity of the spleen and BM was calculated from 10 mice in each genotype. (c) Absolute CD3−NK1.1+ cell number is decreased in p110δD910A/D910A mice. Single-cell suspensions were stained with anti-NK1.1 and anti-CD3ε mAbs. NK cells as specified by CD3−NK1.1+ positivity were gated and analyzed for absolute numbers. Data shown were obtained using cells from seven mice of each genotype. (d) Expression of early developmental markers in fresh p110δD910A/D910A BM NK cells. Percentages of positive NK cells for each marker among CD3−NK1.1+ cells are shown. Gates were set using unstained or nonspecific isotype mAb controls (not depicted). Data presented were means obtained from seven mice of each genotype. Data presented were means and standard deviations from three to five experiments.
p110δ regulates cellularity but not NK cell commitment
To define the role of p110δ in early NK cell development, we analyzed the cell numbers. Total cellularity of both spleen and BM was significantly reduced in p110δD910A/D910A mice (Fig. 1 b). Concurrently, the absolute numbers of the CD3−NK1.1+ NK cell population were also reduced in the spleen of p110δD910A/D910A mice (Fig. 1 c). The numbers of BM-derived CD3−NK1.1+ NK cells were slightly reduced (Fig. 1 c). Recent studies have demonstrated that the process of NK cell development can be defined into five distinct stages based on the expression pattern of cell-surface markers (34). Expression of CD122, which is the β chain of IL-2 and IL-15 receptors, marks the commitment of progenitor cells into the NK lineage in the BM. NKG2D and NK1.1 are the earliest known NK markers (35). Integrins such as CD11b (Mac-1) and CD43 (leukosialin), which can regulate lymphocyte migration and homing, are up-regulated, whereas CD51 (αv) expression declines during the final maturation stage (34). To determine whether functional inactivation of p110δ could affect the maturation of NK cells, we analyzed the expression of developmental markers on fresh NK cells derived from BM (Fig. 1 d) and spleen (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20072327/DC1). Gated CD3−NK1.1+ NK cells were analyzed for the expression of CD122, NKG2D, NKG2A, CD11b, CD244, CD43, CD49b, and CD51. Expression levels of CD122, NKG2D, NKG2A, CD11b, and CD244 were unaffected in p110δD910A/D910A mice (Fig. 1 d). Inactivation of p110δ also did not affect the expression of CD49b, CD51, and CD43 in CD3−NK1.1+ NK cells (Fig. 1 d). Similar results were observed in IL-2–activated NK cells (Fig. S2, a and b).
p110δ regulates the terminal maturation of Ly49C/I NK subsets
Acquisition of distinct Ly49 receptors by CD3−NK1.1+ NK cells is an important developmental step by which the NK “repertoire” is generated. Expression of Ly49C and Ly49I were severely impaired in the fresh NK cells from the BM and spleen of p110δD910A/D910A mice (Fig. 2, a and b). However, expression of inhibitory Ly49A or Ly49G2 and activating Ly49D receptors was unaffected by p110δ inactivation in fresh NK cells (Fig. 2, a and b). Analyses of IL-2–activated BM NK cells further indicated the defective expression of Ly49C and Ly49I receptors (Fig. 2 c). Similar to freshly isolated NK cells, the proportion of Ly49A, Ly49D, and Ly49G2 subsets remained unaffected in IL-2–activated BM NK cells (Fig. 2 c). Comparable results were observed in spleen-derived and IL-2–activated NK cells (Fig. 2 d) from p110δD910A/D910A mice. Thus, a reduction in Ly49C or Ly49I did not depend on the coexpression of other Ly49 receptors, and the adverse effect of p110δ mutation only affected the maturation of Ly49C and Ly49I NK subsets.
Figure 2.

Catalytic inactivation of p110δ results in impaired maturation of Ly49C and Ly49I NK subsets. Expression of maturation markers on p110δD910A/D910A NK cells was analyzed by flow cytometry. Percentages of Ly49C/I+ NK subset are significantly reduced on both fresh BM (a) and splenic (b) p110δD910A/D910A NK cells. Percentages of Ly49C/I+ or Ly49I+ NK subset are significantly reduced in the IL-2–activated BM (c) or splenic (d) p110δD910A/D910A NK cells that were stained with anti–Ly49C/I-PE or anti–Ly49I-PE mAbs in combination with anti–Ly49G2-allophycocyanin, or anti–Ly49A-PE in combination with anti–Ly49D-FITC mAbs. Percentages of each NK subset are shown. NK cells from seven (a and b) or three (c and d) mice of each genotype were used. Data shown were obtained from three to five independent experiments.
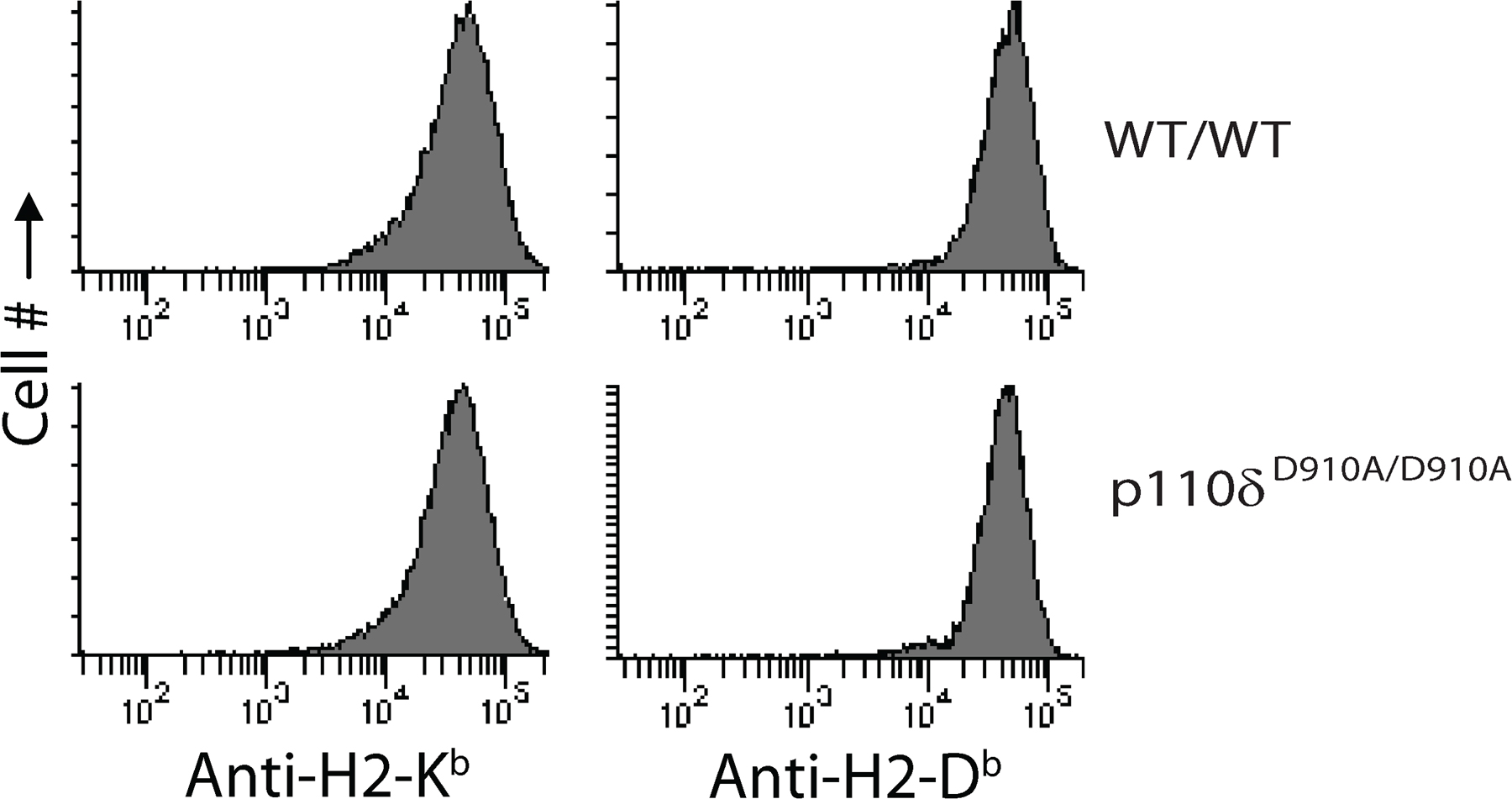
Reduction in the absolute NK cell numbers and in Ly49C/I needs further explanation. PI3K-mediated signaling initiates key prosurvival pathways (36). It is also important to note that only Ly49C/I receptors have the ability to recognize the self–MHC class I molecules, H2-Kb and H2-Db. Expression of these MHCs are not altered because of the inactivation of p110δ, arguing against one possible extrinsic effect during NK maturation (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20072327/DC1). Currently, we cannot exclude the potential possibility that p110δ-mediated signaling is required for “licensing” (37) or “arming” (38). In the absence of p110δ, either the rate of NK cell proliferation or cell survival could have been altered. To differentiate between these two possibilities, spleen-derived IL-2–activated NK cells were labeled with BrdU to analyze their proliferation. Results presented in Fig. 3 (a–c) demonstrate that the overall percentages of proliferating p110δD910A/D910A NK cells were significantly reduced. In particular, proliferation of Ly49C/I subsets were more severely impaired (P = 0.0096) than other Ly49 subsets. To determine the role of apoptosis in the reduction of Ly49C/I subsets, NK cells were cultured with IL-2 for 6 d, and individual subsets were analyzed with 7-aminoactinomycin D (7-AAD) and Annexin V staining. Our data indicate that among NK cells from spleen (Fig. 3 d), there was a consistent, significant but minimal increase of Annexin V–positive Ly49 subsets from p110δD910A/D910A mice (<10% of the total analyzed). However, no major changes were seen between BM-derived WT/WT and p110δD910A/D910A NK cells (Fig. S4). These results indicate that a lack of p110δ most probably affects the proliferative rate of NK cells and that the effect on their survival is minimal.
Figure 3.

Defective proliferation but not increased apoptosis affects the Ly49C/I NK subsets in p110δD910A/D910A mice. IL-2–cultured splenic NK cells were pulsed with BrdU as in Materials and methods. 4 h later, NK cells were stained and analyzed for Ly49A (a), Ly49C/I (b), and Ly49G2 (c) subset proliferation. The percentages of BrdU-positive and -negative Ly49 subsets are also shown as histograms (a–c, right). Data presented are mean values from three mice of each genotype. (d) IL-2–cultured splenic NK cells were stained with Annexin V and 7-AAD for apoptosis analysis of each Ly49 NK subset. Data presented are mean values from three mice of each genotype. One representative of three independent experiments is shown.
CD27HighCD11bHigh NK populations are severely reduced in p110δD910A/D910A mice
Recent studies have shown that the mature CD11b+ NK cells can be further divided into CD27High and CD27Low with distinct functional abilities (39–41). These studies demonstrate that CD27HighCD11bHigh NK cells were superior in their ability to lyse target cells compared with that of CD27LowCD11bHigh cells. Further, CD27HighCD11bHigh NK cells produced considerably higher amounts of IFN-γ (39). Because NK cells from p110δD910A/D910A mice showed a specific defect in Ly49C/I expression, we extended our analyses to CD27-based NK maturation. Fresh BM and splenocytes were gated for CD3−NK1.1+ cells and analyzed for CD27 and CD11b markers. Fig. 4 shows that a lack of functional p110δ altered the ratio of CD27- and CD11b-expressing NK subsets. There was a significant reduction in the CD27HighCD11bLow NK cells in both fresh BM (Fig. 4 a) and spleen (Fig. 4 b) of p110δD910A/D910A mice. The CD27HighCD11bHigh NK cell numbers were also affected in the spleen of p110δD910A/D910A mice. More importantly, the decrease in the CD27HighCD11bHigh population was correlated with a concomitant increase in the CD27LowCD11bHigh NK subsets in both fresh BM and spleen of the p110δD910A/D910A NK population. Earlier studies have also indicated a skewed expression of Ly49C/I receptors in CD27LowCD11bhigh NK cells (39). Because we observed a specific reduction in the Ly49C/I subsets in the absence of functional p110δ, we analyzed the fresh NK cells from p110δD910A/D910A mice for CD27 and Ly49 expressions. Results presented in Fig. 4 c indicate that Ly49A, Ly49I, Ly49D, and Ly49G2 NK subsets with CD27High positivity were reduced more in p110δD910A/D910A NK cells than those of WT/WT. However, the reduction in the Ly49I+CD27High NK subset was more severe compared with other subsets in p110δD910A/D910A NK cells. Based on these results, we conclude that p110δ is needed for both Ly49C/I subset specification and proliferation, and CD27HighCD11bHigh NK maturation.
Figure 4.

Catalytic inactivation of p110δ reduces CD27High NK subsets. BM- (a) or spleen- (b) derived CD3−NK1.1+ NK cells were analyzed for CD11b and CD27 expression. The percentages of CD27HighCD11bLow, CD27HighCD11bHigh, and CD27LowCD11bHigh cells were calculated. Data are means ± SD. (c) Alteration in CD27High NK subsets correlates with the reduction in Ly49C/I NK subsets. Spleen-derived single-cell suspensions were stained with anti-NK1.1, anti-CD3ε, and each of the indicated anti-Ly49 or CD11b and CD27 mAbs. NK cells as specified by CD3−NK1.1+ positivity were gated and analyzed. Data shown were obtained using cells from five to seven mice of each genotype. One representative out of three independent experiments is shown.
Cytotoxicity is marginally impaired in p110δD910A/D910A NK cells
A detailed analysis of the role of PI3K and its subunits in regulating “induced-self” and “missing-self” recognition is lacking. Therefore, we evaluated the ability of p110δD910A/D910A NK cells to mediate cytotoxicity toward several tumor targets in 51Cr-release assays. Fresh NK cells were purified from splenocytes and were tested for their ability to mediate the lysis of EL4H60 target cells (42, 43). Compared with WT/WT, the cytotoxic potential of p110δD910A/D910A NK cells was marginally reduced (Fig. 5 a). Similar to fresh NK cells, IL-2–activated splenic p110δD910A/D910A NK cells also had a reduced ability to lyse EL4H60 compared with WT/WT NK cells (Fig. 5 b). As expected, there is only a minimal lysis of EL4 by either IL-2–activated WT/WT or p110δD910A/D910A NK cells (Fig. 5 b). This cytotoxicity response directed to EL4H60 was primarily mediated through the NKG2D receptor, because an anti-NKG2D mAb (C7) abrogated this response in both WT/WT and p110δD910A/D910A NK cells (Fig. S5, available at http://www.jem.org/cgi/content/full/jem.20072327/DC1). Another tumor cell line, YAC-1, which naturally expresses H60, was also lysed less by p110δD910A/D910A NK cells (Fig. 5 b). Similar observations were made using BM-derived NK cells (unpublished data). Cells that lack or have reduced expression of self–MHC class I molecules are susceptible to NK-mediated cytotoxicity (44). As seen in Fig. 5 b, p110δD910A/D910A NK cells showed reduced cytotoxicity against RMA/S cells, which express lower levels of MHC class I. To test whether p110δ plays a role in Ly49D-mediated cytotoxicity, we used Chinese hamster ovary (CHO) cells that express a hamster homologue of H2-Dd (14). As shown in Fig. 5 b, spleen-derived, IL-2–activated p110δD910A/D910A NK cells had reduced lysis against CHO cells.
Figure 5.

Ex vivo– and IL-2–activated p110δD910A/D910A NK cell cytotoxicity are moderately impaired. (a) Fresh splenic NK cells were purified and used in a 4-h 51Cr-release assay using EL4H60 target cells. (b) IL-2–activated splenic NK cells were tested against 51Cr-labeled target cells at the indicated effector/target (E/T) ratios. Cytotoxicity was tested against EL4, EL4H60, YAC-1, RMA/S, or CHO cells. Three to five mice were used for each genotype. Open and closed circles represent the mean values from WT/WT and p110δD910A/D910A mice, respectively. NKG2D-mediated cytotoxic potential of the Ly49C/I+ NK subset was more severely impaired compared with Ly49C/I− NK subsets. (c) Ly49C/I+Ly49G2±, (d) Ly49C/I−Ly49G2+, and (e) Ly49C/I−Ly49G2− NK subsets were sorted from IL-2–activated splenic NK cells derived from WT/WT and p110δD910A/D910A mice, rested for 12 h, and used in cytotoxicity assays with EL4H60 as target cells. Data presented are means ± SD (a and b) or are representative (c–e) of at least three independent experiments.
Because the terminal maturation of Ly49C and Ly49I NK subsets was significantly impaired in the p110δD910A/D910A mice, we intended to analyze their functional abilities to recognize and lyse tumor cells. Toward this, the IL-2–activated splenic NK cells derived from p110δD910A/D910A mice were stained with anti-Ly49C/I and anti-Ly49G2 mAbs, sorted, and tested against EL4H60. Because proportions of Ly49C and Ly49I subsets are severely decreased and total numbers of spleen and BM cells are significantly reduced in p110δD910A/D910A mice, sorting enough Ly49C/I single-positive NK subsets is difficult. Therefore, we pooled Ly49C/I single-positive and Ly49C/I, Ly49G2 double-positive NK subsets (Ly49C/I+Ly49G2±) for the 51Cr-release assay. Cytotoxicity of Ly49C/I+Ly49G2± NK subsets from p110δD910A/D910A mice (Fig. 5 c) was relatively more impaired compared with sorted Ly49C/I−Ly49G2+ (Fig. 5 d) and Ly49C/I−Ly49G2− (Fig. 5 e) NK subsets. Nevertheless, these results for the first time demonstrate a preferential requirement of p110δ by Ly49C/I NK subsets for cytotoxicity.
Next, we evaluated the ability of p110δD910A/D910A mice to clear in vivo tumor growth. Toward this, WT/WT and p110δD910A/D910A mice were challenged with RMA and RMA/S cells. These tumor cells were labeled with CFSE or SNAF and were injected in combinations into the intraperitoneal cavity; 6 h later, peritoneal exudates were collected and analyzed for the level of tumor clearance and NK cell accumulation. Clearance of RMA/S versus RMA cells was calculated compared with the input ratio, which was normalized to one. RMA cells, which are not cleared by either WT/WT or p110δD910A/D910A mice, acted as an internal control. Our results show that both WT/WT and p110δD910A/D910A mice did not clear RMA cells. Compared with WT/WT, the ability of the p110δD910A/D910A mice to clear the RMA/S cells was marginally impaired (Fig. S6, a and b, available at http://www.jem.org/cgi/content/full/jem.20072327/DC1). Comparable numbers of NK cells accumulated in the peritoneal cavity of both WT/WT and p110δD910A/D910A mice (Fig. S6, c and d). These results confirm our in vitro observations and the earlier reports on these mice (45).
Cytokine/chemokine generation is impaired in p110δD910A/D910A NK cells
NK cells can generate inflammatory cytokines and chemokines such as IFN-γ, GM-CSF, macrophage inflammatory protein (MIP) 1α, MIP-1β, and regulated upon activation, normal T cell expressed and secreted (RANTES) (46). Therefore, we tested CD3−NK1.1+ NK cells for cytokine generation. Freshly purified CD3−NK1.1+ NK cells from WT/WT spleens generated an optimal amount of cytokines and chemokines when activated by plate-bound anti-NKG2D, anti-Ly49D, and anti-NK1.1 mAbs. However, NK cells from p110δD910A/D910A mice were significantly impaired in generating these cytokines and chemokines (Fig. 6 a). Thus, the ex vivo–purified NK cells that use NKG2D–DAP10 complexes for signaling depend on p110δ to generate cytokine response. Next, we tested the role of p110δ in cytokine and chemokine generation by IL-2–activated splenic NK cells. As seen in Fig. 6 b, WT/WT NK cells produced a large amount of IFN-γ or GM-CSF when stimulated with anti-NKG2D, anti-Ly49D, and anti-NK1.1 mAbs. However, p110δD910A/D910A NK cells were severely impaired to produce these cytokines. Further, generation of the chemokines MIP1-α, MIP1-β, and RANTES from p110δD910A/D910A NK cells was also significantly reduced (Fig. 6 b). Similar reductions in the cytokines and chemokines were observed in the BM-derived p110δD910A/D910A NK cells (Fig. S7, available at http://www.jem.org/cgi/content/full/jem.20072327/DC1). This substantial reduction could be caused by the inability of the p110δD910A/D910A NK cells to produce cytokines or simply by a defect in cytokine secretion. To distinguish between these two possibilities, IFN-γ production in response to anti-NKG2D mAb was measured by intracellular staining. The percentage of IFN-γ–positive cells among gated NK1.1+ populations in p110δD910A/D910A NK cells was significantly lower compared with WT/WT NK cells (P = 0.017; Fig. 7 a) Inactivation of p110δ reduced the number and functions of NK cells, particularly, the Ly49C/I subsets. Therefore, to determine the cytokine defects in individual Ly49 subsets, we analyzed the levels of intracellular IFN-γ in each one of them. All of the subsets from p110δD910A/D910A mice generated a significantly less amount of intracellular IFN-γ (P < 0.007; Fig. 7 b), and we did not see any preferential reduction in Ly49C/I subsets. This indicates that although p110δ regulates both of the effector functions, it is critically required for cytokine generation. To further investigate whether the defect is at the transcriptional level, we quantified the amounts of IFN-γ–encoding mRNA before and after plate-bound anti-NKG2D mAb activation. Fig. 8 a demonstrates a significantly lower copy number of IFN-γ–encoding mRNA in both BM- and spleen-derived p110δD910A/D910A NK cells, indicating a defect at the transcriptional level. To determine whether this is generalized hyporesponsiveness or an exclusive defect associated with YINM- and ITAM-containing activation receptors, we stimulated the NK cells with IL-12, IL-18, or both in the presence of a suboptimal concentration of anti-NKG2D mAb (A10). Interestingly, both WT/WT and p110δD910A/D910A-derived NK cells responded equally well to distinct combinations of this activation (Fig. 8 b). Similar results were obtained using IL-12, IL-18, or both without anti-NKG2D mAb (unpublished data). This demonstrates that p110δD910A/D910A NK cells are fully capable of responding through their cytokine receptors.
Figure 6.

Ability of ex vivo– and IL-2–activated p110δD910A/D910A NK cells to generate cytokines or chemokines is severely impaired. (a) 105 freshly purified splenic CD3−NK1.1+ NK cells per well were activated with plate-bound anti-NKG2D (A10), anti-Ly49D (4D11), or anti-NK1.1 (PK136) mAbs, and the supernatants were tested for the indicated cytokines and chemokines in Multiplex assays. (b) 105 IL-2–activated splenic NK cells per well were activated with plate-bound anti-NKG2D (A10), anti-Ly49D (4D11), or anti-NK1.1 (PK136) mAbs, and the resulting supernatants were used in Multiplex assays. Data presented are mean values ± SD from six mice of each genotype from two independent experiments.
Figure 7.

IFN-γ generation by p110δD910A/D910A NK cells were defective in all Ly49 subsets. (a) Levels of intracellular IFN-γ positivity are significantly reduced in p110δD910A/D910A NK cells. IL-2–activated splenic NK cells were stimulated with plate-bound anti-NKG2D (A10) mAb for 8 h, combined with Brefeldin A, and incubated for another 4 h. Activated NK cells stained for intracellular IFN-γ. Cells shown are gated for the CD3−NK1.1+ NK population. (b) Levels of intracellular IFN-γ positivity in different Ly49 subsets from WT and p110δD910A/D910A mice. Percentages of CD3−NK1.1+ NK cells that were positive for intracellular IFN-γ are shown with Ly49 markers. Data presented are representative of three independent experiments.
Figure 8.

Defect in cytokine production occurs at the transcriptional level, and IL-12/IL-18–mediated generation of cytokine/chemokine is intact in p110δD910A/D910A NK cells. (a) IFN-γ–encoding mRNA levels were reduced in p110δD910A/D910A NK cells. NK cells were stimulated with anti-NKG2D mAb for 6 h, and mRNA was extracted and IFN-γ–encoding transcripts were quantified. (b) NK cell cytokine and chemokine production upon stimulation with a suboptimal concentration of anti-NKG2D mAbs in the presence or absence of IL-12, IL-18, or both. Data presented are means ± SD and are representative of three independent experiments.
NK-mediated cytokine production requires p110δ during antiinfluenza response
Influenza virus primarily infects lung epithelial cells that can lead to lung pathogenesis. Hence, in this study, we used mouse lung epithelial cells (LA4) to characterize the role of p110δ in NK cytotoxicity and cytokine generation during influenza virus infection. We detected the cell-surface expression of viral HA on LA4 cells (Fig. 9 a) after they were infected with PR8, a mouse-adapted human influenza virus. A previous study showed that HA functions as a viral ligand for NCR1, and its interaction with NCR1 can initiate NK cytolysis against PR8-infected 1106mel cells (21). To determine the role of p110δ in regulating the NCR1-mediated NK cell recognition of PR8-infected LA4 cells, we first compared the WT and p110δD910A/D910A NK cytotoxicity. Co-incubation of WT/WT NK cells with uninfected LA4 cells led to a basal level of cytotoxicity. PR8 infection of LA4 cells moderately increased the cytotoxic potentials of WT/WT NK cells. However, such an increase in the cytotoxicity was not observed with p110δD910A/D910A NK cells (Fig. 9 b). Further, direct observations of the lysis of PR8-infected LA4 in NK/LA4 co-cultures (Fig. 9 c) validated the reduced ability of the p110δD910A/D910A NK cells to mediate killing of virally infected cells. Because NK cells can generate inflammatory cytokines during viral infection (46), we quantified these soluble mediators in the supernatants of PR8-infected LA4/NK co-cultures. Results presented in Fig. 9 d demonstrate that although the WT/WT NK cells could produce ample levels of IFN-γ and GM-CSF, p110δD910A/D910A NK cells generated significantly lower levels of these two cytokines. Collectively, these results for the first time demonstrate that p110δ plays a crucial role in the NK cell–mediated cytokine generation during antiinfluenza viral responses.
Figure 9.

NK cells from p110δD910A/D910A mice have a decreased ability to mediate cytotoxicity and produce cytokines against influenza virus infection. (a) LA4 cells were infected with PR8 at an MOI of 0.05. 24 h after infection, expression of HA was quantified using anti-HA mAb by flow cytometry. (b) Recognition of PR8-infected LA4 cells by p110δD910A/D910A NK cells is impaired. LA4 was infected with PR8 at an MOI of 0.05 for 24 h, labeled with 51Cr, and used for cytotoxicity assays. Data presented are the mean values from three WT/WT and p110δD910A/D910A mice, respectively. (c) p110δD910A/D910A NK cells have severely impaired ability to lyse infected LA4 cells, as shown by direct observation of cytolysis. LA4 cells were infected with PR8 at an MOI of 0.05 for 1 h, and NK cells were added and co-cultured for 8 h. Lysis of the infected LA4 monolayer was observed under light microscopy and recorded with a mounted camera. Bars, 12 μm. (d) p110δD910A/D910A NK cells generate significantly lower levels of cytokines when co-cultured with PR8-infected LA4 cells. Generation of IFN-γ and GM-CSF was quantified in the supernatants of PR8-infected LA4/NK cell co-cultures collected at 24 h after infection. Data presented were either one representative or means, and mean values of three to five experiments.
JNK1/2 regulates cytokine generation downstream of p110δ
What are the downstream targets of p110δ that regulate the cytotoxicity and cytokine gene transcriptions? Earlier studies have shown that activation of mitogen-activated protein kinases (MAPKs) are required for cytotoxic granule release and cytokine generation in mouse and human NK cells (47–50). Also, the proposed involvement of p38 and JNK1/2 in regulating NK cell effector functions is contentious (49, 50). To identify the target molecules downstream of p110δ, NK cells were activated with plate-bound NKG2D mAb, and their lysates were analyzed for the activation status of multiple MAPKs. Phosphorylation levels of extracellular signal-regulated kinase (ERK) 1/2 were comparable between the activated WT/WT and p110δD910A/D910A NK cells. p38 phosphorylation is slightly but consistently reduced in p110δD910A/D910A NK cells (Fig. 10 a). However, the level of JNK1/2 phosphorylation was significantly reduced in p110δD910A/D910A NK cells (Fig. 10 a). These results indicate that JNK1/2 are major target molecules downstream of p110δ in NK cells. We confirmed the role of JNK1/2 using a specific pharmacological inhibitor of JNK activation, SP600125. Use of this inhibitor significantly reduced NKG2D-mediated lysis of EL4H60 target cells by both WT/WT and p110δD910A/D910A NK cells in a dose-dependent manner. However, the inhibition on p110δD910A/D910A NK cells was significantly less than that on WT/WT NK cells (Fig. 10 b). Blocking the function of p38 with its inhibitor, SB202190, had no significant effect on NKG2D-mediated lysis of the same target cells (Fig. 10 b). To determine their role in cytokine generation, we blocked JNK1/2 and p38 and assayed the ability of NK cells to respond to anti-NKG2D mAb–mediated stimulation. Fig. 10 c shows that inhibiting JNK function abolished the ability of both WT/WT and p110δD910A/D910A NK cells to generate cytokines and chemokines in a dose-dependent manner. Conversely, p38 inhibitor only weakly blocked the cytokine generation, further confirming the crucial role of JNK1/2 in cytokine gene transcription. To avoid the nonspecific inhibition on other kinases, we tested, selected, and used two optimal concentrations of JNK1/2 and p38 inhibitors (51). Further, cells treated under these concentrations were tested for their viability using Annexin V and 7-AAD staining, which excluded cell death as the reason for our observations (unpublished data). Based on these results, we conclude that impairment in the JNK1/2 activation is a probable cause for the defect in cytotoxicity and cytokine generation in p110δD910A/D910A NK cells.
Figure 10.

JNK1/2 regulates cytotoxicity and cytokine generation downstream of p110δ. (a) IL-2–cultured NK cells were stimulated with plate-bound anti-NKG2D mAb for the indicated times. Phosphorylation of ERK1/2, JNK1/2, and p38 was detected by Western blotting. One representative experiment out of three is shown. (b) Inhibition of cytotoxicity against EL4H60 by p38 inhibitor(SB 202190) or JNK1/2 inhibitor (SP600125). (c) Inhibition of cytokine/chemokine generation upon stimulation with plate-bound anti-NKG2D mAb by p38 inhibitor (SB 202190) or JNK1/2 inhibitor (SP600125). Data presented in b and c were means ± SD obtained from three mice of each genotype. Data shown were obtained from three to five independent experiments.
DISCUSSION
In this study, we present genetic evidence that unmasks the critical functions of PI3K-p110δ in NK cells. The important role of PI3K in T and B cell development and function has been extensively characterized (28, 29, 32). Using p110δD910A/D910A mice, we demonstrate that p110δ plays a crucial role in NK cell terminal maturation and cytokine/chemokine generation.
In recent years, multiple p85 and p110 gene KO mice have been generated (28, 29, 33, 52, 53). Interestingly, in these KO mice, altering the expression of one PI3K subunit has affected the expression of others. Cells from p85α KO mice contained increased levels of p85β and decreased expression of p110α, p110β, and p110δ (28, 29). In contrast, the levels of p85α expression were augmented in p110α KO mice (33). Some of these mice have been recently used to characterize the development and functions of NK cells. Tassi et al. shows a significant reduction of p85α subunit in NK cells derived from p110δ KO mice (54). Also, NK cells from these mice have increased expression of p110β and p110γ subunits, although the p110α expression level was not changed. Recently, another independently generated p110δ KO mice were used by Kim et al. in a NK cell study (55), and an earlier characterization of these mice showed decreased expression of p85α, p55α, and p50α subunits in B cells (52). Therefore, in this study, we used mice with a point mutation that completely inactivated the catalytic function of p110δ subunit without altering the normal expression levels of p110δ protein (32). More importantly, this strategy did not result in any compensatory change in the expression levels of total p85, p110α, and p110β subunits in thymocytes (32). In the present study, we show that both BM- and spleen-derived NK cells from p110δD910A/D910A mice did not have any alteration in the levels of either p110δ or other p110 subunits. Further, there were no changes in the levels of total p85 or p85α subunits. These results are in line with our earlier observations (32). However, one of the major concerns in introducing a single amino acid change in p110δ protein is its possible dominant negative effect. Our earlier findings demonstrated a differential sensitivity of distinct receptor systems after p110δ inactivation in p110δD910A/D910A mice (56). For example, PI3K signaling via c-kit and FcεRI receptors was almost fully abolished in p110δD910A/D910A mast cells; this is in contrast to PI3K (Akt/protein kinase B) signaling induced by IL-3, which was only partially blocked (56). The remaining PI3K signaling downstream of the IL-3 receptor could be fully blocked by broad-spectrum PI3K inhibitors (56), indicating that kinase-dead p110δ does not exert a dominant-negative function. Thus, a knock-in mouse with a point mutation in p110δ that abolishes its kinase function but not its protein expression provides an exclusive system to delineate the precise functions of PI3K in NK cells.
Using p110δD910A/D910A mice, we first determined the development and maturation of NK cells. Common lymphoid progenitors that give rise to T, B, and NK cells have been found in the BM (57). Committed NK precursors (NKPs), which exclusively mature into NK cells, have been identified in the fetal thymus and in adult BM (58). This earliest developmental stage of NKPs is defined by the expression of CD122. Thus, the expression of CD122 uniquely marks the committed NKPs (35). The expression of CD122 in p110δD910A/D910A NK cells is comparable to that of WT, indicating that the catalytic activity of p110δ is dispensable for the early NK cell development. In addition, earlier studies from us and others showed that a lack of phospholipase C γ2 did not alter the numbers of CD122+ NKPs (59–61). Thus, it appears that neither p110δ nor phospholipase C γ2 are needed for the commitment of early progenitors into NK lineage. In the second developmental stage, NKPs acquire NK1.1, NKG2D, and CD94/NKG2A/C/E in a sequential developmental process (34). The start of the expression of integrins such as CD49b and CD51 is also part of this second stage (34, 58). Normal expressions of these markers by p110δD910A/D910A NK cells in the BM and spleen demonstrate that NK maturation is not hindered at this stage.
Mature mouse NK cells are defined by the expression of one or more Ly49 receptors. Terminal maturation of NK cells is initiated by a decrease in the expression of CD51, active proliferation, and a concomitant increase in the levels of both CD11b and CD43 (34). At this stage, the “immature” NKPs start acquiring different Ly49s (58). Analyses of Ly49 expression on p110δD910A/D910A NK cells indicated an exclusive reduction in Ly49C and Ly49I subsets. Other NK subsets, such as Ly49A, Ly49G2, and Ly49D were normal. Recently, Tassi et al. has used p110γ, p110δ, or p110γ/δ double KO mice to determine the NK cell development and function (54). These studies showed reductions in Ly49A and Ly49C/I subsets in p110γ and p110γ/p110δ double KO mice; however, no changes were observed in Ly49C/I NK subsets in p110δ KO mice. Conversely, Kim et al., using independently generated KO mice, demonstrated small reductions in the Ly49C/I in p110γ KO mice and Ly49G2 in p110δ KO mice (55). The p110δD910A/D910A mice used in the present study and one of the two p110δ KO mice are of the C57BL/6 background. This suggests that strain differences in mice did not account for the observed variations in the results. Recent studies indicated that the mature CD11b+ NK cells can be further subdivided into CD27High and CD27Low (39). This study demonstrated that CD27High NK cells were superior in their ability to lyse target cells and generated considerably higher amounts of IFN-γ compared with those of CD27Low cells.
Reduction in the Ly49C and Ly49I NK subsets in p110δD910A/D910A mice needs further evaluation. Gene deletion of another lipid phosphatase, PTEN, whose major substrate is phosphatidylinositol (3,4,5)-triphosphate, leads to an increased Ly49C and Ly49I expression in NKT cells (62). Interestingly, this abnormal increase of Ly49C or Ly49I in NKT cells from PTEN KO mice was reversed by backcrossing the PTEN-deficient mice with p110δD910A/D910A mice (62). Collectively, our findings in the reduction of Ly49C and Ly49I expression predict a key role for p110δ in the dynamic interaction between PI3K, phosphatidylinositol (3,4,5)-triphosphate, lipid phosphatases, and inhibitory Ly49 receptors.
NK cells can kill tumor cells without prior sensitization. To study the role of p110δ in lytic signaling, we used different tumor cells representing self, induced-self, and missing-self. Lack of p110δ moderately affected the antitumor responses against these target cells. This marginal impairment in cytotoxicity could be caused by the reduction in the CD27High NK subsets in p110δD910A/D910A mice, because this subset has been demonstrated to play a dominant role in cytotoxic granule release (39). NK cells generate inflammatory cytokines and chemokines as part of innate immunity. Previous studies demonstrated a lack of DAP12 or Syk/ZAP70 but not DAP10, severely impairing NKG2D-mediated cytokine production (7, 11, 63). In contrast, a significant reduction in cytokine generation mediated via NKG2D and Ly49D was observed in p110δD910A/D910A NK cells. Further, similar reductions were also observed when p110δD910A/D910A NK cells were activated via the NK1.1 receptor that uses FcRγ to mediate its signal (24). These results suggest that p110δ plays a pivotal role in the cytokine generation by NK cells. Earlier studies have demonstrated that CD27HighCD11bHigh NK cells generated IFN-γ in response to NKG2D-mediated stimuli (39). Thus, a significant decline in the CD27High NK cells in p110δD910A/D910A mice provides additional cellular explanations for the reduction in cytokine generation. The proinflammatory cytokines IL-12 and IL-18, derived from monocytes, can induce IFN-γ generation in NK cells (64). In combinations, IL-12 and IL-18 can mediate signaling events that are distinct from those of YINM- or ITAM-containing receptor-mediated activations. Stimulation with IL-12 and IL-18, along with a suboptimal concentration of anti-NKG2D mAb, resulted in comparable levels of IFN-γ, GM-CSF, MIP-1α, MIP-1β, and RANTES between WT/WT and p110δD910A/D910A NK cells. Compared with p110δ KO NK cells (55), we did not observe any increased production of IFN-γ upon IL-12 and IL-18 stimulation. We conclude that ITAM-independent pathways are fully operational in p110δD910A/D910A NK cells. An explanation for this observation comes from the comparable levels of p38 phosphorylation between WT/WT and p110δD910A/D910A NK cells, which is crucial for the generation of cytokines downstream of IL-12/IL-18 receptors (65, 66).
MAPKs are potential downstream targets for multiple activation receptors, including NKG2D (67). MAPKs are serine-threonine kinases with a T-X-Y motif in their cytoplasmic domain, and they regulate multiple immune cell effector functions (68). The MAPK family includes ERK1/2, JNK1/2, and p38. The regulatory roles of these MAPKs in NK functions are emerging (47–50, 67, 69). ERK1/2 can be activated by multiple mitogenic stimuli; however, JNK1/2 and p38 are activated by stress-inducing agents or proinflammatory cytokines. Earlier studies using T cells have provided evidence for the role of PI3K in JNK activation (70). In T cells, full activation of the MAPKs that phosphorylate the Jun activation domain of JNK1 and JNK2 needs combined stimulation through TCR and CD28 together; alone, each stimulus resulted in little or no effect (70). Our data indicate that JNK1/2 are critical downstream effectors of P110δ, and that they could regulate cytokine gene transcription and cytotoxic granule release. These findings are in line with the recent observations in an NKG2D-expressing human NKL cell line, where inhibition of JNK activity resulted in the impaired movement of microtubule organizing center, granzyme B, and paxillin to the immune synapse (49). Several transcription factors as substrates have been described for JNK1/2, including activator protein 1 (71). Evidence for this is provided by the activator protein 1–dependent cytokine gene transcription in FcγRIIIA-stimulated human NK cells (72). In conclusion, our current observations establish a strong basis to further explore PI3K-mediated novel NK cell cytokine generation pathways.
NK cells can clear an intracellular infection either by direct killing of the infected cells or through the release of cytokines, e.g., IFN-γ. It has been demonstrated that NK cells can recognize and kill influenza virus–infected cells through the interaction of NCR1 with viral HA (21). Further, functional loss of NCR1 caused lethal influenza virus infection in a mouse model (73). However, the signaling pathway governing NCR1-mediated activation has not been investigated. Influenza virus primarily target respiratory cells. Mouse lung epithelial cells (LA4) can be infected with PR8 and express viral HA. p110δD910A/D910A NK cells showed reduced lysis against PR8-infected cells, which indicates the involvement of p110δ in NCR1-mediated cytotoxicity. Most significantly, the p110δD910A/D910A NK cells were impaired in their ability to produce IFN-γ and GM-CSF upon recognition of PR8-infected LA4, confirming the importance of p110δ in antiviral immunity. Replication of high pathogenic influenza infection usually peaks within 5 d (74). Controlling flu infection during this short time window currently relies on antiflu drugs and vaccines, which usually are not effective for virulent influenza virus. Our results suggest an alternative approach to control emerging pandemic influenza infection by boosting the NK cell effector functions through potential drugs specifically targeted to signaling molecules.
MATERIALS AND METHODS
Mice and cell lines.
Generation of PI3K-p110δD910A/D910A mice was previously described (30). PI3K-p110δD910A/D910A mice used in the present study were backcrossed to C57BL/6 for N12/N13 generations. The mutant and C57BL/6 (WT/WT) mice were maintained in pathogen-free conditions at the Biological Resource Center (BRC) of the Medical College of Wisconsin (MCW). All of the animal protocols used were approved by the Institutional Animal Care and Use Committee, BRC, MCW. Target cells (EL4, EL4H60, RMA/S, and YAC-1) and their culture conditions were previously described (35). CHO cells were cultured similarly to other target cells. Madin-Darby canine kidney (MDCK) cells and a mouse lung epithelial cell line (LA4) were purchased from the American Type Culture Collection and were cultured in RPMI 1640 medium with 10% or 15% FBS, respectively.
Virus and viral infections.
Mouse-adapted human influenza virus A/PR8 (H1N1) was obtained from T. Moran (Mount Sinai School of Medicine, New York, NY). Influenza virus stocks were propagated in MDCK cells, and titers were determined by a plaque-forming assay.
NK cell preparation.
NK cells were purified as previously described (35). In brief, single-cell suspensions from spleen and BM were passed through nylon wool columns to deplete adherent populations consisting of B cells and macrophages. Nylon wool–nonadherent cells were cultured with 1,000 U/ml of IL-2 (NCI BRB Preclinical Repository). Purity of the NK cultures was checked, and preparations with >90% NK1.1+ cells were used. Alternatively, for ex vivo assays, NK cells were purified with DX5 mAb (eBioscience) by magnetic sorting according to manufacture's protocol. Purify of NK cells was usually ∼80%.
Co-culture assays.
2.5 × 105 LA4 cells per well were seeded in 24-well tissue culture plates overnight. Cells were washed twice and inoculated with PR8 at a multiplicity of infection (MOI) of 0.05. After a 1-h incubation at 37°C, the inoculum was removed and LA4 cells were washed twice, followed by the addition of IL-2–activated splenic NK cells at the ratios indicated in the figures. Supernatants were collected at the times indicated in the figures for cytokine quantification.
Flow cytometry.
Cell preparations were stained with fluorescent-labeled mAbs, as previously described (75). mAbs for NK1.1 (PK136), CD3ε (145-2C11), NKG2D (A10), NKG2A (16a11), CD43 (1B11), CD49b (DX5), CD51 (RMV-7), CD69 (H1.2F3), CD122 (5H4), CD11b (M1/70), CD27 (LG.7F9), and Ly49I (YLI-90) were obtained from eBioscience. mAbs for CD244 (2B4), Ly49A (A1), Ly49D (4E5), Ly49C/I (5E6), and Ly49G2 (4D11) were obtained from BD Biosciences. mAbs for mouse H-2Kb and H-2Db were obtained from Invitrogen. Antibody for influenza HA (IVC102) was obtained from GeneTex, Inc. A standard flow cytometry analysis was performed using an LSR II with FACSDiva software (Becton Dickinson). IL-2–activated splenic NK cells derived from p110δD910A/D910A mice were also stained with Ly49C/I (5E6) and Ly49G2 (4D11) mAbs and subjected to cell sorting using a FACSAria (Becton Dickinson). Cytotoxicity of sorted NK subsets was tested against EL4H60 (see the following section).
Cytotoxicity assays.
NK-mediated cytotoxicity was quantified using 51Cr-labeled target cells, including EL4, EL4H60, RMA/S, YAC-1, CHO, and PR8-infected and noninfected LA4 cells at varied effector/target ratios. Anti-NKG2D mAb C7 (eBioscience) were used at the concentrations indicated in the figures to block the recognition of H60 on EL4H60 through the NKG2D receptor. The percentage of specific lysis was calculated using amounts of absolute, spontaneous, and experimental 51Cr release from target cells. For the in vivo tumor clearance assay, RMA and RMA/S cells were labeled with 0.1 μM CFSE and 0.2 μM SNAF (Invitrogen), respectively, for 10 min and were quenched with 10% FBS 1640 medium. Cells were mixed at a concentration of 10 × 106 cells/ml of each in PBS, and 400 μl was injected intraperitoneally into each mouse. Peritoneal exudate was collected 6 h later, and the fluorescence of single-cell suspensions was analyzed by flow cytometry. The ratio of RMA/S to RMA cells was calculated. NK cells in the cell suspensions were analyzed by staining with anti-CD3 and anti-NK1.1 mAbs. A dye swap was also performed.
NK cell proliferation.
Splenic NK cells were cultured for 6 d, harvested, seeded into a 6-well plate (2 × 106 cells/well), and incubated for 2 h. BrdU was then added to a final concentration of 10 μM, and cells were further incubated for 4 h, after which cells were stained with anti-CD3, -NK1.1, -Ly49A, -Ly49C/I, and -Ly49G2 mAbs for 30 min on ice, washed, and fixed with 1% paraformaldehyde–0.01% Tween-20 in PBS at 4°C for 20 min. Cells were washed and resuspended in permeabilization buffer containing 50 Kunitz DNase I/ml and incubated for 30 min at 37°C. After the wash, cells were finally stained with anti–BrdU-FITC (BD Biosciences). For apoptosis analysis, 6-d IL-2–activated NK cells were stained with FITC-conjugated mAbs for Ly49A, Ly49D, Ly49C/I, and Ly49G2; washed; and stained with 7-AAD and Annexin V–PE (BD Biosciences), according to manufacturer's protocol.
Western blotting.
Immunoblots of PI3K-p110 isoforms in NK cells were performed. In brief, 106 NK cell lysates were resolved using 10% SDS-PAGE gels and transferred to polyvinylidene fluoride membranes. The blot was probed with the mAbs indicated in the figures. Anti–rabbit PI3K-p110α, p110β, p110γ, and p110δ, and anti–mouse PI3K-p85α mAbs were purchased from Santa Cruz Biotechnology, Inc. Pan–anti–PI3K-p85 mAb was obtained from Millipore, and anti–β-actin mAb was from Boehringer Mannheim. After incubation with either horseradish peroxidase–conjugated anti–rabbit or anti–mouse mAbs (Santa Cruz Biotechnology, Inc.), membranes were washed and signals were detected using an enhanced chemiluminescence kit (GE Healthcare).
MAPK activation.
Cultured NK cells were serum starved for 2 h in PBS and stimulated with plate-bound anti-NKG2D mAb for the time indicated in the figure. Cells were lysed directly in the 96-well plate by the addition of 1 × RIPA buffer (Boston BioProducts) containing protease inhibitors and phosphatase inhibitor cocktails (EMD). Lysates were analyzed for phospho-p38 (Thr180/Tyr182), phospho-ERK1/2 (Thr202/Tyr204), phospho-JNK1/2 (Thr183/Tyr185), respective total proteins, and β-actin (Cell Signaling Technology) by Western blotting.
Cytokine and chemokine quantification.
Fresh or IL-2–cultured, Fc-blocked NK cells were activated with titrated concentrations of plate-bound anti-NKG2D (A10), anti-Ly49D (4E5), and anti-NK1.1 (PK136) mAbs. Where indicated in the figures, NK cells were activated with 2.5 ng/ml IL-12 and 2.5 ng/ml IL-18, or both, in the presence (Fig. 9) or absence (not depicted) of anti-NKG2D mAb (A10). IFN-γ, GM-CSF, MIP-1α, MIP-1β, and RANTES were quantified using Multiplex kit (Bio-Rad Laboratories). Intracellular cytokines were quantified using established methodologies (75). In brief, NK cells were activated with 10 μg/ml of plate-bound anti-NKG2D (A10) mAb for 12 h in the presence of Brefeldin A for the last 4 h of activation. Activated NK cells were Fc-blocked; stained for NK1.1 alone or in combination with anti-Ly49A, -Ly49D, -Ly49C/I, -Ly49I, or -Ly49G2 mAbs; fixed; permeabilized; and quantified for intracellular IFN-γ using FITC- (eBioscience) or PE- (BD Biosciences) conjugated anti–IFN-γ mAbs through flow cytometry. For IFN-γ encoding mRNA quantification, NK cells were activated for 6 h and harvested for RNA extraction using the RNeasy Mini Kit (QIAGEN). IFN-γ real-time PCR was performed by using a previously published SYBR green protocol with a thermal cycler (model 7900HT; Applied Biosystems) (76). The transcript in each sample was assayed in triplicate, and the mean cycle threshold was used to calculate the x-fold change and control changes for each gene. Three housekeeping genes were used for global normalization in each experiment (Actin, Rps11, and Tubulin). Data validity by modeling of reaction efficiencies and analysis of measurement precision was determined as described previously (76). Primer sequences for IFN-γ were 5′-GACTGTGATTGCGGGGTTGT-3′ (sense) and 5′-GGCCCGGAGTGTAGACATCT-3′ (antisense).
Drug inhibition assay.
p38 and JNK specific inhibitors SB 202190 and SP600125 (Sigma-Aldrich) were dissolved in DMSO and used in a cytotoxicity assay with 51Cr-labeled EL4H60 or a cytokine stimulation assay using plate-bound anti-NKG2D mAb. Starting concentrations for inhibitors were 20 μM, based on the kinase activity curve (51). In brief, cultured NK cells were harvested and seeded into 96-well plates at 105 cells for the cytotoxicity inhibition assay. Before incubation with target cells, NK cells were treated with SB 202190 or SP600125 for 1 h at 37°C, washed, and combined with target cells in a 4-h cytotoxicity assay. For cytokine stimulation, 2 × 105 NK cells/well were seeded directly into 96-well plates that were precoated with anti-NKG2D mAb, followed by the addition of SB 202190 and SP600125 inhibitors. Plates were incubated for 1 h at 37°C, washed, and added with 200 μl RPMI 1640 with 10% FBS per well in an 18-h stimulation assay. Potential apoptosis caused by both inhibitors at each concentration was tested by 7-AAD and Annexin V staining.
Statistics.
Statistical analysis was performed with the two-tailed, unpaired Student's t test using Microsoft Excel 2003 software to compare the differences between WT/WT and PI3K-p110δD910A/D910A mice. P ≤ 0.05 was considered significant.
Online supplemental material.
Fig. S1 shows expression levels of early developmental markers in fresh splenic NK cells. Fig. S2 shows normal expression levels of developmental and activation markers in IL-2–activated NK cells. Fig. S3 shows that catalytic inactivation of PI3K-p110δ does not affect the normal expression of MHC class I molecules. Fig. S4 shows that the reduced Ly49C/I NK subset is not caused by cell death in PI3K-p110δD910A/D910A mice. Fig. S5 shows that the cytotoxicity of NK cells directed toward EL4H60 is predominantly mediated through the NKG2D receptor. Fig. S6 shows that NK cells from PI3K-p110δD910A/D910A mice have decreased abilities to clear tumor cells in vivo. Fig. S7 shows that IL-2–activated BM-derived NK cells from PI3K-p110δD910A/D910A mice have a reduced ability to generate cytokines and chemokines. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20072327/DC1.
Supplementary Material
Acknowledgments
We thank Dr. Thomas Moran for influenza virus and help with real-time PCR. The authors thank R. Kamalakannan for help with CD27/CD11b analyses.
H.Guo is supported by an MCW Cancer Center Postdoctoral Fellowship. This work is supported in part by an American Cancer Society scholar grant (RSG-02-172-LIB), a Roche Organ Transplantation Research Foundation grant (111662730), and a National Institutes of Health grant (R01 A1064826-01 to S. Malarkannan).
The authors claim no financial conflict of interest.
Abbreviations used: 7-AAD, 7-aminoactinomycin D; CHO, Chinese hamster ovary; ERK, extracellular signal-regulated kinase; HA, hemagglutinin; ITAM, immunoreceptor tyrosine-based activation motif; JNK, c-Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; MIP, macrophage inflammatory protein; MOI, multiplicity of infection; NCR, natural cytotoxicity receptor; NKP, NK precursor; PI3K, phosphatidylinositol 3-kinase; PTK, protein tyrosine kinase; RANTES, regulated upon activation, normal T cell expressed and secreted; YINM, Tyr-Ile-Asn-Met.
References
- 1.Lanier, L.L. 2005. NK cell recognition. Annu. Rev. Immunol. 23:225–274. [DOI] [PubMed] [Google Scholar]
- 2.Groh, V., S. Bahram, S. Bauer, A. Herman, M. Beauchamp, and T. Spies. 1996. Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc. Natl. Acad. Sci. USA. 93:12445–12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cosman, D., J. Mullberg, C.L. Sutherland, W. Chin, R. Armitage, W. Fanslow, M. Kubin, and N.J. Chalupny. 2001. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity. 14:123–133. [DOI] [PubMed] [Google Scholar]
- 4.Diefenbach, A., A.M. Jamieson, S.D. Liu, N. Shastri, and D.H. Raulet. 2000. Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nat. Immunol. 1:119–126. [DOI] [PubMed] [Google Scholar]
- 5.Cerwenka, A., A.B. Bakker, T. McClanahan, J. Wagner, J. Wu, J.H. Phillips, and L.L. Lanier. 2000. Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity. 12:721–727. [DOI] [PubMed] [Google Scholar]
- 6.Carayannopoulos, L.N., O.V. Naidenko, D.H. Fremont, and W.M. Yokoyama. 2002. Cutting edge: murine UL16-binding protein-like transcript 1: a newly described transcript encoding a high-affinity ligand for murine NKG2D. J. Immunol. 169:4079–4083. [DOI] [PubMed] [Google Scholar]
- 7.Billadeau, D.D., J.L. Upshaw, R.A. Schoon, C.J. Dick, and P.J. Leibson. 2003. NKG2D-DAP10 triggers human NK cell-mediated killing via a Syk-independent regulatory pathway. Nat. Immunol. 4:557–564. [DOI] [PubMed] [Google Scholar]
- 8.Gilfillan, S., E.L. Ho, M. Cella, W.M. Yokoyama, and M. Colonna. 2002. NKG2D recruits two distinct adapters to trigger NK cell activation and costimulation. Nat. Immunol. 3:1150–1155. [DOI] [PubMed] [Google Scholar]
- 9.Wu, J., H. Cherwinski, T. Spies, J.H. Phillips, and L.L. Lanier. 2000. DAP10 and DAP12 form distinct, but functionally cooperative, receptor complexes in natural killer cells. J. Exp. Med. 192:1059–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McVicar, D.W., L.S. Taylor, P. Gosselin, J. Willette-Brown, A.I. Mikhael, R.L. Geahlen, M.C. Nakamura, P. Linnemeyer, W.E. Seaman, S.K. Anderson, et al. 1998. DAP12-mediated signal transduction in natural killer cells. A dominant role for the Syk protein-tyrosine kinase. J. Biol. Chem. 273:32934–32942. [DOI] [PubMed] [Google Scholar]
- 11.Zompi, S., J.A. Hamerman, K. Ogasawara, E. Schweighoffer, V.L. Tybulewicz, J.P. Di Santo, L.L. Lanier, and F. Colucci. 2003. NKG2D triggers cytotoxicity in mouse NK cells lacking DAP12 or Syk family kinases. Nat. Immunol. 4:565–572. [DOI] [PubMed] [Google Scholar]
- 12.Mason, L.H., S.K. Anderson, W.M. Yokoyama, H.R. Smith, R. Winkler-Pickett, and J.R. Ortaldo. 1996. The Ly-49D receptor activates murine natural killer cells. J. Exp. Med. 184:2119–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coudert, J.D., L. Scarpellino, F. Gros, E. Vivier, and W. Held. 2008. Sustained NKG2D engagement induces cross-tolerance of multiple distinct NK cell activation pathways. Blood. 111:3571–3578. [DOI] [PubMed] [Google Scholar]
- 14.Karlhofer, F.M., R.K. Ribaudo, and W.M. Yokoyama. 1992. MHC class I alloantigen specificity of Ly-49+ IL-2-activated natural killer cells. Nature. 358:66–70. [DOI] [PubMed] [Google Scholar]
- 15.Vitale, M., C. Bottino, S. Sivori, L. Sanseverino, R. Castriconi, E. Marcenaro, R. Augugliaro, L. Moretta, and A. Moretta. 1998. NKp44, a novel triggering surface molecule specifically expressed by activated natural killer cells, is involved in non-major histocompatibility complex-restricted tumor cell lysis. J. Exp. Med. 187:2065–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pessino, A., S. Sivori, C. Bottino, A. Malaspina, L. Morelli, L. Moretta, R. Biassoni, and A. Moretta. 1998. Molecular cloning of NKp46: a novel member of the immunoglobulin superfamily involved in triggering of natural cytotoxicity. J. Exp. Med. 188:953–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pende, D., S. Parolini, A. Pessino, S. Sivori, R. Augugliaro, L. Morelli, E. Marcenaro, L. Accame, A. Malaspina, R. Biassoni, et al. 1999. Identification and molecular characterization of NKp30, a novel triggering receptor involved in natural cytotoxicity mediated by human natural killer cells. J. Exp. Med. 190:1505–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Biassoni, R., A. Pessino, C. Bottino, D. Pende, L. Moretta, and A. Moretta. 1999. The murine homologue of the human NKp46, a triggering receptor involved in the induction of natural cytotoxicity. Eur. J. Immunol. 29:1014–1020. [DOI] [PubMed] [Google Scholar]
- 19.Walzer, T., M. Blery, J. Chaix, N. Fuseri, L. Chasson, S.H. Robbins, S. Jaeger, P. Andre, L. Gauthier, L. Daniel, et al. 2007. Identification, activation, and selective in vivo ablation of mouse NK cells via NKp46. Proc. Natl. Acad. Sci. USA. 104:3384–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Campbell, K.S., S. Yusa, A. Kikuchi-Maki, and T.L. Catina. 2004. NKp44 triggers NK cell activation through DAP12 association that is not influenced by a putative cytoplasmic inhibitory sequence. J. Immunol. 172:899–906. [DOI] [PubMed] [Google Scholar]
- 21.Mandelboim, O., N. Lieberman, M. Lev, L. Paul, T.I. Arnon, Y. Bushkin, D.M. Davis, J.L. Strominger, J.W. Yewdell, and A. Porgador. 2001. Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature. 409:1055–1060. [DOI] [PubMed] [Google Scholar]
- 22.Carlyle, J.R., A. Martin, A. Mehra, L. Attisano, F.W. Tsui, and J.C. Zuniga-Pflucker. 1999. Mouse NKR-P1B, a novel NK1.1 antigen with inhibitory function. J. Immunol. 162:5917–5923. [PubMed] [Google Scholar]
- 23.Iizuka, K., O.V. Naidenko, B.F. Plougastel, D.H. Fremont, and W.M. Yokoyama. 2003. Genetically linked C-type lectin-related ligands for the NKRP1 family of natural killer cell receptors. Nat. Immunol. 4:801–807. [DOI] [PubMed] [Google Scholar]
- 24.Arase, N., H. Arase, S.Y. Park, H. Ohno, C. Ra, and T. Saito. 1997. Association with FcRγ is essential for activation signal through NKR-P1 (CD161) in natural killer (NK) cells and NK1.1+ T cells. J. Exp. Med. 186:1957–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Held, W., J. Roland, and D.H. Raulet. 1995. Allelic exclusion of Ly49-family genes encoding class I MHC-specific receptors on NK cells. Nature. 376:355–358. [DOI] [PubMed] [Google Scholar]
- 26.Fahlen, L., U. Lendahl, and C.L. Sentman. 2001. MHC class I-Ly49 interactions shape the Ly49 repertoire on murine NK cells. J. Immunol. 166:6585–6592. [DOI] [PubMed] [Google Scholar]
- 27.Vanhaesebroeck, B., S.J. Leevers, K. Ahmadi, J. Timms, R. Katso, P.C. Driscoll, R. Woscholski, P.J. Parker, and M.D. Waterfield. 2001. Synthesis and function of 3-phosphorylated inositol lipids. Annu. Rev. Biochem. 70:535–602. [DOI] [PubMed] [Google Scholar]
- 28.Fruman, D.A., S.B. Snapper, C.M. Yballe, L. Davidson, J.Y. Yu, F.W. Alt, and L.C. Cantley. 1999. Impaired B cell development and proliferation in absence of phosphoinositide 3-kinase p85alpha. Science. 283:393–397. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki, H., Y. Terauchi, M. Fujiwara, S. Aizawa, Y. Yazaki, T. Kadowaki, and S. Koyasu. 1999. Xid-like immunodeficiency in mice with disruption of the p85alpha subunit of phosphoinositide 3-kinase. Science. 283:390–392. [DOI] [PubMed] [Google Scholar]
- 30.Vanhaesebroeck, B., J.L. Rohn, and M.D. Waterfield. 2004. Gene targeting: attention to detail. Cell. 118:274–276. [DOI] [PubMed] [Google Scholar]
- 31.Vanhaesebroeck, B., K. Ali, A. Bilancio, B. Geering, and L.C. Foukas. 2005. Signalling by PI3K isoforms: insights from gene-targeted mice. Trends Biochem. Sci. 30:194–204. [DOI] [PubMed] [Google Scholar]
- 32.Okkenhaug, K., A. Bilancio, G. Farjot, H. Priddle, S. Sancho, E. Peskett, W. Pearce, S.E. Meek, A. Salpekar, M.D. Waterfield, et al. 2002. Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science. 297:1031–1034. [DOI] [PubMed] [Google Scholar]
- 33.Bi, L., I. Okabe, D.J. Bernard, A. Wynshaw-Boris, and R.L. Nussbaum. 1999. Proliferative defect and embryonic lethality in mice homozygous for a deletion in the p110alpha subunit of phosphoinositide 3-kinase. J. Biol. Chem. 274:10963–10968. [DOI] [PubMed] [Google Scholar]
- 34.Kim, S., K. Iizuka, H.S. Kang, A. Dokun, A.R. French, S. Greco, and W.M. Yokoyama. 2002. In vivo developmental stages in murine natural killer cell maturation. Nat. Immunol. 3:523–528. [DOI] [PubMed] [Google Scholar]
- 35.Vosshenrich, C.A., T. Ranson, S.I. Samson, E. Corcuff, F. Colucci, E.E. Rosmaraki, and J.P. Di Santo. 2005. Roles for common cytokine receptor gamma-chain-dependent cytokines in the generation, differentiation, and maturation of NK cell precursors and peripheral NK cells in vivo. J. Immunol. 174:1213–1221. [DOI] [PubMed] [Google Scholar]
- 36.Franke, T.F., D.R. Kaplan, and L.C. Cantley. 1997. PI3K: downstream AKTion blocks apoptosis. Cell. 88:435–437. [DOI] [PubMed] [Google Scholar]
- 37.Kim, S., J. Poursine-Laurent, S.M. Truscott, L. Lybarger, Y.J. Song, L. Yang, A.R. French, J.B. Sunwoo, S. Lemieux, T.H. Hansen, and W.M. Yokoyama. 2005. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature. 436:709–713. [DOI] [PubMed] [Google Scholar]
- 38.Fernandez, N.C., E. Treiner, R.E. Vance, A.M. Jamieson, S. Lemieux, and D.H. Raulet. 2005. A subset of natural killer cells achieves self-tolerance without expressing inhibitory receptors specific for self-MHC molecules. Blood. 105:4416–4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hayakawa, Y., and M.J. Smyth. 2006. CD27 dissects mature NK cells into two subsets with distinct responsiveness and migratory capacity. J. Immunol. 176:1517–1524. [DOI] [PubMed] [Google Scholar]
- 40.Silva, A., D.M. Andrews, A.G. Brooks, M.J. Smyth, and Y. Hayakawa. 2008. Application of CD27 as a marker for distinguishing human NK cell subsets. Int. Immunol. 20:625–630. [DOI] [PubMed] [Google Scholar]
- 41.Hayakawa, Y., S.V. Watt, K. Takeda, and M.J. Smyth. 2008. Distinct receptor repertoire formation in mouse NK cell subsets regulated by MHC class I expression. J. Leukoc. Biol. 83:106–111. [DOI] [PubMed] [Google Scholar]
- 42.Malarkannan, S., P.P. Shih, P.A. Eden, T. Horng, A.R. Zuberi, G. Christianson, D. Roopenian, and N. Shastri. 1998. The molecular and functional characterization of a dominant minor H antigen, H60. J. Immunol. 161:3501–3509. [PubMed] [Google Scholar]
- 43.Regunathan, J., Y. Chen, D. Wang, and S. Malarkannan. 2005. NKG2D receptor-mediated NK cell function is regulated by inhibitory Ly49 receptors. Blood. 105:233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karre, K., H.G. Ljunggren, G. Piontek, and R. Kiessling. 1986. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature. 319:675–678. [DOI] [PubMed] [Google Scholar]
- 45.Saudemont, A., K. Okkenhaug, and F. Colucci. 2007. p110delta is required for innate immunity to transplantable lymphomas. Biochem. Soc. Trans. 35:183–185. [DOI] [PubMed] [Google Scholar]
- 46.Dorner, B.G., H.R. Smith, A.R. French, S. Kim, J. Poursine-Laurent, D.L. Beckman, J.T. Pingel, R.A. Kroczek, and W.M. Yokoyama. 2004. Coordinate expression of cytokines and chemokines by NK cells during murine cytomegalovirus infection. J. Immunol. 172:3119–3131. [DOI] [PubMed] [Google Scholar]
- 47.Jiang, K., B. Zhong, D.L. Gilvary, B.C. Corliss, E. Hong-Geller, S. Wei, and J.Y. Djeu. 2000. Pivotal role of phosphoinositide-3 kinase in regulation of cytotoxicity in natural killer cells. Nat. Immunol. 1:419–425. [DOI] [PubMed] [Google Scholar]
- 48.Wei, S., D.L. Gilvary, B.C. Corliss, S. Sebti, J. Sun, D.B. Straus, P.J. Leibson, J.A. Trapani, A.D. Hamilton, M.J. Weber, and J.Y. Djeu. 2000. Direct tumor lysis by NK cells uses a Ras-independent mitogen-activated protein kinase signal pathway. J. Immunol. 165:3811–3819. [DOI] [PubMed] [Google Scholar]
- 49.Li, C., B. Ge, M. Nicotra, J.N. Stern, H.D. Kopcow, X. Chen, and J.L. Strominger. 2008. JNK MAP kinase activation is required for MTOC and granule polarization in NKG2D-mediated NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA. 105:3017–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Trotta, R., K. Fettucciari, L. Azzoni, B. Abebe, K.A. Puorro, L.C. Eisenlohr, and B. Perussia. 2000. Differential role of p38 and c-Jun N-terminal kinase 1 mitogen-activated protein kinases in NK cell cytotoxicity. J. Immunol. 165:1782–1789. [DOI] [PubMed] [Google Scholar]
- 51.Davies, S.P., H. Reddy, M. Caivano, and P. Cohen. 2000. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351:95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Clayton, E., G. Bardi, S.E. Bell, D. Chantry, C.P. Downes, A. Gray, L.A. Humphries, D. Rawlings, H. Reynolds, E. Vigorito, and M. Turner. 2002. A crucial role for the p110δ subunit of phosphatidylinositol 3-kinase in B cell development and activation. J. Exp. Med. 196:753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jou, S.T., N. Carpino, Y. Takahashi, R. Piekorz, J.R. Chao, N. Carpino, D. Wang, and J.N. Ihle. 2002. Essential, nonredundant role for the phosphoinositide 3-kinase p110delta in signaling by the B-cell receptor complex. Mol. Cell. Biol. 22:8580–8591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tassi, I., M. Cella, S. Gilfillan, I. Turnbull, T.G. Diacovo, J.M. Penninger, and M. Colonna. 2007. p110gamma and p110delta phosphoinositide 3-kinase signaling pathways synergize to control development and functions of murine NK cells. Immunity. 27:214–227. [DOI] [PubMed] [Google Scholar]
- 55.Kim, N., A. Saudemont, L. Webb, M. Camps, T. Ruckle, E. Hirsch, M. Turner, and F. Colucci. 2007. The p110delta catalytic isoform of PI3K is a key player in NK-cell development and cytokine secretion. Blood. 110:3202–3208. [DOI] [PubMed] [Google Scholar]
- 56.Ali, K., A. Bilancio, M. Thomas, W. Pearce, A.M. Gilfillan, C. Tkaczyk, N. Kuehn, A. Gray, J. Giddings, E. Peskett, et al. 2004. Essential role for the p110delta phosphoinositide 3-kinase in the allergic response. Nature. 431:1007–1011. [DOI] [PubMed] [Google Scholar]
- 57.Kondo, M., I.L. Weissman, and K. Akashi. 1997. Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell. 91:661–672. [DOI] [PubMed] [Google Scholar]
- 58.Rosmaraki, E.E., I. Douagi, C. Roth, F. Colucci, A. Cumano, and J.P. Di Santo. 2001. Identification of committed NK cell progenitors in adult murine bone marrow. Eur. J. Immunol. 31:1900–1909. [DOI] [PubMed] [Google Scholar]
- 59.Regunathan, J., Y. Chen, S. Kutlesa, X. Dai, L. Bai, R. Wen, D. Wang, and S. Malarkannan. 2006. Differential and nonredundant roles of phospholipase Cgamma2 and phospholipase Cgamma1 in the terminal maturation of NK cells. J. Immunol. 177:5365–5376. [DOI] [PubMed] [Google Scholar]
- 60.Tassi, I., R. Presti, S. Kim, W.M. Yokoyama, S. Gilfillan, and M. Colonna. 2005. Phospholipase C-gamma 2 is a critical signaling mediator for murine NK cell activating receptors. J. Immunol. 175:749–754. [DOI] [PubMed] [Google Scholar]
- 61.Caraux, A., N. Kim, S.E. Bell, S. Zompi, T. Ranson, S. Lesjean-Pottier, M.E. Garcia-Ojeda, M. Turner, and F. Colucci. 2006. Phospholipase C-gamma2 is essential for NK cell cytotoxicity and innate immunity to malignant and virally infected cells. Blood. 107:994–1002. [DOI] [PubMed] [Google Scholar]
- 62.Kishimoto, H., T. Ohteki, N. Yajima, K. Kawahara, M. Natsui, S. Kawarasaki, K. Hamada, Y. Horie, Y. Kubo, S. Arase, et al. 2007. The Pten/PI3K pathway governs the homeostasis of Valpha14iNKT cells. Blood. 109:3316–3324. [DOI] [PubMed] [Google Scholar]
- 63.Colucci, F., E. Schweighoffer, E. Tomasello, M. Turner, J.R. Ortaldo, E. Vivier, V.L. Tybulewicz, and J.P. Di Santo. 2002. Natural cytotoxicity uncoupled from the Syk and ZAP-70 intracellular kinases. Nat. Immunol. 3:288–294. [DOI] [PubMed] [Google Scholar]
- 64.Fehniger, T.A., M.H. Shah, M.J. Turner, J.B. VanDeusen, S.P. Whitman, M.A. Cooper, K. Suzuki, M. Wechser, F. Goodsaid, and M.A. Caligiuri. 1999. Differential cytokine and chemokine gene expression by human NK cells following activation with IL-18 or IL-15 in combination with IL-12: implications for the innate immune response. J. Immunol. 162:4511–4520. [PubMed] [Google Scholar]
- 65.Berenson, L.S., J. Yang, B.P. Sleckman, T.L. Murphy, and K.M. Murphy. 2006. Selective requirement of p38alpha MAPK in cytokine-dependent, but not antigen receptor-dependent, Th1 responses. J. Immunol. 176:4616–4621. [DOI] [PubMed] [Google Scholar]
- 66.Akira, S. 2000. The role of IL-18 in innate immunity. Curr. Opin. Immunol. 12:59–63. [DOI] [PubMed] [Google Scholar]
- 67.Chen, X., P.P. Trivedi, B. Ge, K. Krzewski, and J.L. Strominger. 2007. Many NK cell receptors activate ERK2 and JNK1 to trigger microtubule organizing center and granule polarization and cytotoxicity. Proc. Natl. Acad. Sci. USA. 104:6329–6334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Su, B., and M. Karin. 1996. Mitogen-activated protein kinase cascades and regulation of gene expression. Curr. Opin. Immunol. 8:402–411. [DOI] [PubMed] [Google Scholar]
- 69.Gross, O., C. Grupp, C. Steinberg, S. Zimmermann, D. Strasser, N. Hannesschlager, W. Reindl, H. Jonsson, H. Huo, D.R. Littman, et al. 2008. Multiple ITAM-coupled NK cell receptors engage the Bcl10/Malt1 complex via Carma1 for NF-{kappa }B and MAPK activation to selectively control cytokine production. Blood. 112:2421–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Su, B., E. Jacinto, M. Hibi, T. Kallunki, M. Karin, and Y. Ben-Neriah. 1994. JNK is involved in signal integration during costimulation of T lymphocytes. Cell. 77:727–736. [DOI] [PubMed] [Google Scholar]
- 71.Bogoyevitch, M.A., and B. Kobe. 2006. Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases. Microbiol. Mol. Biol. Rev. 70:1061–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Aramburu, J., L. Azzoni, A. Rao, and B. Perussia. 1995. Activation and expression of the nuclear factors of activated T cells, NFATp and NFATc, in human natural killer cells: regulation upon CD16 ligand binding. J. Exp. Med. 182:801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gazit, R., R. Gruda, M. Elboim, T.I. Arnon, G. Katz, H. Achdout, J. Hanna, U. Qimron, G. Landau, E. Greenbaum, et al. 2006. Lethal influenza infection in the absence of the natural killer cell receptor gene Ncr1. Nat. Immunol. 7:517–523. [DOI] [PubMed] [Google Scholar]
- 74.Doherty, P.C., S.J. Turner, R.G. Webby, and P.G. Thomas. 2006. Influenza and the challenge for immunology. Nat. Immunol. 7:449–455. [DOI] [PubMed] [Google Scholar]
- 75.Malarkannan, S., J. Regunathan, H. Chu, S. Kutlesa, Y. Chen, H. Zeng, R. Wen, and D. Wang. 2007. Bcl10 plays a divergent role in NK cell-mediated cytotoxicity and cytokine generation. J. Immunol. 179:3752–3762. [DOI] [PubMed] [Google Scholar]
- 76.Fernandez-Sesma, A., S. Marukian, B.J. Ebersole, D. Kaminski, M.S. Park, T. Yuen, S.C. Sealfon, A. Garcia-Sastre, and T.M. Moran. 2006. Influenza virus evades innate and adaptive immunity via the NS1 protein. J. Virol. 80:6295–6304. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}