

Abstract
Folates typically have γ-linked polyglutamyl tails that make them better enzyme substrates and worse transport substrates than the unglutamylated forms. The tail can be shortened or removed by the vacuolar enzyme γ-glutamyl hydrolase (GGH). It is known that GGH is active only as a dimer and that plants can have several GGH genes whose homodimeric products differ functionally. However, it is not known whether GGH dimers dissociate under in vivo conditions, whether heterodimers form, or how heterodimerization impacts enzyme activity. These issues were explored using the GGH system of tomato (Solanum lycopersicum). Tomato has three GGH genes that, like those in other eudicots, apparently diverged recently. LeGGH1 and LeGGH2 are expressed in fruit and all other organs, whereas LeGGH3 is expressed mainly in flower buds. LeGGH1 and LeGGH2 homodimers differ in bond cleavage preference; the LeGGH3 homodimer is catalytically inactive. Homodimers did not dissociate in physiological conditions. When coexpressed in Escherichia coli, LeGGH1 and LeGGH2 formed heterodimers with an intermediate bond cleavage preference, whereas LeGGH3 formed heterodimers with LeGGH1 or LeGGH2 that had one-half the activity of the matching homodimer. E. coli cells expressing LeGGH2 showed approximately 85% reduction in folate polyglutamates, but cells expressing LeGGH3 did not, confirming that LeGGH2 can function in vivo and LeGGH3 cannot. The formation of LeGGH1-LeGGH2 heterodimers was demonstrated in planta using bimolecular fluorescence complementation. Plant GGH heterodimers thus appear to form wherever different GGH genes are expressed simultaneously and to have catalytic characteristics midway between those of the corresponding homodimers.
Tetrahydrofolate (THF) and its one-carbon (C1) substituted forms (collectively termed folates) are cofactors in one-carbon transfer reactions that form Ser, Gly, Met, purines, thymidylate, pantothenate, and formyl-methionyl tRNA in nearly all organisms (Lucock, 2000; Scott et al., 2000). Folates generally have a short, γ-linked polyglutamyl tail (Fig. 1) that affects their biological activity. Folate-dependent enzymes typically prefer polyglutamates, whereas folate transporters prefer nonglutamylated forms (Shane, 1989; Matherly and Goldman, 2003). In addition, folates are less prone to oxidative breakdown when protein bound than when free (Suh et al., 2001). Polyglutamylation thus tends to enhance cofactor efficacy, to favor folate retention in cells or subcellular compartments, and to protect folates from breakdown.
Figure 1.

Structure of tetrahydrofolate polyglutamates. One-carbon units at various oxidation levels can be coupled to the N5 and/or N10 position. A γ-linked polyglutamyl tail of up to about six residues is attached to the first Glu. Folates undergo oxidative cleavage of the C9-N10 bond yielding pterin and pABAGlun moieties. The arrows show bonds cleaved by GGH.
The polyglutamyl tail is added, one Glu at a time, by folylpolyglutamate synthase (EC 6.3.2.17), which occurs in nearly all organisms (Shane, 1989; Cossins and Chen, 1997). The tail can be shortened or removed by γ-glutamyl hydrolase (GGH; EC 3.4.19.9), an endo- and/or exopeptidase that occurs only in animals and plants; it is lysosomal (and secreted) in animals and vacuolar in plants (Galivan et al., 2000; Orsomando et al., 2005). Besides folate polyglutamates, GGH attacks polyglutamates of the folate breakdown product p-aminobenzoylglutamate (pABA-Glu). Whereas animals have one GGH gene, plants often have small GGH gene families (Orsomando et al., 2005); these families have been little studied and their biological significance is unclear.
Animal and plant GGHs are known to exist as nondissociating homodimers in solution, and dimerization appears to be essential for catalytic activity (Orsomando et al., 2005; Eisele et al., 2006). But it is not known whether monomers encoded by different genes form heterodimers and, if so, how each monomer contributes to dimer activity. This point is important because recombinant plant GGH homodimers have distinct catalytic properties (Orsomando et al., 2005). The characteristics of such homodimers might not reflect those of GGHs in planta if the latter exist partly as heterodimers. Moreover, in soybean (Glycine max), one GGH gene encodes a protein that lacks catalytically vital residues and is presumably inactive (Huangpu et al., 1996; Orsomando et al., 2005). An inactive monomer might lower the activity of a catalytically competent partner via dominant negative interaction. Also relevant to heterodimerization, animal GGH dimers have been shown not to dissociate (Eisele et al., 2006); it is not known whether plant GGH dimers do so. Nondissociation would mean that only GGH monomers made in the same cell at the same time could heterodimerize; dissociation would remove the time constraint.
We recently introduced tomato (Solanum lycopersicum) as a model to explore folate metabolism and its engineering (Díaz de la Garza et al., 2007; Noiriel et al., 2007). Accordingly, in this study, we characterized tomato GGH genes and their recombinant products, emphasizing heterodimerization and dimer stability, and investigated heterodimer formation in planta. Our data indicated that GGH gene families evolved recently, that heterodimers form if different GGH genes are expressed at the same time and place, and that each monomer in a heterodimer contributes equally to its catalytic properties.
RESULTS
Tomato Has Three GGH Genes
Surveys of the The Institute for Genomic Research (TIGR) and dbEST databases indicated the presence of three tomato GGH genes. Two of these were represented by TIGR contigs (TC167575 and TC165736) comprising ESTs from various tissues; the third was a singleton (BI931176) from flower buds. cDNA clones for each sequence, all 5′ truncated, were acquired from the SOL Genomics Network (SGN) and the missing regions were obtained by 5′-RACE or reverse transcription (RT)-PCR based on recovered genomic sequence. The TC167575-, TC165736-, and BI931176-type DNA sequences were renamed LeGGH1, LeGGH2, and LeGGH3, respectively. The corresponding deduced protein sequences are shown in Supplemental Figure S1.
Southern analysis of tomato cultivar Ailsa Craig was performed to estimate gene copy number, using gene-specific 3′-untranslated region (UTR) probes and a full-length LeGGH2 probe (Fig. 2A). Hybridization patterns were consistent with there being just three GGH genes because all major bands detected by the full-length probe could be reconciled with those detected by the specific probes. Although two minor hybridization signals were detected for LeGGH3, this was likely due to the presence of sequence repeats in the 3′-UTR region of this gene and hence the probe. Mapping using introgression lines and gene-specific 3′-UTR hybridization probes indicated that LeGGH1 and LeGGH3 are located in a 30- to 40-cM region on the long arm of chromosome 7 and that LeGGH2 is on the short arm of chromosome 10 (data not shown).
Figure 2.

Southern-blot analysis and GGH phylogeny. A, Tomato genomic DNA (10 μg) was digested with restriction enzyme BstnI, DraI, EcoRI, EcoRV, HaeIII, or ScaI. The digested samples were separated on a 1% agarose gel, blotted to nitrocellulose, and hybridized to gene-specific 32P-labeled probes corresponding to the 3′-UTR of each GGH gene (top two frames and bottom left frame) or to the coding region of LeGGH2 (bottom right frame). B, Evolutionary relationships of GGH sequences of tomato (Le), Arabidopsis (At), soybean (Gm), Populus trichocarpa (Pt), rice (Os), barley (Hv), and maize (Zm). Sequences were extracted from genome and EST databases. The tree was constructed by the neighbor-joining method. The percentages of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) are shown next to branches. The tree is drawn to scale; evolutionary distances are in units of the number of amino acid substitutions per site.
The LeGGH genes encode proteins that are 60% to 72% identical to each other. Comparison of these proteins with other plant and mammalian GGHs (Supplemental Fig. S1) revealed four features. First, all have predicted signal peptides, consistent with the lysosomal or vacuolar location of GGH (Galivan et al., 2000; Orsomando et al., 2005). Second, catalytically essential residues (Cys-110 and His-220 in the mammalian enzyme) are conserved, with two exceptions: soybean GGH1 lacks both residues (as noted above) and LeGGH3 has Asn instead of Cys. Sequencing the LeGGH3 gene from a second tomato cultivar (MicroTom) confirmed this substitution. Third, the dimer interface region (Li et al., 2002) is conserved in plant GGHs. Last, all GGHs have a conserved N-glycosylation motif near the N terminus, suggesting that plant GGHs, like their mammalian counterparts, are glycoproteins (Galivan et al., 2000).
Phylogenetic analysis placed plant GGHs in two subgroups corresponding to eudicots and monocots (Fig. 2B). The eudicots all had two or three GGHs, whereas monocots had one. The sequences from each eudicot species branched together, generally with high bootstrap values, implying that they diverged within the lineage leading to that species, and that the multiple GGH genes of eudicots are paralogous. Interestingly, the GGHs lacking catalytically essential residues (LeGGH3 and soybean GGH1) are both diverged substantially from their sisters in the same species.
Most Tissues Express LeGGH1 and LeGGH2
The mRNA expression patterns in various tomato tissues were determined by northern-blot analysis using specific 3′-UTR probes for each gene (Fig. 3A). LeGGH1 was expressed in fruits throughout development and in all other tissues examined. LeGGH2 showed a similar pattern, except for higher transcript levels in petals and stamens. LeGGH3 mRNA was barely detectable in any tissues, except flower buds, where it was relatively abundant.
Figure 3.

LeGGH gene expression and enzyme activity. A, Northern analysis. Total RNA (15 μg) was fractionated on a 1% agarose-formaldehyde gel, blotted to a nylon membrane, and hybridized to gene-specific 32P-labeled probes corresponding to the 3′-UTR of each gene. B, GGH activity was assayed in desalted crude tissue extracts using 0.2 mm PteGlu5 as substrate. Specific activity values are the means ± se of three independent experiments using tissue pooled from three to five different plants. MG, Mature green fruit; Br, breaker stage fruit; RR, red ripe fruit; L, leaf; R, root; S, stem; Sh, shoot; P, pedicel; FB, flower bud; F, flower; Se, sepal; Pe, petal; St, stamen; C, carpel.
To accompany the mRNA data, GGH activity was assayed in developing fruit and in other tissues from which sufficient protein could be obtained (Fig. 3B). Activity fell markedly during ripening, red-ripe fruit having only 5% of that in mature green fruit. A mixing experiment established that this decline was not due to factors in mature fruit that inhibit GGH activity or destroy it during extraction, for coextraction of green and red-ripe fruit yielded activities equal to the average of these tissues extracted separately (data not shown). All other tissues had GGH activity, flower buds having less than might be expected from the strength of the northern signals (Fig. 3B).
Characterization of Recombinant Homodimers
The three tomato GGH gene products were expressed in Escherichia coli as the predicted mature proteins with N-terminal His tags, as was done for the mammalian and Arabidopsis (Arabidopsis thaliana) enzymes (Chave et al., 2000; Orsomando et al., 2005). Recombinant proteins were purified by Ni2+ affinity chromatography; in the case of LeGGH1, an additional cation exchange step was required. On SDS-PAGE, the LeGGH1 and LeGGH2 proteins migrated as 40- and 42-kD species, respectively, consistent with their predicted molecular masses (36.4 and 36.7 kD; Fig. 4A). The LeGGH3 protein migrated as a single 39-kD species (data not shown), again consistent with its predicted mass (36.3 kD).
Figure 4.

Recombinant GGH purification, bond cleavage specificity, and in vivo activity. A, Purification of His6-tagged LeGGH1 and LeGGH2 homodimers by Ni2+ affinity chromatography; a subsequent cation exchange step was used for LeGGH1. Proteins were separated by SDS-PAGE and stained with Coomassie Blue. For each protein, lane 1 was loaded with E. coli extract containing 10 μg of total protein, and lane 2 with 2 μg of purified protein. B, Progress curves for hydrolysis of PteGlu5 (0.1 mm) by LeGGH1 and LeGGH2. Data are presented as plots of relative concentration of each reaction product versus extent of reaction and are representative of results obtained in three independent experiments. Symbols and their numbers illustrate the number of Glu residues remaining on the pteroyl moiety following hydrolysis of the polyglutamate tail. C, Progress curves for hydrolysis of pABAGlu5 (0.1 mm) by LeGGH1 and LeGGH2. Data plots are as above. D, Effect of LeGGH2 or LeGGH3 expression on folate polyglutamylation in E. coli. Bars show the extent of polyglutamylation of folates extracted from logarithmically growing E. coli cultures (A600 = 0.5–0.7) harboring pET28b alone or containing LeGGH2 or LeGGH3. Folates were analyzed by HPLC with electrochemical detection; the species were tetrahydrofolate (THF), 5-methyltetrahydrofolate (5-CH3-THF), 5,10-methenyltetrahydrofolate (5,10-CH-THF), and 5-formyltetrahydrofolate (5-CHO-THF). For each species, the data show percentages of total folate that were polyglutamylated (two to eight Glu residues) or were in the monoglutamyl form. Data are mean values from three independent experiments.
The bond cleavage specificities of LeGGH1 and LeGGH2 were determined from the reaction products obtained when folic acid pentaglutamate (PteGlu5) or pABA pentaglutamate (pABAGlu5) was used as substrate (Fig. 4, B and C). LeGGH1 liberated PteGlu1 and, to a lesser extent, PteGlu2 from PteGlu5, and pABAGlu2 and pABAGlu1 from pABAGlu5, indicating that it acts solely as an endopeptidase. In contrast, LeGGH2 produced all possible cleavage products from either PteGlu5 or pABAGlu5, yielding (in order of abundance) PteGlu1 > PteGlu4 > PteGlu3 > PteGlu2, and pABAGlu4 > pABAGlu1 > pABAGlu3 > pABAGlu2, respectively, indicating that LeGGH2 has both endo- and exopeptidase activity.
The kinetic constants for LeGGH1 and LeGGH2 for PteGlu5 and pABAGlu5 as substrates are listed in Table I. The Km values for PteGlu5, 1.20 μm and 1.38 μm for LeGGH1 and LeGGH2, respectively, are comparable to those reported for the mammalian and Arabidopsis enzymes. LeGGH1 and LeGGH2 both cleaved PteGlu5 more efficiently than pABAGlu5, as reflected by the kcat/Km ratios. This is also true of Arabidopsis GGHs (Orsomando et al., 2005). The kcat values for the tomato GGHs were lower than those reported for Arabidopsis.
Table I.
Kinetic parameters of LeGGH1 and LeGGH2 homodimers with PteGlu5 or pABAGlu5 as substrate
Measurements were made at 37°C in 100 mm potassium phosphate buffer, pH 6.0, containing 10% (v/v) glycerol. Reactions were started by adding substrate. Data are the means of three independent determinations ±se.
| Homodimer | PteGlu5
|
pABAGlu5
|
||||
|---|---|---|---|---|---|---|
| Km | kcat | kcat/Km | Km | kcat | kcat/Km | |
| μm | s−1 | s−1m−1 | μm | s−1 | s−1m−1 | |
| LeGGH1 | 1.20 ± 0.09 | 1.69 ± 0.27 | 1.41 × 106 | 0.98 ± 0.05 | 0.96 ± 0.06 | 9.75 × 105 |
| LeGGH2 | 1.38 ± 0.13 | 2.13 ± 0.42 | 1.55 × 106 | 1.31 ± 0.2 | 1.08 ± 0.11 | 8.19 × 105 |
No activity was found in purified LeGGH3, the assay being sensitive enough to detect 0.5% of the activity observed for the other tomato proteins. This negative result is consistent with the absence of the catalytically essential Cys residue in LeGGH3 and with the low GGH activity in flower buds, which express predominantly LeGGH3 (Fig. 3A). To confirm that LeGGH3 has no activity in vivo as well as in vitro, we compared the folate polyglutamylation profile of E. coli cells expressing LeGGH3 with those of cells expressing LeGGH2 or harboring vector alone (Fig. 4D). The three strains had similar doubling times (5–6 h), although the lag phases were longer for cells expressing the GGH proteins. Folates in the vector control were >90% polyglutamylated and folates in cells expressing LeGGH2 were 85% deglutamylated. Cells expressing LeGGH3 had an identical profile to the vector control, indicating total lack of GGH activity.
Tomato GGHs Form Heterodimers in E. coli
The structure of recombinant human GGH reveals two active sites (Li et al., 2002). No residue of either monomer participates directly in the active site of the other, but helix α2 is important in forming both the dimer interface and the active site, suggesting that the active site of each monomer might be affected by the monomer with which it is paired. If so, the catalytic properties of heterodimers would not be predictable from those of homodimers. The conservation of the dimer interface region in tomato GGHs (Fig. 5A) implies that, a priori, heterodimerization is likely.
Figure 5.

Conservation of the GGH dimer interface and evidence for formation of nondissociating LeGGH homo- and heterodimers. A, Conservation of the dimer interface. Amino acids that form the dimer interface comprise two helices (α2 and α9) and a single β-strand (β13). Numbers correspond to those amino acids forming the interface and are based on the human GGH crystal structure (Li et al., 2002). B, Dimerization. LeGGH monomers carrying His6 or FLAG tags were expressed in E. coli singly or in pairs; extracted proteins were then analyzed by western blotting using antibodies to the His6 tag, before (bottom) and after (top) affinity purification on FLAG M2 resin. When differentially tagged LeGGH2 proteins were coexpressed, homodimers formed, as shown by detection of the His6 epitope in FLAG-affinity-purified samples. When LeGGH1-FLAG or LeGGH3-FLAG was coexpressed with LeGGH2-His6, or LeGGH1-FLAG with LeGGH3-His6, heterodimer formation was evident from the presence of the His6 epitope in FLAG-affinity-purified proteins. C, Dimer stability. Equimolar amounts of LeGGH2-FLAG and LeGGH3-His6 homodimers were incubated together at 30°C for up to 4 h in physiological conditions (100 mm potassium phosphate, pH 6.0, 10% glycerol). At intervals, samples were applied to FLAG M2 resin, and the bound fraction was analyzed by western blotting using antibodies to the His6 tag. LeGGH2-His6/LeGGH3-FLAG purified heterodimer standards corresponding to 2% to 10% heterodimer formation were treated the same way. A portion of the homodimer mixture (Input) was analyzed prior to the affinity step as a positive control. The experimental sample blot was stained with Ponceau red to confirm protein recovery from the affinity resin (bottom).
Heterodimer formation was investigated in E. coli by coexpressing pairs of individual GGH proteins, each with a hexahistidine (His6) or FLAG affinity tag (see Supplemental Fig. S2 for constructs). Cell extracts were analyzed by western blotting using antibody to the His6 tag before and after affinity purification using the FLAG tag (Fig. 5B). The system was validated by showing that cells expressing separate LeGGH2 proteins carrying His6 or FLAG tags formed a doubly tagged homodimer (i.e. a protein that was retained by the FLAG affinity resin and recognized by the His6 antibody). Heterodimers were then shown to form between His6-tagged LeGGH2 and FLAG-tagged LeGGH1 or LeGGH3, and between His6-tagged LeGGH3 and FLAG-tagged LeGGH1.
GGH Dimers Do Not Dissociate and Reassociate
A similar approach was used to test whether dimers dissociate under physiological conditions. Equimolar amounts of LeGGH2-FLAG and LeGGH3-His6 homodimers were mixed and incubated at 30°C in 0.1 m potassium phosphate buffer, pH 6.0, containing 10% glycerol. At intervals, samples were applied to the FLAG affinity resin, and the bound fraction was subjected to western analysis using His6 antibody. No His6-labeled protein was detected after as long as 4 h, indicating that the extent of homodimer dissociation and reassociation into heterodimers was negligible (Fig. 5C). Controls showed that 2% heterodimer formation would have been detected (Fig. 5C).
Characterization of GGH Heterodimers
The activities of affinity-purified hetero- and homodimers were compared using PteGlu5 as substrate (Fig. 6A). The coexpression system outlined above enabled isolation of pure preparations of dimers comprised of one His6- and one FLAG-tagged monomer following sequential affinity purification steps on FLAG- and Ni2+-affinity resins. Tests with LeGGH2 homodimers confirmed that the doubly tagged enzyme had the same properties as the singly His6-tagged version described above (Fig. 4B). LeGGH1-LeGGH2 heterodimers had a specific activity similar to that of LeGGH2 homodimers, but heterodimers involving the inactive monomer LeGGH3 (LeGGH2-LeGGH3 and LeGGH1-LeGGH3) showed 50% to 60% less activity than dimers containing active monomers. LeGGH1 homodimers containing His6- and FLAG-tagged monomers were not included in these tests because they could not be adequately purified.
Figure 6.

The impact of heterodimer formation on GGH activity and bond cleavage specificity and evaluation of folate binding by LeGGH3. A, GGH activity. Doubly His6- and FLAG-tagged homodimers of LeGGH2 (2-2) and heterodimers of LeGGH1 and LeGGH2 (1-2), LeGGH2 and LeGGH3 (2-3), or LeGGH1 and LeGGH3 (1-3) were affinity purified and tested for total GGH activity. Data are means and se for three experiments. The purity of the purified proteins was verified by SDS-PAGE and staining with Coomassie Blue (inset). Note that the LeGGH1, LeGGH2, and LeGGH3 proteins are resolved from each other. B, Products formed from PteGlu5 by GGH homo- and heterodimers. Homo- and heterodimers were as above, with the addition of the LeGGH1 homodimer (1-1). C, Effect on GGH activity and cleavage product profile of adding the catalytically inactive LeGGH3 homodimer (0–10 μm) to reaction mixtures containing 1 μm PteGlu5 and 5 ng of LeGGH2 homodimer.
That an inactive monomer roughly halves the activity of the GGH heterodimer suggests that, while dimer formation is necessary for activity, the catalytic function of each monomer is independent of the other. To further dissect the situation, the bond cleavage specificity of each dimer was assessed using PteGlu5 as substrate (Fig. 6B). As in the data of Figure 4, LeGGH1 cleaved PteGlu5 to PteGlu1 and PteGlu2, whereas LeGGH2 gave all possible cleavage products. LeGGH1-LeGGH2 heterodimers also cleaved PteGlu5 to all possible products but the proportion of each product reflected the bond cleavage specificity of the component monomers. Thus, the heterodimer produced PteGlu3 and PteGlu4, which LeGGH1 alone cannot produce, yet in amounts approximately 50% less than produced by the LeGGH2 homodimer (Fig. 6B). As might be expected, when LeGGH1 or LeGGH2 was paired with the inactive LeGGH3 monomer, the bond cleavage specificity of the heterodimer was that of the active component.
The Inactive LeGGH3 Homodimer Does Not Sequester Folate
Given the catalytic incompetence of the LeGGH3 homodimer, we tested whether this protein has folate-binding activity that could make folates unavailable for enzymatic reactions. The activity of LeGGH2 was measured at a subsaturating concentration (1 μm) of PteGlu5 in the presence of various amounts of LeGGH3, so that the molar ratio PteGlu5/LeGGH3 ranged from 0.1 to 10 (Fig. 6C). Tight binding of PteGlu5 by LeGGH3 would limit its availability to LeGGH2 and so reduce product formation. The observed reduction in total cleavage products was modest even at the highest LeGGH3 concentration tested (10 μm, 0.72 mg mL−1), which is at least 1,000-fold greater than that likely to occur in planta. Nor was there much effect on the nature of the cleavage products except that PteGlu4, the result of exopeptidase action, became more prominent at high LeGGH3 levels. These results make it unlikely that LeGGH3 sequesters folates in vivo.
LeGGH1 and LeGGH2 Form Heterodimers in Planta
A bimolecular fluorescence complementation (BiFC) approach was used to investigate whether heterodimers form in plants. The entire open reading frames of LeGGH1 and LeGGH2 (including their signal peptides) were C-terminally fused via a linker to complementary fragments of the yellow fluorescent protein (YFP; see Supplemental Fig. S3 for constructs). When cotransformed into Arabidopsis protoplasts, control LeGGH2 constructs carrying complementary YFP fragments gave fluorescence signals comparable to those obtained with intact YFP, demonstrating homodimer formation as expected (Fig. 7). Similar signals were observed in protoplasts cotransformed with LeGGH1 and LeGGH2 carrying complementary YFP fragments, but not when the region specifying the mature protein was deleted from the LeGGH1construct or when protoplasts were transformed with the complementary YFP fragments alone (Fig. 7). The latter observations exclude the possibility that the fluorescence complementation occurs merely because the complementing fragments are juxtaposed in the same cell or subcellular compartment. Together, these data demonstrate that GGH heterodimers can form in planta.
Figure 7.

BiFC evidence for GGH heterodimer formation in planta. A, Representative fluorescence (i) and bright-field (ii) images of Arabidopsis protoplasts transiently expressing YFP, its N-terminal (YFPN) and C-terminal (YFPC) fragments, or YFPN and YFPC as fusions to LeGGH1, LeGGH2, and/or the signal peptide of LeGGH1 (sp1), as indicated on the right of the images. B, Percentage of the transformed protoplasts above exhibiting BiFC signals from a minimum of three independent experiments. BiFC intensity was visually scored as low, medium, or high (inset) from a minimum of 100 protoplasts viewed for each transformation. Note the lack of high intensity BiFC signals from protoplasts transformed with YFPN/YFPC or GGH1-YFPN/sp1-YFPC combinations.
DISCUSSION
While considered physiologically important, GGH in plants remains poorly understood. First, unlike animals, plants often have families of GGH genes, but the evolutionary origin and functional significance of this diversity are unclear. Second, whereas it is clear that dimerization is necessary for activity, much of what is known about GGH rests on studies of homodimers, although these may not be the only—or even the major—forms in planta. Our results shed light on both of these areas.
Like Arabidopsis, tomato proved to have three GGH genes, although differently arranged; they are concatenated in Arabidopsis (Orsomando et al., 2005) but are on two different chromosomes in tomato. A survey of plant genomes and phylogenetic analysis of plant GGH sequences indicated that small GGH gene families are common among eudicots and that these families most probably arose from recent duplications of a single ancestral gene within various lineages. Multiple GGH gene preservation in the eudicot lineage could be the outcome of subfunctionalization (Ward and Durrett, 2004). Thus, LeGGH1 behaves exclusively as an endopeptidase, whereas LeGGH2 has an additional exopeptidase activity, so that the two enzymes in a sense complement each other. The same is true of Arabidopsis AtGGH1 and AtGGH2 (Orsomando et al., 2005). Hence, it appears that individual eudicot GGH genes have undergone subfunctionalization and, as a result, are somewhat complementary. That LeGGH1 and LeGGH2 are expressed in parallel and that their individual bond cleavage patterns are both reflected in various tomato tissues supports this notion.
Surprisingly, one tomato gene (LeGGH3) was found to specify a nonfunctional protein lacking a catalytic Cys residue. The product of the soybean GGH1 gene (Huangpu et al., 1996) lacks the same residue and is thus also presumably nonfunctional. This coincidence encourages speculation that such GGH genes confer some adaptive advantage, but, if this exists, it seems not to lie either in dominant negative regulatory interactions or in folate sequestration (see below).
All possible heterodimers between LeGGH1, LeGGH2, and LeGGH3 monomers were shown to form when their genes were coexpressed in E. coli, and LeGGH1-LeGGH2 heterodimers were also demonstrated in Arabidopsis protoplasts. Such heterodimer formation has not been shown previously for GGH from any source. The LeGGH1-LeGGH2 heterodimer showed homodimer-like activity levels and a substrate preference midway between those of the homodimers, suggesting that the active site of each monomer functions independently. This notion is supported by the roughly halved activity of heterodimers between LeGGH1 or LeGGH2 and the inactive LeGGH3, and by the retention in these heterodimers of the substrate preference of the active partner. Thus, each monomer in GGH dimers appears to function autonomously and to contribute equally to activity and overall bond cleavage specificity. The halved activity of the heterodimers containing a LeGGH3 subunit argues against the scenario, mooted for mammalian GGHs, that the two active sites alternate in function (i.e. that dimers show half-of-the-sites reactivity; Eisele et al., 2006; Alexander et al., 2008). More generally, the apparent autonomy of each monomer in a GGH dimer suggests that the catalytic activity in a cell expressing both LeGGH1 and LeGGH2 would be unaffected whether these proteins were present in homo- or heterodimeric form.
The absence of a dominant negative effect (i.e. an activity loss of >50%) of LeGGH3 on heterodimer activity excludes the possibility that LeGGH3 (or presumably soybean GGH1) suppresses GGH activity when coexpressed with an active GGH. LeGGH3 expression merely draws active monomers into heterodimeric associations without diminishing their activity. Nor does LeGGH3 appear to bind folate polyglutamates tightly enough to protect them from attack by active GGH, or presumably by other folate-dependent enzymes. Nevertheless, as noted above, the occurrence of similar inactive GGH isoforms in both tomato and soybean warrants conjecture that such isoforms are biologically significant. This view is reinforced by the high expression level of the inactive soybean GGH1 compared to GGH2, as reflected by relative EST abundance (266 versus 26, respectively; http://compbio.dfci.harvard.edu/tgi/cgi-bin/tgi/gimain.pl?gudb=soybean; Soybean Gene Index). Furthermore, of two GGHs in the slime mold Dictyostelium discoideum (GenBank accession nos. XP637673 and XP636287), the latter lacks the Gln residue in the conserved catalytic center motif (QXHPE) of this family of hydrolases. One possibility is that GGH proteins have an undiscovered second catalytic activity that does not depend on residues essential to GGH activity. In this connection, it will be informative to see whether more examples of inactive GGH proteins emerge as genome and EST sequencing progresses.
The lack of dimer dissociation-reassociation implies that only genes that are expressed simultaneously will give rise to heterodimers because sequential expression would produce homodimers. However, as northern analyses indicate that LeGGH1 and LeGGH2 are coexpressed in most organs, it is likely that LeGGH1-LeGGH2 heterodimers occur throughout the plant. That LeGGH1 and LeGGH2 are expressed at similar levels suggests the possibility that these heterodimers are major forms of GGH in planta.
Finally, it is interesting to note that expressing LeGGH2 in E. coli caused massive folate deglutamylation without preventing growth. A similar result has been reported in the bacterium Lactococcus lactis; expressing mammalian GGH led to complete folate deglutamylation, but not to growth reduction (Sybesma et al., 2003). Such findings suggest that the polyglutamyl tail might be less metabolically crucial than generally thought (e.g. Lowe et al., 1993, and refs. therein). However, the role of polyglutamylation may be more prominent in plants due to compartmentalization of folates in various organelles. Massive deglutamylation in planta, where high demands for folate cofactors exist in mitochondria and, to a lesser degree in chloroplasts, might therefore have more severe effects. The presence of the folate tail generally prevents transport of polyglutamates across membranes (Appling, 1991) so that folate polyglutamylation in plant organelles likely favors folate retention, as it does in mammalian mitochondria (Shane, 1989). In this context, impairing vacuolar GGH activity could serve to trap polyglutamylated folates already present in this compartment, where 20% to 60% of the total cellular folate pool resides (Orsomando et al., 2005). With the GGH system in tomato now characterized, metabolic engineering can in principle be used to explore the importance of folate polyglutamylation in this important crop species.
MATERIALS AND METHODS
Plant Materials
Seed for tomato (Solanum lycopersicum ‘Ailsa Craig’) were acquired from the Tomato Genetics Resource Center (UC Davis). In the summer of 2006, eight plants were grown to maturity in soil containing slow-release fertilizer in climate and light-controlled greenhouses (28°C/16-h day/21°C/8-h night) at the Boyce Thompson Institute for Plant Research (Cornell University). Arabidopsis (Arabidopsis thaliana) cell suspension cultures were maintained in Murashige and Skoog medium supplemented with 3% (w/v) Suc, 1 mg L−1 2,4-dichlorophenoxycetic acid, and 1× Murashige and Skoog vitamin solution (Caisson Laboratories) with constant shaking in darkness. Culture medium was replaced every 5 d by dilution of 1:5 (inoculum: fresh medium) and cells required for experiments were taken 3 to 4 d after subculture.
Cloning of Tomato GGH Genes
Public sequence data from the SGN (www.sgn.cornell.edu) were utilized to identify EST sequences and corresponding cDNA clones for three tomato GGH genes via BLAST with Arabidopsis homologs. The cDNA clones that form the Unigene for each GGH gene (LeGGH1, SGN-U318513, SGN-U318514, and SGN-U336541; LeGGH2, SGN-U328054; LeGGH3, SGN-U331140) were sequenced using M13 primers (M13forward, 5′-GGAAACAGCTATGACCATG-3′, M13reverse, 5′-GTAAACGACGGCCAGT-3′). Whereas SGN-U318513 contained a full LeGGH2 predicted open reading frame, the available LeGGH1 and LeGGH3 cDNA harbored truncated cDNAs. The remaining LeGGH1 5′ sequence was recovered from tomato bacterial artificial chromosome clone LeHBaEcoRI-174-D5, which was identified by screening an EcoRI tomato bacterial artificial chromosome library (www.sgn.cornell.edu) arrayed on high-density nylon filters and probed with the 3′-UTR spanning nucleotides 1,290 to 1,451 of the LeGGH1 cDNA sequence (labeling and hybridization method described in Southern-blot analysis). The remaining LeGGH3 5′ sequence was recovered from a 5′-RACE product generated using the SMART RACE cDNA amplification kit (CLONTECH) using primer 5′-TCGACCGGAGTCTCCGTCACCGGGATG-3′. Once full-length cDNA sequence was available for all three GGH genes, RT-PCR was employed to recover and clone full-length cDNAs. RNA was extracted from combined tomato flower buds spanning approximately −3 to +2 d postanthesis. Full-length cDNAs for LeGGH1, LeGGH2, and LeGGH3 were amplified using gene-specific primers designed in their respective 5′- and 3′-UTRs (see Supplemental Table S1) using the FastStart High Fidelity PCR System (Roche). Resulting cDNA fragments were ligated into the pGEM-T Easy Vector System I (Promega) and sequenced to verify integrity.
Southern and Northern Analyses
Tomato genomic DNA (cv M82) and total RNA (cv Ailsa Craig) extractions, gel fractionation, blotting, and hybridizations were performed as described (Barry et al., 2005). PCR-amplified probes from the 3′-UTR of each LeGGH gene (see Supplemental Table S1 for PCR primers) were labeled as described by Feinberg and Vogelstein (1983). Images were obtained by exposing membranes (Hybond N+; Amersham) to a storage phosphor screen (Molecular Dynamics) for 16 h prior to scanning by a Storm 840 Optical Scanner with software packages Storm Control, version 5.02, and ImageQuant, version 5.2 (Molecular Dynamics).
Expression Constructs
To express LeGGHs in E. coli, the signal peptide sequences (Supplemental Fig. S1) were replaced by initiation codons using full-length cDNAs as templates and PCR mediated by Pfu Turbo polymerase (Stratagene); the primers (P1–P6) are given in Supplemental Table S1 and the expression constructs are diagrammed in Supplemental Figure S2. The amplicons were cloned between the NdeI and XhoI sites of pET28b (Novagen), which adds an N-terminal His6 tag; the three constructs were named pHGGH1 to pHGGH3. Using these constructs as templates, the His6 tags were replaced with a FLAG tag by PCR using primer P7 in combination with the reverse primers for each original construct (P2, P4, and P6). The amplicons were cloned between the NcoI and XhoI sites of pET28b, which generated N-terminal FLAG fusions of each GGH; these constructs were named pFGGH1-3. To coexpress individually tagged GGHs, the above constructs were utilized as follows. Using pHGGH2 as template, primer pair P8/P4 was used to amplify the His6-tagged GGH2 open reading frame, which was subsequently cloned into the XhoI site of pFGGH2. The resulting construct (pFHGGH2-2) encodes a bicistronic message composed of FLAG-GGH2 and His6-GGH2 separated by a ribosome binding site. Similarly, the PCR product generated by primer pair P8/P4 was cloned into the XhoI site of pFGGH3, yielding pFHGGH3-2, permitting coexpression of FLAG-GGH3 and His6-GGH2. Using pFGGH1 as template, primer pair P7/P9 generated a PCR product that was cloned between the NcoI and NotI sites of pET28b. This plasmid was then digested with BamHI and NotI and the PCR product generated with primer pair P10/P11 using pHGGH2 as a template was ligated between these sites. The resulting construct (pFHGGH1-2) coexpresses FLAG-GGH1 and His6-GGH2. To coexpress FLAG-GGH1 and His6-GGH3, XbaI-digested pHGGH3 was ligated to the fragment released from XbaI-digested pFHGGH1-2 to create pFHGGH1-3. All constructs were cloned in E. coli DH10B cells and sequence verified.
Recombinant Protein Expression and Purification
The above constructs were electroporated into E. coli BL21-CodonPlus (DE3)-pRIL cells, which were grown at 37°C in Luria-Bertani medium containing 75 μg mL−1 kanamycin and 25 μg mL−1 chloramphenicol. When A600 reached 0.6, isopropyl 1-thio-β-d-galactopyranoside was added to a final concentration of 1 mm and incubation continued for 4 h at 27°C, at which point cells were pelleted, washed in 50 mm potassium phosphate, 150 mm NaCl, pH 8.0, repelleted, and frozen at −80°C. Proteins were extracted as described (Orsomando et al., 2005) and His6-tagged proteins were purified by Ni2+ affinity chromatography. In the case of LeGGH1, partially pure effluents from Ni2+ affinity resin were applied to a Mono S 5/50 GL column (Amersham Biosciences) equilibrated in buffer A (100 mm potassium phosphate, pH 5.5). GGH activity was eluted (2 mL min−1) with a 25-mL linear gradient of 0 to 1.0 m KCl in buffer A, collecting 0.3-mL fractions using an ÄKTApurifier UPC10 FPLC system (GE Healthcare). Active fractions were pooled, desalted on PD-10 columns equilibrated with 100 mm potassium phosphate, pH 6.0, 10% (v/v) glycerol, 10 mm β-mercaptoethanol, and concentrated to 0.3 mL in a Centricon-10 (Millipore). FLAG-tagged proteins were purified using M2-affinity resin according to the manufacturer's instructions (Sigma); bound proteins were eluted with four column volumes of FLAG peptide (150 μg mL−1) in 100 mm potassium phosphate, pH 6.0, 10% glycerol, and immediately desalted on Sephadex G-25 minicolumns (Helmerhorst and Stokes, 1980) equilibrated with the same buffer plus 10 mm β-mercaptoethanol. If proteins were not to be used for activity assays, they were eluted from the M2-affinity resin with 0.1 m Gly, pH 3.5, and rapidly equilibrated with one-tenth the elution volume of 0.5 m Tris-HCl, 1.5 m NaCl, pH 7.4. Protein concentration was estimated by the method of Bradford (1976) using bovine serum albumin as the standard.
SDS-PAGE and Western Analysis
Proteins were separated by SDS-PAGE using 10% (w/v) polyacrylamide gels (Sambrook et al., 1989). For western analysis, proteins were electrophoretically transferred to Immobilon-P membranes (Millipore) using a Trans-Blot Semi-Dry transfer system (Bio-Rad). Blotted membranes were incubated overnight in Tris-buffered saline (TBS; 20 mm Tris-HCl, 150 mm NaCl, pH 7.5) containing 5% (w/v) nonfat milk powder, washed with TBS containing 0.1% (v/v) Tween 20, and incubated for 1 h with anti-His5 antibody (1:2,000; Qiagen) in TBS containing 5% (w/v) nonfat milk powder. Membranes were then washed in TBS containing 0.1% (v/v) Tween 20 and incubated with alkaline phosphatase-conjugated goat anti-mouse IgG (1:3000) (Bio-Rad) in TBS containing 5% (w/v) nonfat milk powder. Alkaline phosphatase activity was detected by incubating for 10 min in 100 mm Tris-HCl, pH 9.5, 100 mm NaCl, 5 mm MgCl2, 0.04% (w/v) nitro blue tetrazolium, 0.02% (w/v) 5-bromo-4-chloroindolyl phosphate.
Tomato Protein Extraction and GGH Assays
Tomato tissue was ground in liquid N2 to a fine powder and proteins were extracted in 100 mm potassium phosphate, pH 6.0, 10% (v/v) glycerol, 10 mm β-mercaptoethanol, and 3% (w/v) polyvinylpolypyrrolidone. Extracts were centrifuged (20,000g, 20 min, 4°C) and supernatants were desalted on PD-10 columns in 100 mm potassium phosphate, pH 6.0, 10% (v/v) glycerol, 10 mm β-mercaptoethanol (GGH buffer). GGH activity was measured as described (Orsomando et al., 2005). Reactions were performed in GGH buffer and were initiated by adding PteGlu5 or pABAGlu5 at a final concentration of 0.1 mm. Following incubation at 37°C for up to 8 h, reactions were stopped by boiling for 3 min and clarified by centrifugation. Reaction products were separated by HPLC using a 5-μm, 25- × 0.46-cm Discovery C18 column (Supelco), eluted with a 10-min linear gradient from 20% to 50% methanol in 50 mm sodium phosphate, pH 6.0, containing 8 mm tetrabutylammonium bisulfate. PteGlun or pABAGlun products were detected by absorption at 282 nm or by fluorescence (270-nm excitation, 350-nm emission), respectively, and quantified relative to standards obtained from Schirks Laboratories. For kinetic studies, initial rate of formation of total products were measured when no more than 15% of the substrate was consumed. Bond cleavage specificity was determined by plotting the extent of hydrolysis versus the relative concentration of the various PteGlun or pABAGlun products, as described (Bhandari et al., 1990). These terms are defined as follows:
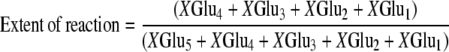 |
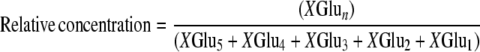 |
where XGlun = PteGlun or pABAGlun. The slope of the line corresponding to each product measures the relative extent of its formation and therefore indicates the γ-glutamyl bond cleavage specificity of each enzyme.
Folate Analyses of E. coli Cells Expressing GGH
E. coli BL21-CodonPlus (DE3)-pRIL cells were transformed with pHGGH2, pHGGH3, and/or the pET28b vector alone, as described above. The vector alone was digested with XbaI and XhoI, blunted with T4 DNA polymerase, and religated to stop expression of the 52-residue peptide encoded by the multiple cloning site. Cells were grown in 100 mL of M9 minimal medium supplemented with 0.1% thiamine (w/v) at room temperature in the presence of 1 mm isopropylthio-β-galactoside until A600 was between 0.5 and 0.7 at which point cells were pelleted and stored at −80°C. Folates were extracted and quantified by HPLC with electrochemical detection as described (Naponelli et al., 2007). Cell pellets were resuspended in 5 mL of 50 mm HEPES/CHES, pH 7.8, 2% (w/v) sodium ascorbate, 10 mm β-mercaptoethanol, and boiled for 10 min followed by centrifugation (13,000g, 10 min). The pellets were reextracted in the same way and the extracts were combined, split in two, and treated plus or minus rat plasma conjugase to determine total folate or degree of folate polyglutamylation, respectively. Folates were purified on 2.5-mL folate affinity columns prior to HPLC analysis (Gregory and Toth, 1988). The folate content of cells harboring vector, pHGGH2, and pHGGH3 were 1.10 ± 0.12, 0.94 ± 0.15, and 2.23 ± 0.06 nmol mg−1 protein, respectively. These values are all in the normal range for E. coli (Rohlman and Matthews, 1990).
BiFC
BiFC vectors were constructed using the pBS-YFP vector (GenBank accession no. AY189981) as a template. The primers (P12-P20) used are listed in Supplemental Table S1 and the BiFC vectors are illustrated in Supplemental Figure S3. The N-terminal YFP fragment (YFPN, amino acids 1–155) was amplified with primer pair P12/P13 and ligated into the NdeI/XhoI sites of pET28b, whereas the C-terminal YFP fragment (YFPC, amino acids 156–239) was amplified with primer pair P14/P15 and ligated into the NdeI/XhoI sites of pFGGH2. This generated N-terminal His6- and FLAG-tags fused via a linker to YFPN and YFPC, respectively. These tagged YFP fragments were released by digestion with NcoI and XbaI. The pBS-YFP vector was digested with NcoI/XbaI to release the full YFP sequence and replaced with either tagged YFPN or YFPC to generate pHNYFP or pFCYFP, respectively. pHNYFP and pFCYFP were each digested with NcoI, blunted, and ligated to create an NsiI site immediately upstream of the tagged YFP fragments. The full-length LeGGH2 sequence was amplified with primer pair P16/P17, digested with PstI, and ligated into NsiI-digested pHNYFP and pFCYFP creating pG2NYFP and pG2CYFP, respectively. The full-length LeGGH1 sequence was amplified with primer pair P18/P19, digested with NcoI, and ligated into the NcoI site of pHNYFP creating pG1NYFP. The signal peptide region of LeGGH1 (amino acids 1–32) was amplified with primer pair P18/P20, digested with NcoI, and ligated into the NcoI site of pFCYFP creating pSPG1CYFP.
BiFC experiments were performed in Arabidopsis protoplasts isolated from cell suspension culture. Cells were incubated in 1% (w/v) cellulose R-10, 0.25% (w/v) macerozyme R-10 (Yakult), 0.4 m mannitol, 8 mm CaCl2, 5 mm MES-KOH, pH 5.7, for 4 h with gentle agitation. Digested cells were passed through a 70-μm nylon mesh (Spectrum Laboratories) and washed twice with washing buffer (0.4 m mannitol, 70 mm CaCl2, 5 mm MES-KOH, pH 5.7). Protoplasts were purified by flotation on a 0.5 m Suc solution, washed twice in washing buffer, and incubated in W5 solution (154 mm NaCl, 5 mm KCl, 125 mm CaCl2, 5 mm Glc, 2 mm MES-KOH, pH 5.7) for 30 min on ice prior to transfection. Protoplasts were transfected according to the methods of Sheen and colleagues (http://genetics.mgh.harvard.edu/sheenweb) and fluorescence was visualized with an Olympus BX51 fluorescence microscope as described (Paul et al., 2005).
Sequence data from this article can be found in the GenBank/EMBL data libraries under accession numbers EU621369 (LeGGH1), EU621370 (LeGGH2), and EU621371 (LeGGH3).
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure S1. Alignment of GGH amino acid sequences.
Supplemental Figure S2. E. coli expression constructs.
Supplemental Figure S3. Bimolecular fluorescence complementation constructs.
Supplemental Table S1. Synthetic oligonucleotides used in this study.
Supplementary Material
Acknowledgments
We thank Dr. Albrecht von Arnim for providing the pBS-YFP vector, Dr. Anna-Lisa Paul for technical guidance on BiFC imaging, Dr. Rob Ferl and Beth Laughner for providing the Arabidopsis cell suspension culture, Dr. Thomas Ryan for advice on GGH construct design, and Dr. D.L. Purich for suggesting the folate sequestration experiment.
This work was supported by the National Science Foundation (grant no. MCB–0443709) and by an endowment from the C.V. Griffin, Sr. Foundation. Work in the J.J.G. laboratory was supported by the National Science Foundation (grant no. DBI–0501778) and the TRIAD Foundation.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (www.plantphysiol.org) is: Andrew D. Hanson (adha@ufl.edu).
The online version of this article contains Web-only data.
Open Access articles can be viewed online without a subscription.
References
- Alexander JP, Ryan TJ, Ballou DP, Coward JK (2008) γ-Glutamyl hydrolase: kinetic characterization of isopeptide hydrolysis using fluorogenic substrates. Biochemistry 47 1228–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appling DR (1991) Compartmentation of folate-mediated one-carbon metabolism in eukaryotes. FASEB J 5 2645–2651 [DOI] [PubMed] [Google Scholar]
- Barry CS, McQuinn RP, Thompson AJ, Seymour GB, Grierson D, Giovannoni JJ (2005) Ethylene insensitivity conferred by the Green-ripe and Never-ripe 2 ripening mutants of tomato. Plant Physiol 138 267–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandari SD, Gregory JF III, Renuart DR, Merritt AM (1990) Properties of pteroylpolyglutamate hydrolase in pancreatic juice of the pig. J Nutr 120 467–475 [DOI] [PubMed] [Google Scholar]
- Bradford MM (1976) A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein dye binding. Anal Biochem 72 248–254 [DOI] [PubMed] [Google Scholar]
- Chave KJ, Auger IE, Galivan J, Ryan TJ (2000) Molecular modeling and site-directed mutagenesis define the catalytic motif in human gamma-glutamyl hydrolase. J Biol Chem 275 40365–40370 [DOI] [PubMed] [Google Scholar]
- Cossins EA, Chen L (1997) Folates and one-carbon metabolism in plants and fungi. Phytochemistry 45 437–452 [DOI] [PubMed] [Google Scholar]
- Díaz de la Garza R, Gregory JF III, Hanson AD (2007) Folate biofortification of tomato fruit. Proc Natl Acad Sci USA 104 4218–4222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele LE, Chave KJ, Lehning AC, Ryan TJ (2006) Characterization of human γ-glutamyl hydrolase in solution demonstrates that the enzyme is a non-dissociating homodimer. Biochim Biophys Acta 1764 1479–1486 [DOI] [PubMed] [Google Scholar]
- Feinberg AP, Vogelstein B (1983) A technique for radiolabelling DNA restriction endonuclease fragments to high specific activity. Anal Biochem 132 6–13 [DOI] [PubMed] [Google Scholar]
- Galivan J, Ryan TJ, Chave K, Rhee M, Yao R, Yin D (2000) Glutamyl hydrolase. Pharmacological role and enzymatic characterization. Pharmacol Ther 85 207–215 [DOI] [PubMed] [Google Scholar]
- Gregory JF III, Toth JP (1988) Chemical synthesis of deuterated folate monoglutamate and in vivo assessment of urinary excretion of deuterated folates in man. Anal Biochem 170 94–104 [DOI] [PubMed] [Google Scholar]
- Helmerhorst E, Stokes GB (1980) Microcentrifuge desalting: a rapid, quantitative method for desalting small amounts of protein. Anal Biochem 104 130–135 [DOI] [PubMed] [Google Scholar]
- Huangpu J, Pak JH, Graham MC, Rickle SA, Graham JS (1996) Purification and molecular analysis of an extracellular gamma-glutamyl hydrolase present in young tissues of the soybean plant. Biochem Biophys Res Commun 228 1–6 [DOI] [PubMed] [Google Scholar]
- Li H, Ryan TJ, Chave KJ, Van Roey P (2002) Three-dimensional structure of human gamma-glutamyl hydrolase. A class I glutamine amidotransferase adapted for a complex substrate. J Biol Chem 277 24522–24529 [DOI] [PubMed] [Google Scholar]
- Lowe KE, Osborne CB, Lin BF, Kim JS, Hsu JC, Shane B (1993) Regulation of folate and one-carbon metabolism in mammalian cells. II. Effect of folylpoly-γ-glutamate synthetase substrate specificity and level on folate metabolism and folylpoly-γ-glutamate specificity of metabolic cycles of one-carbon metabolism. J Biol Chem 268 21665–21673 [PubMed] [Google Scholar]
- Lucock M (2000) Folic acid: nutritional biochemistry, molecular biology, and role in disease processes. Mol Genet Metab 71 121–138 [DOI] [PubMed] [Google Scholar]
- Matherly LH, Goldman DI (2003) Membrane transport of folates. Vitam Horm 66 403–456 [DOI] [PubMed] [Google Scholar]
- Naponelli V, Hanson AD, Gregory JF III (2007) Improved methods for the preparation of [3H]folate polyglutamates: biosynthesis with Lactobacillus casei and enzymatic synthesis with Escherichia coli folylpolyglutamate synthetase. Anal Biochem 371 127–134 [DOI] [PubMed] [Google Scholar]
- Noiriel A, Naponelli V, Gregory JF III, Hanson AD (2007) Pterin and folate salvage. Plants and Escherichia coli lack capacity to reduce oxidized pterins. Plant Physiol 143 1101–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsomando G, de la Garza RD, Green BJ, Peng M, Rea PA, Ryan TJ, Gregory JF III, Hanson AD (2005) Plant gamma-glutamyl hydrolases and folate polyglutamates: characterization, compartmentation, and co-occurrence in vacuoles. J Biol Chem 280 28877–28884 [DOI] [PubMed] [Google Scholar]
- Paul AL, Sehnke PC, Ferl RJ (2005) Isoform-specific subcellular localization among 14-3-3 proteins in Arabidopsis seems to be driven by client interactions. Mol Biol Cell 16 1735–1743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohlman CE, Matthews RG (1990) Role of purine biosynthetic intermediates in response to folate stress in Escherichia coli. J Bacteriol 172 7200–7210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T (1989) Molecular Cloning: A Laboratory Manual, Ed 2. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, p 18
- Scott J, Rébeillé F, Fletcher J (2000) Folic acid and folates: the feasibility for nutritional enhancement in plant foods. J Sci Food Agric 80 795–824 [Google Scholar]
- Shane B (1989) Folylpolyglutamate synthesis and role in the regulation of one-carbon metabolism. Vitam Horm 45 263–335 [DOI] [PubMed] [Google Scholar]
- Suh JR, Herbig AK, Stover PJ (2001) New perspectives on folate catabolism. Annu Rev Nutr 21 255–282 [DOI] [PubMed] [Google Scholar]
- Sybesma W, Van Den Born E, Starrenburg M, Mierau I, Kleerebezem M, De Vos WM, Hugenholtz J (2003) Controlled modulation of folate polyglutamyl tail length by metabolic engineering of Lactococcus lactis. Appl Environ Microbiol 69 7101–7107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward R, Durrett R (2004) Subfunctionalization: How often does it occur? How long does it take? Theor Popul Biol 66 93–100 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.